

A novel class of gluten-degrading enzymes were isolated from Rothia bacteria, which are natural colonizers of the oral cavity. The enzymes were identified as subtilisins belonging to the S8 family of peptidases. Food-grade Bacillus species also produce such subtilisins, and these were also able to cleave and abolish gluten immunogenic epitopes. Subtilisins, cleaving after XPX↓, represent an as yet overlooked class of enzymes with great potential for enzyme therapeutic applications in celiac disease.
Keywords: Rothia, therapy, Bacillus, gluten, celiac disease
Abstract
Gluten are proline- and glutamine-rich proteins present in wheat, barley, and rye and contain the immunogenic sequences that drive celiac disease (CD). Rothia mucilaginosa, an oral microbial colonizer, can cleave these gluten epitopes. The aim was to isolate and identify the enzymes and evaluate their potential as novel enzyme therapeutics for CD. The membrane-associated R. mucilaginosa proteins were extracted and separated by DEAE chromatography. Enzyme activities were monitored with paranitroanilide-derivatized and fluorescence resonance energy transfer (FRET) peptide substrates, and by gliadin zymography. Epitope elimination was determined in R5 and G12 ELISAs. The gliadin-degrading Rothia enzymes were identified by LC-ESI-MS/MS as hypothetical proteins ROTMU0001_0241 (C6R5V9_9MICC), ROTMU0001_0243 (C6R5W1_9MICC), and ROTMU0001_240 (C6R5V8_9MICC). A search with the Basic Local Alignment Search Tool revealed that these are subtilisin-like serine proteases belonging to the peptidase S8 family. Alignment of the major Rothia subtilisins indicated that all contain the catalytic triad with Asp (D), His (H), and Ser (S) in the D-H-S order. They cleaved succinyl-Ala-Ala-Pro-Phe-paranitroanilide, a substrate for subtilisin with Pro in the P2 position, as in Tyr-Pro-Gln and Leu-Pro-Tyr in gluten, which are also cleaved. Consistently, FRET substrates of gliadin immunogenic epitopes comprising Xaa-Pro-Xaa motives were rapidly hydrolyzed. The Rothia subtilisins and two subtilisins from Bacillus licheniformis, subtilisin A and the food-grade Nattokinase, efficiently degraded the immunogenic gliadin-derived 33-mer peptide and the immunodominant epitopes recognized by the R5 and G12 antibodies. This study identified Rothia and food-grade Bacillus subtilisins as promising new candidates for enzyme therapeutics in CD.
NEW & NOTEWORTHY
A novel class of gluten-degrading enzymes were isolated from Rothia bacteria, which are natural colonizers of the oral cavity. The enzymes were identified as subtilisins belonging to the S8 family of peptidases. Food-grade Bacillus species also produce such subtilisins, and these were also able to cleave and abolish gluten immunogenic epitopes. Subtilisins, cleaving after XPX↓, represent an as yet overlooked class of enzymes with great potential for enzyme therapeutic applications in celiac disease.
sensitivity to gluten-containing foods is widespread, and manifests predominantly in the form of celiac disease (CD). The increased awareness for CD in the medical community and the general public has led to improved diagnosis and earlier initiation of preventive strategies. The prevalence of CD ranges between ∼1:100 and 1:200 in most populations (2). The development of CD is dependent on exogenous and host-associated factors, whereby ingested gluten is the direct trigger of the disease, and the presence of HLA-DQ2 or HLA-DQ8 alleles, and tissue transglutaminase activity are the major contributing host-associated factors (21, 31). The disease is characterized by inflammation and flattening of the duodenal and jejunal villi, with a broad spectrum of symptoms, ranging from a clinically silent disease to severe malabsorption and a high risk for secondary autoimmune diseases (1).
To date, a strict gluten-free diet is the only treatment option for CD, which is difficult to maintain, and this poses a significant social and psychological burden to the patient. Traces of gluten are present in nearly all refined foods, and the felt quality of life of many patients equals that of hemodialysis patients (32). Therefore, novel therapies that would relieve patients from the need to adhere to the restrictive gluten-free diet are highly desired (27, 30).
The major therapies currently being pursued for CD target the immunogenic gluten peptides and the immune system, e.g., using a vaccine-based strategy, gluten-degrading enzymes, luminal gluten binders, and inhibitors of the body's enzyme tissue transglutaminase, which potentiates gluten antigenicity by deamidation (39). Enzyme therapies are aimed at cleaving gluten and abolishing immunogenic epitopes before they reach the lamina propria of the small intestine where T cell activation occurs (18, 19). This approach has distinct advantages. First, enzyme therapeutics are targeting the most upstream trigger, i.e., antigenic gluten peptides, rather than endogenous molecules or cells downstream in the cascade of intestinal inflammation; second, the enzymes are amenable to optimization of substrate specificity and pH activity (4).
Bacterial and barley-derived gluten-degrading enzymes have been isolated (34, 41) and are being explored for clinical application (19, 35, 42). Furthermore, gluten-degrading bacteria, mostly lactobacilli and bifidobacteria, have been found potentially useful for CD treatment (10, 20, 29), with a few reports indicating actual gluten digestion (6, 7). We have found that exceptionally high gluten-degrading enzyme activities are naturally associated with bacteria that colonize the oral cavity (15). Thus Rothia bacteria from human saliva can hydrolyze gluten domains that are highly immunogenic and resistant to mammalian digestive enzymes (44, 50). This discovery has identified these natural microbes as novel sources of gluten-degrading enzymes (9). Here we isolated the gluten-degrading enzymes from R. mucilaginosa, of which the complete genome sequence is available, and identified the proteinases as subtilisin family members. The discoveries highlight this group of enzymes, with cleavage specificities after Xaa-Pro-Xaa↓, and some with proven safety in the food industry, as hitherto unrecognized candidates for dietary enzyme therapeutics for CD.
METHODS
Bacterial culturing.
R. mucilaginosa ATCC 25296 was routinely grown on Brucella-agar plates (Hardy Diagnostics, Santa Maria, CA) at 37°C for 24 h under aerobic conditions. Individual bacterial colonies were transferred to 100 ml of Todd-Hewitt broth (Becton-Dickinson, Sparks, MD) supplemented with 0.5% Tween 80 (THT) and subcultured into 4 liters of THT. All incubations were carried out in Erlenmeyer flasks, while shaking at 200 revolutions/min for 48 h at 37°C.
Preparation of a R. mucilaginosa cell extract.
Cells were harvested from the 4-liter THT culture by centrifugation at 16,000 g at 4°C for 30 min, washed two times with 20 mM Tris·HCl buffer, pH 7.5, and then resuspended in 150 ml of 20 mM Tris·HCl buffer. Ready-Lyze lysozyme solution was added according to the manufacturer’s instructions for gram-positive bacteria (Epicentre, Madison, WI). After incubation at 37°C for 1 h, 25 mg/ml n-octyl-β-d-glucopyranoside were added (Thermo Fisher, Waltham, MA) as well as 0.3 mg/ml of l-cycteine. The cell suspension was frozen at −20°C, defrosted, and then sonicated on ice using a sonifier with a macro tip (Branson sonifier 450; VWR Scientific, Bridgeport, NJ). The chemical pretreatment of the cells combined with the sonication reduced the optical density at 620 nm of the suspension by 60%. The suspension was centrifuged at 31,209 g for 30 min at 4°C. The cloudy supernatant was harvested and then recentrifuged at 151,243 g for 1 h at 4°C. The resulting pellet, containing most of the activity, was resuspended in 4 ml of 20 mM Tris·HCl, pH 7.5. The protein concentration was determined using the bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Samples were stored at −80°C.
DEAE anion exchange chromatography.
Four milliliters of the resuspended pellet sample were thawed and solubilized by the addition of 40 mg/ml n-octyl-β-d-glucopyranoside. A 6-ml volume of 50 mM Tris·HCl containing 0.3 M NaCl, pH 7.0 (DEAE buffer A), was added. The 10-ml sample was then loaded on an anion-exchange DEAE Sepharose fast-flow column with a column size of 2.6 cm diameter × 27 cm length with a column volume (CV) of 143 ml. The resin used was a cross-linked agarose with the diethylaminoethyl exchange group [-O-CH2CH2N+H(CH2CH3)2; GE Healthcare, Bjorkgatan, Sweden]. The column was coupled to a FPLC system (AKTApurifier 10; GE Healthcare), and the flow rate applied was 1 ml/min. The buffers employed for protein elution were buffer A, containing 50 mM Tris·HCl, 0.3 M NaCl, pH 7.0, and buffer B, containing 50 mM Tris·HCl, 1 M NaCl, pH 7.0. Proteins were separated using a two-step gradient of 0% buffer B for 2.0 CV (286 ml) (isocratic conditions), followed by 100% elution buffer B for 1.5 CV (215 ml). The absorbance was monitored at 219 nm, and the eluate was collected in 10-ml fractions.
Analytical SDS-PAGE.
The protein content of individual DEAE fractions (100 μl) and pooled DEAE fractions (400 μl) was determined using precasted discontinuous 4–12% SDS-PAGE (NuPAGE; Thermofisher, Cambridge, MA) under reducing conditions. After electrophoresis gels were silver stained as described (49).
Gliadin zymography.
Pooled DEAE fractions F1–F7 were desalted using centrifugal tubes with 30-kDa molecular mass cut-off membranes (Amicon Ultra-15; EMD Millipore, Billerica, MA), and aliquots of 200 μl were analyzed for gluten-degrading enzyme activities on a 6% gliadin zymogram gel. The zymogram gel composition and renaturing and developing conditions were reported previously (14, 50).
PAGE and casein zymography.
The F2 fraction, containing the highest enzyme activity (hereafter called R. mucilaginosa enzyme preparation or Rmep), was applied in amounts ranging from 2 to 32 μg on a 6% PAGE under nonreducing conditions. The composition of the gel was the same as the 6% gliadin zymogram gel, but without the incorporated gliadin. After electrophoresis the gel was divided in half. One-half of the gel was silver stained, the other half was developed as a zymogram gel using externally added casein as the enzyme substrate, as described (49).
LC-ESI-MS/MS.
Proteins of interest were excised from the silver-stained gel half and in-gel digested with sequencing-grade trypsin (Promega, Madison, WI), as described (49). The peptides were eluted from the gel, separated by in-line C18 chromatography, and sequenced using an LTQ Orbitrap mass spectrometer (ThermoFinnigan, San Jose, CA). The obtained b- and y-ion spectra were searched against a database of R. mucilaginosa ATCC 25296 containing 1,737 Rothia protein entries and 132 non-Rothia decoy proteins. Applied filtering criteria were X-corr values >1.5, 2.2, and 3.5 for Z = 1, 2, and 3, respectively. The deltaCn and peptide probability values selected were >0.1 and <0.01, respectively.
Hydrolysis of paranitroanilide-derivatized substrates.
Paranitroanilide (pNA)-derivatized tripeptide substrates were chemically synthesized at >90% purity (21st Century Biochemicals, Marlborough, MA). The substrates obtained were Z-YPQ-pNA, Z-QQP-pNA, Z-LPY-pNA, Z-PFP-pNA, and Z-PPF-pNA, where Z is benzyloxycarbonyl, Y is tyrosine, P is proline, Q is glutamine, L is leucine, and F is phenylalanine. Suc-AAPF-pNA was obtained from Sigma, where Suc is N-succinyl and A is alanine. The peptides were dissolved in 75–100% DMSO at 10 mM and were used at a final concentration of 200 μM in 50 mM Tris·HCl, pH 8.0. Purified Rothia enzyme or subtilisin A from Bacillus licheniformis (Sigma) or Nattokinase (extracted from a dietary food supplement) were tested at final concentrations of 1 μg/ml. Some experiments were conducted in the presence of inhibitors 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), aprotinin, E-64, EDTA, phenylmethanesulfonyl fluoride (PMSF), and eglin C, which were tested at final concentrations of 10 mM, 0.08 mM, 0.1 mM, 1.5 mM, 1 mM, and 0.06–1.2 μM, respectively. Enzymes were preincubated with the inhibitors for 20 min before adding Suc-AAPF-pNA. Substrate hydrolysis was monitored for 10 h at 405 nm using a Genios microtiter plate reader (Tecan, Männedorf, Switzerland) in the kinetic mode at 37°C.
Fluorescence resonance energy transfer substrate hydrolysis.
Three fluorescence resonance energy transfer (FRET) substrates comprising the hexapeptides QPQLPY, PQPQPQ, and QGSFQP were synthesized at the NH2-terminus with the HiLyte Fluor 488 label and at the COOH-terminus with K(QXL520) (Anaspec, Fremont, CA). Rmep (1 μg/ml) and subtilisin A (0.5 μg/ml) were incubated at 37°C with the substrates (100 μM) in 50 mM Tris·HCl, pH 8.0. Fluorescence increase, indicating substrate hydrolysis, was measured every 5 min for 30 min, and every 10 min for the next 30 min, at λex 485 nm and λem 520 nm, using a Genios microtiter plate reader in the kinetic mode.
Degradation of mixed gliadins and the 33-mer peptide.
Mixed gliadins were obtained from Sigma. A synthetic highly immunogenic α-gliadin-derived 33-mer peptide (33) was synthesized at a purity of >90% (21st Century Biochemicals). Mixed gliadins or the 33-mer peptide (both at final concentration 250 μg/ml) was incubated with Rmep or subtilisin A (each at 57 μg/ml) in 50 mM Tris·HCl, pH 8.0. After time 0 and 15 min, 30 min, and 2 h, 100-μl sample aliquots were removed and boiled. Gliadin degradation was assessed by 4–12% SDS-PAGE followed by Coomassie staining. Degradation of the 33-mer was determined by reversed-phase (RP)-HPLC.
RP-HPLC.
Separation of the 33-mer peptide and its fragments was achieved by RP-HPLC, using buffer A (0.1% trifluoroacetic acid) and buffer B (0.1% trifluoroacetic acid in 80% acetonitrile) at a gradient of 0–55% buffer B over the 75-min time interval. The equipment used was an HPLC model 715 (Gilson, Middleton, WI) and a C-18 column (TSK-GEL 5 mm, ODS-120T; TOSOHaas, Montgomeryville, PA) (14, 24, 50).
R5 and G12 ELISA assays.
Gluten epitope elimination in the gliadin-Rmep and gliadin-subtilisin A digests was assessed using two sandwich ELISAs employing the R5 monoclonal antibody (RIDASCREEN Gliadin; R-Biopharm, Darmstadt, Germany) or the G12 monoclonal antibody (AgraQuant ELISA Gluten G12; Romer labs, Union, MO). To prevent high background values, the R5 plate wells were blocked with 1% skim milk in PBS before incubation with the samples. The assays were performed according to the manufacturers' instructions and as described (44, 49).
RESULTS
Purification of the enzyme from R. mucilaginosa.
We observed that the gluten-degrading enzyme activity of R. mucilaginosa was primarily cell associated. Therefore, to isolate the enzyme(s), the cells were treated with lysozyme, an N-acetylmuramidase, to break down the peptidoglycan layer and then sonicated and centrifuged. The supernatant, which was turbid and contained microscopic vesicles, was ultracentrifuged. Enzyme activities, monitored with the substrate Z-YPQ-pNA, were primarily localized in the vesicular pellet. The pellet was dissolved to clarity with the mild detergent n-octyl-β-d-glucopyranoside and then subjected to DEAE chromatography applying an isocratic gradient. Figure 1A shows the DEAE chromatogram, Fig. 1B the protein content in fraction aliquots analyzed by SDS-PAGE, and Fig. 1C the enzyme activity, determined with the substrate Z-YPQ-pNA. Most activity was contained in fractions 7 and 8 with some activity trailing in fractions 9–14. Fractions 7 and 8 contained proteins migrating between 75 and 150 kDa. Based on protein patterns and enzyme activities, the fractions were pooled into seven fractions as follows: F1, fractions 1–5; F2, fractions 6–8; F3, fractions 9–12; F4, fractions 13–20; F5, fractions 21–52; F6, fractions 53–56; and F7, fractions 57–65.
Fig. 1.

Isolation of Rothia mucilaginosa gluten-degrading enzymes by DEAE chromatography. R. mucilaginosa cells were lysed and sonicated, the supernatant was ultracentrifuged, and the pellet was dissolved in n-octyl-β-d-glucopyranoside. A: separation of proteins by DEAE chromatography applying an isocratic gradient containing 0.3 M NaCl and 50 mM Tris·HCl, pH 7.0. B: protein content in 100-μl fraction aliquots investigated by 4–12% SDS-PAGE. C: enzyme activity in 50-μl fraction aliquots investigated with Z-YPQ-paranitroanilide (pNA) as the substrate. The data shown are representative of 2 independent experiments.
The protein composition in F1–F7 is shown in Fig. 2A. F2, containing the enzyme activity and hereafter called R. mucilaginosa enzyme preparation (Rmep), displayed a major band between 100 and 150 kDa and a double band between 75 and 100 kDa. Gliadin zymography of F1–F7 showed evidence for a double enzyme band in F2 of ∼75–80 kDa (Fig. 2B). As expected, the highest specific activity was associated with F2 (data not shown). Rmep was subsequently analyzed at four different concentrations on a 6% SDS gel under nonreducing conditions. After electrophoresis, one-half of the gel was silver stained (Fig. 2C) and the other one-half was developed as a zymogram with externally added casein as the substrate (Fig. 2D). This permitted a true comparison in electrophoretic mobilities of proteins in both gel halves. Enzyme activities were again associated with the ∼75- to 80-kDa double band, and not with the also prominent ∼125-kDa band. Several bands were excised from the SDS and the zymogram gel for protein identification by LC-ESI-MS/MS. The inactive 125-kDa band was labeled as band a, the active ∼80-kDa band as band b/d, and the active ∼75-kDa band as band c/e. The major proteins identified in bands a–e were ROTMU0001_0241 (C6R5V9_9MICC), ROTMU0001_0243 (C6R5W1_9MICC), and ROTMU0001_0240 (C6R5V8_9MICC) (Fig. 2E). The relative abundances of the proteins in the respective bands, determined at the MS1 level, are indicated. It shows that the ∼125-kDa band contains a mixture of primarily C6R5V9_9MICC and C6R5W1_9MICC, in a 32:67% ratio. The ∼80-kDa protein is a fragment of C6R5V9_9MICC, and the ∼75-kDa band is derived from C6R5W1_9MICC. Identifications were made based on 49 and 62 matching tryptic peptides, respectively, and >50% sequence coverage. As a control, three other bands were excised from the gel shown in Fig. 1B (fraction 13, lowest band, and fraction 14, two middle bands), yielding identifications other than subtilisin enzymes (data not shown).
Fig. 2.
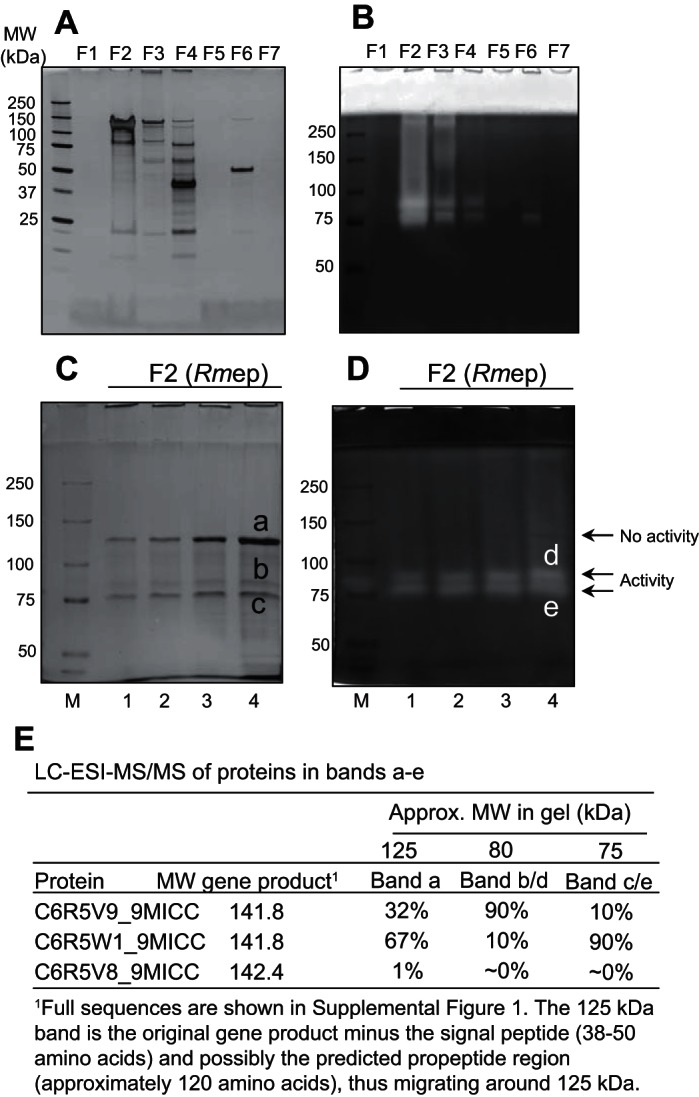
Identification of the gluten-degrading enzymes of R. mucilaginosa. DEAE fractions 1–65 were pooled into 7 fractions designated F1–F7 (see text for details). A: protein content in 400-μl aliquots of F1–F7 analyzed on a 4–12% SDS gel. B: gliadin-degrading enzyme activity in 200-μl desalted aliquots of F1–F7 analyzed on a 6% gliadin zymogram gel. C: PAGE under nonreducing conditions of R. mucilaginosa enzyme preparation (Rmep) (F2), loaded amounts of 4, 8, 16, and 32 μg protein in lanes 1–4, respectively. D: casein zymogram of F2, loaded amounts of 2, 4, 8, and 16 μg in lanes 1–4, respectively. E: proteins identified in excised gel bands labeled a–e. The data shown are representative of at least 4 repeated experiments.
The amino acid sequences of the identified subtilisins ROTMU0001_0241, ROTMU0001_0243, and ROTMU0001_0240 are shown in Supplemental Fig. S1 (Supplemental data for this article are available on the journal website.). They are listed in UniProt as uncharacterized proteins. A search with the Basic Local Alignment Search Tool (BLAST) revealed that all are members of the S8 peptidase family, which share a catalytic triad comprising Asp (D), His (H), and Ser (S). The signal peptide cleavage sites and the catalytic triads are indicated. The protein sequences of all subtilisin genes in the three known members of the Rothia genus, R. aeria, R. mucilaginosa, and R. dentocariosa, were aligned using Clustal Omega (Supplemental Fig. S2). The three variants identified by LC-ESI-MS/MS are highlighted. A phylogenetic tree was drawn using Clustal Omega, showing evidence for eight to nine different Rothia subtilisin sequence types based on sequence conservation around the catalytic residues D, H, and S (data not shown).
Enzymatic characteristics of Rmep.
Rmep was further tested for enzyme activities and specificities relevant for CD. The enzyme was active over a pH range of 6.0–10.0, with low or negligible activities at pH values <5.0 or >11.0 (Fig. 3A). Activity was strongly inhibited by PMSF and AEBSF (serine protease inhibitors), with partial inhibition by aprotinin, but not by E-64 (cysteine protease inhibitor) or EDTA (metalloprotease inhibitor) (Fig. 3B). Eglin C, which is an effective inhibitor of B. licheniformis subtilisin A (Fig. 3C) only partly inhibited Rmep, at high concentrations (Fig. 3D). This difference in sensitivity is likely due to the significant overall structural differences between Rmep and subtilisin A, including in domains flanking their active sites that participate in binding of the inhibitor (16).
Fig. 3.

Effect of pH and inhibitors on Rmep activity. A: activity of Rmep in 0.1 M citric acid-0.2 M phosphate buffer mixtures, pH 2–12, measured with Suc-AAPF-pNA (200 μM) as the substrate. B: inhibitor profile of Rmep. Inhibitors 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), aprotinin, E-64, EDTA, phenylmethanesulfonyl fluoride (PMSF), and eglin C were tested at final concentrations of 10 mM, 0.08 mM, 0.1 mM, 1.5 mM, 1 mM, and 0.06 μM, respectively. Rmep was added at a final concentration of 5.7 μg/ml. The percent inhibition was determined from the ratio of the initial velocities of hydrolysis of Suc-AAPF-pNA in the absence and presence of inhibitor. C and D: inhibition of subtilisin A and Rmep, respectively, at higher eglin C concentrations (1.2 μM).
To investigate enzyme specificities relevant for gliadin degradation, the substrates Z-YPQ-pNA, Z-QQP-pNA, Z-LPY-pNA, Z-PFP-pNA, and Z-PPF-pNA were employed in which the tripeptides are all contained with high frequency in the immunogenic gliadin domains. Suc-AAPF-pNA was also used, since it is a preferred substrate for subtilisins. Z-QQP-pNA and Z-PFP-pNA were not hydrolyzed by either subtilisin A or Rmep (data not shown). In contrast, substrates with a P residue in the P2 position were cleaved, albeit at quite different rates; Suc-AAPF-pNA was hydrolyzed most rapidly, followed by Z-YPQ-pNA (Fig. 4, A and B). The rapid hydrolysis of Suc-AAPF-pNA by Rmep further supports the identification of subtilisin-like enzymes. LPY was hydrolyzed by Rmep at 5-fold increased enzyme concentrations and longer incubation times (data not shown). PPF was hydrolyzed by subtilisin A at 50-fold increased enzyme concentrations, but not by Rmep (data not shown).
Fig. 4.

Cleavage specificities of Rmep and subtilisin A (subt A). A and B: hydrolysis of Suc-AAPF-pNA, Z-YPQ-pNA, and Z-LYP-pNA, each at 200 μM, by Rmep (A) and subtilisin A (B), each at 1 μg/ml. C and D: hydrolysis of gliadin-derived fluorescence resonance energy transfer (FRET) substrates. FRET substrates containing QPQLPY, PQPQPQ, and QGSFQP, each at 100 μM, were incubated with Rmep (C) and subtilisin A (D), at 1 and 0.5 μg/ml, respectively. Controls were boiled Rmep and subtilisin A incubated with each of the three FRET substrates (no activity, baseline).
Activities were also determined toward three gliadin-derived FRET substrates (Fig. 4, C and D). The substrates were QPQLPY, contained in three immunogenic gliadin epitopes; PQPQPQ, a theoretical substrate for Rothia enzymes; and QGSFQP, contained in two gliadin epitopes. The first two peptides, containing XPX motifs, were rapidly hydrolyzed by Rmep (Fig. 4C). In contrast, QGSFQP, not containing XPX, was not cleaved. Subtilisin A showed the highest activity toward the PQPQPQ substrate (Fig. 4D).
Elimination of immunogenic epitopes by Rmep.
Hydrolysis of the FRET substrates would suggest CD-relevant epitope degradation. To further investigate this, the time courses of degradation of mixed gliadins and of the highly immunogenic 33-mer α-gliadin peptide were determined. Epitope elimination was monitored by two ELISAs employing the R5 antibody recognizing QQPFP and related pentapeptides (28, 46) and the G12 antibody recognizing QPQLPY contained in the 33-mer peptide (24, 25). Data obtained with Rmep are shown in Fig. 5 and with subtilisin A in Fig. 6.
Fig. 5.

Degradation of mixed gliadins by Rmep and abolishment of immunogenic epitopes. A: gliadins (250 μg/ml) in 50 mM Tris·HCl, pH 8.0, were incubated with Rmep at 57 μg/ml. After time 0 and 15 min, 30 min, and 2 h incubation, 100-μl aliquots were removed, boiled, and analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue. The bold arrow (left) points to the position of the major band in the gliadin preparation, the thin arrow (right) points to the 140-kDa band in the Rmep preparation, and the dashed arrows (right) point to the gliadin degradation fragments. B: reversed-phase (RP)-HPLC of degradation of the immunogenic 33-mer peptide from α-gliadin. Arrow (top) points to the intact 33-mer; C and D: assessment of the survival of epitopes in the gliadin-Rmep degradation mixture determined with the R5 ELISA (C) or G12 ELISA (D).
Fig. 6.

Degradation of mixed gliadins by subtilisin A and abolishment of immunogenic epitopes. A: gliadins (250 μg/ml) in 50 mM Tris·HCl, pH 8.0, were incubated with subtilisin A at 57 μg/ml. Experiments were conducted as described in the legend of Fig. 5 except using subtilisin A instead of Rmep.
Rmep rapidly degraded mixed gliadins within 15 min incubation and completed 33-mer peptide degradation within 30 min of incubation (Fig. 5, A and B, respectively). Already in the time 0 sample 46% reduction in antibody binding could be observed compared with the control, corrected for enzyme only values. After 15 min virtually all R5 epitopes were eliminated (Fig. 5C). After 30 min the G12 antibody binding had decreased by 65% and showed a time-dependent decline to 85% reduction after 2 h of incubation (Fig. 5D). Subtilisin A degraded gliadins even faster, with near complete degradation in the time 0 sample (Fig. 6A). However, the 33-mer peptide was not as efficiently cleaved, and after 2 h of incubation residual 33-mer still remained (74% degradation; Fig. 6B). The R5 epitopes in the gliadin-subtilisin A digest were rapidly eliminated, by 90% at time 0 and by 100% within 15 min of incubation (Fig. 6C). In contrast, the G12 epitopes were quite resistant to subtilisin A and were degraded by 22% at time 0 and 45% at 2 h (Fig. 6D). This agrees with the modest activity of subtilisin A toward the 33-mer.
Last, we tested Nattokinase, which is a subtilisin from B. subtilis. It is available commercially as a dietary supplement for human consumption. Like Rmep and subtilisin A, Nattokinase degraded gliadins effectively, down to nanogram per milliliter concentrations (Fig. 7, A and B). Nattokinase hydrolyzed Z-AAPF-pNA (Fig. 7C) and degraded the 33-mer peptide (Fig. 7D), although a large fragment eluting just before the 33-mer in the RP-HPLC chromatogram remained. It abolished the R5 epitopes (Fig. 7E) but was less effective in eliminating the G12 epitopes, consistent with observations made with subtilisin A (Fig. 7F).
Fig. 7.

Gliadin degradation and epitope abolishment by nattokinase (NattoK) from B. subtilis. A: mixed gliadins (G, 250 μg/ml) were incubated with NattoK (57 μg/ml) for 0, 15, 30, and 120 min. Four lanes on left, controls without enzyme or gliadins, respectively, each at time 0 and 120 min. B: dilution series of NattoK (3.5-0.06 μg/ml) incubated for 30 min with mixed gliadins. Lanes on right, gliadin (G) and NattoK (NK) control. C: dilution series of NattoK incubated with Suc-AAPF-pNA. Hydrolysis was measured at 405 nm. D: RP-HPLC analysis of the gliadin-derived 33-mer (250 μg/ml) incubated for 0, 15, 30, and 120 min with NattoK (57 μg/ml). E and F: epitope abolishment in mixed gliadins (250 μg/ml) incubated for 0, 15, 30, and 120 min with NattoK (57 μg/ml) assessed with the R5 ELISA (E) and G12 ELISA (F).
DISCUSSION
The gluten-degrading enzymes of Rothia were identified as members of the subtilisin protease family. The gluten-degrading activities of this class of enzymes were shown to extend beyond the Rothia genus. The cleavage specificity of subtilisins is XPX↓, with X in the P1 position preferably being a hydrophobic amino acid. In accordance, gluten substrates with Q in the P1 position were found to be highly susceptible to cleavage by Rmep and subtilisin A. XPQ is present in the majority of the antigenic gluten epitopes relevant in CD (40). A subset of such Q residues in gliadins is deamidated by the enzyme tissue transglutaminase in the lamina propria, a key step in the pathogenesis of CD (47). This deamidation increases the peptides' affinity for HLA-DQ2 and HLA-DQ8 expressed on antigen-presenting cells and triggers the destructive mucosal T cell response. Glutamine residues in the XPQXP context are particularly prone to such deamidation (38, 45). Selective cleavage of this sequence will prevent Q deamidation, and thus both Rothia and Bacillus subtilisins can be anticipated to be efficient in preventing T cell activation in the lamina propria. This is supported by our finding of the elimination of the major gliadin epitopes in two independent ELISA assays, even without predigesting gliadins with the mammalian digestive enzymes pepsin and trypsin.
The three major Rothia species are R. aeria, R. mucilaginosa, and R. dentocariosa. Analysis of the gluten-degrading Rothia enzymes showed that they belong to the D-H-S class of subtilisins (36, 37) and not to the family of kexins (an S8 protease subfamily). All contain a signal peptide for secretion, which is presumably cleaved off, since most contain the consensus sequence AxA|A, which is recognized by a signal peptidase. With the use of NCBI BlastP (or InterPro) analysis, it was found that none of the sequences contain a COOH-terminal LPxTG-type peptidoglycan anchor. However, most Rothia subtilisins did contain two to three COOH-terminal surface layer homology (SLH) domains, called Pfam00395 (22). Such bacterial SLH domain proteins are noncovalently anchored to the cell surface via a conserved mechanism involving cell wall polysaccharide pyruvylation. The theoretical cell-envelope association agrees with our finding that Rothia subtilisins could be isolated from cell-derived vesicles that were harvested by ultracentrifugation.
The mass spectrometric identification of subtilisins prompted us to explore subtilisins from other microbes, specifically Bacillus species. Like Rmep, subtilisins A and Nattokinase degraded gluten efficiently. Like Rothia, the Bacillus genomes encode for several subtilisins. In general, bacteria that express multiple extracellular proteases reside in a protein-rich environment and use proteases to degrade proteins into peptides that can subsequently be used for growth. This is the case for the natural habitat of Rothia species, the oral cavity, where XPQ-containing substrates are prevalent. Interestingly, in the oral cavity, the XPQ-rich proteins are not only represented by ingested gluten proteins but also by salivary proline-rich proteins that are produced constitutively by the salivary glands (3, 12, 17, 43). In this context it is of interest to note that, like Rothia, B. subtilis also colonizes the oral cavity (8). Salivary proline-rich proteins undergo extensive proteolytic fragmentation in the oral cavity, with primary cleavage after XPQ↓ (13, 23, 48). Based on the data presented here, these cleavages are carried out most likely by the oral bacterial subtilisins.
Some interesting observations were made when the substrate specificities of subtilisin A and Rmep were compared. First, both enzymes cleaved AAPF rapidly, but not PPF. This suggests that a P in the P3 position interferes with substrate recognition by both enzymes. For gluten degradation this is less of a concern since in most gluten immunogenic domains containing the XPF sequence, X is represented by a Q, and not by a P and the FRET results showed that QPF is efficiently cleaved. Also abundant in gliadin immunogenic domains is the sequence LPY, which was hydrolyzed. Overall, R. mucilaginosa cleaves the 33-mer at LPY↓, QPQ↓, and YPQ↓ (44) and it is very likely that all cleavages are carried out by the same subtilisin enzyme.
An important observation made was that the molecular mass of the active subtilisins, migrating in the ∼75- to 80-kDa gel area, was substantially shorter than the parent gene product of ∼140 kDa. It is well known that subtilisins from Bacillus species undergo autocatalytic activation to produce a shorter mature enzyme (11). In the Rothia subtilisins the catalytic triad is located at the NH2-terminal portion of the protein. We postulate that the parent inactive precursor proteins encoded by C6R5V9_9MICC and C6R5W1_9MICC are processed at the NH2-termini to remove the inhibitory propeptide and at the COOH-terminus between the Ig-like segment and SLH domains, generating the active ∼75- and ∼80-kDa mature enzymes, as outlined in Fig. 8. Further structural analysis should reveal the exact processing sites and the secondary and tertiary structures of the active Rothia subtilisins.
Fig. 8.

Domain composition of C6R5V9_9MICC. The protein of 1,328 amino acids contains a peptidase S8 propeptide domain (also called proteinase inhibitor I9), a peptidase S8/S53 family domain, an immunoglobulin-like fold domain, and 3 surface layer homology domains. Figure based on InterPro analysis.
B. subtilis is food safe and has been consumed for decades, e.g., in a product called natto, a Japanese fermented soy bean dish. In natto the active enzyme is Nattokinase, a 27.7-kDa subtilisin enzyme (26) that was used in our studies. Despite a long history of consumption of B. subtilis and its products, there are very few reports of adverse events. The food-grade status of B. subtilis, and the already widely consumed natto products, open new avenues for potential therapeutic applications of the subtilisin enzymes. The R. mucilaginosa subtilisins have superior epitope-detoxifying capacities compared with Bacillus subtilisins, and they are predicted to be more effective in neutralizing gluten epitopes, as based on our in vitro assessments. The usefulness and safety of subtilisins in vivo should be further explored. These include assessment of enzyme activity in buffers and systems that mimic gastroduodenal conditions and that take into account the variable pH conditions in the stomach and duodenum, the presence of endogenous digestive enzymes, and of competing food proteins. In addition, functional assays should reveal if the biochemical observations of epitope abolishment in vitro can be validated in cell-based assays, e.g., in CD biopsy-derived T cell lines and in in vivo mouse models for CD. Furthermore, it should be established if subtilisin doses required for in vivo gluten digestion are safe for long term consumption. Going forward, because gluten-degrading enzymes are the preferred therapy of choice for CD (5), and given the exceptional activity of the subtilisins and their association with natural human microbial colonizers, they are worthy of further exploration for clinical applications in CD and potentially other gluten-intolerance disorders.
GRANTS
These studies were supported by National Institute of Allergy and Infectious Diseases Grants AI-087803 and AI-101067 to E. J. Helmerhorst.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.W., N.T., D.S., and E.J.H. conception and design of research; G.W., N.T., and E.J.H. performed experiments; G.W., N.T., and E.J.H. analyzed data; G.W., N.T., D.S., and E.J.H. interpreted results of experiments; G.W., N.T., R.S., and E.J.H. prepared figures; G.W., N.T., R.S., D.S., and E.J.H. approved final version of manuscript; R.S., D.S., and E.J.H. edited and revised manuscript; E.J.H. drafted manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Maram Zamakhchari for contributions to the project and Ross Tomaino for conducting the mass spectrometric analyses.
REFERENCES
- 1.Abadie V, Discepolo V, Jabri B. Intraepithelial lymphocytes in celiac disease immunopathology. Semin Immunopathol 34: 551–566, 2012. [DOI] [PubMed] [Google Scholar]
- 2.Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol 29: 493–525, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Azen EA, Amberger E, Fisher S, Prakobphol A, Niece RL. PRB1, PRB2, and PRB4 coded polymorphisms among human salivary concanavalin-A binding, II-1, and Po proline-rich proteins. Am J Hum Genet 58: 143–153, 1996. [PMC free article] [PubMed] [Google Scholar]
- 4.Bethune MT, Khosla C. Oral enzyme therapy for celiac sprue. Methods Enzymol 502: 241–271, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Branchi F, Tomba C, Ferretti F, Norsa L, Roncoroni L, Bardella MT, Conte D, Elli L. Celiac disease and drug-based therapies: inquiry into patients demands. Digestion 93: 160–166, 2016. [DOI] [PubMed] [Google Scholar]
- 6.Caminero A, Herran AR, Nistal E, Perez-Andres J, Vaquero L, Vivas S, Ruiz de Morales JM, Albillos SM, Casqueiro J. Diversity of the cultivable human gut microbiome involved in gluten metabolism: isolation of microorganisms with potential interest for coeliac disease. FEMS Microbiol Ecol 88: 309–319, 2014. [DOI] [PubMed] [Google Scholar]
- 7.De Angelis M, Rizzello CG, Fasano A, Clemente MG, De Simone C, Silano M, De Vincenzi M, Losito I, Gobbetti M. VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for Celiac Sprue. Biochim Biophys Acta 1762: 80–93, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. The human oral microbiome. J Bacteriol 192: 5002–5017, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Feo M, Wei G, Blumenkranz G, Dewhirst FE, Schuppan D, Oppenheim FG, Helmerhorst EJ. The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clin Microbiol Infect 19: E386–E394, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freitag TL, Loponen J, Messing M, Zevallos V, Andersson LC, Sontag-Strohm T, Saavalainen P, Schuppan D, Salovaara H, Meri S. Testing safety of germinated rye sourdough in a celiac disease model based on the adoptive transfer of prolamin-primed memory T cells into lymphopenic mice. Am J Physiol Gastrointest Liver Physiol 306: G526–G534, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher T, Gilliland G, Wang L, Bryan P. The prosegment-subtilisin BPN' complex: crystal structure of a specific ‘foldase.’ Structure 3: 907–914, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Helmerhorst EJ, Oppenheim FG. Saliva: a dynamic proteome. J Dental Res 86: 680–693, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Helmerhorst EJ, Sun X, Salih E, Oppenheim FG. Identification of Lys-Pro-Gln as a novel cleavage site specificity of saliva-associated proteases. J Biol Chem 283: 19957–19966, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helmerhorst EJ, Wei G. Experimental strategy to discover microbes with gluten-degrading enzyme activities. Proc SPIE 9112: 91120D91121–91120D 91111, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helmerhorst EJ, Zamakhchari M, Schuppan D, Oppenheim FG. Discovery of a novel and rich source of gluten-degrading microbial enzymes in the oral cavity. PloS one 5: e13264, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hipler K, Priestle JP, Rahuel J, Grutter MG. Active site binding loop stabilization in the subtilisin inhibitor eglin c: structural and functional studies on specifically designed mutants in complex with subtilisin and the uncomplexed inhibitor. Adv Exp Med Biol 379: 43–47, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Inzitari R, Cabras T, Rossetti DV, Fanali C, Vitali A, Pellegrini M, Paludetti G, Manni A, Giardina B, Messana I, Castagnola M. Detection in human saliva of different statherin and P-B fragments and derivatives. Proteomics 6: 6370–6379, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Kaukinen K, Lindfors K. Novel treatments for celiac disease: glutenases and beyond. Dig Dis 33: 277–281, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Lahdeaho ML, Kaukinen K, Laurila K, Vuotikka P, Koivurova OP, Karja-Lahdensuu T, Marcantonio A, Adelman DC, Maki M. The glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 146: 1649–1658, 2014. [DOI] [PubMed] [Google Scholar]
- 20.Lindfors K, Blomqvist T, Juuti-Uusitalo K, Stenman S, Venalainen J, Maki M, Kaukinen K. Live probiotic Bifidobacterium lactis bacteria inhibit the toxic effects induced by wheat gliadin in epithelial cell culture. Clin Exp Immunol 152: 552–558, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundin KE, Qiao SW, Snir O, Sollid LM. Coeliac disease: from genetic and immunological studies to clinical applications. Scand J Gastroenterol 50: 708–717, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Mesnage S, Fontaine T, Mignot T, Delepierre M, Mock M, Fouet A. Bacterial SLH domain proteins are non-covalently anchored to the cell surface via a conserved mechanism involving wall polysaccharide pyruvylation. EMBO J 19: 4473–4484, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messana I, Cabras T, Pisano E, Sanna MT, Olianas A, Manconi B, Pellegrini M, Paludetti G, Scarano E, Fiorita A, Agostino S, Contucci AM, Calo L, Picciotti PM, Manni A, Bennick A, Vitali A, Fanali C, Inzitari R, Castagnola M. Trafficking and postsecretory events responsible for the formation of secreted human salivary peptides: a proteomics approach. Mol Cell Proteomics 7: 911–926, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Moron B, Bethune MT, Comino I, Manyani H, Ferragud M, Lopez MC, Cebolla A, Khosla C, Sousa C. Toward the assessment of food toxicity for celiac patients: characterization of monoclonal antibodies to a main immunogenic gluten peptide. PloS one 3: e2294, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moron B, Cebolla A, Manyani H, Alvarez-Maqueda M, Megias M, Thomas Mdel C, Lopez MC, Sousa C. Sensitive detection of cereal fractions that are toxic to celiac disease patients by using monoclonal antibodies to a main immunogenic wheat peptide. Am J Clin Nutr 87: 405–414, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura T, Yamagata Y, Ichishima E. Nucleotide sequence of the subtilisin NAT gene, aprN, of Bacillus subtilis (natto). Biosci Biotechnol Biochem 56: 1869–1871, 1992. [DOI] [PubMed] [Google Scholar]
- 27.Norsa L, Tomba C, Agostoni C, Branchi F, Bardella MT, Roncoroni L, Conte D, Elli L. Gluten-free diet or alternative therapy: a survey on what parents of celiac children want. Int J Food Sci Nutr 66: 590–594, 2015. [DOI] [PubMed] [Google Scholar]
- 28.Osman AA, Uhlig HH, Valdes I, Amin M, Mendez E, Mothes T. A monoclonal antibody that recognizes a potential coeliac-toxic repetitive pentapeptide epitope in gliadins. Eur J Gastroenterol Hepatol 13: 1189–1193, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Rollan G, De Angelis M, Gobbetti M, de Valdez GF. Proteolytic activity and reduction of gliadin-like fractions by sourdough lactobacilli. J Appl Microbiol 99: 1495–1502, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA, and American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol 108: 656–677, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology 137: 1912–1933, 2009. [DOI] [PubMed] [Google Scholar]
- 32.Shah S, Akbari M, Vanga R, Kelly CP, Hansen J, Theethira T, Tariq S, Dennis M, Leffler DA. Patient perception of treatment burden is high in celiac disease compared with other common conditions. Am J Gastroenterol 109: 1304–1311, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shan L, Molberg O, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Structural basis for gluten intolerance in celiac sprue. Science 297: 2275–2279, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Siegel M, Bethune MT, Gass J, Ehren J, Xia J, Johannsen A, Stuge TB, Gray GM, Lee PP, Khosla C. Rational design of combination enzyme therapy for celiac sprue. Chem Biol 13: 649–658, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Siegel M, Garber ME, Spencer AG, Botwick W, Kumar P, Williams RN, Kozuka K, Shreeniwas R, Pratha V, Adelman DC. Safety, tolerability, and activity of ALV003: results from two phase 1 single, escalating-dose clinical trials. Dig Dis Sci 57: 440–450, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Siezen RJ, Leunissen JA. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 6: 501–523, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siezen RJ, Renckens B, Boekhorst J. Evolution of prokaryotic subtilases: genome-wide analysis reveals novel subfamilies with different catalytic residues. Proteins 67: 681–694, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2: 647–655, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Sollid LM, Khosla C. Novel therapies for coeliac disease. J Intern Med 269: 604–613, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 64: 455–460, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stepniak D, Spaenij-Dekking L, Mitea C, Moester M, de Ru A, Baak-Pablo R, van Veelen P, Edens L, Koning F. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol 291: G621–G629, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Tack GJ, van de Water JM, Bruins MJ, Kooy-Winkelaar EM, van Bergen J, Bonnet P, Vreugdenhil AC, Korponay-Szabo I, Edens L, von Blomberg BM, Schreurs MW, Mulder CJ, Koning F. Consumption of gluten with gluten-degrading enzyme by celiac patients: a pilot-study. World J Gastroenterol 19: 5837–5847, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian N, Messana I, Leffler DA, Kelly CP, Hansen J, Cabras T, D'Alessandro A, Schuppan D, Castagnola M, Helmerhorst EJ. Salivary proline-rich proteins and gluten: Do structural similarities suggest a role in celiac disease? Proteomics 9: 953–964, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian N, Wei G, Schuppan D, Helmerhorst EJ. Effect of Rothia mucilaginosa enzymes on gliadin (gluten) structure, deamidation, and immunogenic epitopes relevant to celiac disease. Am J Physiol Gastrointest Liver Physiol 307: G769–G776, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vader LW, de Ru A, van der Wal Y, Kooy YM, Benckhuijsen W, Mearin ML, Drijfhout JW, van Veelen P, Koning F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med 195: 643–649, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valdes I, Garcia E, Llorente M, Mendez E. Innovative approach to low-level gluten determination in foods using a novel sandwich enzyme-linked immunosorbent assay protocol. Eur J Gastroenterol Hepatol 15: 465–474, 2003. [DOI] [PubMed] [Google Scholar]
- 47.van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol 161: 1585–1588, 1998. [PubMed] [Google Scholar]
- 48.Vitorino R, Barros A, Caseiro A, Domingues P, Duarte J, Amado F. Towards defining the whole salivary peptidome. Proteomics 3: 528–540, 2009. [Google Scholar]
- 49.Wei G, Tian N, Valery AC, Zhong Y, Schuppan D, Helmerhorst EJ. Identification of pseudolysin (lasB) as an aciduric gluten-degrading enzyme with high therapeutic potential for celiac disease. Am J Gastroenterol 110: 899–908, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zamakhchari M, Wei G, Dewhirst F, Lee J, Schuppan D, Oppenheim FG, Helmerhorst EJ. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PloS one 6: e24455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.