

Abstract
Glutamine-derived carbon becomes available for anabolic biosynthesis in cancer cells via the hydrolysis of glutamine to glutamate, as catalyzed by GAC, a splice variant of kidney-type glutaminase (GLS). Thus, there is significant interest in understanding the regulation of GAC activity, with the suggestion being that higher order oligomerization is required for its activation. We used x-ray crystallography, together with site-directed mutagenesis, to determine the minimal enzymatic unit capable of robust catalytic activity. Mutagenesis of the helical interface between the two pairs of dimers comprising a GAC tetramer yielded a non-active, GAC dimer whose x-ray structure displays a stationary loop (“activation loop”) essential for coupling the binding of allosteric activators like inorganic phosphate to catalytic activity. Further mutagenesis that removed constraints on the activation loop yielded a constitutively active dimer, providing clues regarding how the activation loop communicates with the active site, as well as with a peptide segment that serves as a “lid” to close off the active site following substrate binding. Our studies show that the formation of large GAC oligomers is not a pre-requisite for full enzymatic activity. They also offer a mechanism by which the binding of activators like inorganic phosphate enables the activation loop to communicate with the active site to ensure maximal rates of catalysis, and promotes the opening of the lid to achieve optimal product release. Moreover, these findings provide new insights into how other regulatory events might induce GAC activation within cancer cells.
Keywords: enzyme mechanism, glutaminase, metabolism, mutagenesis, protein structure
Introduction
To accommodate the cellular changes underlying tumor establishment and maintenance, metabolic remodeling occurs, resulting in dramatic increases in aerobic glycolysis (see Refs. 1–5; i.e. the “Warburg effect”). This is often accompanied by an acquired reliance on glutamine as a carbon source for anabolic processes such as fatty acid and nucleotide synthesis, as well as serving as a fuel for the tricarboxylic acid cycle. The marked increases in glutamine metabolism in tumor cells represent a critical difference in the physiology of normal and transformed tissues that may offer novel therapeutic targets for the treatment of cancer. Transcriptional and post-translational responses to transformation that determine the glutamine dependence of certain tumors are therefore of great interest as they identify microscopic changes that give rise to “glutamine addicted” neoplasms (6).
One such change is the increased expression and catalytic activity of mitochondrial kidney-type glutaminase (GLS)2 (7, 8). Glutamine is the most abundant amino acid in blood serum, thus providing a ready precursor for macromolecular synthesis after conversion to glutamate, as catalyzed by GLS. Thus, GLS helps to provide a molecular gateway to glutamine-derived biosynthesis within cells undergoing Warburg glycolysis, and in doing so offers a potentially attractive target for inhibiting cancer cell growth (9, 10). The two alternatively spliced isoforms of GLS, most often designated as KGA (kidney-type glutaminase) and GAC, account for the majority of glutamine to glutamate conversion in cells, with the overexpression of GAC being observed in a number of cancer cell types (11). The elevated activity of GLS has been linked to oncogenes such as Myc (12, 13) and Ras (14), as well as to the hyper-activation of Rho GTPases (9, 15).
It has been reported that oligomerization is both necessary and sufficient for activating GAC, with the formation of GAC tetramers and higher order oligomers being suggested to be essential for full enzymatic activity (16–18). To further examine the mechanistic basis for the activation of this key metabolic enzyme, we have made use of structural analysis, determining the x-ray crystal structures for both wild-type GAC and a stable GAC dimer, together with multiangle light scattering size determinations, mutagenesis, and biochemical assays of enzyme activity. By combining these approaches, we were able to design a GAC dimer that was constitutively active, thus demonstrating that higher order oligomerization is not an absolute requirement for maximal catalysis. Closer examination of this constitutively active GAC dimer allowed us to introduce specific mutations that revealed the intramolecular coupling of 1) an activation loop that mediates phosphate-stimulated activity and represents the binding site for a small molecule allosteric inhibitor (19–21), and 2) a peptide “lid” that governs product release from the GAC catalytic site. Together these coupled sites provide for a tiered regulation of this important metabolic enzyme, which offers new possibilities for targeted drug therapy of glutamine-addicted cancer cells.
Results
Disruption of the GAC Helical Interface Results in an Inactive Dimer
Previous studies have suggested that mammalian glutaminases exist as dimers in their inactive state and need to oligomerize into tetramers and higher order oligomers to be capable of catalytic activity (16–18). Several x-ray crystal structures of wild-type glutaminase, as well as our own analysis of human GAC, reveal the presence of four molecules in the asymmetric unit (Fig. 1A). This quaternary assembly is formed by the association of two inactive GAC dimers and involves two sets of interfaces. One interface is made up of the contacts between monomers i and ii, and those between monomers iii and iv (Fig. 1A), which buries considerably more surface area than the second interface between monomers i and iii, and monomers ii and iv. The latter pairing of dimers to form a tetramer has been suggested to be a critical step in the stimulation of glutaminase activity by the allosteric activator, inorganic phosphate (21). The GAC dimer-dimer interface results from the interaction between two pairs of anti-parallel α-helices contributed by two monomers that are twisted 180° relative to one another. Closer inspection of the interactions between the two helices, referred to as the interface helices, reveals the presence of a salt bridge between aspartic acid 391 and lysine 401 (Fig. 1A, inset). The anti-parallel configuration of the helices therefore consists of four of these interactions for each GAC tetramer. This observation led us to hypothesize that the disruption of this salt bridge by mutagenesis might destabilize the tetrameric assembly and yield a GAC species trapped as a dimer.
FIGURE 1.
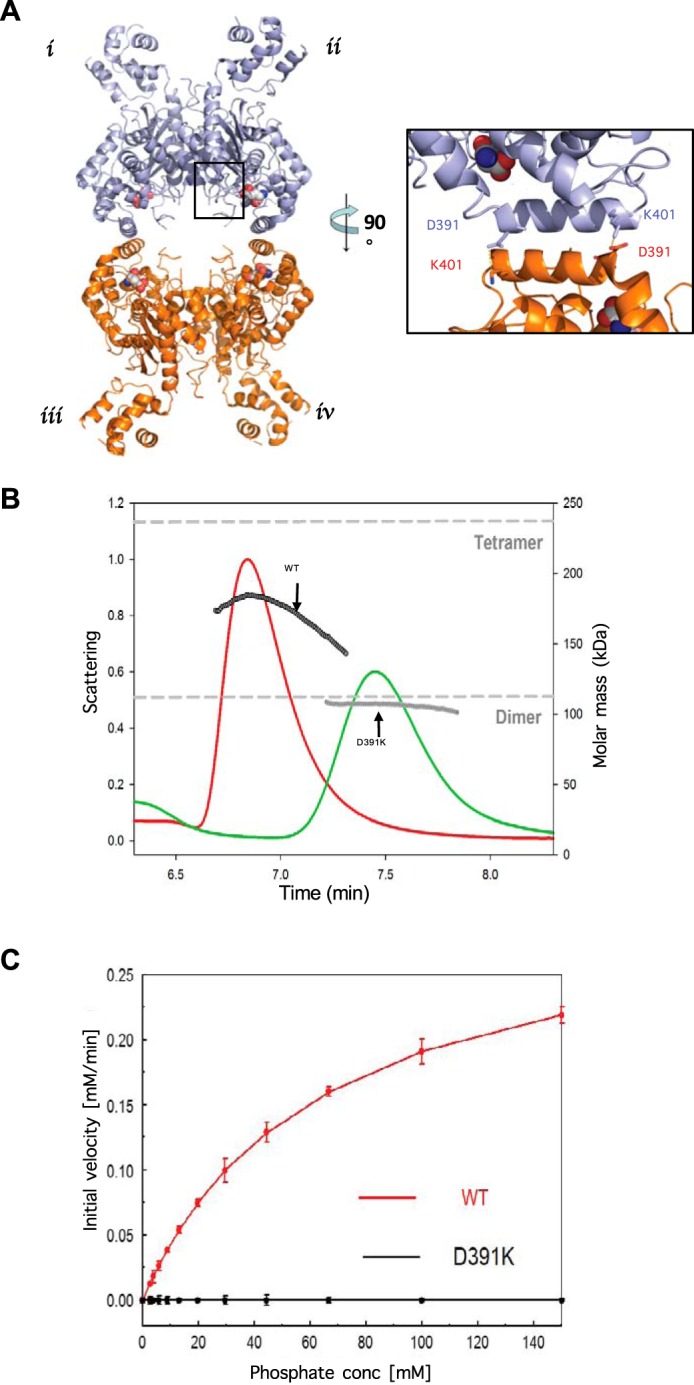
A single mutation in the helical interface of glutaminase results in an inactive, dimeric form of the enzyme. A, x-ray crystal structure of wild-type human GAC (PDB 5D3O) showing the anti-parallel salt bridge at the helical interface of two dimers (inset). B, SEC-MALS analysis of wild-type GAC and GAC(D391K). The signal from the 90° scattering detector is shown as red (wild-type GAC) and green (GAC(D391K)) lines (left, y axis). Arrows indicate the average molecular weight as calculated (each second) across the protein elution peak (right, y axis). Theoretical molecular weights based on the primary sequences for the GAC dimer and the tetramer are indicated as horizontal dashed lines. Protein samples (100 μm) were injected into the SEC-MALS system for analysis. C, comparison of specific activities of 50 nm wild-type GAC or GAC(D391K) with increasing phosphate concentration. The GAC catalyzed conversion of glutamine to glutamate was assayed as described under ”Experimental Procedures.“ Results are representative of three independent titrations.
To test this idea, we mutated aspartic acid 391 to a lysine residue (GAC(D391K)), thereby introducing an electrostatic repulsion between the newly introduced lysine 391 residue and the wild-type lysine 401 residue. The GAC(D391K) mutant was expressed in Escherichia coli to levels comparable with that of wild-type GAC and its oligomeric status was examined using SEC-MALS. Initial SEC-MALS analysis of wild-type GAC indicated the presence of a heterogeneous species with a calculated molecular mass that was intermediate between that of a dimer (∼115 kDa) and a tetramer (∼230 kDa; Fig. 1B). The apparent molecular mass for wild-type GAC, as determined by SEC-MALS, increased with increasing protein concentration suggesting the rapid inter-conversion between the dimeric and tetrameric GAC species with respect to the time scale of the gel filtration experiment. In contrast, the GAC(D391K) mutant behaved as a homogeneous species with a molecular mass of ∼115 kDa, corresponding to that of a GAC dimer (Fig. 1B), which persisted at high concentrations of GAC(D391K) (>100 μm).
Wild-type GAC, at a concentration of 50 nm, exhibits low enzymatic activity in the absence of any activators such as inorganic phosphate; although, its activity increases with the addition of inorganic phosphate in a dose-dependent manner (Fig. 1C, red). However, the GAC(D391K) mutant fails to exhibit detectable activity, even when assayed at inorganic phosphate concentrations as high as 100 mm (Fig. 1C, black).
The X-ray Crystal Structure of the GAC(D391K) Mutant Reveals a Stationary Activation Loop in Contrast to Wild-type GAC Structures
Crystals of GAC(D391K) were grown under conditions that were markedly different from those for the crystallization of wild-type GAC, and were observed to diffract to 2.3-Å resolution (see Table 1). The structure for GAC(D391K) was solved by molecular replacement, using the x-ray structure for wild-type GAC as a search model (Fig. 2A). Unlike the structure for wild-type GAC, only two molecules of GAC(D391K) were present in the asymmetric unit, consistent with the homogeneous dimeric species observed in SEC-MALS experiments (Fig. 1B). The overall three-dimensional structures of the individual monomers in GAC(D391K) are highly similar to those for wild-type GAC, with the calculated r.m.s. deviation value being 0.55 Å. Of particular interest was the essentially identical positioning of the active site residues in wild-type GAC and the dimeric, inactive GAC(D391K) mutant (Fig. 2B), suggesting that wild-type GAC whereas a tetramer in the x-ray crystal structure, apparently is in a catalytically inactive state.
TABLE 1.
Data collection and refinement statistics
| GAC(D391K) | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 85.2, 101.4, 145.2 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 2.3 |
| I/σI | 7.2/3.9 |
| Completeness (%) | 92.87 |
| Redundancy | 7.3 |
| Refinement | |
| Resolution (Å) | 50.0–2.30 |
| No. reflections | 51,086 |
| Rwork/Rfree | 0.19641/0.22761 |
| No. atoms | |
| Protein | 6,669 |
| Water | 172 |
| Average B factor (Å 2) | 30.712 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.001 |
| Bond angles (°) | 1.4 |
FIGURE 2.
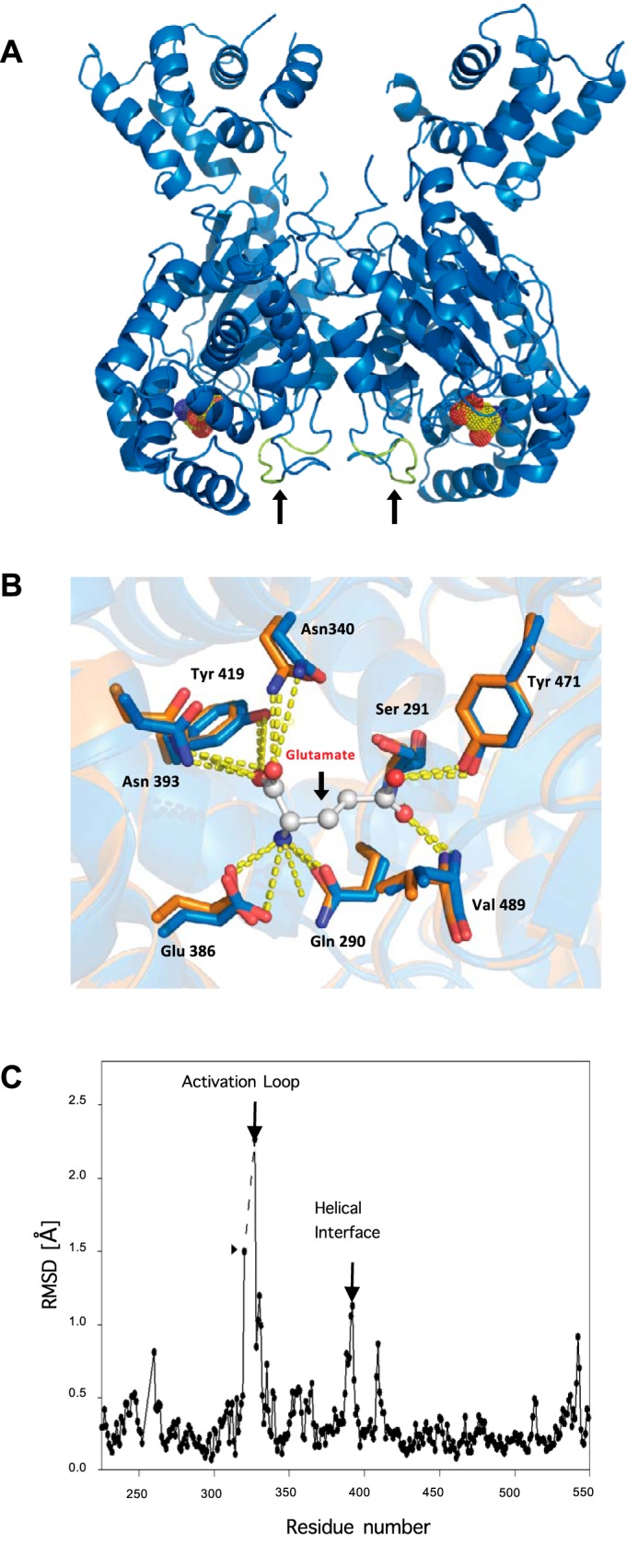
Structure determination of GAC(D391K) and comparison with wild-type GAC coordinates. A, ribbon depiction of GAC(D391K) showing the location of the active site based on the structure for the product of the GAC-catalyzed reaction, glutamate (yellow), bound to wild-type GAC (PDB 3SS5), and the resolved activation loop (colored in green indicated with black arrows) near the helical interface. B, details of the glutamine/glutamate binding site (bound glutamate is shown) where wild-type GAC-tetramer (orange) and GAC(D391K)-dimer (blue) contact residues are superimposed. C, r.m.s. deviation plot of carbon atom coordinates in the two crystalline forms of GAC illustrating the apparent flexibility of the activation loop in wild-type but not GAC(D391K) dimeric GAC. The dashed line indicates the region lacking resolvable electron density in the tetrameric structure. Differences in the backbone structures also occur at the helical interface and the substrate lid (see text).
In the x-ray crystal structure of the GAC(D391K) mutant, a peptide loop was observed that extends from glycine 315 to methionine 338, with the central portion (leucine 321 to 326) being unresolved in the structure of wild-type GAC. Interestingly, an allosteric inhibitor of GAC, BPTES, was recently co-crystallized with GAC (20, 22) and shown to interact with this loop, forming a bridge between two dimers that stabilizes an inactive tetramer (18, 23). When comparing the backbone structures of wild-type GAC and the GAC(D391K) mutant, the calculated r.m.s. deviation plot revealed that the largest difference between these structures exists for residues glycine 320 and phenylalanine 327, which are present at either end of the unresolved stretch of residues (designated with the dashed line in Fig. 2C). Thus, the structure of the GAC(D391K) mutant revealed that this loop, henceforth referred to as the activation loop, is less flexible in the dimeric structure, much like the case for the BPTES inhibited structure, whereas, this region of GAC is not resolvable in the wild-type enzyme crystal structures.
Mutation of Lysine 325 in the GAC(D391K) Background Results in a Constitutively Active Enzyme
The fact that the binding of BPTES to the activation loop of GAC results in an inactive enzyme suggests that conformational changes in this loop are important for enzymatic activity. In an effort to identify the individual loop residues important for activity, alanine-scanning mutagenesis was carried out on residues leucine 321 to phenylalanine 327 (Fig. 3A). Each loop mutation was made in the GAC(D391K) background, and then assayed for enzyme activity in the presence and absence of 50 mm inorganic phosphate. Alanine substitution of arginine 322, phenylalanine 323, asparagine 324, and leucine 326 within the GAC(D391K) background yielded GAC double mutants that were catalytically inactive (data not shown). Mutation of either leucine 321 or phenylalanine 327 to alanine resulted in an enzyme with enhanced basal activity (i.e. measured in the absence of inorganic phosphate), compared with GAC(D391K), that was ∼50 and 90% of the basal activity measured for wild-type GAC (Fig. 3B). However, the mutation of lysine 325 to alanine within the GAC(D391K) background yielded an enzyme, GAC(K325A/D391K), with basal activity comparable with that for wild-type GAC measured in the presence of 50 mm inorganic phosphate, with no further stimulation occurring upon phosphate addition (Fig. 3B).
FIGURE 3.

Alanine scanning of the activation loop reveals lysine 325 as a critical residue for GAC enzyme activity. A, numbered residues constituting the activation loop and their proximity to the active site. B, alanine substitution in the inactive dimeric GAC(D391K) background and the resulting effects on glutaminase activity in the presence or absence of 50 mm phosphate. The specific activity of all recombinant proteins (50 nm) with or without 50 mm phosphate was tested and the relative activity normalized with respect to wild-type GAC. Results are the average of three independent determinations with error bars representing standard error. C, SEC-MALS determination of GAC(K325A/D391K) oligomer size distribution demonstrates that the activation loop mutant induces the formation of a heterogeneous population of tetramers. The signals from the 90° scattering detector are shown as red (GAC(K325A/D391K)) and green (GAC(D391K)) lines (left, y axis). Arrows indicate the average molecular weight as calculated (each second) across the protein elution peak (right, y axis). Theoretical molecular weights based on the primary sequence for the dimer and the tetramer are indicated as horizontal dashed lines. D, subcellular localization of V5-tagged GAC(K325A/D391K) (green, upper panel) compared with the mitochondrial marker, DLST (red, lower panel) in SKBR3 cells. Arrows highlight macroscopic oligomeric forms of GAC(K325A/D391K) or the distribution of the mitochondrial marker DLST.
Ferreira et al. (18) had previously reported that mutating lysine 325 to alanine in a wild-type GAC background resulted in an enzyme that was active in the absence of inorganic phosphate, but formed a large higher order oligomer via the interactions of the N-terminal domains of GAC tetramers. The formation of this higher order GAC oligomer was suggested to be a prerequisite for full enzyme activity. Analysis of the GAC(K325A/D391K) double mutant at a concentration of ∼100 μm by SEC-MALS revealed a polydisperse species with a molecular mass intermediate between the predicted size of a dimer and a tetramer (Fig. 3C). Upon ectopically expressing the GAC(K325A/D391K) double mutant in the SKBR3 human breast cancer cell line, immunofluorescence microscopy showed the presence of rod-like species (Fig. 3D, top panel) that did not show an exact co-localization with the mitochondrial marker DLST (Fig. 3D, bottom panel). These micron size structures may provide a “sink” for endogenous GAC as well, because we observed less total GAC in the mitochondria of cells ectopically expressing the GAC(K325A/D391K) double mutant (data not shown). Attempts at isolating cell clones that stably expressed the constitutively active forms of GAC were unsuccessful, suggesting that the higher oligomeric form of GAC is toxic or at least inhibitory for cell growth.
Generation of a Constitutively Active GAC Dimer
The mutation of lysine 316 to glutamine in GAC(K325A) was suggested to prevent the formation of large oligomers of GAC (18). Therefore, we introduced the K316Q substitution into the GAC(K325A/D391K) background and found that it exhibited a level of activity comparable with that of either the GAC(K325A/D391K) double mutant or wild-type GAC in the presence of 50 mm inorganic phosphate (Fig. 4A). Analysis of the GAC(K316Q/K325A/D391K) triple mutant by SEC-MALS showed the presence of a single monodisperse species with a molecular mass corresponding to a dimer (Fig. 4B, triple). Moreover, the triple mutant is able to localize properly to the mitochondria in SKBR3 cells, similar to what we observe for wild-type GAC (compare top to bottom micrographs in Fig. 4C, left panel). Thus, we have been able to engineer a dimeric form of GAC, which exhibits constitutive enzyme activity that is completely uncoupled from the formation of higher oliogomeric species.
FIGURE 4.

Uncoupling the necessity of GAC oligomerization for enzyme activation and the connection between the activation loop and the glutaminase active site. A, engineering a constitutively active dimeric form of GAC. The specific activity of each recombinant GAC (50 nm) was assayed with or without 50 mm phosphate. B, SEC-MALS analysis of GAC(D391K), GAC(K325A/D391K), and the triple mutant GAC(K316Q/K325A/D391K). The signals from the 90° scattering detector are shown as red (GAC(K325A/D391K)), green (GAC(D391K)), and blue (GAC(K316Q/K325A/D391K)) lines (left, y axis). Arrows are pointing to the average molecular weight, which is calculated (each second) across the protein elution peak (right, y axis). Theoretical molecular weight based on primary sequence for the dimer and tetramer are indicated as horizontal dashed lines. C, subcellular localization of V5-tagged wild-type GAC or the triple mutant GAC(K316Q/K325A/D391K) (green, left panels) compared with the mitochondrial marker, DLST (red, right panels) in SKBR3 cells. The K316Q substitution restores wild-type-like mitochondrial localization to GAC. Right panel, Dbl-induced focus formation is enhanced by co-expression of either GACWT or GAC(K316Q/K325A/D391K). NIH-3T3 cells were co-transfected with the indicated amounts of Dbl plasmid and 1 μg of either GACWT or mutant plasmid. Cells were grown for 12 days, fixed with 3.7% formaldehyde in PBS, and stained with 1% crystal violet in methanol to visualize foci. D, primary sequence alignment of several bacterial glutaminases whose structures are known illustrating the conservation of the transducing peptide containing the active site serine 286 (black arrow), glycine 320 (red arrow), and lysine 325 in mammalian glutaminase (green arrow). E, relative positions of the catalytic site serine 291 to the activation loop residue lysine 325, and glycine 320 within the connecting peptide between serine 291 and lysine 325. Colored arrowheads point to residues and correspond to those indicated in the sequence alignment shown in D. F, glycine 320 is critical for activation loop communication to serine 291 in the GAC active site. The specific activity of all the proteins at 50 nm was assayed with and without added phosphate (50 mm) and standard errors are based on three independent experiments.
We compared the ability of the constitutively active GAC(K316Q/K325A/D391K) triple mutant, versus wild-type GAC, to enhance the transforming activity induced by the hyper-activation of Rho GTPases due to the expression of the oncogenic Rho-GEF, Dbl (for diffuse B-cell lymphoma) (24). Previously, we showed that co-expression of wild-type GAC with the activated Cdc42(F28L) mutant displayed a marked synergy in promoting NIH-3T3 cells to form foci (9). Similar results are obtained upon co-expressing wild-type GAC with oncogenic Dbl (Fig. 4C, right panel). However, surprisingly, given the increased glutamine dependence induced by Dbl expression (15), the constitutively active GAC triple mutant did not significantly enhance the transformation potential of the oncogenic form of Dbl relative to wild-type GAC (Fig. 4C, right panel).
Communication between the Activation Loop and the Enzyme Catalytic Site
Serine 291 and lysine 294 are part of a conserved SXXK motif, which is present in the active sites of all glutaminases and are essential for enzymatic activity (9). Therefore, we examined whether communication between these active site residues and the activation loop could be altered by mutating residues within an intervening connecting peptide segment (i.e. serine 291 to lysine 325; Fig. 4D shows this segment for mouse GAC aligned with sequences from additional solved x-ray crystal structures of glutaminase enzymes). Within this connecting segment is a highly conserved glycine (residue 320 indicated by the red arrow in Fig. 4, D and E), which we changed to a proline to restrict the rotational freedom of the connecting segment, to see whether flexibility at this site is necessary for enzyme activation. When this substitution was made within a wild-type GAC background, there was a complete loss of both basal and phosphate-stimulated enzymatic activity (Fig. 4F). Similarly, the G320P substitution within the constitutively active GAC(K325A) mutant markedly reduced enzymatic activity, both in the presence and absence of the allosteric activator, inorganic phosphate (Fig. 4F). Similar results were obtained with a more conservative substitution (G320A) (data not shown), confirming a critical role for this peptide segment in linking the activation loop to the site of catalysis.
Cooperation between the Activation Loop and a Conserved Motif That Encloses the Catalytic Site Upon Substrate Binding
Analysis of the available x-ray crystal structures of the complex between wild-type GAC and its catalytic product, glutamate (25), reveals that a highly conserved YIP motif (Fig. 5A, boxed residues) provides contacts for the bound substrate and product (Fig. 5B). Moreover, superimposing the x-ray crystal structures of wild-type GAC, in the presence and absence of bound glutamate, revealed movement in this conserved motif, such that it would form a lid over the catalytic site when it is occupied (i.e. by substrate), as illustrated by the comparison of Fig. 5, B and C. We therefore examined whether mutations introduced into this active site lid could influence enzymatic activity by affecting accessibility to the catalytic site. Indeed, a Y254F substitution, within a wild-type GAC background, yielded an enzyme with markedly enhanced basal activity (i.e. in the absence of inorganic phosphate) compared with the wild-type protein, whereas the same substitution, when introduced within the GAC(D391K) dimer background, failed to activate enzyme activity (Fig. 5D). GAC was also inactive when the Y254F substitution was made within the GAC(G320P) background and only minimally active when the K325A substitution was added (see column pairs 4 and 5 in Fig. 5D). Thus, the ability of the Y254F substitution to confer an increased basal activity is dependent upon the proper orientation of the activation loop, which in turn depends on tetramer formation. Interestingly, the tyrosine 254 to phenylalanine substitution, within the wild-type GAC background, caused a significant shift in the dose-response for inorganic phosphate (Fig. 5E), indicating that disrupting the active site lid with a conservative Tyr to Phe substitution can significantly enhance the binding of inorganic phosphate and alter the specific activity of the enzyme (Fig. 5F, also see “Discussion”). As will be described further below, these results, and those presented in the preceding sections, now offer a fresh picture of how the enzymatic activity of GAC is activated in response to allosteric activators such as inorganic phosphate.
FIGURE 5.

Phosphate activation of GAC influences substrate (glutamine) accessibility and product (glutamate) release. A, primary sequence alignment guided by the available glutaminase structures as described in the legend to Fig. 4A highlighting the conservation of the YIP motif constituting the substrate pocket lid. B, proximal relationship of the activation loop and the substrate accessibility lid when either glutamate or glutamine is bound (PDB 3SS5). C, ligand free depiction of GAC where the dashed line segments designate unresolved regions in the crystal structure (PDB 3SS3). D, effect of YIP motif disruption on the activity of recombinant GAC. The specific activity of all purified proteins at 50 nm was assayed with and without added inorganic phosphate (50 mm). E, YIP motif lid disruption results in a higher apparent affinity of inorganic phosphate for GAC(Y254F). Dose dependence of phosphate activation of 50 nm Y254F versus GACWT. The titrations and fits are representative of three independent experiments. F, initial velocity analysis showing the Vmax difference between Y254F versus GACWT. GAC was assayed at 300 nm with increasing substrate (glutamine) concentrations. Michaelis-Menten parameters for the best fits shown by the solid curves are: GAC(WT) Km = 5.6 mm, Vmax = 0.09 mm/min, and for GAC(Y254F) Km = 9.5 mm, Vmax = 0.30 mm/min. Results are representative of three independent trials.
Discussion
Members of the glutaminase family of mitochondrial enzymes are responsible for catalyzing the key first step in glutamine metabolism, specifically, the conversion of glutamine to glutamate with the generation of ammonia. This becomes an especially important reaction in a number of cancer cells undergoing Warburg glycolysis. It provides a pathway by which glutamine-derived carbon enters the TCA cycle as α-ketoglutarate, thereby compensating for the majority of pyruvate being converted to lactic acid rather than to citrate, which normally starts the cycle (5). Depending on the degree to which a given cancer cell requires glutamine-dependent anaplerosis (i.e. becomes “glutamine-addicted”), glutamine withdrawal, or small molecule inhibition of glutaminase activity, slows or halts the growth of cancer cells and inhibits tumor growth in mouse models (9, 12, 15, 27, 28). Cancer cells often exhibit high levels of GLS expression, which is especially the case for the GAC splice variant, as well as providing signals that ensure its maximal activation (29). Thus, the potential for targeting GAC as a clinical strategy makes it of great importance to understand the molecular and mechanistic aspects of its activation.
Previous studies have suggested that the transition of the enzyme from a dimeric to a tetrameric species was tightly correlated with enzyme activation (21). However, GAC was observed to undergo even higher order oligomerization under conditions where it was catalytically active, leading to the proposal that the formation of super-aggregates of the enzyme is a pre-requisite for full catalytic activity (18). If this were the case, there would be a number of questions that would need to be considered. For example, how is the generation of super-aggregates of GAC achieved in cancer cells and what are the consequences of forming such large structures to mitochondrial and cellular function? Therefore with these issues in mind, we set out to learn more about what is the minimal enzyme unit capable of full activity, and what might this suggest regarding the regulation of GAC activation.
An examination of the existing x-ray crystal structures of members of the glutaminase family, including that for human GAC (Fig. 1), suggested that the allosteric activator, inorganic phosphate, by stabilizing the tetrameric helical interface of GAC, might help to drive enzyme activation by inducing conformational changes within what we refer to as the “activation loop” (residues 320–327). Indeed, we show that the introduction of a charge-reversal at the helical interface (i.e. GAC(D391K)) effectively traps GAC as an inactive dimeric species. Importantly, the structure of GAC(D391K) shared a key feature with that for GAC bound to an allosteric inhibitor, BPTES, namely, a resolvable activation loop. This then suggested that the flexibility of this loop might represent a critical element for enzyme activation.
We, and others (21, 23), have found that mutation of the activation loop residue lysine 325, within a wild-type GAC background, resulted in a constitutively active enzyme. Introducing this same change into the activation loop of the GAC(D391K) mutant yielded constitutive enzymatic activity. This double mutant (GAC(K325A/D391K)), unlike GAC(D391K), is not stabilized in the dimeric state, but is capable of forming tetramers and higher aggregates through N-terminal dimer-dimer interactions.
By taking advantage of an earlier observation that substitutions at lysine 316 prevented GAC from forming super-aggregates (18), we made an important discovery. Specifically, when this same substitution was introduced into the GAC(K325A/D391K) background, it was possible to generate a constitutively active GAC triple mutant (GAC(K316Q/K325A/D391K)) that was trapped in a dimeric state. Perhaps most important, the ectopically expressed GAC(K316Q/K325A/D391K) triple mutant did not give rise to higher order oligomeric structures in cells, unlike other activated forms of GAC, nor was it cytotoxic, but localized normally to the mitochondria.
To examine the effects of enhanced GAC activity on cell growth, we compared the ability of wild-type GAC versus constitutively active GAC to augment the transformation of NIH-3T3 cells. In particular, we examined the abilities of wild-type GAC and the constitutively active GAC triple mutant to potentiate the focus forming activity of low concentrations of oncogenic Dbl. No significant difference was observed between these two forms of GAC in their effectiveness to synergize with Dbl in driving cellular transformation. This suggests that overexpressing wild-type GAC is sufficient to provide the enhanced glutaminase activity that magnifies the effects of oncogene-driven focus formation, such that the supportive role that glutamine metabolism appears to play can be accomplished equally well by both wild-type and mutant forms of GAC. Although more subtle effects (e.g. subcellular localization, complex formation) may yet be identified for glutaminase point mutations occurring in the cancer genome (curated in the CbioPortal), the simple up-regulation of intrinsic GLS activity does not appear to impact cell growth by itself.
Thus having established that the activation loop of GAC can induce an activated state, independent of the need for the enzyme to form higher order oligomers, or even simply tetramers, we wanted to better understand how this loop was able to communicate with the catalytic active site. An attractive candidate for making this connection was a peptide segment (residues 291–325), and indeed, mutagenesis analysis showed that changing a highly conserved glycine at position 320 to proline uncoupled the communication between the activation loop and the active site. By comparing the structures of GAC in the presence and absence of the product glutamate, we were led to examine a conserved tyrosine-isoleucine-proline (YIP; Fig. 5A) motif that forms a lid over the substrate binding pocket upon the binding of substrate (Fig. 5, B and C). Changing a highly conserved tyrosine to phenylalanine within this lid increased the basal activity of wild-type GAC (Fig. 5D). However, a particularly interesting finding was that the Y254F substitution resulted in an apparent increase in the binding affinity of the enzyme for inorganic phosphate, i.e. shifting the activating doses of inorganic phosphate to lower concentrations (Fig. 5E). The increase in glutaminase activity observed for the Y254F mutant appears to be a result of more rapid substrate (glutamine)/product (glutamate) exchange (i.e. due to the opening of the lid), as suggested by an initial velocity analysis where the Vmax of the Y254F mutant glutaminase was 5-fold higher without a change in Km (Fig. 5F).
When taken together, these different observations now lead to a plausible scheme, regarding how allosteric regulators like inorganic phosphate activate GAC (Fig. 6). In this model, the binding of inorganic phosphate is proposed to enhance interactions between residues within the tetrameric helical interface, thus stabilizing the formation of activated tetramers. Specifically, this would enable the activation loop to assume the proper orientation in order for it to communicate with the connecting peptide segment (containing glycine 320) and thereby optimally position the active site residues for catalysis. Upon the binding of glutamine, additional interactions between the substrate and the lid increase the residence time of glutamine as it awaits the proper orientation of the active site residues for catalysis. The binding of inorganic phosphate, and the resulting changes in the activation loop and connecting peptide segment, would then not only be important for ensuring that the proper orientation of the catalytic residues occurs, but would also weaken the association of the lid with the active center, so as to ensure optimal product release. Note that although inorganic phosphate would weaken the binding of the lid to the active center, in a reciprocal fashion, the interaction of the lid with the active site would weaken the binding of inorganic phosphate to the enzyme. Thus, the Y254F substitution, by weakening the interaction of the lid with the active site, would also give rise to an enhanced interaction between the enzyme and phosphate, resulting in the observed shifting of the dose-response curve for phosphate shown in Fig. 5E.
FIGURE 6.

Proposed mechanism of GAC activation illustrating critical points of up-regulation (green) and inhibition (red). 1) The dimeric GAC species has an activation loop orientation that does not support catalysis. 2) Tetrameric GAC has an activation loop that supports catalysis and an open active site lid that facilitates more rapid substrate binding and product release. 3) Close-up of a monomeric species within the tetramer: BPTES or the G320P substitution stabilizes the activation loop in an orientation that does not support catalysis, whereas the K325A mutant or the presence of inorganic phosphate results in the positioning of the activation loop required for catalysis. 4) Close-up of a monomeric species within the tetramer. Upon the binding of glutamine, the active site lid closes over the substrate. The Y254F substitution promotes catalysis by facilitating the opening of the active site lid allowing for accelerated substrate binding and release of product.
Our identification of a minimal enzymatic dimeric unit capable of full activity demonstrates that the formation of higher oligomers is not an absolute pre-requisite for full enzyme activation. These findings now also raise some interesting possibilities regarding how specific types of post-translational modifications within GAC might trigger enzyme activation. Such changes might include the modification of lysine residues through the addition of acetyl and succinyl moieties (30, 31). Future studies will be directed toward understanding how such modifications of GAC within cancer cells might help to mediate the communication between its activation loop and active center, and/or possibly enhance the release of its active site lid in a manner that significantly accelerates product release, thus providing multiple mechanisms by which the activation of this important metabolic enzyme might be achieved.
Experimental Procedures
Recombinant GAC Preparations
Human GAC (residues 73 to 598) was cloned into the pQE80 vector (Qiagen) containing an N-terminal histidine (His)-tag. The recombinant plasmid was transformed into the E. coli strain BL21(DE3) and then purified using Ni2+-affinity and gel filtration chromatography. The purified protein was snap frozen and stored in gel filtration buffer (5 mm Tris-HCl, pH 7.5, and 0.15 m NaCl) at −80 °C.
A mouse kidney type isoform 2 (GAC, NP_001106854.1) plasmid (residues 73–603 for GAC) was cloned into a pET28a vector containing an N-terminal His tag and thrombin cleavage site. The protein was expressed in E. coli and then purified using Co2+ affinity beads (Clontech), followed by His tag cleavage with human thrombin (Haemetologic Technologies) overnight at 4 °C, and further purified by anion exchange (GE Healthcare) and gel filtration chromatography. Purified GAC was snap frozen and stored in a high salt-containing buffer (20 mm Tris-HCl, pH 8.5, 500 mm NaCl, 1 mm NaN3) at −80 °C. The human wild-type GAC and the mouse GAC(D391K) mutant were used for crystallization and structural analyses. The mouse GAC was used for all in vitro assays, whereas the human GAC was used for cell transfections and immunocytochemistry. The mouse numbering is used throughout to minimize confusion.
Crystallization, X-ray Data Collection, and Structure Determinations
Crystals of human GAC (20 mg/ml) were grown using the hanging drop method at 18 °C by mixing 2 μl of the protein solution with 2 μl of the reservoir solution (12% PEG6000 (w/v), 1.0 m LiCl, 1% DMSO (v/v) and 0.1 m Tris-HCl buffer, pH 8.5). Crystals appeared after 24 h and reached a size of 100 × 100 × 200 μm3 after 7 days. Crystals were cryofrozen under high pressure before data collection (26).
For the GAC(D391K) mutant (5 mg/ml), the primary screening was performed on the Phoenix liquid handling system using the Qiagen ComPAS screening kit. Crystallization was performed in 50 mm Tris-HCl, pH 9.0, 9% PEG 10,000, and 100 mm NaCl. The sitting drop vapor diffusion technique was used (4 μl of 5 mg/ml of protein plus 4 μl of precipitant). Rod-shaped crystals appeared in about 3–4 days, and grew for ∼10 days.
All datasets were collected at the Cornell High Energy Synchrotron Source (CHESS, Ithaca, NY) on beamlines A1 and F1. The coordinates of the glutaminase domain of human GAC (PDB code 3CZD) were used as a search model for molecular replacement for human GAC, and those of chain A from mouse glutaminase (PDB 3SS5) were used for GAC(D391K). Data reduction was performed prior to phasing and refinement, which were carried out with the software packages HKL2000, CCP4, and COOT.
Glutaminase Activity Assays
Assays used to evaluate the activity of GAC mutants followed a two-step protocol as described previously (17). Typically, GAC was added at the indicated concentrations to 100 μl of a solution containing 20 mm glutamine, 65 mm Tris acetate (pH 8.6), and 0.2 mm EDTA, in either the presence or absence of the anionic activator K2HPO4, and incubated at room temperature for 10 min. The reaction was quenched by the addition of ice-cold hydrochloric acid to a final concentration of 0.3 m. An aliquot of this was added to a buffer (glutamate dehydrogenase assay buffer: 160 mm Tris-HCl (pH 9.4)) containing 0.35 mm adenosine diphosphate, 1.7 mm nicotinamide adenine dinucleotide (NAD), and 6.3 units/ml of glutamate dehydrogenase) in a UV-transparent Costar 96-well plate (Corning), and incubated at room temperature for 50 min. The absorbance at 340 nm was measured and converted to glutamate concentrations using the extinction coefficient for the conversion of NAD to NADH of 6220 m−1 cm−1.
Cell Culture and Immunocytochemistry
NIH-3T3 cells were transformed by oncogenic Dbl as previously described (24). The cells were transfected with full-length wild-type human GAC (reference sequence NM_001256310.1) or with the constitutively active GAC triple mutant (GAC(K316Q/K325A/D391K)) using Lipofectamine (Life Technologies). SKBR3 human breast cancer cells were obtained from ATTC and cultured according to supplier specifications. The breast cancer cells were transfected with full-length human GAC, C terminally tagged with the V5 epitope, using Lipofectamine. Immunocytochemistry was performed 48 h post-transfection by fixation (10 min) in 3.7% CH2O in PBS, followed by 5 min of permeabilization in PBS supplemented with 0.05% Triton X-100. Cells were rinsed 3 times in PBS and incubated with mouse anti-V5 (Thermo-Fisher) 1:100 in PBS and rabbit anti-DLST (Cell Signaling; D2281) at 1:50 for 3 h at room temperature, followed by PBS washing (3 times). Secondary fluorescently conjugated antibodies (Invitrogen) were used to visualize protein localization with a Zeiss Axioplot fluorescence microscope.
SEC-MALS
Purified GAC and GAC mutants were examined using multiangle light scattering (MALS) as previously described by Møller et al. (17). Briefly, 50-μl samples of 5 or 10 mg/ml of GAC were injected onto a WTC-030S5 size exclusion column (Wyatt Technology) coupled to a static 18-angle light scattering detector (DAWN HELEOS-II) and a refractive index detector (OptiLab T-rEX, Wyatt Technology) kept at 23 °C. The size exclusion column was equilibrated with 20 mm Tris-HCl (pH 8.5), and 200 mm NaCl, with or without 50 mm K2HPO4. The flow rate was normally kept at 1 ml/min for all MALS analyses. R.m.s. radius and mass distribution (polydispersity) were analyzed using the ASTRA software and monomeric BSA (Sigma) was used to normalize the light scattering signal.
Author Contributions
All authors participated in the conception and design of the experiments. Y. L., J. W. E., C. A. S., W. P. K., and S. R. performed the experiments and critiqued the manuscript. Q. H. and Y. L. performed crystallization screens and collected and analyzed the raw x-ray crystallography data. J. W. E., S. R., and R. A. C. wrote the manuscript.
Acknowledgments
We thank Cindy Westmiller for excellent secretarial assistance and David J. Schuller and MacCHESS (National Institutes of Health Grant P41 GM103485) for help with x-ray data analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM040654 and R01 CA163255. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GLS
- glutaminase isoform 1
- GAC
- glutaminase C
- SEC-MALS
- size exclusion chromatography-multiangle light scattering
- BPTES
- bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide
- r.m.s.
- root mean square deviation
- PDB
- Protein Data Bank.
References
- 1. DeBerardinis R. J., Lum J. J., Hatzivassiliou G., and Thompson C. B. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20 [DOI] [PubMed] [Google Scholar]
- 2. Cantor J. R., and Sabatini D. M. (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hensley C. T., Wasti A. T., and DeBerardinis R. J. (2013) Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 5. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wise D. R., and Thompson C. B. (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiang Y., Stine Z. E., Xia J., Lu Y., O'Connor R. S., Altman B. J., Hsieh A. L., Gouw A. M., Thomas A. G., Gao P., Sun L., Song L., Yan B., Slusher B. S., Zhuo J., et al. (2015) Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. Clin. Investig. 125, 2293–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., and Thompson C. B. (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J. B., Erickson J. W., Fuji R., Ramachandran S., Gao P., Dinavahi R., Wilson K. F., Ambrosio A. L., Dias S. M., Dang C. V., and Cerione R. A. (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Erickson J. W., and Cerione R. A. (2010) Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 1, 734–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levine A. J., and Puzio-Kuter A. M. (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344 [DOI] [PubMed] [Google Scholar]
- 12. Gao P., Tchernyshyov I., Chang T. C., Lee Y. S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., and Dang C. V. (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wise D. R., DeBerardinis R. J., Mancuso A., Sayed N., Zhang X. Y., Pfeiffer H. K., Nissim I., Daikhin E., Yudkoff M., McMahon S. B., and Thompson C. B. (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., et al. (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lukey M. J., Greene K. S., Erickson J. W., Wilson K. F., and Cerione R. A. (2016) The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 7, 11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Godfrey S., Kuhlenschmidt T., and Curthoys P. (1977) Correlation between activation and dimer formation of rat renal phosphate-dependent glutaminase. J. Biol. Chem. 252, 1927–1931 [PubMed] [Google Scholar]
- 17. Møller M., Nielsen S. S., Ramachandran S., Li Y., Tria G., Streicher W., Petoukhov M. V., Cerione R. A., Gillilan R. E., and Vestergaard B. (2013) Small angle x-ray scattering studies of mitochondrial glutaminase C reveal extended flexible regions, and link oligomeric state with enzyme activity. PLoS ONE 8, e74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferreira A. P., Cassago A., Gonçcalves, Kde A., Dias M. M., Adamoski D., Ascenção C. F., Honorato R. V., de Oliveira J. F., Ferreira I. M., Fornezari C., Bettini J., Oliveira P. S., Paes Leme A. F., et al. (2013) Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J. Biol. Chem. 288, 28009–28020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson M. M., McBryant S. J., Tsukamoto T., Rojas C., Ferraris D. V., Hamilton S. K., Hansen J. C., and Curthoys N. P. (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeLaBarre B., Gross S., Fang C., Gao Y., Jha A., Jiang F., Song J. J., Wei W., and Hurov J. B. (2011) Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770 [DOI] [PubMed] [Google Scholar]
- 21. Stalnecker C. A., Ulrich S. M., Li Y., Ramachandran S., McBrayer M. K., DeBerardinis R. J., Cerione R. A., and Erickson J. W. (2015) Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc. Natl. Acad. Sci. U.S.A. 112, 394–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thangavelu K., Pan C. Q., Karlberg T., Balaji G., Uttamchandani M., Suresh V., Schüler H., Low B. C., and Sivaraman J. (2012) Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McDonald C. J., Acheff E., Kennedy R., Taylor L., and Curthoys N. P. (2015) Effect of lysine mutations on the phosphate activation and BPTES inhibition of glutaminase. Neurochem. Int. 88, 10–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hart M. J., Eva A., Zangrilli D., Aaronson S. A., Evans T., Cerione R. A., and Zheng Y. (1994) Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J. Biol. Chem. 269, 62–65 [PubMed] [Google Scholar]
- 25. Cassago A., Ferreira A. P., Ferreira I. M., Fornezari C., Gomes E. R., Greene K. S., Pereira H. M., Garratt R. C., Dias S. M., and Ambrosio A. L. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. U.S.A. 109, 1092–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim C. U., Hao Q., and Gruner S. M. (2006) Solution of protein crystallographic structures by high-pressure cryocooling and noble-gas phasing. Acta Crystallogr. D Biol. Crystallogr. 62, 687–694 [DOI] [PubMed] [Google Scholar]
- 27. Lukey M. J., Wilson K. F., and Cerione R. A. (2013) Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med. Chem. 5, 1685–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gross M. I., Demo S. D., Dennison J. B., Chen L., Chernov-Rogan T., Goyal B., Janes J. R., Laidig G. J., Lewis E. R., Li J., Mackinnon A. L., Parlati F., Rodriguez M. L., Shwonek P. J., et al. (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 [DOI] [PubMed] [Google Scholar]
- 29. van den Heuvel A. P., Jing J., Wooster R. F., and Bachman K. E. (2012) Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 13, 1185–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong Y., and Guan K. L. (2012) Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hirschey M. D., and Zhao Y. (2015) Metabolic regulation by lysine malonylation, succinylation and glutarylation. Mol. Cell Proteomics 14, 2308–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]