

Abstract
Several mitochondrial tRNA mutations have been associated with maternally inherited diabetes and deafness. However, the pathophysiology of these tRNA mutations remains poorly understood. In this report, we identified the novel homoplasmic 14692A→G mutation in the mitochondrial tRNAGlu gene among three Han Chinese families with maternally inherited diabetes and deafness. The m.14692A→G mutation affected a highly conserved uridine at position 55 of the TΨC loop of tRNAGlu. The uridine is modified to pseudouridine (Ψ55), which plays an important role in the structure and function of this tRNA. Using lymphoblastoid cell lines derived from a Chinese family, we demonstrated that the m.14692A→G mutation caused loss of Ψ55 modification and increased angiogenin-mediated endonucleolytic cleavage in mutant tRNAGlu. The destabilization of base-pairing (18A-Ψ55) caused by the m.14692A→G mutation perturbed the conformation and stability of tRNAGlu. An approximately 65% decrease in the steady-state level of tRNAGlu was observed in mutant cells compared with control cells. A failure in tRNAGlu metabolism impaired mitochondrial translation, especially for polypeptides with a high proportion of glutamic acid codons such as ND1, ND6, and CO2 in mutant cells. An impairment of mitochondrial translation caused defective respiratory capacity, especially reducing the activities of complexes I and IV. Furthermore, marked decreases in the levels of mitochondrial ATP and membrane potential were observed in mutant cells. These mitochondrial dysfunctions caused an increasing production of reactive oxygen species in the mutant cells. Our findings may provide new insights into the pathophysiology of maternally inherited diabetes and deafness, which is primarily manifested by the deficient nucleotide modification of mitochondrial tRNAGlu.
Keywords: diabetes, hearing, mitochondrial disease, mtDNA, mutant, pathogenesis, posttranslational modification (PTM), tRNA
Introduction
Deafness is one of the major public health problems, affecting 360 million persons worldwide. Deafness can be grouped into non-syndromic deafness (where hearing loss is the only obvious medical problem) and syndromic deafness (where hearing loss is coupled with other medical problems, such as diabetes) (1). Among syndromic manifestation, maternally inherited diabetes and deafness (MIDD)3 is characterized by the onset of sensorineural hearing loss and diabetes in adulthood (2, 3). Mutation(s) in mtDNA is the most important cause of MIDD (4–7). In fact, the human mtDNA encodes 13 subunits of the oxidative phosphorylation system, two rRNAs, and 22 tRNAs required for mitochondrial protein synthesis (8). Among these tRNAs, eight tRNAs (such as tRNAGlu and tRNAA1a) reside at the cytosine-rich light (L) strand. The remaining tRNAs, including tRNALys and tRNAHis, are located at the guanine-rich heavy (H) strand (9, 10). These MIDD-associated mtDNA mutations are often present in heteroplasmy (a mixture of wild-type and mutated molecules) (4). Of these, large deletions or duplications in mtDNA have been associated with diabetes and deafness in some pedigrees (5, 11). The most prevalent tRNA point mutation associated with MIDD is the heteroplasmic m.3243A→G mutation in the tRNALeu(UUR) gene, accounting for over 1% of diabetic patients in some populations (6, 12–14). The other MIDD-associated mtDNA mutations were the tRNALeu(UUR) 3264T→C, tRNAGlu 14709T→C and tRNAGly 10003T→C mutations (15–17). These mutations may lead to the structural and functional consequences of tRNAs, including processing of the RNA precursor, aminoacylation, and nucleotide modification of tRNAs (15–20). In particular, the m.3243A→G mutation caused the loss of τm5U, τm5s2U, and taurine modifications in the mutant tRNALeu(UUR) (20–22). A failure in tRNALeu(UUR) metabolism impaired mitochondrial protein synthesis and subsequently reduced the activities of oxidative phosphorylation (18, 19, 23, 24). However, the pathophysiology of these mtDNA mutations remains poorly understood.
As part of a genetic screening program for deafness in a cohort of 2651 Han Chinese hearing-impaired subjects (25, 26), we identified the U-to-C transition at position 14692 (m.14692A→G) in the tRNAGlu gene in three genetically unrelated probands whose families exhibited the maternal transmission of deafness and diabetes. As shown in Fig. 1, the m.14692A→G mutation is localized at a highly conserved nucleotide (U55) of the T-loop of tRNAGlu (27). The uridine at position 55 (U55) of tRNAGlu is modified to pseudouridine (Ψ55), which plays an important role in the structure and function of this tRNA (27–28). Therefore, substitution of U with C at position 55 of tRNAGlu may cause loss of pseudouridine (Ψ55) and destabilize the A18-Ψ55 base pair between the T-loop and D-loop of tRNAGlu, thereby altering the tertiary structure and function of tRNAGlu. In particular, the mutation may affect the stability of this tRNA, thus impairing mitochondrial protein synthesis. It was also proposed that an impairment of mitochondrial translation caused by the tRNA mutation altered respiration and production of ATP and reactive oxygen species (ROS). To investigate the pathophysiology of this mutation, we generated lymphoblastoid cell lines derived from two affected matrilineal relatives carrying the m.14692A→G mutation and two control subjects lacking the mtDNA mutation but belonging to the same mtDNA haplogroup. First we examined whether the m.14692A→G mutation perturbs the Ψ55 modification of tRNAGlu by using CMCT modification/reverse transcription (29) and in vitro angiogenin cleavage assays (30). We then investigated whether the m.14692A→G mutation alters the conformation and stability of RNAGlu. This m.14692A→G mutation was further assessed for the effect on mitochondrial translation, respiration, production of ATP, mitochondrial membrane potential, and the generation of ROS through the use of mutant and control lymphoblastoid cell lines.
FIGURE 1.

Pseudouridine sequencing of mitochondrial tRNAGlu. A, schematic of pseudouridine sequencing shown in the cloverleaf structures of the human mitochondrial tRNAGlu (WT and mutant (MT). Solid lines represent the DIG-labeled oligonucleotide probe specific for tRNAGlu. Broken lines represent the potential stops of reverse transcription reaction caused by base modification, such as CMC-pseudouridine and m1A. B, 2 μg of mitochondrial RNA from control (C3 and C4) and mutant (III-6 and III-9) cells was incubated with CMCT for CMC modification of Ψ residues. Reverse transcription was carried out to identify the stops caused by CMC-pseudouridine. The tRNAGlu transcript was used as a control without base modification. Marker, DIG-labeled oligonucleotides of variable length.
Results
Identification of the tRNAGlu 14692A→G Mutation in a Large Cohort of Hearing-impaired Subjects
The m.14692A→G mutation in the tRNAGlu gene was identified in three genetically unrelated probands among 2651 Chinese hearing-impaired probands but absent in 574 Chinese control subjects. As shown in Fig. 1, the m.14692A→G mutation is localized at a highly conserved nucleotide (U55) of the T-loop of the tRNAGlu (31, 32). The uridine at this position (U55) of tRNAGlu is modified to pseudouridine (Ψ55), which forms a tertiary base pair with the A18 in the D-loop and stabilizes the L-shaped tRNA structure (32–34). Thus, it was anticipated that the U-to-C substitution at position 55 by the m.14692A→G mutation would perturb the Ψ55 modification and destabilize the base pairing (18A-Ψ55) of tRNAGlu. The sequence analysis of the entire mtDNA in these three probands exhibited the identical m.14692A→G mutation and distinct sets of polymorphisms belonging to the Eastern Asian haplogroups B5 and D4, respectively (supplemental Table S1) (35). However, there were no other functional significant variants in their mtDNAs. Further analysis showed that the m.14692A→G mutation was present in homoplasmy in all matrilineal relatives but not in other members of three Chinese families (supplemental Fig. S1).
Clinical Presentation of Three Chinese Families
All available members of three Han Chinese families carrying the m.14692A→G mutation, as shown in supplemental Table S2, underwent comprehensive evaluations of their medical histories and physical examination with the aim to identify any clinical abnormalities, genetic factors related to the deafness, and diabetes. The audiological examination was performed as detailed elsewhere (36). The diagnosis of diabetes was based on the criteria of the American Diabetes Association (37). Of 11 matrilineal relatives of pedigree WZD81, as shown in supplemental Fig. S2, six (two male and four female) individuals suffered from diabetes (three subjects with only diabetes and three subjects with both diabetes and hearing impairment). The average ages at onset of diabetes and deafness were 60 (from 42–72) and 27 years, respectively. In pedigree WZ82, only one matrilineal relative (WZD82-II-3) had diabetes at the age of 50, and one matrilineal relative (WZD82-III-1) exhibited severe hearing loss at the age of 26. Among seven matrilineal relatives of pedigree WZD83, two members (WZD83-III-2, and WZD83-IV-1) suffered from moderate hearing loss, whereas two subjects (WZD83-II-2 and III-3) exhibited both deafness and diabetes. The average ages at onset of diabetes and deafness were 50 (from 40–60) and 19 (from 16–22) years, respectively. Moreover, these matrilineal relatives showed no other clinical abnormalities, including cardiac failure, muscular diseases, visual failure, and neurological disorders. On the other hand, none of the other members in these families exhibited diabetic or other clinical abnormalities. Therefore, these familial histories indicate the maternal inheritance of diabetes and deafness.
Deficient Pseudouridinylation at Position 55 of Mitochondrial tRNAGlu
To determine whether the m.14692A→G mutation alters the pseudouridinylation of tRNAGlu, we subjected mitochondrial RNAs from mutant and control lymphoblastoid cell lines to CMCT/reverse transcription with a DIG-labeled oligonucleotide probe specific for tRNAGlu (Fig. 1). This approach involved carbodiimide (CMCT) adduct formation with U, G, and pseudouridine, followed by mild alkali to remove the adduct from U and G but not from N3-[N-cyclohexyl-N′-β-(4-methyl-morpholinium) ethylcarbodiimide]-Ψ (N3-CMC-Ψ) (29). This results in the attenuation of primer reverse transcription, causing a stop band one residue 3′ to the pseudouridine on the sequence gel. As shown in Fig. 1B, the Ψ55 modification was not detected in tRNAGlu derived from the mutant cell lines (III-6, III-9), whereas the Ψ55 modification was present in the tRNAGlu derived from the control cell lines (C3, C4). However, Ψ40, Ψ28, and m1A9 modifications were detected in tRNAGlu obtained from both control and mutant cell lines.
We further examined whether the m.14692A→G mutation perturbs the pseudouridinylation of tRNAGlu by analyzing the sensitivity of wild-type and mutant tRNAGlu to digestion with angiogenin (ANG), a tRNA-specific enzyme of the RNase A superfamily (38). In fact, the loss of certain tRNA modifications increases the angiogenin-medicated cleavage of tRNAs. For this purpose, the wild-type (Ψ55) and mutant (C55) tRNAGlu obtained from in vitro transcription as well as from control and mutant cell lines were digested by angiogenin and followed by Northern blotting analysis. As shown in Fig. 2, the mutant (C55) tRNAGlu transcript and mutant tRNAGlu obtained from the mutant cells (III-9) were more sensitive to angiogenin-mediated digestion than those in the wild-type (U55) tRNAGlu transcript and tRNAGlu obtained from the control cell line (C3), respectively.
FIGURE 2.

In vitro angiogenin cleavage assays. A, angiogenin digestion pattern of in vitro transcripts of wild-type (55U) and 14692A→G (55C) mutant tRNAGlu. 2 μg of purified tRNA transcripts was used for the ANG cleavage reaction at various lengths (from 0–45 min). Cleavage products of tRNA transcripts were electrophoresed through a denaturing polyacrylamide gel and stained with methylene blue. Broken squares indicate the differences between wild-type (U55) and mutant tRNAGlu (C55). B, angiogenin digestion pattern of mt-tRNAGlu purified from the control cell line (C3) and mutant cell line (III-9). 2 μg of human mitochondrial RNAs was used for the ANG cleavage reaction at various lengths (from 0–36 h). Cleavage products of mitochondrial tRNAs were resolved in 15% denaturating polyacrilamide gels with 8 m urea, electroblotted, and hybridized with a DIG-labeled oligonucleotide probe specific for tRNAGlu. Broken squares indicate the differences between RNAs from control (C3) and mutant (III-9) cells, respectively.
Altered Conformation and Stability of tRNAGlu
It was anticipated that the destabilization of base pairing (18A-Ψ55) by the m.14692A→G mutation leads to structural alterations of tRNAGlu. The stability of the transcripts of wild-type and mutant tRNAGlu (72 nt) as well as tRNAPhe (74 nt), tRNAAsp (71 nt), and tRNAThr (69 nt) were first examined by the measurement calculating the derivatives of the absorbance against a temperature curve. As shown in supplemental Fig. S3, the Tm values for wild-type (U55) and mutant (C55) tRNAGlu transcripts were 50 °C and 46 °C, respectively. This suggested that the global folding of mutant tRNAGlu was less stable at high temperature than that of wild-type tRNAGlu. These transcripts were then assessed for conformation change by PAGE analysis under denaturing and native conditions. As shown in Fig. 3, there was no migration difference between wild-type (U55) and mutant (C55) tRNAGlu transcripts under denaturing conditions, whereas the wild-type (U55) tRNAGlu transcript migrated slightly faster than the mutant (C55) tRNAGlu transcript under the native condition. To further test whether the m.14692A→G mutation affects the conformation of tRNAGlu, total mitochondrial RNA was electrophoresed through a 10% polyacrylamide gel (native condition) in Tris borate-EDTA buffer and then electroblotted onto a positively charged nylon membrane for hybridization analysis with oligodeoxynucleotide probes for tRNAGlu and tRNAHis, respectively. As shown in Fig. 3, the electrophoretic patterns showed that the tRNAGlu in two cell lines carrying the m.14692A→G mutation migrated much slower than those of control cell lines lacking this mutation. These data indicated that the m.14692A→G mutation changed the conformation of tRNAGlu.
FIGURE 3.

Analysis of the conformation and stability of tRNAGlu. A, cloverleaf structure of mutant human mitochondrial tRNAGlu. Broken lines represent the anticipated tertiary base pairings such as A18:U55. B, schematic of the tertiary structure of tRNAGlu derived from Suzuki et al. (27). The broken square represents the elbow region (D- and T-loops) of tRNA. C, assessment of conformation changes by PAGE analysis under denaturing and native conditions. The transcripts of wild-type and mutant tRNAsGlu, tRNAPhe (74 nt), tRNAAsp, and tRNAThr were electrophoresed through native or denaturing polyacrylamide gel stained with ethidium bromide. D, Northern blotting analysis of mitochondrial tRNA under native conditions. 2 μg of total mitochondrial RNA from various cell lines was electrophoresed through native polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for tRNAGlu and tRNAHis, respectively.
Marked Decrease in the Steady-state Levels of tRNAGlu
To further evaluate whether the m.14692A→G mutation impairs tRNA metabolism, we subjected mitochondrial RNAs from mutant and control lymphoblastoid cell lines to Northern blotting with probes for tRNAGlu, tRNAAla as representatives of the whole L-strand transcription unit and tRNAHis, tRNAIle, and tRNAThr derived from the H-strand transcription unit, as well as a nucleus-encoded mitochondrial 5S RNA (under denaturing conditions) (8, 9, 39). As shown in Fig. 4A, the amount of tRNAGlu in two mutant cell lines was markedly decreased compared with those in two control cell lines. For comparison, the average levels of each tRNA in control or mutant cell lines were normalized according to the level of the 5S rRNA. As shown in Fig. 4B, the steady-state levels of tRNAGlu in two mutant cell lines were 35.5% (p < 0.01) of the average values of two control cell lines lacking the mtDNA mutation. However, the average steady-state levels of tRNAHis, tRNAIle, tRNAThr, and tRNAAla in two mutant cell lines were comparable with those in control cell lines.
FIGURE 4.

Northern blotting analysis of mitochondrial tRNA under a denaturing condition. A, equal amounts (2 μg) of total mitochondrial RNA from various cell lines were electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with DIG-labeled oligonucleotide probes specific for tRNAGlu, tRNAAla, tRNAHis, tRNAIle, tRNAThr, and 5S rRNA. B, quantification of mitochondrial tRNA levels. Shown is the average relative tRNA content per cell normalized to the average content per cell of 5S rRNA in cells derived from two affected subjects (WZD81-III-6 and III-9) carrying the m.14692A→G mutation and two Chinese controls belonging to the same haplogroup (C3 and C4) lacking the mutation. The values for the latter are expressed as percentages of the average values for the control cell lines. The calculations were based on three independent determinations. The error bars indicate 2 standard errors of the means. p indicates the significance, according to a t test, of the differences between mutant and control cell lines. The horizontal dashed lines represent the average value for each group.
Mitochondrial Protein Synthesis Defect
To assess whether the m.14692A→G mutation affects mitochondrial translation, a Western blotting analysis was carried out to examine the steady-state levels of seven mtDNA-encoded respiratory complex subunits in mutant and control cells with GAPDH as a loading control. As shown in Fig. 5, the levels of CO2 (subunit II of cytochrome c oxidase); ND1, ND4, ND5, and ND6 (subunits 1, 4, 5, and 6 of NADH dehydrogenase); ATP6 (subunit 6 of the H+-ATPase); and CYTB (apocytochrome b) exhibited the variable reductions in two mutant cell lines, with an average of 28.8% (p = 0.033), relative to the mean value measured in the control cell lines. In the mutant cells, the average levels of ND1, ND6, and CO2 with a higher proportion of glutamic acid residues were much lower than those of ND4, ND5, ATP6, and CYTB with relatively less proportion of glutamic acid residues with respect to the average values of control cells. However, the levels of polypeptide synthesis in mutant cells relative to those in control cells showed no significant correlation with either the number of codons or the proportion of glutamic acid residues (supplemental Table S3).
FIGURE 5.

Western blotting analysis of mitochondrial proteins. A, 20 μg of total cellular proteins from various cell lines was electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with seven respiratory complex subunits in mutant and control cells with GAPDH as a loading control. B, quantification of mitochondrial protein levels. The levels of mitochondrial proteins in two mutant cell lines and two control cell lines were determined as described elsewhere (26). C, quantification of seven respiratory complex subunits. The levels of ND1, ND4, ND5, ND6, CO2, CYTB, and ATP6 in two mutant cell lines and two control cell lines were determined as described elsewhere (26). The graph details and symbols are explained in the legend for Fig. 4.
Reduced Activities of Complex I and IV
To investigate the effect of the m.14692A→G mutation on oxidative phosphorylation, we measured the activities of respiratory complexes by isolating mitochondria from two mutant and two control cell lines. Complex I (NADH ubiquinone oxidoreductase) activity was determined by following the oxidation of NADH with ubiquinone as the electron acceptor (40, 41, 42). Complex III (ubiquinone cytochrome c oxidoreductase) activity was measured as the reduction of cytochrome c (III) using d-ubiquinol-2 as the electron donor. The activity of complex IV (cytochrome c oxidase) was monitored by following the oxidation of cytochrome c (II). As shown in Fig. 6, the activity of complexes I, II, III, and IV in the mutant cells carrying the m.14692A→G mutation were 53.2%, 94%, 96.7%, and 75.4% of the mean value measured in two control cell lines, respectively.
FIGURE 6.

Enzymatic activities of respiratory chain complexes. The activities of respiratory complexes were investigated by enzymatic assay on complexes I, II, III, and IV in mitochondria isolated from various cell lines. The calculations were based on four independent determinations. The graph details and symbols are explained in the legend for Fig. 4.
Respiration Deficiency
To evaluate whether the m.14692A→G mutation alters cellular bioenergetics, we examined the oxygen consumption rates (OCRs) of two mutant cell lines carrying the m.14692A→G mutation and two control cell lines. As shown in Fig. 7, the basal OCR in mutant cell lines was ∼59.5% (p = 0.005) relative to the mean value measured in the control cell lines. To investigate which of the enzyme complexes of the respiratory chain was affected in the mutant cell lines, the OCR was measured after sequential addition of oligomycin (to inhibit ATP synthase), FCCP (to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the electron transfer chain), rotenone (to inhibit complex I), and antimycin A (to inhibit complex III). The difference between the basal OCR and the drug-insensitive OCR yields the amount of ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR. As shown in Fig. 7, the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR in mutant cell lines were∼60%, 56%, 64%, 67%, and 71.5% relative to the mean value measured in the control cell lines (p = 0.014, 0.058, 0.017, 0.024, and 0.194), respectively.
FIGURE 7.

Respiration assays. A, an analysis of O2 consumption in the various cell lines using different inhibitors. The OCRs were first measured on 2 × 104 cells of each cell line under basal condition and then sequentially added to oligomycin (1.5 μm), FCCP (0.5 μm), rotenone (1 μm), and antimycin A (1 μm) at the indicated times to determine different parameters of mitochondrial functions. B, the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and non-mitochondrial OCR in mutant and control cell lines. The non-mitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. The basal OCR was determined as the OCR before oligomycin minus OCR after rotenone/antimycin A. The ATP-lined OCR was determined as the OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. The maximal OCR was determined as the OCR after FCCP minus non-mitochondrial OCR. The reserve capacity was defined as the difference between maximal OCR after FCCP minus basal OCR. The average values of five determinations for each cell line are shown. The horizontal dashed lines represent the average value for each group. The graph details and symbols are explained in the legend for Fig. 4.
Reduced Production of Mitochondrial ATP
The capacity of oxidative phosphorylation in mutant and wild-type cells was examined by measuring the levels of cellular and mitochondrial ATP using a luciferin/luciferase assay. Populations of cells were incubated in media in the presence of glucose or 2-deoxy-d-glucose with pyruvate (26). The levels of ATP production in mutant cells in the presence of glucose (total cellular levels of ATP) were comparable with those measured in the control cell lines (Fig. 8A). By contrast, the levels of ATP production in mutant cell lines in the presence of pyruvate and 2-deoxy-d-glucose to inhibit glycolysis (mitochondrial levels of ATP) were only 61% (p < 0.001) relative to the mean value measured in the control cell lines (Fig. 8B).
FIGURE 8.
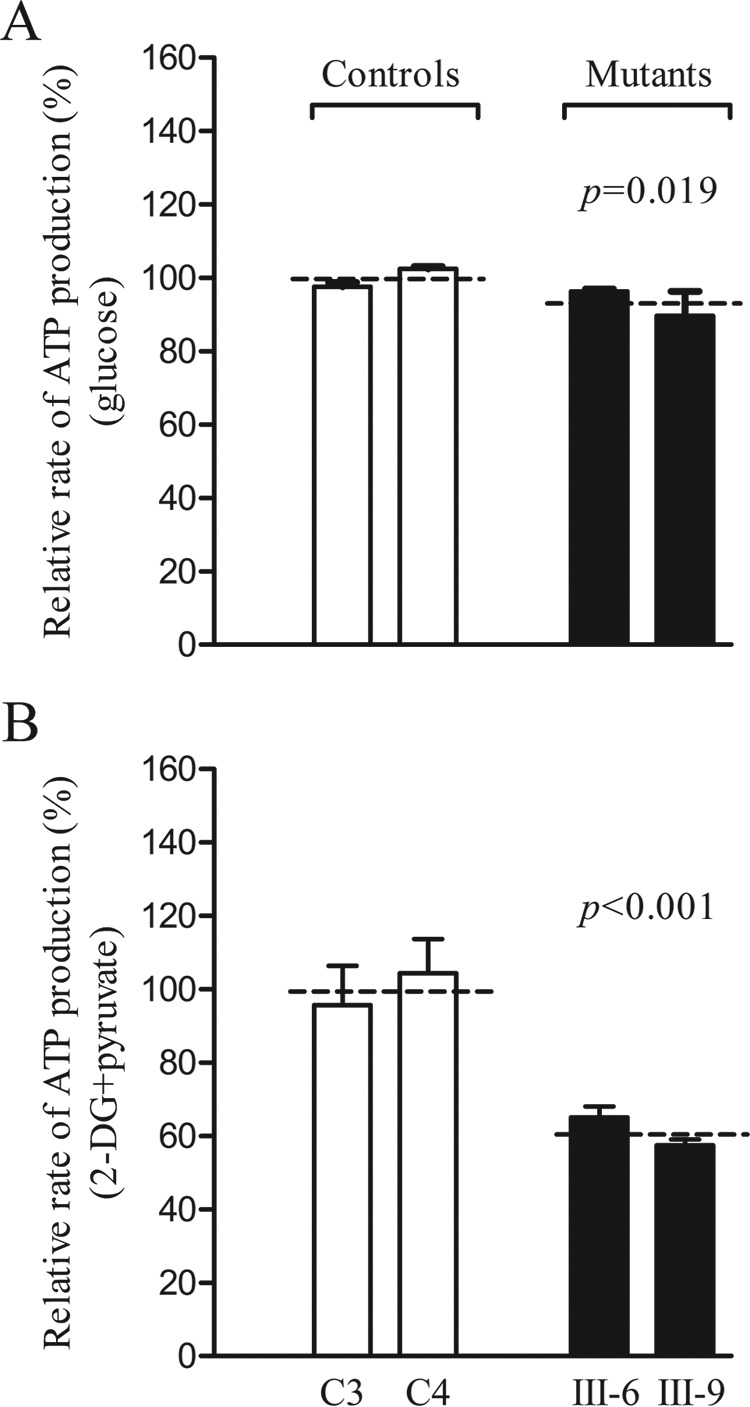
Measurement of cellular and mitochondrial ATP levels using a bioluminescence assay. Cells were incubated with 10 mm glucose or 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line and are shown. A, ATP level in total cells. B, ATP level in mitochondria. Four to five determinations were made for each cell line. The graph details and symbols are explained in the legend for Fig. 4.
Decrease of Mitochondrial Membrane Potential
The mitochondrial membrane potential (ΔΨm) is the central bioenergetic parameter that controls respiratory rate, ATP synthesis, and the generation of ROS and is itself controlled by electron transport and proton leaks. The levels of ΔΨm were measured in two mutant and two control cell lines using a fluorescence probe JC-10 assay system. The ratios of fluorescence intensity Ex/Em = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate theΔΨm level of each sample. The relative ratios of the FL590/FL530 geometric mean between mutant and control cell lines were calculated to represent the level of ΔΨm. As shown in Fig. 9, the levels of ΔΨm in the mutant cell lines carrying the m.14692A→G mutation ranged from 57 % to 73%, with an average of 65% (p < 0.001) of the mean value measured in the control cell lines. In contrast, the levels of ΔΨm in mutant cells in the presence of FCCP were comparable with those measured in the control cell lines.
FIGURE 9.
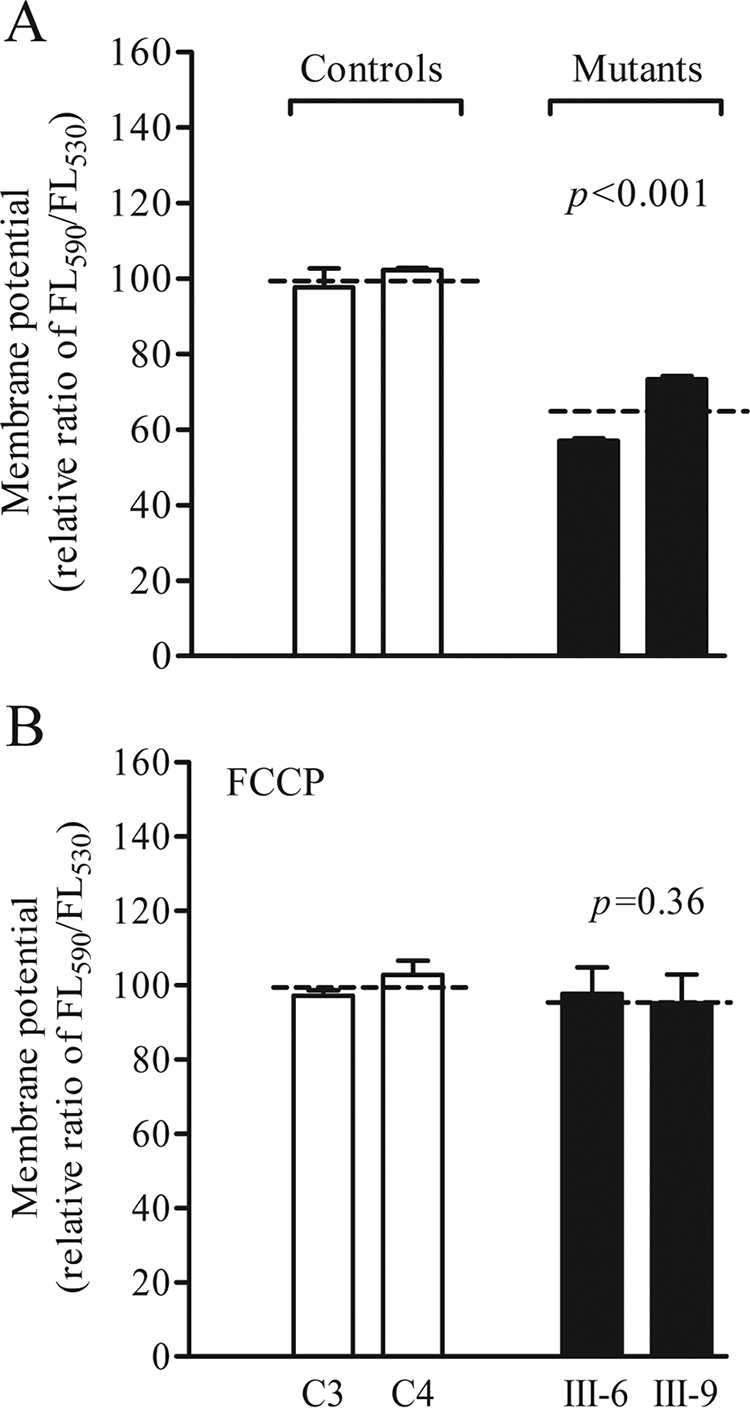
Mitochondrial membrane potential analysis. The mitochondrial membrane potential was measured in two mutant and two control cell lines using a fluorescence probe JC-10 assay system. The ratio of fluorescence intensities Ex/Em = 490/590 and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. The relative ratios of FL590/FL530 geometric mean between mutant and control cell lines were calculated to reflect the level of ΔΨm. Shown is the relative ratio of JC-10 fluorescence intensities at Ex/Em = 490/530 and 490/590 nm in the absence (A) and presence (B) of 10 μm FCCP. The average of four to five determinations for each cell line is shown. The graph details and symbols are explained in the legend for Fig. 4.
The Increase in ROS Production
The levels of ROS generation in viable cells, derived from two matrilineal relatives carrying the m.14692A→G mutation and two control individuals lacking the mutation, were measured with flow cytometry under normal and H2O2 stimulation (42, 43). Geometric mean intensity was recorded to measure the rate of ROS of each sample. The ratio of geometric mean intensity between unstimulated and stimulated with H2O2 in each cell line was calculated to delineate the reaction upon increasing levels of ROS under oxidative stress. As shown in Fig. 10, the levels of ROS generation in mutant cells ranged from 124.7% to 140.9%, with an average of 132.9% (p < 0.001) of the mean value measured in the control cells.
FIGURE 10.
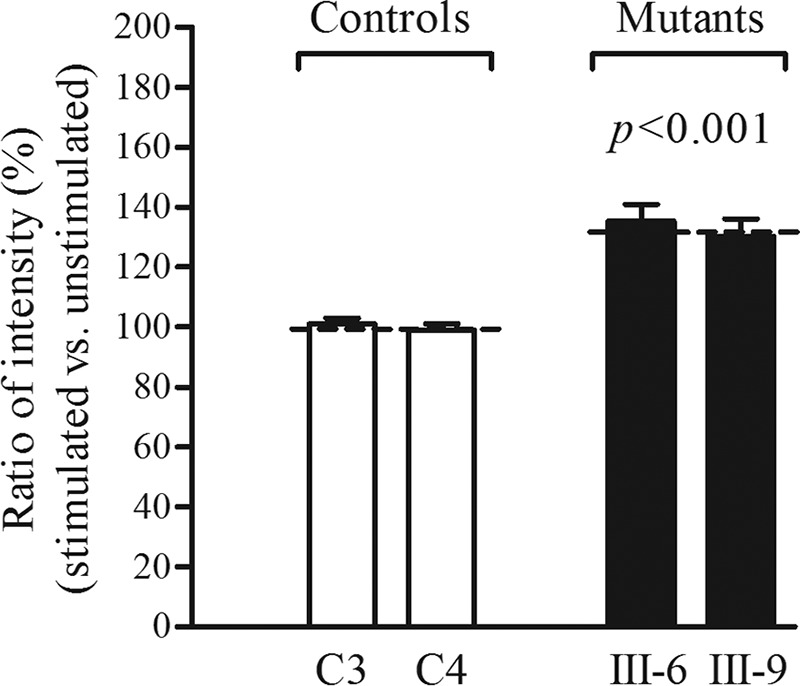
Measurement of ROS. The rates of production in ROS from two affected matrilineal relatives and two control individuals were analyzed by a BD-LSR II flow cytometer system with or without H2O2 stimulation. The relative ratio of intensity (stimulated versus unstimulated with H2O2) was calculated. The average of three determinations for each cell line is shown. The graph details and symbols are explained in the legend for Fig. 4.
Discussion
In this study, we investigated the pathogenic mechanism of the deafness and diabetes-associated m.14692A→G mutation in the mitochondrial tRNAGlu gene. The occurrence of the m.14692A→G mutation in three genetically unrelated Chinese families affected by deafness and diabetes strongly indicates that this mutation is involved in the pathogenesis of these disorders. The m.14692A→G mutation affected a highly conserved nucleotide (U55) of the T-loop that is important for the interaction of the tRNA with several components of translation machinery (27, 31). The uridine at this position (U55) of tRNAGlu is modified to pseudouridine (Ψ55), catalyzed by TRUB2, whereas the adenine at this position (A55) of tRNALys is not modified (31, 44, 45). The Ψ55 forms a tertiary base pair with the A18 in the D-loop and stabilizes the L-shaped tRNA structure (33, 34). In this study, we demonstrated that the m.14692A→G mutation caused the loss of Ψ55 in mutant tRNAGlu. The U55-to-C55 substitution may destabilize the base pairing (18A-Ψ55) of tRNAGlu, cause improper folding, and then alter its structure and function, as in the case of the m.12315G→A mutation in the tRNALeu(CUN) (46). In particular, the perturbed tertiary structure then makes the mutant tRNAGlu more unstable and subject to degradation. Here the decreased melting temperature and altered conformation were observed in mutant tRNAGlu transcript (C55) compared with the wild-type transcript (U55). Alternatively, the altered tertiary structure may affect the aminoacylated efficiency of tRNAGlu (27, 31). Notably, mutant cell lines carrying the m.14692A→G mutation exhibited ∼65% decrease in the steady-state level of tRNAGlu in contrast with a 40% reduction in tRNALys in cells carrying the m.8344A→G mutation at position 55 in tRNALys (47, 48). However, the reduced level of tRNAGlu in mutant cells carrying the m.14692A→G mutation was above the proposed threshold level (>70%) to produce a clinical phenotype associated with a mitochondrial tRNA mutation (9, 26, 49, 50). This suggests that the m.14692A→G mutation alone is insufficient to produce a clinical phenotype, as in the case of deafness-associated 12S rRNA 1555A→G and m.1494C→T mutations (36, 51).
The aberrant tRNA metabolism, including shortage of tRNAGlu, contributed to the impairment of mitochondrial protein synthesis. Alternatively, the mutant tRNAGlu may not interact correctly with the translational machinery, thereby altering mitochondrial protein synthesis (47, 48). In this study, reduced levels of mitochondrial proteins (an average decrease of ∼29%) were comparable with the reduced rate of mitochondrial translation observed in lymphoblastoid cell lines carrying the m.4263A→G and m.4435A→G mutations (50, 52). Variable decreases in the levels of seven mtDNA-encoded polypeptides were observed in the mutant cell lines. As shown in supplemental Table S3, mutant cell lines carrying the m.14692A→G mutation exhibited marked reductions (34%-66%) in the levels of three polypeptides (ND1, ND6, and CO2) harboring 3.5%-5.7% of glutamic acid codons, relative mild reductions (20%-30%) in the levels of ATP6, ND4, and ND5 carrying 1.3%-2% of glutamic acid codons, but a mildly increased level (21%) in CYTB with only 1.1% of glutamic acid codons. In contrast to what was shown previously in cells carrying the tRNALys 8344A→G mutation (48), polypeptide levels in mutant cell lines, relative to those in control cell lines, did not significantly correlate with the number of glutamic acid codons. Thus, the impaired synthesis of ND1, ND4, ND5, and ND6, the subunits of complex I and CO2, and the subunit of complex IV was responsible for the reduced activities of complex I and complex IV but not those of complex III in these mutant cell lines carrying the m.14692A→G mutation. Furthermore, the impairment of mitochondrial translation led to reduced rates in the basal OCR or ATP-linked OCR, reserve capacity, and maximal OCR in the mutant cell lines. This observation is clearly consistent with the critical role of tRNAGlu metabolic failure in producing their respiratory phenotypes, as in the cases of cell lines carrying the deafness-associated tRNASer(UCN) 7445A→G, 7511T→C, and tRNAHis 12201T→C mutations (9, 26, 49).
The respiratory deficiency caused by the m.14692A→G mutation may result in the uncoupling of the oxidative pathway for ATP synthesis, oxidative stress, and subsequent failure of the cellular energetic process (53). In this study, a 39% drop in mitochondrial ATP production in cell lines carrying the m.14692A→G mutation, which may be caused by the defective activity of complex I and IV, was much lower than those in cell lines carrying the tRNALys 8344A→G and tRNALeu(UUR) 3243A→G mutations (54, 55). Furthermore, the deficient activities of respiratory chain complexes caused by tRNA mutations often alter mitochondrial membrane potentials, which is a key indicator of cellular viability (26, 56). Indeed, mitochondrial membrane potentials reflect the pumping of hydrogen ions across the inner membrane during the process of electron transport and oxidative phosphorylation (57, 58). In this study, a 35% reduction in mitochondrial membrane potential in lymphoblastoid cell lines carrying the m.14692A→G mutation was much lower than those in cell lines carrying the m.12201T→C mutation (26). The abnormal oxidative phosphorylation and mitochondrial membrane potential resulted in increased production of reactive oxygen species and the subsequent failure of cellular energetic processes in mutant cells carrying the m.14692A→G mutation. In particular, the overproduction of ROS can establish a vicious cycle of oxidative stress in the mitochondria, thereby damaging mitochondrial and cellular proteins, lipids, and nucleic acids and increasing apoptotic signaling (59–60). Hair cells and neurons in the cochlea or β cells in the pancreas may be preferentially affected because they are somehow exquisitely sensitive to subtle imbalances in the cellular redox state or increased level of free radicals (61–64). This would lead to the dysfunction or death of hair cells in the cochlea or/and β cells in the pancreas carrying the m.14692A→G mutation.
The variable phenotypes of deafness and diabetes among the maternal lineage indicated the involvement of modifier factors in the phenotypic manifestation. Of 23 matrilineal relatives in three Chinese families, three members exhibited only deafness, four individuals only exhibited diabetes, five subjects suffered from both deafness and diabetes, and 11 other matrilineal relatives did not have any clinical phenotype. In contrast to the low penetrance of hearing loss in families carrying the m.7445A→G and m.1555A→G mutations (65, 66), the average penetrances of deafness in these Chinese families were relatively high (30.4%). The striking feature in these families was that matrilineal relatives developed hearing loss at the average age of 22 years but not congenital hearing loss. However, some of the matrilineal relatives in certain families carrying the m.1555A→G mutation exhibited profound congenital hearing loss (66, 67). Furthermore, the average age at onset of diabetes in matrilineal relatives of three families carrying the m.14692A→G mutation was 57 years, whereas the average ages at onset of diabetes in subjects carrying the heteroplasmic tRNALeu(UUR) 3243A→G and homoplasmic tRNAGly 10003T→C mutations were 37 and 50 years, respectively (13, 14, 17). In addition to diabetes and hearing loss, some subjects harboring the m.3243A→G mutation developed other symptoms, including neurological disorders, cardiac failure, and renal failure (13, 68). This discrepancy of clinical features between the m.14692A→G and m.3243A→G mutations may be attributed to the nature of the mutations. In fact, the m.14692A→G mutation only caused a relatively mild decrease (27.8%) in the levels of mitochondrial translation. By contrast, the heteroplasmic tRNALeu(UUR) 3243A→G and tRNALys 8344A→G mutations were severe mtDNA mutations causing profound mitochondrial defects (19, 48). In this regard, the heteroplasmic levels of these mtDNA mutations in tissues such as the endocrine pancreas or cochlea determined the phenotypic manifestations (69). In this investigation, the homoplasmy, mild mitochondrial dysfunction, late onset, and incomplete penetrance of diabetes and deafness observed in these Chinese families suggested that the m.14692A→G mutation is an inherited risk factor for the development of deafness and diabetes. Thus, the other genetic, epigenetic, and environmental modifier factors may be involved in the phenotypic manifestation of m.14692A→G mutation in these Chinese pedigrees (13, 60, 68, 70). Alternatively, tissue-specific differences in tRNA metabolism may also contribute to the development of deafness and diabetes (71, 72).
In summary, our findings convincingly demonstrate the pathogenic mechanism leading to impaired oxidative phosphorylation in cells carrying the deafness- and diabetes-associated m.14692A→G mutation in the tRNAGlu gene. The m.14692A→G mutation altered the structure and function of tRNAGlu. This included the loss of Ψ55 modification, improper folding of tRNAGlu, and a decrease of the steady-state level in tRNAGlu. A failure in tRNA metabolism impaired mitochondrial translation and respiration. Therefore, the respiratory deficiency impaired mitochondrial ATP production, membrane potentials, and production of oxidative reactive species. Therefore, mitochondrial dysfunction caused by the m.14692A→G mutation manifests as MIDD. However, the tissue specificity of this pathogenic mtDNA mutation is likely due to the involvement of nuclear modifier genes or tissue-specific differences in tRNA metabolism. Thus, our findings may provide new insights into the pathophysiology of MIDD, which is manifested by the deficient modification of mitochondrial tRNAGlu.
Experimental Procedures
Subjects
A total of 2651 Chinese hearing-impaired probands were recruited from the Otology Clinics, Wenzhou Medical University, China, as detailed previously (25, 26). This study was in compliance with the Declaration of Helsinki. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members under protocols approved by the Ethics Committees of Zhejiang University and Wenzhou Medical University. Audiological and neurological examinations of hearing impairment were defined as detailed previously (36). Diagnosis of diabetes was based on the criteria of the American Diabetes Association (37). All available members of three pedigrees carrying the m.14692A→G mutation were evaluated at length to identify both personal and family medical histories of deafness, diabetes, and other clinical abnormalities. The 574 control subjects were obtained from a panel of unaffected subjects of Han Chinese ancestry from the same region.
Mutational Analysis of Mitochondrial DNA
Genomic DNA was isolated from whole blood of 2651 probands and 574 Chinese control subjects by using the QIAamp DNA Blood Mini Kit (Qiagen, 51104). Subject DNA fragments spanning the mitochondrial tRNAGlu gene were amplified by PCR using oligodeoxynucleotides corresponding to mtDNA at positions 14448–14747 (8). Each fragment was purified and subsequently analyzed by direct sequencing. These sequence results were compared with the updated consensus Cambridge sequence (GenBank accession number NC_012920) (8). The entire mitochondrial genomes of three probands (WZD81-III-9, WZD82-III-1, and WZD83-III-2) carrying the m.14693A→G mutation and two control subjects (C3 and C4) were PCR-amplified in 24 overlapping fragments using sets of the light (L) strand and the heavy (H) strand oligonucleotide primers, as described previously (73). These sequence results were compared with the updated consensus Cambridge sequence as described above. To quantify the m.14692A→G mutation, the PCR segment (300 bp) was amplified using genomic DNA as the template and oligodeoxynucleotides corresponding to mtDNA at positions 14448–14747. Equal amounts of various digested samples were then analyzed by electrophoresis through 3% agarose gel. After ethidium bromide staining, an Image-Quant program was used to determine the proportions of digested and undigested PCR product to determine whether the m.14692A→G mutation is in homoplasmy in these subjects.
Cell Lines and Culture Conditions
Lymphoblastoid cell lines were immortalized by transformation with the Epstein-Barr virus as described elsewhere (74). Cell lines derived from two affected subjects (WZD81-III-6 andWZD81-III-9) carrying the m.14692A→G mutation and two Chinese control subjects (C3 and C4) lacking the mutation were grown in RPMI 1640 medium supplemented with 10% FBS.
Detection of Pseudouridine Residues in tRNAGlu Using CMCT Modification/Reverse Transcription
Total mitochondrial RNA was obtained from mitochondria isolated from lymphoblastoid cell lines (∼1.0 × 108 cells) (75). 2 μg of mitochondrial RNA was incubated with 160 mm CMCT for 20 min at 37 °C to allow for CMCT modification of Ψ residues. The reaction contained 7 m urea, 4 mm EDTA, and 50 mm Bicine (pH 8.5). Then the modified RNAs were precipitated by adding 2 μl of pellet paint co-precipitant, 50 μl of 3 m sodium acetate (pH 5.5), and a triple volume of ethanol and incubated for at least 2 h at −20 °C before centrifuging at 12,000 rpm for 30 min. Then we dissolved the RNA pellets in 1 m sodium carbonate (pH 10.4), incubated for 4 h at 37 °C, and precipitated the RNA again as described above. The Primescript II First Strand cDNA Synthesis Kit (TAKARA) was used for reverse transcription with a digoxigenin (DIG)-labeled oligodeoxynucleotide (5′-TGGTATTCTCGCACG-3′) probe specific for tRNAGlu. RNase A was added to the extension reaction to remove the mitochondrial RNA. Then the DNA was precipitated by ethanol at −20 °C overnight after phenol extraction. 2 μg of DNA samples was applied onto 15% polyacrylamide, 7 m urea electrophoresis gel and electroblotted onto a positively charged nylon membrane. The quantification of density in each band is described elsewhere (29).
In Vitro Angiogenin Cleavage Assay
In vitro transcriptions of human mitochondrial tRNAGlu (wild-type and mutant) were performed as described previously (76). 2 μg of purified tRNA transcripts or human mitochondrial RNAs were used for the cleavage reaction with 2.5 μg/ml recombinant angiogenin in the buffer containing 30 mm HEPES (pH 7.4), 30 mm NaCl, and 0.01% bovine serum albumin. Mixtures were incubated at 37 °C for the indicated times and quenched by adding 5 μl of gel loading buffer. Cleavage products of tRNA transcripts were electrophoresed through a denaturing polyacrylamide gel and stained with methylene blue. Cleavage products of human mitochondrial RNAs were resolved in 15% denaturating polyacrilamide gels with 8 m urea, electroblotted, and hybridized with a DIG-labeled oligonucleotide probe specific for the tRNAGlu (30).
Mitochondrial tRNA Analysis
The tRNA concentrations from in vitro transcription were measured by UV absorbance at 260 nm, and the extinction coefficient was calculated according to the sequence of each tRNA (77). 1 μg of each tRNA transcript was electrophoresed through a 10% polyacrylamide gel (native gel) or with 8 m urea (denaturing gel). The UV melting studies for tRNA transcripts were performed as described elsewhere (46).
For the tRNA Northern blotting analysis, 2 μg of total mitochondrial RNA was electrophoresed through a 10% polyacrylamide/8 m urea gel in Tris borate-EDTA buffer after heating the sample at 65 °C for 10 min and then electroblotted onto a positively charged nylon membrane for the hybridization analysis with oligodeoxynucleotide probes. Oligodeoxynucleosides used for DIG-labeled probes of tRNAGlu, tRNAHis, tRNAIle, tRNAAla, tRNAThr, and 5S rRNA were described as elsewhere (26, 42, 78–80). The hybridization and the quantification of density in each band was made as detailed previously (42, 79).
Western Blotting Analysis
Western blotting analysis was performed as detailed previously (26, 42). The antibodies used for this investigation were from Abcam (anti GAPDH (ab72655), ND1 (ab74257), ND5 (ab92624), ATP6 (ab101908), and CO2 (ab110258)), Santa Cruz Biotechnology (ND4 (sc-20499-R) and ND6 (sc-20667)), and Proteintech (CYTB (55090-1-AP)). Peroxidase Affini Pure goat anti-mouse IgG and goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) were used as secondary antibodies, and protein signals were detected using the ECL system (CWBIO). Quantification of density in each band was performed as detailed previously (26, 42).
Assays of Activities of Respiratory Complexes
The enzymatic activities of complexes I, II, III, and IV were assayed as detailed elsewhere (40–42).
Measurements of Oxygen Consumption
The rates of OCR in lymphoblastoid cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer as detailed previously (26, 42, 81).
ATP Measurements
The CellTiter-Glo® luminescent cell viability assay kit (Promega) was used for the measurement of cellular and mitochondrial ATP levels according to the modified instructions of the manufacturer (26, 42).
Assessment of Mitochondrial Membrane Potential
Mitochondrial membrane potential was assessed with the JC-10 Assay Kit-Microplate (Abcam) following the general recommendations of the manufacturer, with some modifications, as detailed elsewhere (26, 58).
Measurement of ROS Production
ROS measurements were performed following procedures detailed previously (26, 42, 43, 58).
Author Contributions
M. X. G. designed the experiments and wrote the main manuscript text. M. W., H. L., M. Z., and X. L. performed biochemical assays. J. Z., L. W., and H. W. performed the mutational screening of mitochondrial tRNA genes. B. C. and W. F. collected clinical data and performed the evaluation. X. Z., G. E., and P. J. performed data analysis. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
We thank the patients and their family members for participation. We also thank Dr. Tsutomu Suzuki from the University of Tokyo for valuable suggestions.
This work was supported by National Basic Research Priorities Program of China Grant 2014CB541700 and Chinese National Natural Science Foundation Grants 81330024 (to M. X. G.) and 81500611 (to M. W.), and Fundamental Research Funds for the Central Universities Grant 2016QNA7022 (to M. W.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figures S1–S3 and Tables S1–S3.
- MIDD
- maternally inherited diabetes and deafness
- ROS
- reactive oxygen species
- ANG
- angiogenin
- nt
- nucleotide(s)
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- Ex/Em
- excitation/emission
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- DIG
- digoxigenin
- CMCT
- 1-cyclohexyl-3-(2-(4-morpholinyl)ethyl) carbodiimide tosylate
- CYTB
- apocytochrome b.
References
- 1. Morton C. C. (2002) Genetics, genomics and gene discovery in the auditory system. Hum. Mol. Genet. 11, 1229–1240 [DOI] [PubMed] [Google Scholar]
- 2. Thomas F., Balkau B., Vauzelle-Kervroedan F., and Papoz L. (1994) Maternal effect and familial aggregation in NIDDM: the CODIAB study: CODIAB-INSERM-ZENECA Study Group. Diabetes 43, 63–67 [DOI] [PubMed] [Google Scholar]
- 3. Maassen J. A., 'T Hart L. M., Van Essen E., Heine R. J., Nijpels G., Jahangir Tafrechi R. S., Raap A. K., Janssen G. M., and Lemkes H. H. (2004) Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes 53, S103–S109 [DOI] [PubMed] [Google Scholar]
- 4. Guillausseau P.-J., Massin P., Dubois-LaForgue D., Timsit J., Virally M., Gin H., Bertin E., Blickle J.-F., Bouhanick B., Cahen J., Caillat-Zucman S., Charpentier G., Chedin P., Derrien C., Ducluzeau P. H., et al. (2001) Maternally inherited diabetes and deafness: a multicenter study. Ann. Intern. Med. 134, 721–728 [DOI] [PubMed] [Google Scholar]
- 5. Ballinger S. W., Shoffner J. M., Hedaya E. V., Trounce I., Polak M. A., Koontz D. A., and Wallace D. C. (1992) Maternally transmitted diabetes and deafness associated with a 10.4 kb mitochondrial DNA deletion. Nat. Genet. 1, 11–15 [DOI] [PubMed] [Google Scholar]
- 6. van den Ouweland J. M., Lemkes H. H., Ruitenbeek W., Sandkuijl L. A., de Vijlder M. F., Struyvenberg P. A., van de Kamp J. J., and Maassen J. A. (1992) Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1, 368–371 [DOI] [PubMed] [Google Scholar]
- 7. Ruiz-Pesini E., Lott M. T., Procaccio V., Poole J. C., Brandon M. C., Mishmar D., Yi C., Kreuziger J., Baldi P., and Wallace D. C. (2007) An enhanced mitomap with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andrews R. M., Kubacka I., Chinnery P. F., Lightowlers R. N., Turnbull D. M., and Howell N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147. [DOI] [PubMed] [Google Scholar]
- 9. Guan M. X., Enriquez J. A., Fischel-Ghodsian N., Puranam R. S., Lin C. P., Maw M. A., and Attardi G. (1998) The deafness-associated mtDNA 7445 mutation, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase ND6 subunit gene expression. Mol. Cell Biol. 18, 5868–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng J., Ji Y., and Guan M. X. (2012) Mitochondrial tRNA mutations associated with deafness. Mitochondrion 12, 406–413 [DOI] [PubMed] [Google Scholar]
- 11. Negrier M.-L. M., Coquet M., Moretto B. T., Lacut J.-Y., Dupon M., Bloch B., Lestienne P., and Vital C. (1998) Partial triplication of mtDNA in maternally transmitted diabetes mellitus and deafness. Am. J. Hum. Genet. 63, 1227–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Odawara M., Sasaki K., and Yamashita K. (1995) Prevalence and clinical characterization of Japanesediabetesmellitus with an A-to-G mutation at nucleotide 3243 of the mitochondrial tRNALeu(UUR) gene. J. Clin. Endocrinol. Metab. 80, 1290–1294 [DOI] [PubMed] [Google Scholar]
- 13. Murphy R., Turnbull D. M., Walker M., and Hattersley A. T. (2008) Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. 25, 383–399 [DOI] [PubMed] [Google Scholar]
- 14. Lu J., Wang D., Li R., Li W., Ji J., Zhao J., Ye W., Yang L., Qian Y., Zhu Y., and Guan M. X. (2006) Maternally transmitted diabetes mellitus associated with the mitochondrial tRNALeu(UUR) A3243G mutation in a four-generation Han Chinese family. Biochem. Biophys. Res. Commun. 348, 115–119 [DOI] [PubMed] [Google Scholar]
- 15. Mezghani N., Mkaouar-Rebai E., Mnif M., Charfi N., Rekik N., Youssef S., Abid M., and Fakhfakh F. (2010) The heteroplasmic m.14709T>C mutation in the tRNAGlu gene in two Tunisian families with mitochondrial diabetes. J. Diabetes Complications 24, 270–277 [DOI] [PubMed] [Google Scholar]
- 16. Suzuki Y., Suzuki S., Hinokio Y., Chiba M., Atsumi Y., Hosokawa K., Shimada A., Asahina T., and Matsuoka K. (1997) Diabetes associated with a novel 3264 mitochondrial tRNALeu(UUR) mutation. Diabetes Care 20, 1138–1140 [DOI] [PubMed] [Google Scholar]
- 17. Liu H., Li R., Li W., Wang M., Ji J., Zheng J., Mao Z., Mo J. Q., Jiang P., Lu J., and Guan M. X. (2015) Maternally Inherited diabetes caused by a homoplasmic T10003C mutation in the mitochondrial tRNAGly gene. Mitochondrion 21, 49–57 [DOI] [PubMed] [Google Scholar]
- 18. Li R., and Guan M. X. (2010) Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell Biol. 30, 2147–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chomyn A., Enriquez J. A., Micol V., Fernandez-Silva P., and Attardi G. (2000) The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem. 275, 19198–19209 [DOI] [PubMed] [Google Scholar]
- 20. Kirino Y., Yasukawa T., Ohta S., Akira S., Ishihara K., Watanabe K., and Suzuki T. (2004) Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 101, 15070–15075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yasukawa T., Suzuki T., Ueda T., Ohta S., and Watanabe K. (2000) Modification defect at anticodon wobble nucleotide of mitochondrial tRNAsLeu(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J. Biol. Chem. 275, 4251–4257 [DOI] [PubMed] [Google Scholar]
- 22. Kirino Y., Goto Y., Campos Y., Arenas J., and Suzuki T. (2005) Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 102, 7127–7132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Picard M., Zhang J., Hancock S., Derbeneva O., Golhar R., Golik P., O'Hearn S., Levy S., Potluri P., Lvova M., Davila A., Lin C. S., Perin J. C., Rappaport E. F., Hakonarson H., et al. (2014) Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. U.S.A. 111, E4033–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Andrade P. B., Rubi B., Frigerio F., van den Ouweland J. M., Maassen J. A., and Maechler P. (2006) Diabetes-associated mitochondrial DNA mutation A3243G impairs cellular metabolic pathways necessary for beta cell function. Diabetologia 49, 1816–1826 [DOI] [PubMed] [Google Scholar]
- 25. Tang X., Zheng J., Ying Z., Cai Z., Gao Y., He Z., Yu H., Yao J., Yang Y., Wang H., Chen Y., and Guan M. X. (2015) Mitochondrial tRNASer(UCN) variants in 2651 Han Chinese subjects with hearing loss. Mitochondrion 23, 17–24 [DOI] [PubMed] [Google Scholar]
- 26. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T., and Guan M. X. (2014) A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 42, 8039–8048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki T., Nagao A., and Suzuki T. (2011) Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu. Rev. Genet. 45, 299–329 [DOI] [PubMed] [Google Scholar]
- 28. Allnér O., and Nilsson L. (2011) Nucleotide modifications and tRNA anticodon-mRNA codon interactions on the ribosome. RNA 17, 2177–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ofengand J., Del Campo M., and Kaya Y. (2001) Mapping pseudouridines in RNA molecules. Methods 25, 365–373 [DOI] [PubMed] [Google Scholar]
- 30. Martínez-Zamora A., Meseguer S., Esteve J. M., Villarroya M., Aguado C., Enríquez J. A., Knecht E., and Armengod M. E. (2015) Defective expression of the mitochondrial tRNA modifying enzyme GTPBP3 triggers AMPK-mediated adaptive responses involving complex I assembly factors, uncoupling protein 2, and the mitochondrial pyruvate carrier. PLoS ONE 10, e0144273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Florentz C., Sohm B., Tryoen-Tóth P., Pütz J., and Sissler M. (2003) Human mitochondrial tRNAs in health and disease. Cell Mol. Life Sci. 60, 1356–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki T., and Suzuki T. (2014) A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 42, 7346–7357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sekine S., Nureki O., Shimada A., Vassylyev D. G., and Yokoyama S. (2001) Structural basis for anticodon recognition by discriminating glutamyl-tRNA synthetase. Nat. Struct. Biol. 8, 203–2066 [DOI] [PubMed] [Google Scholar]
- 34. Roovers M., Hale C., Tricot C., Terns M. P., Terns R. M., Grosjean H., and Droogmans L. (2006) Formation of the conserved pseudouridine at position 55 in archaeal tRNA. Nucleic Acids Res. 34, 4293–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kong Q. P., Bandelt H. J., Sun C., Yao Y. G., Salas A., Achilli A., Wang C. Y., Zhong L., Zhu C. L., Wu S. F., Torroni A., and Zhang Y. P. (2006) Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum. Mol. Genet. 15, 2076–2086 [DOI] [PubMed] [Google Scholar]
- 36. Zhao H., Li R., Wang Q., Yan Q., Deng J. H., Han D., Bai Y., Young W. Y., and Guan M. X. (2004) Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am. J. Hum. Genet. 74, 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33, S62–S69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamasaki S., Ivanov P., Hu G. F., and Anderson P. (2009) Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 185, 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia Z., Wang X., Qin Y., Xue L., Jiang P., Meng Y., Shi S., Wang Y., Qin Mo J., and Guan M. X. (2013) Coronary heart disease is associated with a mutation in mitochondrial tRNA. Hum. Mol. Genet. 22, 4064–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y., D'Aurelio M., Deng J. H., Park J. S., Manfredi G., Hu P., Lu J., and Bai Y. (2007) An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 282, 17557–17562 [DOI] [PubMed] [Google Scholar]
- 41. Zhang J., Jiang P., Jin X., Liu X., Zhang M., Xie S., Gao M., Zhang S., Sun Y. H., Zhu J., et al. (2014) Leber's hereditary optic neuropathy caused by the homoplasmic ND1 m.3635G>A mutation in nine Han Chinese families. Mitochondrion 18, 18–26 [DOI] [PubMed] [Google Scholar]
- 42. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M., Zhao F., Sun Y. H., Zhang M., Zhou X., Chen Y., et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596 [DOI] [PubMed] [Google Scholar]
- 43. Mahfouz R., Sharma R., Lackner J., Aziz N., and Agarwal A. (2009) Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil. Steril. 92, 819–827 [DOI] [PubMed] [Google Scholar]
- 44. Becker H. F., Motorin Y., Planta R. J., and Grosjean H. (1997) The yeast gene YNL292w encodes a pseudouridine synthase (Pus4) catalyzing the formation of psi55 in both mitochondrial and cytoplasmic tRNAs. Nucleic Acids Res. 25, 4493–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ishida K., Kunibayashi T., Tomikawa C., Ochi A., Kanai T., Hirata A., Iwashita C., and Hori H. (2011) Pseudouridine at position 55 in tRNA controls the contents of other modified nucleotides for low-temperature adaptation in the extreme-thermophilic eubacterium Thermus thermophilus. Nucleic Acids Res. 39, 2304–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang M., Zhou X. L., Liu R. J., Fang Z. P., Zhou M., Eriani G., and Wang E. D. (2013) Multilevel functional and structural defects induced by two pathogenic mitochondrial tRNA mutations. Biochem. J. 453, 455–465 [DOI] [PubMed] [Google Scholar]
- 47. Masucci J. P., Davidson M., Koga Y., Schon E. A., and King M. P. (1995) In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNALys gene: two genotypes produce similar phenotypes. Mol. Cell Biol. 15, 2872–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Enriquez J. A., Chomyn A., and Attardi G. (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nat. Genet. 10, 47–55 [DOI] [PubMed] [Google Scholar]
- 49. Li X., Fischel-Ghodsian N., Schwartz F., Yan Q., Friedman R. A., and Guan M. X. (2004) Biochemical characterization of the mitochondrial tRNASer(UCN) T7511C mutation associated with nonsyndromic deafness. Nucleic Acids Res. 32, 867–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S., Li R., Fettermann A., Li Z., Qian Y., Liu Y., Wang X., Zhou A., Mo J. Q., Yang L., Jiang P., Taschner A., Rossmanith W., and Guan M. X. (2011) Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ. Res. 108, 862–870 [DOI] [PubMed] [Google Scholar]
- 51. Guan M. X., Fischel-Ghodsian N., and Attardi G. (2001) Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 10, 573–580 [DOI] [PubMed] [Google Scholar]
- 52. Qu J., Li R., Zhou X., Tong Y., Lu F., Qian Y., Hu Y., Mo J. Q., West C. E., and Guan M. X. (2006) The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Invest. Ophthalmol. Vis. Sci. 47, 475–483 [DOI] [PubMed] [Google Scholar]
- 53. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. James A. M., Sheard P. W., Wei Y. H., and Murphy M. P. (1999) Decreased ATP synthesis is phenotypically expressed during increased energy demand in fibroblasts containing mitochondrial tRNA mutations. Eur. J. Biochem. 259, 462–469 [DOI] [PubMed] [Google Scholar]
- 55. Pallotti F., Baracca A., Hernandez-Rosa E., Walker W. F., Solaini G., Lenaz G., Melzi D'Eril G. V., Dimauro S., Schon E. A., and Davidson M. M. (2004) Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations. Biochem. J. 384, 287–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szczepanowska J., Malinska D., Wieckowski M. R., and Duszynski J. (2012) Effect of mtDNA point mutations on cellular bioenergetics. Biochim. Biophys. Acta. 1817, 1740–1746 [DOI] [PubMed] [Google Scholar]
- 57. Chen Y. B., Aon M. A., Hsu Y. T., Soane L., Teng X., McCaffery J. M., Cheng W. C., Qi B., Li H., Alavian K. N., Dayhoff-Brannigan M., Zou S., Pineda F. J., O'Rourke B., Ko Y. H., et al. (2011) Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J. Cell Biol. 195, 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang P., Wang M., Xue L., Xiao Y., Yu J., Wang H., Yao J., Liu H., Peng Y., Liu H., et al. (2016) A hypertension-associated tRNAAla mutation alters the tRNA metabolism and mitochondrial function. Mol. Cell Biol. 36, 1920–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hayashi G., and Cortopassi G. (2015) (2015) Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 88, 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sena L. A., and Chandel N. S. (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Raimundo N., Song L., Shutt T. E., McKay S. E., Cotney J., Guan M. X., Gilliland T. C., Hohuan D., Santos-Sacchi J., and Shadel G. S. (2012) Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell 148, 716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guan M. X. (2004) Molecular pathogenetic mechanism of maternally inherited deafness. Ann. N.Y. Acad. Sci. 1011, 259–271 [DOI] [PubMed] [Google Scholar]
- 63. Patti M.-E., and Corvera S. (2010) The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 31, 364–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ma Z. A., Zhao Z., and Turk J. (2012) Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp. Diabetes Res. 2012, 703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reid F. M., Vernham G. A., and Jacobs H. T. (1994) A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum. Mutat. 3, 243–247 [DOI] [PubMed] [Google Scholar]
- 66. Lu J., Qian Y., Li Z., Yang A., Zhu Y., Li R., Yang L., Tang X., Chen B., Ding Y., Li Y., You J., Zheng J., Tao Z., Zhao F., et al. (2010) Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A>G mutation. Mitochondrion 10, 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bykhovskaya Y., Shohat M., Ehrenman K., Johnson D., Hamon M., Cantor R. M., Aouizerat B., Bu X., Rotter J. I., Jaber L., and Fischel-Ghodsian N. (1998) Evidence for complex nuclear inheritance in a pedigree with nonsyndromic deafness due to a homoplasmic mitochondrial mutation. Am. J. Med. Genet. 77, 421–426 [DOI] [PubMed] [Google Scholar]
- 68. Maassen J. A., 'T Hart L. M., Van Essen E., Heine R. J., Nijpels G., Jahangir Tafrechi R. S., Raap A. K., Janssen G. M., and Lemkes H. H. (2004) Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes 53, S103–109 [DOI] [PubMed] [Google Scholar]
- 69. Craven L., Elson J. L., Irving L., Tuppen H. A., Lister L. M., Greggains G. D., Byerley S., Murdoch A. P., Herbert M., and Turnbull D. (2011) Mitochondrial DNA disease: new options for prevention. Hum. Mol. Genet. 20, R168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen C., Chen Y., and Guan M. X. (2015) A peep into mitochondrial disorder: multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 6, 862–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dittmar K. A., Goodenbour J. M., and Pan T. (2006) Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2, e221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen D., Li F., Yang Q., Tian M., Zhang Z., Zhang Q., Chen Y., and Guan M. X. (2016) The defective expression of gtpbp3 related to tRNA modification alters the mitochondrial function and development of zebrafish. Int. J. Biochem. Cell Biol. 77, 1–9 [DOI] [PubMed] [Google Scholar]
- 73. Rieder M. J., Taylor S. L., Tobe V. O., and Nickerson D. A. (1998) Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 26, 967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Miller G., and Lipman M. (1973) Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U.S.A. 70, 190–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. King M. P., and Attardi G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 268, 10228–10237 [PubMed] [Google Scholar]
- 76. Hao R., Yao Y. N., Zheng Y. G., Xu M. G., and Wang E. D. (2004) Reduction of mitochondrial tRNALeu(UUR) aminoacylation by some MELAS-associated mutations. FEBS Lett. 578, 135–139 [DOI] [PubMed] [Google Scholar]
- 77. Puglisi J. D., and Tinoco I. Jr. (1989) Absorbance melting curves of RNA. Methods Enzymol. 180, 304–325 [DOI] [PubMed] [Google Scholar]
- 78. Wang X., Lu J., Zhu Y., Yang A., Yang L., Li R., Chen B., Qian Y., Tang X., Wang J., Zhang X., and Guan M. X. (2008) Mitochondrial tRNAThr G15927A mutation may modulate the phenotypic manifestation of ototoxic 12S rRNA A1555G mutation in four Chinese families. Pharmacogenet. Genomics. 18, 1059–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yan X., Wang X., Wang Z., Sun S., Chen G., He Y., Mo J. Q., Li R., Jiang P., Lin Q., Sun M., Li W., Bai Y., Zhang J., Zhu Y., et al. (2011) Maternally transmitted late-onset non-syndromic deafness is associated with the novel heteroplasmic T12201C mutation in the mitochondrial tRNAHis gene. J. Med. Genet. 48, 682–690 [DOI] [PubMed] [Google Scholar]
- 80. Guan M. X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E., Suzuki T., del Castillo I., Peters J. L., Li R., Qian Y., et al. (2006) Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 79, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.