

Abstract
The basal ganglia have a central role in motor patterning, habits, motivated behaviors, and cognition as well as in numerous neuropsychiatric disorders. Receptors for histamine, especially the H3 receptor (H3R), are highly expressed in the striatum, the primary input nucleus of the basal ganglia, but their effects on this circuitry have been little explored. H3R interacts with dopamine (DA) receptors ex vivo; the nature and functional importance of these interactions in vivo remain obscure. We found H3R activation with the agonist R-(-)-α-methylhistamine to produce a unique time- and cell type-dependent profile of molecular signaling events in the striatum. H3 agonist treatment did not detectably alter extracellular DA levels or signaling through the cAMP/DARPP-32 signaling pathway in either D1- or D2-expressing striatal medium spiny neurons (MSNs). In D1-MSNs, H3 agonist treatment transiently activated MAPK signaling and phosphorylation of rpS6 and led to phosphorylation of GSK3β-Ser9, a novel effect. Consequences of H3 activation in D2-MSNs were completely different. MAPK signaling was unchanged, and GSK3β-Ser9 phosphorylation was reduced. At the behavioral level, two H3 agonists had no significant effect on locomotion or stereotypy, but they dramatically attenuated the locomotor activation produced by the D1 agonist SKF82958. H3 agonist co-administration blocked the activation of MAPK signaling and the phosphorylation of rpS6 produced by D1 activation in D1-MSNs, paralleling behavioral effects. In contrast, GSK3β-Ser9 phosphorylation was seen only after H3 agonist treatment, with no interactive effects. H3R signaling has been neglected in models of basal ganglia function and has implications for a range of pathophysiologies.
Keywords: Akt PKB, dopamine, dopamine receptor, histamine, MAPK, H3R, medium spiny neuron, striatum
Introduction
The basal ganglia play a critical role in the regulation of cognition, motor control, motivated behavior, and habit learning as well as in the pathophysiology of a range of neuropsychiatric conditions, including Parkinson's disease, Tourette syndrome, and drug addiction (1–6). Factors that modulate information flow through the basal ganglia circuitry are therefore of broad importance. The roles of dopamine (DA)4 and acetylcholine in the regulation of striatal information processing have been extensively studied. Histamine (HA) is produced in the tuberomamillary nucleus of the posterior hypothalamus; these neurons project broadly throughout the brain, including to the striatum (7). HA receptors are prominent throughout the basal ganglia (7, 8), but their function in modulating this circuitry is not well understood, and very few studies have examined their functional importance in vivo (9).
The striatum (equivalent to the caudate and putamen in primates) is the largest nucleus of the basal ganglia and the primary site of cortical and thalamic input. Its principal cells, the medium spiny neurons (MSNs), can be divided into two subpopulations based on their projections. Striatonigral or “direct pathway” neurons preferentially express the D1 DA receptor and have a net disinhibitory effect on the thalamus, which processes most output from the basal ganglia. Striatopallidal or “indirect pathway” neurons preferentially express the D2 DA receptor and have a net inhibitory effect on the thalamus (3, 6, 10). Modulatory DA critically regulates plasticity and relative activity in D1- and D2-expressing MSNs.
The molecular processes regulated by DA in D1- and D2-expressing MSNs have been a focus of intense research (11, 12). The importance of striatal regulation by DA is highlighted by the striking consequences of DA receptor stimulation, which produces locomotor activation and stereotypy (13). In D1-MSNs, D1 receptor activation leads to increased cAMP, phosphorylation of DARPP-32, and activation of the MAPK signaling pathway. In D2-MSNs, in contrast, D2 receptor activation leads to decreased cAMP and reduced phosphorylation of DARPP-32 (14) as well as β-arrestin-dependent dephosphorylation, and thus deactivation, of the kinase Akt, and consequent reduced phosphorylation, and thus activation, of GSK3β (15).
Recent clinical data have focused new attention on modulation of the basal ganglia by HA (9). A dominant-acting loss-of-function mutation in the HA biosynthetic enzyme histidine decarboxylase (Hdc) has been implicated as a rare cause of Tourette syndrome (16); other genetic analyses have suggested abnormalities in histaminergic signaling in Tourette syndrome more broadly (17, 18). HDC protein is particularly concentrated in the striatum, suggesting an important role for HA there (19). We have shown loss of Hdc to lead to elevated striatal DA, increased tic-like stereotypies, and alterations in MAPK and Akt signaling (1, 20, 21).
HA binds to four G protein-coupled receptors, H1R-H4R (7). Of these, H3R is expressed only in the nervous system; it is highly enriched in the basal ganglia (7, 8). H3R has a well established role as a presynaptic inhibitor of neurotransmitter release on both histaminergic and other neurons (7, 22). In particular, it has been shown to reduce DA release in some brain regions (23). However, this may not be its primary role in the striatum (22). A series of ex vivo studies have suggested that postsynaptic H3R in MSNs forms physical complexes with both D1 and D2 receptors that modulate the signaling properties of both (24–27). In striatal slices, H3R complexed with D1R activates MAPK signaling (25–27). Effects on Akt signaling in the striatum are less clear. H3R activation can inhibit the Akt1 target GSK3β in immortalized cells and cultured cortical neurons (28, 29), but as these systems are not known to recapitulate the unique β-arrestin-dependent mechanism by which Akt and GSK3β are regulated in D2-MSNs, it is unclear whether this regulation will generalize to the striatum in vivo.
We characterized the regulation of striatal signaling by H3R in vivo and examined functional interactions between H3R and D1R activation. We find a unique temporal and cell-specific profile of H3 effects on MAPK and GSK3β signaling and striking nonlinear interactions between H3 and D1 effects at both molecular and behavioral levels. These interactions are likely to be an important and underappreciated regulator of basal ganglia function in vivo.
Results
H3R Agonist Treatment Does Not Regulate the cAMP/PKA Pathway in Striatal MSNs
Activation of the H3R has been linked to numerous molecular events that we seek to better characterize in vivo. H3R is coupled to Gαi and thereby can reduce cAMP-mediated signaling, at least in some cells (7, 30, 31). This could oppose the effects of D1R activation on the cAMP/PKA/DARPP-32 signaling pathway. We therefore first sought to characterize whether H3R activation regulates cAMP-mediated signaling in the striatum in vivo. We used phosphorylation of DARPP-32, which is responsive to changes in cAMP levels in striatal MSNs, as a surrogate marker for changes in cAMP.
We characterized the effects of RAMH (45 mg/kg), an H3 agonist, on DARPP-32 phosphorylation using transgenic mice that express FLAG- and Myc-taggd DARPP-32 in D1- and D2-MSNs, respectively, permitting DARPP-32 phosphorylation to be disociably quantified in these two cell types (14). Increased cAMP and PKA activity lead to increased phosphorylation of DARPP-32 at Thr34 (11). PKA-dependent activation of protein phosphatase 2A (PP-2A) leads to dephosphorylation of DARPP-32 at Thr75 (32, 33). However, RAMH did not produce significant changes in these phosphorylation sites in either D1- or D2-MSNs (Fig. 1, A and B). Thus, H3 activation does not appear to significantly regulate the cAMP/PKA signaling pathway in either D1- or D2-MSNs in vivo, at least at this time point.
FIGURE 1.
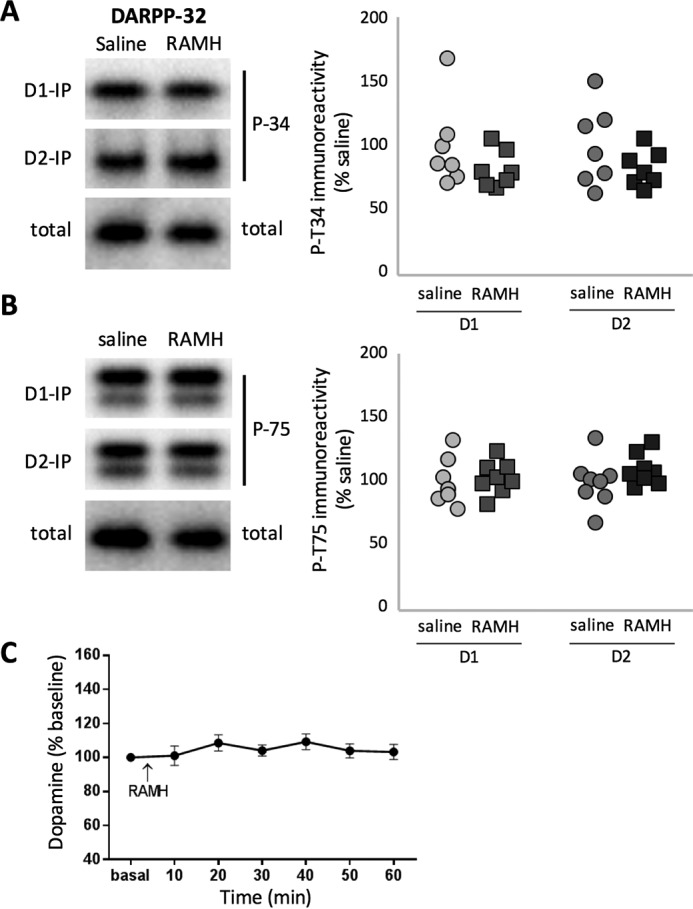
Lack of effect of RAMH on cAMP/DARPP-32 signaling. A and B, the transgenic reporter DARPP-32 was specifically immunoprecipitated from either D1- or D2-MSNs, and phosphorylation of Thr34 and Thr75 was analyzed by immunoblotting as described previously (14) 15 min following systemic RAMH. Left panels, representative immunoblots. Right panels, summary data for DARPP-32 phosphorylation in D1-MSNs (left) or D2-MSNs (right). Signals for either Thr(P)34 or Thr(P)75 were normalized to total DARPP-32 level. Representative blots of total DARPP-32 from D1-MSN immunoprecipitations are shown. A, phosphorylation of DARPP-32 at Thr34 was not affected by in vivo RAMH challenge (45 mg/kg) in either MSN population. n = 7–8/group. Student's t test: D1, t[13] = 1.07, p = 0.3; D2, t[12] = 1.33, p = 0.21. B, phosphorylation of DARPP-32 at Thr75 was not affected by in vivo RAMH challenge (45 mg/kg) in either MSN population. n = 7–8/group. Student's t test: D1: t[14] = 0.49, p = 0.63; D2: t[13] = 1.27, p = 0.2. C, RAMH (45 mg/kg) did not affect dorsal striatal DA as measured using in vivo microdialysis. RM-ANOVA: F[17,14] = 0.97, p > 0.4.
Activation of H3Rs on DA terminals has been shown ex vivo to regulate DA release (23); this represents an alternative way in which the H3R might modulate striatal function. Whether this regulation occurs in the striatum in vivo has not been established. We measured striatal DA before and after systemic administration of RAMH (45 mg/kg) using in vivo microdialysis. There was no detectable change in striatal DA levels after RAMH treatment (Fig. 1C).
Thus, these two well established mechanisms of H3R signaling appear not to be operative in vivo in the striatum under these conditions. H3R activation does not significantly regulate either extracellular DA levels or cAMP-mediated signaling.
Differential and Time-dependent Regulation of MAPK in D1-MSNs
H3 activation has been shown to activate MAPK in D1-positive cells ex vivo (26). We examined the effect of RAMH on MAPK activation in vivo using immunoblotting of striatal extracts collected after systemic RAMH challenge. Phosphorylation of ERK1/2 MAPK at Thr202/Tyr204 was strongly increased 15 min after RAMH administration but returned to normal by 45 min (Fig. 2A).
FIGURE 2.
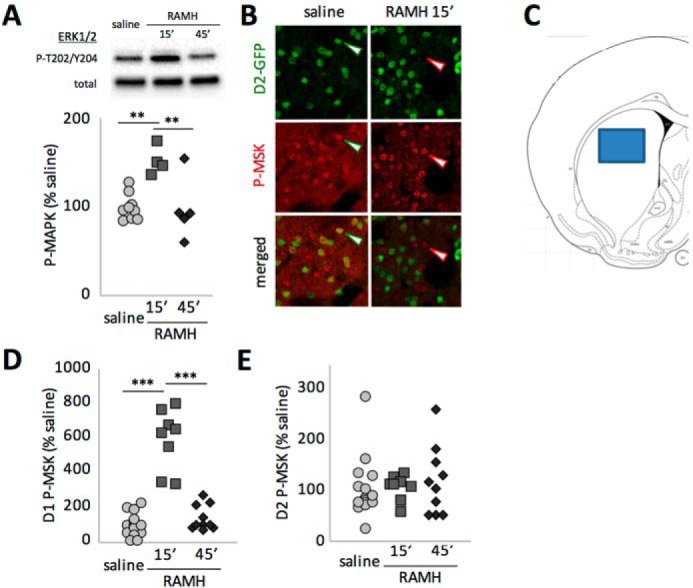
Regulation of MAPK signaling after H3R activation. A, phosphorylation of ERK1/2 MAPK was assayed in total striatal extract 15 and 45 min after RAMH (45 mg/kg) or saline injection. ERK phosphorylation was increased at 15 min but returned to baseline at 45 min. n = 9 SAL, 4 RAMH-15, and 5 RAMH-45. One-way ANOVA: F[2,15] = 9.1, p = 0.0026. B, phosphorylation of the MAPK target MSK was assessed in D2-GFP transgenic mice in GFP-negative (presumptive D1-MSNs, red arrowheads) and GFP-positive cells (presumptive D2-MSNs, green arrowheads). C, placement of a typical confocal image for cell counting. Similar fields from the central dorsal striatum were collected in five coronal sections from each mouse. D, phospho-MSK positive cells in confocal Z stacks were counted and categorized as D1- or D2-MSNs. Values were normalized to the corresponding saline control. Phosphorylation in D1-MSNs paralleled the activation of ERK seen in total striatal extract (n = 15 SAL, 8 RAMH-15, and 10 RAMH-45; one-way ANOVA: F[2,30] = 63.4). E, there were no effects in D2-MSNs (n = 16 SAL, 8 RAMH-45, and 10 RAMH-45; one-way ANOVA: F[2,30] = 0.33, p > 0.7). **, p < 0.01; ***, p < 0.005; Sidak's post-hoc test.
We next examined the cell specificity of the RAMH effect on MAPK activation using D2-EGFP transgenic mice. We found immunohistochemistry for P-MAPK to produce inadequately specific cell body signal to permit categorization and counting of positive cells. We therefore used phosphorylation of the ERK target mitogen- and stress-activated kinase 1 (MSK1) at Thr581 as a marker of MAPK signaling (Fig. 2B) (34). (MSK1 is a target of both the ERK1/2 and p38 MAPK signaling pathways (35)).
P-MSK1-positive cells were visualized in a single confocal image from the central dorsal striatum in each slice (Fig. 2C) and categorized as either co-localizing or not with GFP. In GFP-negative striatal neurons, the vast majority of which are expected to be D1-MSNs, P-MSK-Thr581 was strongly but transiently modulated by RAMH, with immunopositive cells increasing nearly 6-fold at 15 min and returning to baseline at 45 min, paralleling the effects on MAPK seen by immunoblotting (Fig. 2D). In contrast, in GFP-positive cells, the vast majority of which are D2-MSNs, there was no modulation of MSK1 by RAMH (Fig. 2E).
We next examined phosphorylation of ribosomal protein S6 (rpS6). rpS6 is a key regulator of inducible translation in neurons (36). Its phosphorylation at Ser235/Ser236 is regulated by numerous signaling pathways (we have shown it to depend on MAPK signaling in these neurons under some circumstances (20)), but it is also regulated by PKA, S6 kinase, and other pathways (36). A nearby pair of phosphorylation sites, Ser240/Ser244, is regulated by S6 kinase but not by MAPK signaling (36).
Immunoblotting revealed an increase in phosphorylation of rpS6 at the Ser235/Ser236 site 15 min after RAMH treatment; this returned to baseline by 45 min, paralleling ERK 1/2 phosphorylation (Fig. 3A, compare with Fig. 2A). We again discriminated between D1- and D2-MSN contributions to this effect using immunostaining in D2-GFP transgenic mice. In D1-MSNs, RAMH treatment modulated Ser(P)235/Ser(P)236-rpS6 (Fig. 3B), with an increase at 15 min that returned nearly to baseline by 45 min, paralleling MSK1 activation. In D2-MSNs, there was no statistically significant effect of RAMH on Ser(P)235/Ser(P)236-rpS6, although there was nominally increased phosphorylation at 45 min that did not reach statistical significance. There was no effect of RAMH treatment on Ser(P)240/Ser(P)244-rpS6 in either MSN subtype at either time point (Fig. 3C).
FIGURE 3.
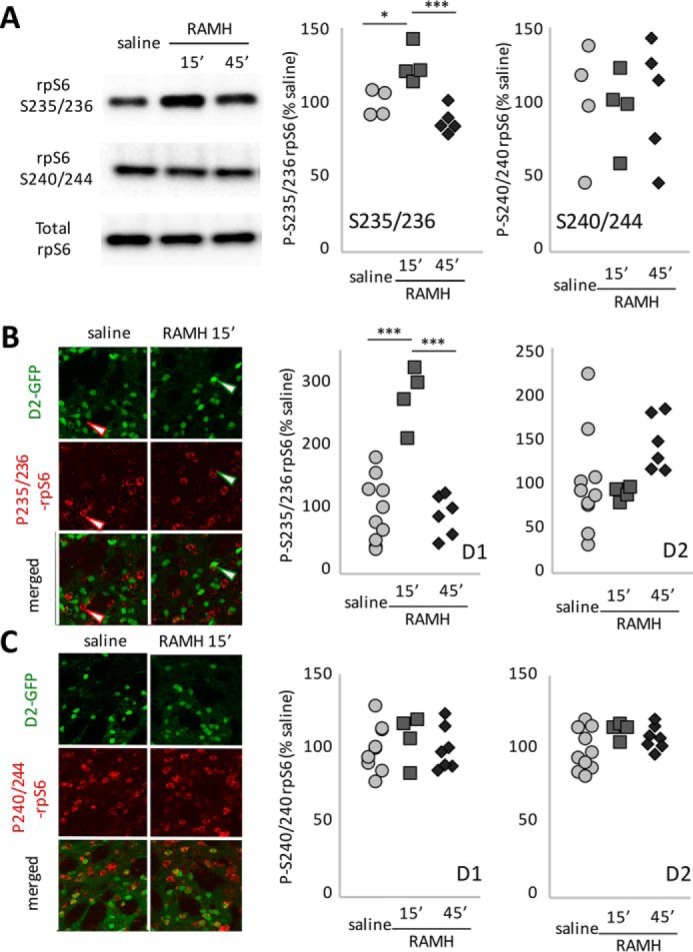
Regulation of rpS6 after H3R activation. A, immunoblotting for phospho-rpS6 shows modulation of the Ser235/Ser236 phosphorylation site but not the Ser240/Ser244 site after RAMH treatment in vivo. n = 4 saline, 4 RAMH-15 min, and 4 RAMH-45 min. Ser235/Ser236, one-way ANOVA: F[2,10] = 15.91, p = 0.0008. Ser240/Ser244, one-way ANOVA: F[2,10] = 0.36, p = 0.7. B, phosphorylation of rpS6 at Ser235/Ser236 was quantified in D2-GFP transgenic mice in GFP-negative (presumptive D1-MSNs) and GFP-positive (presumptive D2-MSNs) cells after acute RAMH challenge (45 mg/kg). Phosphorylation increased markedly in D1-MSNs at 15 min but returned to baseline by 45 min (n = 10 SAL, 4 RAMH-15, and 7 RAMH-45; D1: one-way ANOVA: F[2,18] = 27.5, p < 0.0001; D2: one-way ANOVA: F[2,17] = 1.4, p = 0.27). C, there were no changes in rpS6 phosphorylation at Ser240/Ser244 (n = 10 SAL, 4 RAMH-15, and 7 RAMH-45; one-way ANOVA: D1: F[2,18] = 0.34, p > 0.7; D2: F[2,18] < 2.4, p = 0.12). *, p < 0.05; ***, p < 0.005; Tukey's multiple comparisons test.
H3 Regulation of Akt-GSK in D1- and D2-MSNs
Akt-GSK3 signaling is regulated by D2 activation in D2-MSNs (15); its regulation by the H3 receptor in MSNs has not been investigated previously. We quantified Akt phosphorylation at Thr308 by immunoblotting of striatal extracts (20). This revealed a significant modulation of Thr(P)308 Akt in vivo (Fig. 4A); there was little change at 15 min but a marked reduction 45 min after RAMH injection. There was no modulation of the other regulatory site, Ser473. We also examined the Akt target GSK3β-Ser9 and found that it, too, was modulated by RAMH treatment (Fig. 4B). In the case of GSK3β, Ser9 phosphorylation was increased at 15 min but decreased at 45 min, indicating complex temporal regulation.
FIGURE 4.

Akt and GSK3β phosphorylation after H3R agonist treatment. A, in total striatal lysate, Akt phosphorylation at Thr308 was reduced 45 min after RAMH (45 mg/kg; n = 13 SAL, 5 RAMH-15 min, and 8 RAMH-45 min; one-way ANOVA: F[2,23] = 8.9, p = 0.0014). Phosphorylation at Ser473 was not altered (F[2,23] = 0.59, p > 0.5). B, in total striatal lysate, GSK3β, an Akt target, showed increased phosphorylation 15 min after RAMH treatment but decreased phosphorylation (and thus presumptive activation) at 45 min (n = 9 SAL, 5 RAMH-15 min, and 4 RAMH-45 min; one-way ANOVA: F[2,23] = 10.5, p = 0.0014). C, Ser(P)9 GSK3β was visualized by immunofluorescence as before. Positive cells were counted and categorized as presumptive D1-MSNs (red arrowheads) or D2-MSNs (green arrowheads). D, in D2-GFP transgenic mice, Ser(P)9-GSK3β increased at 15 min and then returned to baseline at 45 min in GFP-negative cells (presumptive D1-MSNs; n = 10 SAL, 4 RAMH-15 min; and 6 RAMH-45 min; one-way ANOVA: F[2,30] = 4.0, p = 0.038). In GFP-positive cells (presumptive D2-MSNs), Ser(P)9-GSK3β decreased over time (one-way ANOVA: F[2,30] = 8.3, p = 0.003). *, p < 0.05; **, p < 0.01; Sidak's post-hoc test.
To examine the cell specificity of these effects, we again turned to immunostaining in D2-GFP transgenic mice. We focused on GSK3β (Fig. 4C), as anti-P-Akt antibodies showed a high background on tissue in our hands. In D1-MSNs, there was a clear but transient effect of RAMH on GSK3β, with increased Ser9 phosphorylation at 15 min that returned to baseline at 45 min (Fig. 4D). In D2-MSNs, in contrast, there was an equally clear but slower and qualitatively opposite modulation of Ser9 phosphorylation, with decreasing Ser(P)9 over time that became significant at 45 min. This dissociation explains the biphasic pattern of GSK3β phosphorylation seen by immunoblotting (Fig. 4B).
H3R Activation Attenuates D1 Agonist-induced Locomotor Activation
H3 activation has been reported to attenuate the locomotor activation seen after D1 stimulation in reserpinized mice (24); whether this interaction is present in intact animals has not been demonstrated. We tested the effects of the H3 agonist RAMH (45 mg/kg) and the D1 agonist SKF82958 (5 mg/kg), alone and in combination, on locomotor activity and stereotypy. RAMH injection had no effect on locomotion. SKF82958 challenge, on the other hand, produced locomotor activation as expected (37). Strikingly, the locomotor effect of D1 activation was completely eliminated by RAMH co-administration. (Fig. 5A).
FIGURE 5.

H3R agonist treatment mitigates hyperlocomotion after D1R agonist challenge. A, locomotor activity over 1 h after injection of SKF82958 and/or RAMH (45 mg/kg). n = 11 saline, 10 in other groups. Two-way ANOVA: main effect of SKF, F[1,37] = 19.85, p < 0.0001; main effect of RAMH, F[1,37] = 16.15, p = 0.0003; interaction, F[1,37] = 5.26, p = 0.028. B, stereotypy after SKF and RAMH. n = 8/group; three-way RM-ANOVA: main effect of SKF, F[1,28] = 12.0, p = 0.0017; main effect of RAMH, F[1,28] = 2.7, p = 0.10; interaction, F[1,28] = 0.43, p > 0.5. C, locomotion over 1 h after injection of SKF82958 (45 mg/kg) and/or immepip (20 mg/kg). n = 8 saline, 9 immepip, 9 SKF82958, and 8 immepip + SKF82958. Two-way ANOVA: main effect of SKF, F[1,30] = 22.6, p < 0.0001; main effect of immepip, F[1,30] = 1.07, p = 031; interaction, F[1,30] = 10.1, p = 0.0034. *, p < 0.05; **, p < 0.01; ***, p < 0.005, Sidak's post-hoc test.
Locomotion after psychostimulant treatment can be reduced by an increase in stereotypy. This is seen, for example, at high psychostimulant doses or after repeated injections (1, 38). Specific D1 activation with agents such as SKF82958 does not typically produce high levels of stereotypy, but it is possible that simultaneous RAMH administration might potentiate stereotypy and reduce locomotion by this mechanism. To examine this, we tested behavioral stereotypy after challenge with the two drugs alone and in combination. SKF82958 (5 mg/kg) produced a modest elevation in stereotypy that was not enhanced by RAMH co-administration (45 mg/kg, Fig. 5B).
We repeated the experiment with another H3 agonist, immepip (39). Administration of SKF82958 and immepip (20 mg/kg) produced the same pattern of results as RAMH. Immepip had no effect on locomotion by itself, but it blocked the increase in locomotion seen after SKF82958 (Fig. 5C).
D1-H3 Interaction in the Regulation of MAPK Signaling
We tested whether H3 and D1 agonists also interact at the level of signaling in D1- and D2-MSNs. D1 and H3 agonists have been shown previously ex vivo to interact in the regulation of MAPK in D1-MSNs (25, 26). We replicated this interactive effect in vivo. Both RAMH and SKF82958 treatment increased phosphorylation of MSK (at 15 min, as shown in Fig. 3), but co-administration of both agents completely abolished this effect (Fig. 6A). There were no effects of either agent in D2-MSNs.
FIGURE 6.

H3R-D1R functional interaction on MSK1, rpS6, and GSK3β. A, Thr(P)581-MSK was increased in GFP-negative cells (presumptive D1-MSNs) 15 min after both RAMH and SKF but not when both were given together. Two-way ANOVA: main effect of RAMH: F[1,16] = 0.25, p > 0.6; main effect of SKF, F[1,16] = 1.0, p > 0.3; interaction, F[1,16] = 57, p < 0.0001. No effect was seen in GFP-positive cells (presumptive D2-MSNs). n = 5/group. B, Ser(P)235/Ser(P)236-rpS6 was increased by both RAMH and SKF. Co-administration reduced phosphorylation levels but not all the way to baseline. D1, two-way ANOVA: main effect of RAMH, F[1,16] = 1.0, p = 0.33; main effect of SKF, F[1,16] = 18.3, p = 0.003; interaction: F[1,16] = 18.3, p = 0.0006. No effect was seen in GFP-positive cells. n = 5/group. C, Ser(P)9-GSK3β was increased 15 min after RAMH in GFP-negative cells (presumptive D1-MSNs). SKF82958 had no effect on Ser(P)9-GSK3β and did not modulate the effect of RAMH (two-way ANOVA: main effect of RAMH: F[1,14] = 23.1, p = 0.0003; main effect of SKF: F[1,14] = 0.09, p > 0.7; interaction, F[1,14] = 1.4, p > 0.25). There was no effect of either drug (at this time point) on Ser(P)9-GSK3β in GFP-positive cells (presumptive D2-MSNs). n = 6 SAL, 4 in other groups. *, p < 0.05; **, p < 0.01; Sidak's post-hoc test.
H3 and D1 agonists also interacted in the regulation of rpS6. Both RAMH and SKF82958 led to increased phosphorylation of rpS6, but SKF82958-induced phosphorylation was significantly greater (at least at these doses). Co-administration reduced phosphorylation back to the level of RAMH alone but not (as in the case of MSK) all the way to baseline (Fig. 6B). Again, there were no effects in D2-MSNs.
D1 Stimulation Does Not Regulate Akt
We administered RAMH (45 mg/kg) and SKF82958 (5 mg/kg) alone and in combination and examined GSK3β-Ser9 phosphorylation 15 min later in D1- and D2-MSNs. As before, RAMH led to increased GSK3β phosphorylation. In this case, however, there was no interactive effect. SKF treatment did not influence GSK3β-Ser9 phosphorylation either alone or in conjunction with RAMH (Fig. 6C). There were no detectable effects in D2-MSNs (note that these samples were collected at 15 min; the effects on GSK3β in D2-MSNs documented in Fig. 5 were not prominent until 45 min).
Discussion
HA receptors are prominent in the striatum, but how HA modulates the basal ganglia circuitry is not well understood (9). We found treatment with H3 histamine receptor agonists to attenuate the locomotor effects of the D1 agonist SKF82958. The H3 agonist RAMH modulates signaling in D1- and D2-MSNs in distinct and even opposite ways. In D1-MSNs, H3 activation transiently activates MAPK signaling, recapitulating a prominent effect of D1 activation, but it does not detectably affect cAMP/DARPP-32 signaling, and it triggers a previously uncharacterized transient phosphorylation (and thus presumptive deactivation) of GSK3β. In D2-MSNs, H3 activation has no effect on MAPK or cAMP/DARPP-32, but it regulates Akt-GSK3β, producing a more slowly developing dephosphorylation (and thus presumptive activation) of GSK3β: qualitatively, the opposite of its effect in D1-MSNs. This unique profile of cellular effects suggests an important and largely unappreciated role for HA in the regulation of striatal function.
H3R Regulation of MAPK, MSK, and rpS6
H3 receptors can couple to Gαi (7). Gαi-mediated inhibition of adenylyl cyclase may explain the inhibition of transmitter release by presynaptic H3 homoreceptors and heteroreceptors (7, 23, 40). However, Gαi-mediated signaling mechanisms do not explain the signaling in MSNs that we see in vivo. We find no significant modulation of cAMP/DARPP-32 signaling in either D1- or D2-MSNs.
Recent ex vivo data suggest that H3Rs heterodimerize with D1 and D2 dopamine receptors and thereby acquire unique signaling characteristics in MSNs (24–26). Specifically, activation of H3-D1 heterodimers has been shown to activate MAPK signaling (25, 26), which is consistent with the transient effect we see in D1-MSNs (Fig. 3). H3-D2 heteromers have also been described (24); their effect on downstream signaling has not been characterized. We found no effect of RAMH treatment in vivo on MAPK or cAMP/DARPP-32 signaling in D2-expressing MSNs.
rpS6 is transiently phosphorylated at Ser235/Ser236 after H3R activation in D1-MSNs (Fig. 3). Several observations suggest that this is a MAPK-dependent event. rpS6 is regulated by phosphorylation at two pairs of sites, Ser235/Ser236 and Ser240/Ser244 (41). Both can be phosphorylated by p70S6K via a mammalian target of rapamycin (mTOR)-dependent signaling cascade (42). MAPK signaling, on the other hand, phosphorylates Ser235/Ser236 but not Ser240/Ser244 via ribosomal protein S6 kinase and MSK activation (43). This parallels the pattern of phosphorylation we see here (Fig. 3). The fact that the kinetics of rpS6 phosphorylation parallel those of ERK and MSK activation in D1-MSNs with a marked but transient increase at 15 min that entirely returned to baseline by 45 min further supports the conclusion that this is a MAPK-dependent phenomenon. On the other hand, H3/D1 co-stimulation completely abolishes activation of MSK but only partially attenuates phosphorylation of rpS6 (Fig. 6, A and B). rpS6 may also be phosphorylated at Ser235/Ser236 through a distinct mechanism.
H3R Regulation of Akt-GSK3β Signaling
Our most striking finding is the opposite regulation of Akt-GSK3β signaling by H3R in D1- and D2-MSNs. We observe Ser9 phosphorylation of GSK3β specifically in D1-MSNs 15 min after RAMH administration (Fig. 4). This is consistent with a PI3K-dependent mechanism documented previously in culture and ex vivo (28, 29). D1R activation with SKF82958 does not detectably alter GSK3β phosphorylation or modulate the effects of H3 activation (Fig. 6C). GSK3β can be phosphorylated by MAPK (44–48), but this dissociation between MAPK regulation and GSK3β phosphorylation after D1/H3 co-stimulation argues against this being the primary mechanism at play here.
The effect of H3R activation in D2-MSNs is quite different. RAMH leads to dephosphorylation of GSK3β with a slower time course (Fig. 4). This is similar to the effect of D2R stimulation, which leads to Akt deactivation and thus to GSK3β dephosphorylation via a β-arrestin-dependent mechanism in these cells (15). Whether H3 receptors are coupled to Akt-GSK3β signaling by a similar molecular mechanism remains to be determined. Regardless, this dephosphorylation (and therefore presumptive activation) of GSK3β after H3R stimulation has not, to our knowledge, been reported previously in any cell type. Importantly, H3R activation on D2-MSNs does not recapitulate all effects of D2R stimulation; there is no detectable regulation of cAMP/DARPP-32 signaling (Fig. 1). Therefore, H3R activation leads to a unique pattern of signaling in both D1- and D2-expressing MSNs. The qualitative dissociation of molecular effects of stimulation of a single receptor indicates that H3R signaling is critically modulated by its interaction with other cellular components—either direct interactions with D1 and D2 receptors (25–27) or some other cell type-specific factor.
Phosphorylation of GSK3β in D1-MSNs is transient, peaking at 15 min and reverting to baseline by 45 min. In D2-MSNs, dephosphorylation of GSK3β develops more gradually, with only a trend apparent at 15 min but a robust effect at 45 min. Examination of GSK3β phosphorylation in total striatal extracts (Fig. 4B) combines these two effects into a biphasic pattern of GSK3β phosphorylation, with an increase at 15 min followed by a decrease at 45 min. Examination of these effects with cell type specificity, as we do here, is essential to reveal the dynamics that underlie this complex pattern.
Functional Interaction between H3 and D1 Agonists
Treatment with the H3R agonists RAMH or immepip affects neither locomotion nor stereotypy, but it eliminates the locomotor activation produced the D1 agonist SKF82958 (Fig. 5). A similar effect has been described previously in reserpinized mice (24). The mechanisms underlying this interaction, however, have remained unclear. H3R activation has no effect on intrastriatal DA (Fig. 1C), and H3-D1 co-stimulation does not produce elevated stereotypy (Fig. 5B).
A candidate molecular mechanism for this interaction is the inactivation by phosphorylation of GSK3β at Ser9 that we see in D1-MSNs after RAMH treatment (Figs. 4 and 6). GSK3β has been shown to be necessary for the locomotor stimulatory effects of D1 agonist treatment (49). GSK3β inhibition by H3 agonist treatment, specifically in D1-MSNs, may produce a similar effect.
This implies that intrastriatal HA levels in vivo may regulate susceptibility to locomotor stimulation after DA agonist or psychostimulant challenge. Because striatal HA fluctuates slowly, with a circadian pattern (1, 7), this would predict circadian variation in the sensitivity to D1 agonist effects. Diurnal variation in psychostimulant effects has indeed been documented (50). Of note, H3Rs exhibit a high level of basal activity, even in the absence of agonist (51). These receptors may therefore provide a source of constitutive cell type-specific GSK3β regulation partially independent of fluctuations in local HA concentration.
It is striking that H3R antagonists have also been reported to mitigate locomotor and other actions of psychostimulants (52–54) although not, to our knowledge, of D1-specific agonists. The ability of H3R agonist and antagonist co-administration to produce similar effects on DA agonist-induced hyperlocomotion speaks to the complexity of HA regulation of the basal ganglia circuitry and the importance of analytic approaches that isolate effects on discrete cell types.
Conclusions
The presence and importance of postsynaptic H3Rs on striatal MSNs has only been appreciated recently (22, 24–27). The mechanisms whereby they regulate intracellular signaling and basal ganglia function remain incompletely understood. We observe complex, temporally distinct, and cell type-specific MAPK and Akt-GSK3β regulation in D1- and D2-MSNs. These cell type-specific effects, especially those on GSK3β, are not apparent in analysis of whole striatum extracts; they become clear only when analysis is restricted to specific MSN subtypes. The functional importance of H3R activation is apparent in its ability to mitigate hyperlocomotion because of aberrant striatal function.
H3 receptors are increasingly studied as a pharmacological target under a range of conditions (55–57). Appreciation of their unique regulation of intracellular signaling events in MSNs will be critical to understanding their potential as a therapeutic target. The disparate regulation of GSK3β and rpS6 in D1- and D2-MSNs, in particular, indicates multiple points of interaction between DA and HA signaling.
Materials and Methods
Animals
Adult (2.5- to 5-month-old) male mice were used for all experiments. BAC-D2EGFP mice, which were used to analyze signaling specifically in D2-positive and D2-negative cell populations in most experiments, were originally produced by the GENSAT program at Rockefeller University and were generously provided, backcrossed extensively onto C57Bl/6, by Dr. James Surmeier (58). Double-transgenic D1-DARPP-32-FLAG/D2-DARPP-32-Myc (D1F/D2M) mice, which were used to characterize DARPP-32 signaling, have been described previously (14). All transgenic strains were backcrossed with C57BL/6J to ≥ N9. Wild-type C57BL/6J mice were purchased from The Jackson Laboratory. The care and use of animals were supervised by the Yale University Institutional Animal Care and Use Committee.
Drugs
SKF82958 (5 mg/kg) was from Sigma-Aldrich. This dose produced robust locomotor activation in previous studies (37) and in our own pilot experiments. R-(-)-α-methylhistamine (RAMH, 45 mg/kg) was from Tocris. This dose was found in pilot experiments to modulate DA-mediated locomotor effects (Fig. 5). Immepip (20 mg/kg) was from Tocris. This dose of immepip has been shown to modulate cocaine-induced hyperlocomotion in mice (39). All drugs were dissolved in saline solution (Braun, 0.9% NaCl) and injected i.p. (0.1 ml/10 g).
Locomotor Activity and Stereotypic Behavior
Mice were placed into the behavioral room in their home cages to habituate for 20 min. Next, animals were placed individually into a clean cage with fresh bedding (18.5 inches long × 14.5 inches wide × 8 inches high) in a locomotor monitoring apparatus (Omnitech Electronics) and monitored for 30 min to establish a baseline. Animals were injected with saline, SKF82958, RAMH, SKF82958 + RAMH, immepip, or SKF82958 + immepip (8 animals/group). Locomotor activity was recorded for an additional 1 h and quantified as the number of beam breaks using Fusion software (Omnitech Electronics).
For quantification of stereotypy, mice were placed alone in a plexiglass cage to habituate for 20 min, injected i.p. with saline or drugs as above, and then videotaped individually for 1 h (11 inches long × 8 inches wide × 8 inches high). Various stereotypic behaviors (grooming, cage mouthing, feces mouthing, focused sniffing, digging, and forepaw flailing) were scored off-line from video by a trained observer blind to experimental conditions in four 2-min time bins spaced evenly over 1 h. The reported stereotypy score was calculated as the sum of the total time spent doing any of the measured stereotypic behaviors during these scored intervals.
Immunoprecipitation and Immunoblotting for DARPP-32
D1F/D2M mice were sacrificed by focused microwave irradiation 15 min after RAMH administration. Tissue preparation was carried out as described previously (14). Striata were dissected, sonicated in 500 μl of homogenization buffer, and centrifuged for 20 min at 13,000 rpm. An aliquot of total striatal homogenate was separated from each sample and set aside (the “total” sample). The remaining homogenate was added to 50 μl of anti-FLAG-conjugated agarose beads (Sigma, EZView red) and rotated overnight at 4 °C. FLAG beads were washed twice and eluted in 2× sample buffer. β-Mercaptoethanol was added to the flow-through from each sample, which was then transferred to a 50-μl slurry of anti-Myc-conjugated Sepharose beads (Cell Signaling Technology) and rotated overnight at 4 °C. Myc beads were washed three times and eluted in 3× sample buffer with β-mercaptoethanol. Flow-through was collected, and all samples were frozen at −80 °C until processing. Samples from total, FLAG, and Myc extracts were electrophoresed through 4–20% Tris/glycine midi gels (Life Technologies) and electroblotted overnight onto PVDF membranes. Membranes were blocked for 1 h with 5% nonfat dry milk, incubated overnight with Thr(P)34 DARPP-32 Ab (1:1000, Greengard laboratory, Ab cc500), and then washed in the same buffer. HRP-conjugated anti-rabbit secondary (1:2000, Pierce) was added, and incubation was continued for 1 h at room temperature. Blots were developed with Pico ECL solution (Pierce) and imaged using the Bio-Rad Chemi-doc XRS+ imaging system. Blots were immediately stripped for 3 h in stripping buffer (25 mm glycine, 2% SDS (pH 2)) at 50 °C. Blocker was added for 1 h, followed by total DARPP-32 Ab (1:2000, Greengard laboratory, mAb 6a) overnight at 4 °C. Membranes were incubated with HRP-conjugated anti-mouse secondary Ab (1:2000, Pierce) for 1 h at room temperature and developed with Pico ECL solution using the Bio-Rad Imager. Thr75 blots were processed according to the same protocol using primary Thr(P)75 DARPP-32 Ab (1:1000, Ab cc911) and secondary HRP-conjugated anti-rabbit (1:1000, Pierce).
Immunoblotting
C57BL/6J mice were sacrificed by decapitation 15 or 45 min after RAMH or saline injection, and brains were quickly removed and cooled in ice-cold artificial cerebrospinal fluid. The striata of both hemispheres were dissected on an ice-cold surface and then sonicated in radioimmunoprecipitation assay buffer (Cell Signaling Technology) supplemented with protease inhibitor (cOMPLETE, Roche), phosphatase inhibitors (phosSTOP, Roche), and 2 μm okadaic acid (Millipore). Protein concentration was quantified by BCA protocol (Pierce) in accordance with the instructions of the supplier. Protein extracts (25 μg) were separated on 7.5% SDS gels (Bio-Rad) and then transferred to PVDF membranes overnight at 4 °C (Bio-Rad). Next, membranes were incubated overnight at 4 °C with one the following antibodies (all purchased from Cell Signaling Technology): diphospho-Thr202/Tyr204 ERK1/2 (1:2000, 9101S), Thr(P)308-Akt (1:1000, 2965P), Ser(P)473-Akt (1:1000, 4060S), and Ser(P)9-GSK3β (1:1000, 9323S). As a control for basal levels of the total amount of protein, membranes were stripped and reprobed using primary antibodies (all purchased from Cell Signaling Technology) against Akt (1:1000, 4691S), ERK1/2 (1:2000, 9102S), and GSK3β (1:1000, 9315S). A protein loading control was performed by restripping the membranes and then incubating them with an anti-GADPH primary antibody (1:10,000, Millipore, EPR5266). All proteins were detected by using a goat secondary antibody against rabbit or mouse IgG coupled to peroxidase (1:3000, Vector Laboratories) and developed by ECL (SuperSignal West-Pico, Pierce). Images and data were acquired with Chemidoc XRS system (Bio-Rad). Analysis was performed using ImageJ software.
Immunoblot Analysis
All bands were quantified and normalized to corresponding total respective non-phospho-protein per sample. Normalized values were adjusted to the average of normalized control values on each blot. Values were excluded as outliers when they varied by more or less than 2 standard deviations from the overall mean for the experiment.
Confocal Imaging and Cell Counting
BAC-D2EGFP mice were deeply anesthetized with ketamine (100 kg/kg)/xylazine (10 mg/kg) after 15 or 45 min of RAMH or saline administration and then transcardially perfused with 4% paraformaldehyde supplemented with 0.1 mm NaF. Brains were fixed overnight in paraformaldehyde 4% + NaF and then stored at 4° in 30% sucrose. Brains were cut at 20 μm on a cryostat (Leica). Slices were stored in a solution containing 30% glycerin, 30% ethylene glycol, and 1× TBS supplemented with 0.1 mm NaF.
For staining, slices were washed three times for 10 min in TBS (pH 7.4), incubated for 1 h at room temperature in TBS with 0.2% Triton X-100 and 5% donkey serum, and then incubated overnight at room temperature in the same solution with one of the following antibodies: rabbit anti-phospho-MSK1-Thr581 (1:500, Cell Signaling Technology, 9595P), rabbit anti-Ser(P)235/Ser(P)236-rpS6 (1:400, Cell Signaling Technology, 4858S), and rabbit anti-Ser(P)240/Ser(P)244-rpS6 (1:400, Cell Signaling Technology, 5364P). Detection was performed by incubating for 1 h at room temperature with an Alexa Fluor 555-labeled donkey anti-rabbit secondary antibody (1:400, Life Technologies) in blocking solution. For detection of Ser(P)9 GSK3β, before blocking and incubation with the primary antibody, slices were incubated with Bloxall at room temperature for 20 min (Vector Laboratories). Then secondary detection was performed with a tyramide signal amplification-Cy3 kit (PerkinElmer Life Sciences) according to the instructions of the supplier. Sections were washed three times with PBS and then mounted in Vectashield (Vector Laboratories).
Confocal imaging was performed on an Olympus Fluoview FV-1000 confocal microscope. Z stacks (15 μm) were collected from the dorsal striatum from five sections from each mouse. Images were taken from the central dorsal striatum (see Fig. 2C for placement of a typical confocal image within the dorsal striatum).
For each of the phosphorylated proteins (P-MSK, P-rpS6, and P-GSK3β), we counted cells throughout the Z stack that showed clear immunostaining above background in the cell body. Each cell was categorized as either co-expressing EGFP (thus a presumptive D2-MSN) or not co-expressing EGFP (thus a presumptive D1-MSN). All clearly stained cells in each Z stack were categorized and quantified. Immunopositive cells that could not be unambiguously categorized as D1- or D2-MSNs were excluded. All counting was done blind to pharmacological treatment and time point. The total number of cells was averaged for each mouse for analysis. n for each experiment thus reflects the number of mice, not slices. Tissue at the 15- and 45-min time points was collected in separate cohorts of mice, each with a matched saline control group. Counts of GFP-positive and GFP-negative cells were normalized to the corresponding saline group for analysis.
Microdialysis
DA microdialysis was performed as described previously (1). Mice were surgically implanted unilaterally with guide cannulae targeted to the dorsal striatum (anterior-posterior, +0.5 mm; medial-lateral, 2.0 mm; dorsal-ventral, −2.2 mm) (59) under ketamine/xylazine anesthesia using standard stereotaxic technique. Guide cannulae were affixed to the skull using bilateral skull screws and Cerebond skull fixture adhesive (Plastics One, Roanoke, VA). Dummy cannulae were inserted into the guide cannulae during recovery to ensure patency. C57BL/6J mice recovered from surgery for 3–5 days before continuing with microdialysis. Following the recovery period, a microdialysis probe (2 mm, CMA-7, 6-kDa cutoff; CMA Microdialysis, Stockholm, Sweden) was inserted through the guide cannula to a depth of −4 mm relative to the bregma (i.e. 2 mm below the tip of the guide cannula), and mice were left in the home cage for 20–24 h. The next day, the probe was connected to a 2.5-ml Hamilton syringe and continuously perfused with artificial cerebrospinal fluid (Harvard Apparatus, Holliston, MA) at a rate of 2 μl/min using a programmable infusion pump (CMA Microdialysis). Dialysate was collected in 20 μl/10 min fractions on ice and stored at −80 °C for later analysis. Microdialysis was performed in the home cage. DA was measured using HPLC with electrochemical detection as described previously (1). After microdialysis, a small amount of toluidine blue was injected through the cannula. Mice were euthanized, and their brains were removed, sliced, and examined to confirm cannula placement. All cannulae were successfully targeted to the dorsal striatum as intended (n = 8).
Data Analysis
Statistical analysis was performed using Graph Pad 6.0, except for three-way RM-ANOVA of locomotor activity (Fig. 5), which was performed using SPSS 21 (IBM). Values are expressed as mean ± S.E. and compared using Student's t test, one-way ANOVA, two-way or repeated measures two-way ANOVA, followed by Sidak's post-hoc test, as indicated in each case under “Results” and in the figure legends. Differences among experimental conditions were considered statistically significant when p < 0.05.
Author Contributions
M. R. designed and performed experiments, analyzed data, and wrote and edited the manuscript. L. R. F., K. D. H., R. S., and V. P. performed experiments and analyzed data. A. C. N. contributed unique reagents and edited the manuscript. C. P. designed and supervised experiments, analyzed data, and wrote and edited the manuscript.
Acknowledgments
We thank Paul Greengard for antibodies, James Surmeier for D2-GFP transgenic mice, and Stacey Wilber and Jessica André for assistance with mouse genotyping and management.
This work was supported by the Allison Family Foundation (to C. P.), the Tourette Syndrome Association (to L. F.), National Institutes of Health Grants R01MH091861 (to C. P.) and DA10044 and MH090963 (to A. C. N.), and the State of Connecticut through its support of the Ribicoff Research Facilities at the Connecticut Mental Health Center. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DA
- dopamine
- HA
- histamine
- MSN
- medium spiny neuron
- EGFP
- enhanced GFP
- MSK
- mitogen- and stress-activated kinase
- RAMH
- R-(-)-α-methylhistamine
- Ab
- antibody/antibodies
- RM-ANOVA
- repeated measures analysis of variance
- SAL
- saline
- SKF
- H3R
- H3 receptor.
References
- 1. Castellan Baldan L., Williams K. A., Gallezot J. D., Pogorelov V., Rapanelli M., Crowley M., Anderson G. M., Loring E., Gorczyca R., Billingslea E., Wasylink S., Panza K. E., Ercan-Sencicek A. G., Krusong K., Leventhal B. L., et al. (2014) Histidine decarboxylase deficiency causes Tourette syndrome: parallel findings in humans and mice. Neuron 81, 77–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crittenden J. R., and Graybiel A. M. (2011) Basal ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front. Neuroanat. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Graybiel A. M. (2008) Habits, rituals, and the evaluative brain. Annu. Rev. Neurosci. 31, 359–387 [DOI] [PubMed] [Google Scholar]
- 4. Langen M., Durston S., Kas M. J., van Engeland H., and Staal W. G. (2011) The neurobiology of repetitive behavior: of men. Neurosci. Biobehav. Rev. 35, 356–365 [DOI] [PubMed] [Google Scholar]
- 5. Langen M., Kas M. J., Staal W. G., van Engeland H., and Durston S. (2011) The neurobiology of repetitive behavior: of mice. Neurosci. Biobehav. Rev. 35, 345–355 [DOI] [PubMed] [Google Scholar]
- 6. Macpherson T., Morita M., and Hikida T. (2014) Striatal direct and indirect pathways control decision-making behavior. Front. Psychol. 5, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haas H. L., Sergeeva O. A., and Selbach O. (2008) Histamine in the nervous system. Physiol. Rev. 88, 1183–1241 [DOI] [PubMed] [Google Scholar]
- 8. Pillot C., Heron A., Cochois V., Tardivel-Lacombe J., Ligneau X., Schwartz J. C., and Arrang J. M. (2002) A detailed mapping of the histamine H(3) receptor and its gene transcripts in rat brain. Neuroscience 114, 173–193 [DOI] [PubMed] [Google Scholar]
- 9. Bolam J. P., and Ellender T. J. (2015) Histamine and the striatum. Neuropharmacology 106, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calabresi P., Picconi B., Tozzi A., Ghiglieri V., and Di Filippo M. (2014) Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030 [DOI] [PubMed] [Google Scholar]
- 11. Greengard P.(2001) The neurobiology of dopamine signaling. Biosci. Rep. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 12. Girault J. A. (2012) Integrating neurotransmission in striatal medium spiny neurons. Adv. Exp. Med. Biol. 970, 407–429 [DOI] [PubMed] [Google Scholar]
- 13. Sharp T., Zetterström T., Ljungberg T., and Ungerstedt U. (1987) A direct comparison of amphetamine-induced behaviours and regional brain dopamine release in the rat using intracerebral dialysis. Brain Res. 401, 322–330 [DOI] [PubMed] [Google Scholar]
- 14. Bateup H. S., Svenningsson P., Kuroiwa M., Gong S., Nishi A., Heintz N., and Greengard P. (2008) Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci. 11, 932–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beaulieu J. M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., and Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 16. Ercan-Sencicek A. G., Stillman A. A., Ghosh A. K., Bilguvar K., O'Roak B. J., Mason C. E., Abbott T., Gupta A., King R. A., Pauls D. L., Tischfield J. A., Heiman G. A., Singer H. S., Gilbert D. L., Hoekstra P. J., et al. (2010) L-histidine decarboxylase and Tourette's syndrome. N. Engl. J. Med. 362, 1901–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandez T. V., Sanders S. J., Yurkiewicz I. R., Ercan-Sencicek A. G., Kim Y. S., Fishman D. O., Raubeson M. J., Song Y., Yasuno K., Ho W. S., Bilguvar K., Glessner J., Chu S. H., Leckman J. F., King R. A., et al. (2012) Rare copy number variants in Tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry 71, 392–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karagiannidis I., Dehning S., Sandor P., Tarnok Z., Rizzo R., Wolanczyk T., Madruga-Garrido M., Hebebrand J., Nöthen M. M., Lehmkuhl G., Farkas L., Nagy P., Szymanska U., Anastasiou Z., Stathias V., et al. (2013) Support of the histaminergic hypothesis in Tourette syndrome: association of the histamine decarboxylase gene in a large sample of families. J. Med. Genet. 50, 760–764 [DOI] [PubMed] [Google Scholar]
- 19. Krusong K., Ercan-Sencicek A. G., Xu M., Ohtsu H., Anderson G. M., State M. W., and Pittenger C. (2011) High levels of histidine decarboxylase in the striatum of mice and rats. Neurosci. Lett. 495, 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rapanelli M., Frick L. R., Pogorelov V., Ota K. T., Abbasi E., Ohtsu H., and Pittenger C. (2014) Dysregulated intracellular signaling in the striatum in a pathophysiologically grounded model of Tourette syndrome. Eur. Neuropsychopharmacol. 24, 1896–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu M., Li L., Ohtsu H., and Pittenger C. (2015) Histidine decarboxylase knockout mice, a genetic model of Tourette syndrome, show repetitive grooming after induced fear. Neurosci. Lett. 595, 50–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Panula P., and Nuutinen S. (2013) The histaminergic network in the brain: basic organization and role in disease. Nat. Rev. Neurosci. 14, 472–487 [DOI] [PubMed] [Google Scholar]
- 23. Schlicker E., Malinowska B., Kathmann M., and Göthert M. (1994) Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam. Clin. Pharmacol. 8, 128–137 [DOI] [PubMed] [Google Scholar]
- 24. Ferrada C., Ferré S., Casadó V., Cortés A., Justinova Z., Barnes C., Canela E. I., Goldberg S. R., Leurs R., Lluis C., and Franco R. (2008) Interactions between histamine H3 and dopamine D2 receptors and the implications for striatal function. Neuropharmacology 55, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferrada C., Moreno E., Casadó V., Bongers G., Cortés A., Mallol J., Canela E. I., Leurs R., Ferré S., Lluís C., and Franco R. (2009) Marked changes in signal transduction upon heteromerization of dopamine D1 and histamine H3 receptors. Br. J. Pharmacol. 157, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moreno E., Hoffmann H., Gonzalez-Sepúlveda M., Navarro G., Casadó V., Cortés A., Mallol J., Vignes M., McCormick P. J., Canela E. I., Lluís C., Moratalla R., Ferré S., Ortiz J., and Franco R. (2011) Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J. Biol. Chem. 286, 5846–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreno E., Moreno-Delgado D., Navarro G., Hoffmann H. M., Fuentes S., Rosell-Vilar S., Gasperini P., Rodríguez-Ruiz M., Medrano M., Mallol J., Cortés A., Casadó V., Lluís C., Ferré S., Ortiz J., et al. (2014) Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling: sigma1-D1-H3 receptor complexes as key targets for reducing cocaine's effects. J. Neurosci. 34, 3545–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bongers G., Sallmen T., Passani M. B., Mariottini C., Wendelin D., Lozada A., Marle A., Navis M., Blandina P., Bakker R. A., Panula P., and Leurs R. (2007) The Akt/GSK-3β axis as a new signaling pathway of the histamine H(3) receptor. J. Neurochem. 103, 248–258 [DOI] [PubMed] [Google Scholar]
- 29. Mariottini C., Scartabelli T., Bongers G., Arrigucci S., Nosi D., Leurs R., Chiarugi A., Blandina P., Pellegrini-Giampietro D. E., and Passani M. B. (2009) Activation of the histaminergic H3 receptor induces phosphorylation of the Akt/GSK-3 β pathway in cultured cortical neurons and protects against neurotoxic insults. J. Neurochem. 110, 1469–1478 [DOI] [PubMed] [Google Scholar]
- 30. Ellenbroek B. A. (2013) Histamine H(3) receptors, the complex interaction with dopamine and its implications for addiction. Br. J. Pharmacol. 170, 46–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Torrent A., Moreno-Delgado D., Gómez-Ramírez J., Rodríguez-Agudo D., Rodríguez-Caso C., Sánchez-Jiménez F., Blanco I., and Ortiz J. (2005) H3 autoreceptors modulate histamine synthesis through calcium/calmodulin- and cAMP-dependent protein kinase pathways. Mol. Pharmacol. 67, 195–203 [DOI] [PubMed] [Google Scholar]
- 32. Beaulieu J. M., and Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 [DOI] [PubMed] [Google Scholar]
- 33. Svenningsson P., Nairn A. C., and Greengard P. (2005) DARPP-32 mediates the actions of multiple drugs of abuse. AAPS J. 7, E353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park H. Y., Kang Y. M., Kang Y., Park T. S., Ryu Y. K., Hwang J. H., Kim Y. H., Chung B. H., Nam K. H., Kim M. R., Lee C. H., Han P. L., and Kim K. S. (2014) Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson's disease. J. Neurosci. 34, 11744–11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deak M., Clifton A. D., Lucocq L. M., and Alessi D. R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 17, 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biever A., Valjent E., and Puighermanal E. (2015) Ribosomal protein S6 phosphorylation in the nervous system: from regulation to function. Front. Mol. Neurosci. 8, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Desai R. I., Terry P., and Katz J. L. (2005) A comparison of the locomotor stimulant effects of D1-like receptor agonists in mice. Pharmacol. Biochem. Behav. 81, 843–848 [DOI] [PubMed] [Google Scholar]
- 38. Norman D. A., and Shallice T. (1986) in Consciousness and Self-Regulation: Advances in Research and Theory (Davidson R., Schwarte G., and Shapiro D., eds) pp. 1–8, Plenum, New York [Google Scholar]
- 39. Brabant C., Alleva L., Grisar T., Quertemont E., Lakaye B., Ohtsu H., Lin J. S., Jatlow P., Picciotto M. R., and Tirelli E. (2009) Effects of the H3 receptor inverse agonist thioperamide on cocaine-induced locomotion in mice: role of the histaminergic system and potential pharmacokinetic interactions. Psychopharmacology 202, 673–687 [DOI] [PubMed] [Google Scholar]
- 40. Ellender T. J., Huerta-Ocampo I., Deisseroth K., Capogna M., and Bolam J. P. (2011) Differential modulation of excitatory and inhibitory striatal synaptic transmission by histamine. J. Neurosci. 31, 15340–15351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meyuhas O. (2008) Physiological roles of ribosomal protein S6: one of its kind. Int. Rev. Cell Mol. Biol. 268, 1–37 [DOI] [PubMed] [Google Scholar]
- 42. Pende M. (2006) mTOR, Akt, S6 kinases and the control of skeletal muscle growth. Bulletin du cancer 93(5):E39–43 [PubMed] [Google Scholar]
- 43. Roux P. P., Shahbazian D., Vu H., Holz M. K., Cohen M. S., Taunton J., Sonenberg N., and Blenis J. (2007) RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 282, 14056–14064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Casas-Terradellas E., Tato I., Bartrons R., Ventura F., and Rosa J. L. (2008) ERK and p38 pathways regulate amino acid signalling. Biochim. Biophys. Acta 1783, 2241–2254 [DOI] [PubMed] [Google Scholar]
- 45. Frödin M., and Gammeltoft S. (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 151, 65–77 [DOI] [PubMed] [Google Scholar]
- 46. Hauge C., and Frödin M. (2006) RSK and MSK in MAP kinase signalling. J. Cell Sci. 119, 3021–3023 [DOI] [PubMed] [Google Scholar]
- 47. Roux P. P., and Topisirovic I. (2012) Regulation of mRNA translation by signaling pathways. Cold Spring Harb. Perspect. Biol. 4, a012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendoza M. C., Er E. E., and Blenis J. (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36, 320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller J. S., Tallarida R. J., and Unterwald E. M. (2010) Inhibition of GSK3 attenuates dopamine D1 receptor agonist-induced hyperactivity in mice. Brain Res. Bull. 82, 184–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Webb I. C., Baltazar R. M., Wang X., Pitchers K. K., Coolen L. M., and Lehman M. N. (2009) Diurnal variations in natural and drug reward, mesolimbic tyrosine hydroxylase, and clock gene expression in the male rat. J. Biol. Rhythms 24, 465–476 [DOI] [PubMed] [Google Scholar]
- 51. Morisset S., Rouleau A., Ligneau X., Gbahou F., Tardivel-Lacombe J., Stark H., Schunack W., Ganellin C. R., Schwartz J. C., and Arrang J. M. (2000) High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature 408, 860–864 [DOI] [PubMed] [Google Scholar]
- 52. Banks M. L., Manvich D. F., Bauzo R. M., and Howell L. L. (2009) Effects of histamine H(3) receptor activation on the behavioral-stimulant effects of methamphetamine and cocaine in mice and squirrel monkeys. Pharmacology 83, 164–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mahmood D., Khanam R., Pillai K. K., and Akhtar M. (2012) Protective effects of histamine H3-receptor ligands in schizophrenic behaviors in experimental models. Pharmacol. Rep. 64, 191–204 [DOI] [PubMed] [Google Scholar]
- 54. Vanhanen J., Kinnunen M., Nuutinen S., and Panula P. (2015) Histamine H3 receptor antagonist JNJ-39220675 modulates locomotor responses but not place conditioning by dopaminergic drugs. Psychopharmacology 232, 1143–1153 [DOI] [PubMed] [Google Scholar]
- 55. Brabant C., Alleva L., Quertemont E., and Tirelli E. (2010) Involvement of the brain histaminergic system in addiction and addiction-related behaviors: a comprehensive review with emphasis on the potential therapeutic use of histaminergic compounds in drug dependence. Prog. Neurobiol. 92, 421–441 [DOI] [PubMed] [Google Scholar]
- 56. Rapanelli M., and Pittenger C. (2015) Histamine and histamine receptors in Tourette syndrome and other neuropsychiatric conditions. Neuropharmacology 106, 85–90 [DOI] [PubMed] [Google Scholar]
- 57. Schwartz J. C. (2011) The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br. J. Pharmacol. 163, 713–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chan C. S., Peterson J. D., Gertler T. S., Glajch K. E., Quintana R. E., Cui Q., Sebel L. E., Plotkin J. L., Shen W., Heiman M., Heintz N., Greengard P., and Surmeier D. J. (2012) Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J. Neurosci. 32, 9124–9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paxinos G., and Franklin K. B. J. (2004) The Mouse Brain in Stereotaxic Coordinates, Elsevier Academic Press, Amsterdam [Google Scholar]