

Abstract
Proneural basic helix-loop-helix transcription factor, Atoh1, plays a key role in the development of sensory hair cells. We show here that the level of Atoh1 must be accurately controlled by degradation of the protein in addition to the regulation of Atoh1 gene expression to achieve normal cellular patterning during development of the cochlear sensory epithelium. The stability of Atoh1 was regulated by the ubiquitin proteasome system through the action of Huwe1, a HECT-domain, E3 ubiquitin ligase. An interaction between Huwe1 and Atoh1 could be visualized by a proximity ligation assay and was confirmed by co-immunoprecipitation and mass spectrometry. Transfer of a lysine 48-linked polyubiquitin chain to Atoh1 by Huwe1 could be demonstrated both in intact cells and in a cell-free system, and proteasome inhibition or Huwe1 silencing increased Atoh1 levels. The interaction with Huwe1 and polyubiquitylation were blocked by disruption of casein kinase 1 (CK1) activity, and mass spectrometry and mutational analysis identified serine 334 as an important phosphorylation site for Atoh1 ubiquitylation and subsequent degradation. Phosphorylation by CK1 thus targeted the protein for degradation. Development of an extra row of inner hair cells in the cochlea and an approximate doubling in the number of afferent synapses was observed after embryonic or early postnatal deletion of Huwe1 in cochlear-supporting cells, and hair cells died in the early postnatal period when Huwe1 was knocked out in the developing cochlea. These data indicate that the regulation of Atoh1 by the ubiquitin proteasome pathway is necessary for hair cell fate determination and survival.
Keywords: E3 ubiquitin ligase, hair cell, protein degradation, protein kinase, protein stability, Atoh1, casein kinase, inner ear
Introduction
Loss of mammalian cochlear hair cells, caused by genetic mutations, autoimmune disease, ototoxic medications, exposure to noise, and aging, is usually permanent. Mammals show limited ability to regenerate hair cells (1, 2). Hair cell differentiation is dependent on basic helix-loop-helix (bHLH; transcription factor, Atoh1 (3, 4). Overexpression of Atoh1 via gene transfer results in the generation of new hair cells from inner ear progenitors in the organ of Corti (5).
Increasing information about the transcriptional regulation of the Atoh1 gene has shown that expression of Atoh1 is regulated strictly by overlapping pathways (6–10). We were interested in the downstream regulation of Atoh1 because of the importance of Atoh1 levels for its function in cells of the ear. Posttranslational control of Atoh1 is largely unknown. The ubiquitin-proteasome pathway plays an important role in post-translational regulation of proteins in eukaryotic cells (11). The system not only degrades misfolded or damaged proteins but is also essential for the regulation of cell-signaling pathways, determining the half-lives of proteins (12). Cells utilize spatial distribution of ubiquitin conjugation to regulate local abundance of proteins and compartmentalization of different subcellular domains.
E3 ubiquitin ligases transfer ubiquitin to internal lysine residues of specific proteins to form mono- or polyubiquitin chains after activation by E1 and conjugation by E2. E3 ubiquitin ligases are classified by the occurrence of HECT or RING domains, based on the identity of the domain involved in E2 enzyme interaction (13, 14). More than 600 E3 ligases control levels of eukaryotic proteins. Before ubiquitylation, substrates of the ubiquitin E3 ligases undergo post-translational modification, including phosphorylation, methylation, or acetylation to produce a modified protein containing a “degron” that can be recognized by E3 ubiquitin ligase and targets a protein for ubiquitylation and degradation.
Here, we describe a pathway regulating Atoh1 stability. We show that silencing of HECT-domain E3 ligase Huwe1 decreases the degradation of Atoh1 in the cochlea and in cell lines, which agrees with a previous study identifying Huwe1 as an E3 ligase for Atoh1 (15). We identify a phosphorylated serine that appears to be essential for Atoh1 degradation. The degron is created by phosphorylation of serine 334 by CK1.2 Formation of the phosphodegron affects the interaction of Atoh1 with Huwe1 and subsequent ubiquitylation by the E3 ligase. We also find that disruption of the Huwe1-Atoh1 pathway not only stabilizes Atoh1 but, depending on the time and cell type of Huwe1 deletion, can lead to overproduction of sensory hair cells or to hair cell death. We conclude that proteasomal regulation of Atoh1 determines its level and plays an essential role in cochlear development.
Results
Lys-48-linked Polyubiquitin Targets Atoh1 for Proteasomal Degradation
We assessed the half-life of Atoh1 with and without proteasome inhibition. Half-life was determined using cycloheximide to prevent new protein synthesis and following the time course of Atoh1 disappearance during a chase. Atoh1 protein was almost completely degraded within 2 h of inhibition of new protein synthesis. The half-life, as measured by densitometry in three experiments, was 35.3 min (95% confidence interval: 26.0 to 56.1 min; Fig. 1, A and B). When treated with MG132, the half-life of Atoh1 was significantly extended (Fig. 1, A and B).
FIGURE 1.

Huwe1 interacts with and ubiquitylates Atoh1. A, following treatment with MG132 (10 μm) in the presence of cycloheximide (CHX; 100 μg/ml), lysates of FLAG-HA-Atoh1 293T cells were processed for Western blotting with FLAG antibody (Atoh1) or β-actin antibody (loading control). Labels indicate the time of chase. DMSO was a vehicle control. B, values for the level of Atoh1 determined by densitometry were plotted to determine half-life. Error bars indicate S.E. Data from three experiments are shown. C, FLAG-HA-Atoh1 293T cells were treated with MG132 (10 μm) or DMSO (Control) for 6 h, and the level of protein was measured both before (5% input) and after (IP: Atoh1) immunoprecipitation. Lysates were immunoprecipitated with anti-FLAG antibody and immunoblotted (IB) with antibody to ubiquitin. β-actin is a loading control. D, HEK 293T cells were co-transfected with FLAG-Atoh1 and either wild-type HA-ubiquitin (WT), HA-ubiquitin with all lysines except Lys-48 mutated, or empty vector (Control). FLAG-Atoh1 was immunoprecipitated and blotted with antibodies against HA (ubiquitin) and FLAG (Atoh1). FLAG antibody was used to confirm the immunoprecipitation of Atoh1 (lower panel). E, FLAG-HA tagged Atoh1 was purified from whole cell extracts of FLAG-HA-Atoh1 293T cells. Associated proteins were detected by Coomassie blue staining. The areas indicated by the arrows were excised for mass spectrometry (Tables 2 and 3) and Western blotting. F, FLAG-HA-Atoh1 293T lysates were immunoprecipitated with the indicated antibodies (IgG and HA) and subjected to immunoblotting with an antibodies to Huwe1 and HA. G, FLAG-HA-Atoh1 293T cell lysates were subjected to immunoprecipitation using IgG or Huwe1 antibodies followed by immunoblotting with antibodies to HA and Huwe1. H, Atoh1-Huwe1 interaction was assessed by in situ proximity ligation assay (Duolink) in HEK cells transfected with Atoh1 and Huwe1 together or independently. The scale bar is 10 μm.
Western blotting of immunoprecipitated FLAG-Atoh1 revealed high molecular weight forms of Atoh1, indicating polyubiquitylation (Fig. 1C). Increased density of these high molecular weight forms was seen in samples treated with proteasome inhibitor, MG132, indicating accumulation of polyubiquitylated Atoh1.
We tested whether the ubiquitin attached to Atoh1 used the Lys-48 linkage, which targets proteins to the proteasome. When co-transfection was performed with FLAG-Atoh1 and ubiquitin with mutations in all lysines except Lys-48, high molecular weight bands were indicative of Lys-48 Atoh1 ubiquitylation (Fig. 1D). Thus, Atoh1 was degraded through the ubiquitin-proteasome pathway, and proteasome inhibition extended the half-life of Atoh1 by causing an accumulation of Lys-48-ubiquitylated Atoh1.
Identification of Huwe1 as an Atoh1 Binding Partner
To identify binding partners of Atoh1 for E3 ubiquitin ligases, we performed immunoprecipitation followed by mass spectrometry (14). Lysates of FLAG-HA-Atoh1 293T cells immunoprecipitated with HA antibody were subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to identify proteins associated with Atoh1. We used CompPASS, an unbiased comparative approach for identifying high confidence candidate interacting proteins, to interrogate datasets for parallel mass spectral studies (14, 16). The scoring metric, WDN, which incorporates the frequency of observed interacting partner, its abundance, and the reproducibility of technical replicates, was used to assign scores to each protein. Proteins with a WDN score greater than one were considered high confidence interacting partners (Table 1). HECT domain E3 ubiquitin ligase, Huwe1, was observed and was the only ligase that passed the criteria for WDN score among the Atoh1 interacting proteins. E-proteins, E12, and E47 (TCF3), which are known, interacting partners of Atoh1, were also high confidence candidates, indicating the validity of the immunoprecipitation/mass spectrometry approach. Another E-protein, TCF12, which has not been reported to bind Atoh1, was also found as an Atoh1 binding partner.
TABLE 1.
Identification of Atoh1-interacting proteins
Lysates of FLAG-HA-Atoh1 293T cells were immunoprecipitated with HA antibody and subjected to LC-MS/MS. CompPASS, an unbiased comparative approach for identifying high confidence candidate-interacting proteins, was utilized to interrogate data sets and assign the WDN scoring metrics. Proteins with a WDN score >1 fulfilled these criteria. E3 ligase, Huwe1, is in bold.
| Protein category | Symbol | Gene ID | WDN score |
|---|---|---|---|
| ATOH1 | ATOH1 | 474 | 7.21 |
| E3 ligase | HUWE1 | 10075 | 3.44 |
| Ubiquitin-related enzymes (deubiquitylases) | USP11 | 8237 | 1.41 |
| USP47 | 55031 | 2.00 | |
| E-protein | TCF3 | 6929 | 3.16 |
| TCF12 | 6938 | 2.00 | |
| Cell death and survival | AHSA1 | 10598 | 5.03 |
| ATP2A1 | 487 | 1.41 | |
| MAPK3 | 5595 | 2.00 | |
| PRDX4 | 10549 | 1.23 | |
| PRPS1 | 5631 | 1.00 | |
| SRI | 6717 | 1.41 | |
| Cell movement | ENAH | 55740 | 1.41 |
| FLNC | 2318 | 1.15 | |
| GFAP | 2670 | 2.45 | |
| PDE4D | 5144 | 1.41 | |
| SLC3A2 | 6520 | 1.41 | |
| TUBA1C | 84790 | 1.39 | |
| TUBA4A | 7277 | 3.00 | |
| TUBB2B | 347733 | 2.58 | |
| ZYX | 7791 | 1.58 | |
| Others | AAMP | 14 | 1.00 |
| ACOX1 | 51 | 1.72 | |
| BCKDHA | 593 | 2.00 | |
| CCDC124 | 115098 | 1.00 | |
| CPOX | 1371 | 2.00 | |
| CTPS2 | 56474 | 2.00 | |
| EIF2C1 | 26523 | 1.00 | |
| EIF2C2 | 27161 | 2.45 | |
| EIF2C3 | 192669 | 1.00 | |
| FHL3 | 2275 | 1.00 | |
| NEK9 | 91754 | 2.83 | |
| NT5DC2 | 64943 | 1.41 | |
| PDXK | 8566 | 1.41 | |
| SEC23A | 10484 | 1.23 | |
| SEC23B | 10483 | 1.00 | |
| SERPINH1 | 871 | 1.00 | |
| TNRC6C | 57690 | 2.00 | |
| YARS2 | 51067 | 1.41 |
We then asked whether Huwe1 formed a physical complex with Atoh1. First, mass spectrometric analysis of a Coomassie Blue-stained band at 450 kDa from a lysate immunoprecipitated with an HA antibody identified Huwe1 as an interacting protein (Fig. 1E and Table 2). Mass spectrometry of a 45-kDa band from the same gel confirmed that the immunoprecipitated protein was FLAG-HA-Atoh1 (Fig. 1E, Table 3). Based on reciprocal co-immunoprecipitation analysis with antibodies against either FLAG-HA-Atoh1 or endogenous Huwe1, Atoh1 interacted with endogenous Huwe1 (Fig. 1, F and G).
TABLE 2.
Analysis of Huwe1
Coomassie Blue-stained band of 450 kDa indicated in Fig. 1E was excised and subjected to mass spectrometric analysis to validate Huwe1 (shown in bold) as a binding partner of Atoh1.
| Gene symbol | Total peptide | Unique peptide | Average peptide score | Protein coverage |
|---|---|---|---|---|
| % | ||||
| HUWE1 | 279 | 122 | 3.5367 | 28.78 |
| PRKDC | 161 | 112 | 3.3479 | 25.48 |
| MAP1B | 133 | 78 | 3.7349 | 37.68 |
| DYNC1H1 | 87 | 75 | 3.2275 | 18.02 |
| KIF11 | 79 | 46 | 3.3049 | 37.97 |
| SRRM2 | 53 | 38 | 3.7266 | 20.75 |
| MACF1 | 51 | 50 | 3.2235 | 8.57 |
| MYCBP2 | 51 | 47 | 3.3464 | 13.64 |
| MDN1 | 41 | 37 | 3.3880 | 8.79 |
| HERC2 | 36 | 34 | 3.2990 | 9.08 |
| RNF213 | 32 | 30 | 3.0488 | 6.38 |
| UBR5 | 31 | 26 | 3.3120 | 12.54 |
| UTRN | 30 | 30 | 3.4022 | 10.81 |
| MGA | 29 | 27 | 3.6467 | 13.52 |
| AKAP9 | 26 | 26 | 2.9820 | 8.08 |
| PRRC2C | 26 | 21 | 3.2923 | 8.08 |
| CEP350 | 23 | 21 | 3.1575 | 11.08 |
| SON | 22 | 20 | 3.6486 | 9.66 |
| PLEC | 17 | 16 | 2.8815 | 3.86 |
| MAP1A | 15 | 14 | 3.2701 | 8.46 |
| DMXL1 | 13 | 12 | 3.1873 | 5.48 |
| TRRAP | 12 | 12 | 3.1620 | 3.91 |
| SACS | 12 | 12 | 2.5812 | 2.88 |
| GOLGB1 | 12 | 10 | 2.8166 | 3.28 |
TABLE 3.
Analysis to confirm immunoprecipitation of Atoh1
Coomassie Blue-stained band of 45 kDa indicated in Fig. 1E was excised and subjected to mass spectrometry. ATOH1 and CK1 isoforms are in bold.
| Gene symbol | Total peptide | Unique peptide | Average peptide score | Protein coverage |
|---|---|---|---|---|
| % | ||||
| TUBB2A | 159 | 40 | 3.0903 | 55.5 |
| ATOH1 | 155 | 19 | 2.1144 | 34.18 |
| TUBA1A | 112 | 34 | 3.1383 | 66.08 |
| RPL4 | 106 | 36 | 2.4767 | 53.63 |
| EEF1A1 | 103 | 28 | 2.6454 | 58.87 |
| TUFM | 72 | 38 | 3.2781 | 68.36 |
| RPL3 | 67 | 29 | 2.5399 | 45.66 |
| SSB | 65 | 30 | 2.8608 | 5.43 |
| RBMX | 65 | 27 | 2.5009 | 47.06 |
| ENO1 | 62 | 34 | 3.1674 | 58.29 |
| PSMC5 | 52 | 31 | 3.5295 | 64.29 |
| PSMC2 | 50 | 29 | 2.9925 | 63.74 |
| YARS2 | 11 | 8 | 3.3677 | 29.77 |
| TUBA1C | 7 | 11 | 3.6062 | 15.14 |
| SERPINH1 | 9 | 7 | 2.5123 | 20.81 |
| CSNK1D | 7 | 4 | 3.1615 | 12.5 |
| CSNK1E | 6 | 3 | 2.8233 | 9.86 |
| ACOX1 | 5 | 4 | 2.8799 | 10.30 |
| BCKDHA | 5 | 3 | 2.6629 | 9.44 |
| PRPS1 | 3 | 3 | 2.6642 | 18.46 |
| TUBB2B | 2 | 1 | 3.0318 | 2.70 |
| TUBA4A | 1 | 1 | 3.4258 | 31.31 |
| PRDX4 | 1 | 1 | 3.1432 | 7.01 |
| AAMP | 1 | 1 | 2.3377 | 2.53 |
| PDE4B | 1 | 1 | 2.2331 | 2.04 |
Additional evidence for an Atoh1-Huwe1 interaction came from an in situ proximity ligation assay to visualize protein-protein interaction at a single molecule level (17). Analysis of the interaction showed fluorescent labeling in HEK cells co-transfected with Atoh1 and Huwe1 (Fig. 1H).
Huwe1 Ubiquitylates Atoh1
To determine whether Huwe1 affected the stability of Atoh1, FLAG-HA-Atoh1 293T cells were transfected with Huwe1 or Huwe1 shRNA in gain- and loss-of-function experiments, respectively. Huwe1 overexpression reduced Atoh1 levels, whereas Huwe1 knockdown stabilized Atoh1 (Fig. 2, A and B). Treatment of FLAG-HA-Atoh1 293T cells with Huwe1 shRNA extended the half-life of Atoh1 in an assay using a cycloheximide chase (Fig. 2, C and D). These results support the hypothesis that Huwe1 degrades Atoh1 through the ubiquitin-proteasome pathway.
FIGURE 2.

Huwe1 plays a role in the degradation of Atoh1. A, FLAG-HA-Atoh1 293T cells were transfected with Huwe1 (1 μg/ml) for 48 h. Quantification of Atoh1, Huwe1, and loading control (β-actin) was performed by Western blotting (IB) and densitometry (Huwe1 and Atoh1 levels are shown below each lane). B, FLAG-HA-Atoh1 293T cells were transfected with Huwe1 shRNA (100 nm) for 72 h. Quantification of Atoh1 and Huwe1 was performed by Western blotting and densitometry (Huwe1 and Atoh1 levels are shown below each lane) and normalized to a loading control (β-actin). C and D, FLAG-HA-Atoh1 293T cells were transfected with either control or Huwe1 shRNA for 72 h and treated with cycloheximide (CHX, 100 μm) for the indicated times. Atoh1 (FLAG) and Huwe1 were analyzed by Western blotting, and densitometry was normalized to a loading control in three experiments. The ratio of Atoh1 to β-actin was plotted. Error bars indicate S.E. E, recombinant Atoh1 (100 nm) was incubated with ubiquitin, E1 (UBE1), E2 (UbcH5), and full-length Huwe1 in an ATP-regenerating buffer. The reactions were immunoblotted for ubiquitin. F, Coomassie Blue staining of purified E1 (UBE1), E2 (UbcH5), and E3 (Huwe1) proteins used in E. G, HeLa cells were co-transfected with FLAG-Atoh1, HA-ubiquitin (WT) or mutant Lys-48 ubiquitin, and Myc-Huwe1. FLAG-Atoh1 was immunoprecipitated (IP) and subjected to Western blotting with an anti-FLAG antibody for ubiquitylated Atoh1 and an anti-Myc antibody for Huwe1. H, re-blotting of G with an anti-HA (ubiquitin) antibody to show wild-type or Lys-48 polyubiquitin chain conjugated to Atoh1. I, input control for G. J, HeLa cells, co-transfected with FLAG-Atoh1, Myc-Huwe1 (WT) or mutant C4341S Huwe1, and HA-ubiquitin (WT), were lysed and analyzed with an anti-Myc antibody for Huwe1, an anti-FLAG antibody for Atoh1, and an anti-β-actin antibody in the same experiment as shown in I. The mutant Huwe lane is shown with the same Huwe control lanes as in I.
Because polyubiquitylation is required for proteasomal degradation, we tested whether Huwe1 catalyzed Atoh1 polyubiquitylation. Huwe1 ubiquitylated Atoh1 in a cell-free ubiquitylation assay in the presence of recombinant E1, E2 (UbcH5), and full-length Huwe1, as shown by immunoblotting with anti-ubiquitin (Fig. 2, E and F). The fraction of Huwe1 polyubiquitylated in the cell-free system appeared to be small based on Coomassie Blue staining (data not shown).
Increased ubiquitylation of Atoh1 was observed after co-transfecting Huwe1 into HeLa cells transfected with FLAG-Atoh1, HA-ubiquitin, or ubiquitin with all lysines mutated except lysine 48 (Fig. 2, G–I). The decrease in Atoh1 observed after Huwe1 overexpression (Fig. 2I) was not seen when wild-type Huwe1 was replaced with Huwe1 containing a HECT domain mutation in critical cysteine (18, 19), C4341 (Fig. 2J), indicating that Huwe1 activity was required for the ubiquitylation of Atoh1.
CK1 Regulates Atoh1 Stability through the Ubiquitin-Proteasome Pathway
To ask how Huwe1 interacted with Atoh1 we first tested whether phosphorylation of Atoh1 had an influence on Huwe1 binding. Phosphorylation-dependent binding was suggested by a decrease in Huwe1 interaction with Atoh1 after treatment with λ-phosphatase (Fig. 3, A and B). Our list of Atoh1 interacting proteins obtained by mass spectrometry included two isoforms (CSNK1D and CSNK1E) of CK1, and we initiated studies of a potential role for CK1 in Atoh1 degradation (Table 3).
FIGURE 3.

Atoh1 phosphorylation by CK1 is essential for Huwe1-mediated degradation. A and B, HEK cells were transfected with FLAG-Atoh1 for 24 h. Cell lysates were treated with or without λ-phosphatase before immunoprecipitation with a FLAG antibody. Atoh1 and associated proteins were detected by Coomassie Blue staining (A). The areas indicated (40 and 450 kDa) were excised for mass spectrometry and Western blotting (IB) with FLAG (Atoh1) and Huwe1 antibodies (B). IP, immunoprecipitation, C, HEK cells were co-transfected with FLAG-Atoh1 and Myc-CK1 (including CK1α, CK1δ, and CK1ϵ). Immunoprecipitation was performed under non-denaturing conditions with a FLAG antibody. CK1 and Atoh1 were detected with antibodies to Myc and FLAG. Immunoblotting of 5% input is shown below. D, HEK cells were transfected with FLAG-Atoh1 and treated with CK1 inhibitor, D4476 (10 μm), for 18 h. Lysates were processed for Western blotting with FLAG (Atoh1) or β-actin (loading control) antibodies. E, HEK cells were transfected with FLAG-Atoh1 for 40 h and treated with either vehicle (DMSO) or D4476 (10 μm) for 18 h followed by cycloheximide (100 μg/ml) incubation for the indicated times. β-Actin served as a loading control for input protein. F, the ratio from E of Atoh1 to β-actin based on densitometry was plotted. Error bars indicate S.E. Data from three experiments are shown. G, HEK cells were co-transfected with HA-ubiquitin, FLAG-Atoh1, and/or Myc-CK1∂ for 48 h and treated with MG132 (10 μm) for 4 h and λ-phosphatase before immunoprecipitation with a FLAG antibody. Endogenous Huwe1 was detected with an antibody to Huwe1, and Atoh1 was detected with anti-FLAG. Immunoblotting of 5% input is shown below. H, HEK cells were co-transfected with HA-ubiquitin and FLAG-Atoh1 for 48 h and treated with CK1 inhibitors (D4476) and/or proteasome inhibitor MG132 (10 μm) for 4 h. Immunoprecipitation was performed with anti-FLAG antibody. Endogenous Huwe1 was detected with an antibody to Huwe1. Atoh1 was detected with anti-FLAG, and ubiquitin was detected with an antibody to HA. Immunoblotting of 5% input is shown below.
Co-immunoprecipitation showed that CK1α, CK1δ, and CK1ϵ bound to Atoh1 (Fig. 3C). CK1 overexpression decreased steady-state Atoh1 level, whereas inactivation of CK1 by small-molecule inhibitor, D4476, resulted in Atoh1 accumulation (Fig. 3D). CK1 inhibition extended the half-life of Atoh1 to >2 h, compared with about 30 min for a control treated with DMSO in a cycloheximide-chase assay (Fig. 3, E and F), indicating a role in regulation of Atoh1 stability.
Because Huwe1 bound to Atoh1 in a phosphorylation-dependent manner and overexpression of CK1 affected the steady-state Atoh1 level, we asked whether CK1∂ exerted its effect on Atoh1 stability through the ubiquitin-proteasome pathway. Overexpression of CK1 increased the interaction between Atoh1 and Huwe1, whereas CK1-mediated Huwe1 binding was abolished by λ-phosphatase (Fig. 3G). Moreover, CK1 inhibitor, D4476, diminished Atoh1-Huwe1 interaction and ubiquitylation (Fig. 3H), suggesting that CK1 specified Atoh1 for degradation by the ubiquitin-proteasome pathway.
Serine 334 Phosphorylation by CK1 Targets Atoh1 for Huwe1-catalyzed Proteasomal Degradation
To assess which regions affected degradation of Atoh1, we next generated a panel of two N-terminal (Δ10–93 for deletion 1 and Δ94–156 for deletion 2) and two C-terminal (Δ214–305 for deletion 3 and Δ306–347 deletion 4) deletions of Atoh1, retaining the basic bHLH domain (Fig. 4A, right panel). Atoh1-deletion 4 had the longest half-life in a cycloheximide chase assay, suggesting that motifs affecting the half-life of Atoh1 fell between amino acids 306 and 347 (Fig. 4B). In addition, we also found that Atoh1 deletion 4 was not ubiquitylated and showed decreased Huwe1 binding (Fig. 4C), suggesting that a degron motif resided in this region.
FIGURE 4.

Serine 334 is a critical residue for Atoh1 degradation. A, HEK cells were transfected with either wild-type or a truncated FLAG-Atoh1 (4 mutants as shown) for 48 h and incubated with cycloheximide (CHX; 100 μg/ml) for the indicated times. β-Actin served as a loading control for input protein. B, the ratio of Atoh1 to β-actin based on densitometry was plotted. Data from three experiments are shown. Error bars indicate S.E. C, HEK cells were co-transfected with wild-type or truncated FLAG-Atoh1 and HA-ubiquitin for 48 h. Immunoprecipitation (IP) was performed with anti-FLAG antibody. Ubiquitin and Atoh1 were detected with antibodies to HA and FLAG. Endogenous Huwe1 was detected with an anti-Huwe1 antibody. Immunoprecipitation with IgG was used for the control. Blotting of 5% of total inputs was performed with antibodies against FLAG (Atoh1) and Huwe1. IB, immunoblot. D, conserved serines at 325, 328, 331, and 334 are marked with asterisks in an alignment of the Atoh1 C-terminal region. E, HEK cells were transfected with wild-type or mutated FLAG-Atoh1 for 40 h and treated with either vehicle (DMSO) or proteasome inhibitor (MG132, 10 μm). β-Actin served as a loading control for input protein. F, HEK cells were transfected with wild-type or mutated FLAG-Atoh1 for 40 h and treated with MG132 (10 μm). After 6 h of treatment S334A had the smallest increase in Atoh1 (vehicle treatment is marked with a minus sign) compared with wild-type or other Atoh1 mutants. β-Actin served as a loading control for input protein. The ratio of Atoh1 to β-actin based on densitometry was plotted. Data from three experiments are shown. Error bars indicate S.E. G, HEK cells were transfected with either wild-type or mutated FLAG-Atoh1 for 40 h and incubated with cycloheximide (100 μg/ml) for the indicated times. The ratio of Atoh1 to β-actin based on densitometry was plotted. Data from three experiments are shown. H and I, HEK cells were transfected with either wild-type or S339A FLAG-Atoh1 for 48 h and incubated with cycloheximide (100 μg/ml) for the indicated times. β-Actin served as a loading control (H). The ratio of Atoh1 to β-actin based on densitometry was plotted (I). Data from three experiments are shown. Error bars indicate S.E. J, HEK cells were co-transfected with ubiquitin with all lysines except Lys-48 mutated and wild type (WT) or mutated (S334A) FLAG-Atoh1 for 48 h. Immunoprecipitation was performed with an antibody to FLAG. Huwe1, Atoh1, and ubiquitin were detected with antibodies against Huwe1, HA, and FLAG. K, HEK cells were transfected with wild-type (WT) or mutated FLAG-Atoh1 (deletion 4 (del4) or S334A) for 40 h and treated with either vehicle (DMSO, marked with a minus sign) or D4476 (10 μm). β-Actin served as a loading control for input protein. Additional bands shown in the β-actin panel are from Atoh1 remaining after blot stripping.
Cross-species sequence comparison of Atoh1 by MegaAlign (DNAstar, Madison, WI) indicated that serines 309, 325, 328, 331, and 334 within the Atoh1-deletion-4 region were conserved across species (Fig. 4D). To assess biological function, we generated mutated Atoh1 with alanine in place of each serine. The S334A mutant was protected from degradation based on its higher level of expression and the diminished effect of proteasome inhibition with MG132, compared with wild-type Atoh1 and the other mutants (Fig. 4E). Mutations at positions 328 and 331 modestly prolonged Atoh1 half-life, whereas mutation at position 334 had a dramatic effect (Fig. 4F). Another potential site in the C terminus, serine 339 (15), as well as N-terminal serines 52 and 56 (20), were also reported to be responsible for Atoh1 stability. However, none of the mutations increased the stability as efficiently as serine 334 (Fig. 4, G–I). Decreased Huwe1 binding and ubiquitylation were observed when Atoh1 mutated at serine 334 was transfected into HEK cells followed by immunoprecipitation (Fig. 4J). Therefore, we conclude that serine 334 in the C terminus of Atoh1 contains a degron that specifies Atoh1 for proteasomal degradation.
We detected phosphorylation of serine 334 by mass spectrometry after CK1 overexpression (Fig. 5; mass spectrometry data in Table 4). We suspect that rapid degradation of Ser-334-phosphorylated Atoh1 under control conditions precludes detection, and we only detect Ser-334 phosphorylation with CK1 overexpression. We next tested whether serine 334 was involved in CK1-mediated degradation of Atoh1 using CK1 inhibitor, D4476, and observed stabilization of wild-type Atoh1 but not Atoh1 containing the truncated degron-containing region (Atoh1-deletion 4) or mutated serine 334 (Fig. 4K). We, therefore, conclude that CK1 phosphorylates serine 334 of Atoh1 to form a phosphodegron, thus targeting Atoh1 for Huwe1-catalyzed ubiquitylation and proteasomal degradation.
FIGURE 5.

Atoh1 phosphorylation sites were detected by mass spectrometry. Phosphorylated residues are shown in red (CK1∂ overexpression) or green (control).
TABLE 4.
Summary of Atoh1 Phosphorylation
| Positiona | Atoh1b | Atoh1 + CK1c | Conservationd | Play a role in Atoh1 degradatione | Peptidef |
|---|---|---|---|---|---|
| 309 | x | x | N | No | DLSPSLPGGILQPVQEDNSK |
| 311 | x | N** | N/A | DLSPSLPGGILQPVQEDNSK | |
| 325 | x | Y | No | DLSPSLPGGILQPVQEDNSKTSPR | |
| 328 | x | Y | Low | DLSPSLPGGILQPVQEDNSKTSPR | |
| 331 | x | x | Y | Low | SHRSDGEFSPHSHYSDSDEAS |
| 334 | x | Y | High | SDGEFSPHSHYSDSDEAS | |
| 339 | x | x | N* | No | SDGEFSPHSHYSDSDEAS |
| 345 | x | x | N* | N/A | SDGEFSPHSHYSDSDEAS |
| 347 | x | x | N* | N/A | SDGEFSPHSHYSDSDEAS |
a Position of the amino acid.
b Peptide found after Atoh1 overexpression only.
c Peptide found after Atoh1 and CK1 overexpression.
d Cross-species sequence comparison. Y, conserved; N*, non-conserved in one tested species; N**, non-conserved in two tested species; N, non-conserved.
e Cycloheximide-chase assay results shown in Fig. 4.
f Phosphorylated peptide sequence with the phosphorylated residue underlined.
Disruption of the Ubiquitin-Proteasome Pathway Up-regulates Atoh1 in the Cochlea
To define the role of the ubiquitin-proteasome pathway in the regulation of Atoh1 in the cochlea, we tested if proteasome inhibition stabilized cochlear Atoh1. Atoh1 levels were increased after MG132 treatment of the organ of Corti in newborn mice (Fig. 6A). To test whether Huwe1 was involved in this pathway, we treated organ of Corti explants from newborn mice with 100 nm Huwe1 siRNA for 72 h, suppressing Huwe1 mRNA by 59.5% (Fig. 6B) and protein by 55% (Fig. 6C). Depletion of Huwe1 caused a marked accumulation of Atoh1 in the cochlea. Hair cell marker, myosin VIIa, was also up-regulated by Huwe1 siRNA, indicating a potential increase in the expression of hair cell-related genes after stabilization of Atoh1 by knockdown of Huwe1 (Fig. 6C).
FIGURE 6.

Huwe1 knockdown increased the stability of Atoh1 in organ of Corti explants. A, organs of Corti were treated with proteasome inhibitor MG132 (10 μm) for 6 h. Atoh1 and β-actin (loading control) were quantified by Western blotting (IB) and densitometry (Atoh1 levels are shown below each lane). B, P1 organ of Corti was treated with the indicated siRNA (100 nm) for 72 h. Experiments were done in triplicate. Error bars indicate S.E. (n = 3 experiments). C, organs of Corti were treated with the indicated siRNA (100 nm) for 72 h. Huwe1, myosin VIIa, Atoh1, and HSC70 (loading control) were quantified by Western blotting, and densitometry (Huwe1, myosin VIIa, and Atoh1) levels are shown below each lane).
Deletion of Huwe1 Affects Cochlear Hair Cell Development
To assess the role of Huwe1 in cochlear development, we induced the conditional deletion of Huwe1 using a Cre driven by Sox2 expression, which is specific for the prosensory progenitors in the developing cochlea and is subsequently expressed in the developing spiral ganglion and cochlear supporting cells. Disruption of Huwe1 at E16 stimulated generation of a single extra row of inner hair cells at E19 (Fig. 7A compared with Fig. 7B). The extra hair cells were found in the inner pillar cell region, where extra hair cells are also produced upon stimulation of Wnt signaling in cochlear supporting cells (21). We were unable to examine the phenotype in the mature animal after deletion of Huwe1 in Sox2-expressing cells, however, as the mice did not reach maturity. Mice with Huwe1 knocked out at P1 survived to adulthood, and assessment of their phenotype at P30 revealed an extra row of inner hair cells, similar to the E15-deleted animals. Further examination revealed that the extra hair cells were normally innervated (16.1 ± 0.4 synapses per hair cell in the modiolar row, 15.6 ± 0.6 in the pillar cell row versus 16.4 ± 0.4 in control animals), indicating an effective doubling of the total number of afferent synapses in the cochlea (Fig. 7C compared with 7D). Auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) thresholds remained normal at P30 in Huwe1 knock-out ears (Fig. 7, E and F). Thus, the alterations in cochlear cells (development of extra inner hair cells that were fully innervated) had no obvious effects on function.
FIGURE 7.

Huwe1 conditional knock-out gives rise to an extra row of inner hair cells. A, yellow arrows indicate additional inner hair cells in the organ of Corti at E19 after knock-out of Huwe1 in Sox2-positive supporting cells (Huwe1flox/y) at E16. Myosin VIIa labels hair cells and Sox2 labels supporting cells; DAPI is a nuclear marker. The white line marks the location of the orthogonal view shown beneath the surface view, and yellow and white brackets indicate inner and outer hair cells, respectively. The scale bar is 25 μm. B, E19 organ of Corti from Sox2-Cre control mice contains a single row of inner hair cells. C and D, Z (left) and X (right) projection confocal images of the inner hair cell region in the P30 organ of Corti from a Huwe1 conditional knock-out (C) or control (D) ear is shown after deletion of Huwe1 at P1. The cochleae were immunostained with antibodies against myosin VIIa, CtBP2, and GluR2. Arrows indicate extra inner hair cells. Yellow and orange dotted lines delineate extra inner hair cells (IHC) on the pillar or modiolar side of the organ, respectively (C, right). E and F, DPOAE (E) and ABR (F) thresholds recorded at P30 from Huwe1 conditional knock-out (n = 3) and control (n = 5) ears are shown. Error bars indicate S.E. SPL, sound pressure level.
Hair cells at P2 in ears with Huwe1 knocked out in the developing cochlea by the hair cell-specific Atoh1-Cre were enlarged and had irregular shapes (Fig. 8, A and B). By P5, Huwe1 knock-out had resulted in overt hair cell loss (Fig. 8, C and D). The alterations in hair cells were correlated to an increased level of Atoh1 (Fig. 8E). Thus, deletion of Huwe1 in supporting cells led to extra hair cell generation, whereas Huwe1 knock-out in hair cells caused hair cell loss. The results indicate that the function of Huwe1 is important for proper cellular patterning of the cochlea and to prevent cell death.
FIGURE 8.

Huwe1 conditional knock-out causes hair cell death. A, enlarged inner hair cells can be seen throughout the cochlea at P2 after knock-out of Huwe1 in Atoh1-positive hair cells. The white line marks the location of the orthogonal view shown beneath the surface view, and yellow and white brackets indicate inner and outer hair cells, respectively. Arrows point to enlarged inner hair cells. Myosin VIIa labels hair cells and Sox2 labels supporting cells; DAPI is a nuclear marker. B, P2 organ of Corti from Atoh1-Cre control mice. C, inner hair cells (arrows) are lost, and outer hair cells (asterisks) are enlarged in a P5 organ of Corti from an Atoh1-Cre;Huwe1flox/y mouse. D, P5 organ of Corti from Atoh1-Cre mice. E, organ of Corti from P5 Atoh1-Cre and Atoh1-Cre-Huwe1flox/y. Atoh1 and β-actin (loading control) were quantified by Western blotting (IB; average Atoh1 levels based on densitometry from two experiments are shown below the blot).
Discussion
Atoh1 is a controlling factor in hair cell differentiation. It is shut off after birth, and the timing is highly regulated at the transcriptional level (7, 22, 23). An effect on Atoh1 levels in the cochlea is expected to influence hair cell genesis because of this central role in prosensory progenitor cell development. Its absence results in deafness and a lack of hair cells (3), and its overexpression in neonatal ears results in extra hair cells (5). Our understanding of the regulation of these transcriptional events has progressed rapidly. Notch signaling comes up early in development of the inner ear and is needed for the maintenance of the sensory domain (24, 25). Atoh1 increases when Notch signaling is down-regulated by lateral inhibition in cells destined to become hair cells. Wnt is also depressed by Notch, and Wnt mediator, β-catenin, increases when Notch is extinguished, thus increasing Atoh1 expression (9). Atoh1 not only initiates differentiation but is also necessary for critical phases of hair cell maturation and stability (22).
In addition to the multiple pathways that regulate expression of this powerful cell-fate regulator, we expected that pathways for its down-regulation would limit its expression. We show here that Atoh1 levels are indeed regulated by its stability, and proteasome-mediated degradation results in a short half-life. Atoh1 is targeted for ubiquitylation by E3 ubiquitin ligase, Huwe1. The polyubiquitylation of Atoh1 activity by Huwe1 could be demonstrated in a cell-free system. Ubiquitylation was a signal for proteasomal degradation. Consistent with the role of Huwe1 in the cochlea and its influence on Atoh1, inhibition of Atoh1 degradation altered hair cell development. We confirmed the interaction of Huwe1 with Atoh1 through immunoprecipitation-mass spectrometry and found that Atoh1 was degraded through Lys-48-linked ubiquitylation. Its ubiquitylation was initiated by Atoh1 phosphorylation achieved by the action of CK1 on specific serines in the C terminus of Atoh1. Accordingly, the half-life of Atoh1 could be extended by pharmacological inhibition of CK1 activity or direct proteasomal inhibition via silencing of Huwe1 with siRNA. Silencing stabilized Atoh1 protein in cochlear tissue. The inner hair cell phenotype of the Huwe1 knock-out was consistent with an increase in Atoh1, and terminating Atoh1 activity after hair cell differentiation and maturation appeared to be critical for normal cellular patterning in the cochlea, although the hair cells formed by Atoh1 stabilization were innervated normally at ribbon synapses. The pathways regulating the stability of Atoh1 are thus important in the determination of cell fate in the cochlea. However, hair cell stability was affected after knock-out of Huwe1 in developing hair cells and suggests that the extra Atoh1 is toxic to the cells, consistent with previous reports (26). The long term survival of the hair cells coming from Huwe1 deletion in supporting cells may be a result of insufficient effects of prolonged Atoh1 half-life at the decreased level of gene expression at P2. These functions of Huwe1 may account for the hearing loss associated with intellectual disability in humans with a mutation that results in Huwe1 overexpression (27).
Although there have been reports of neural transcription factor regulation by ubiquitylation and proteasomal degradation, there is no example of proteasomal regulation of transcription factors in auditory system development. Huwe1 affects the stability of several transcription factors, including N-Myc (19), c-Myc (28), p53 (29), Mcl-1 (30), Cdc6 (31), and MyoD (32) as well as tumor suppressor, BRCA1 (33), DNA polymerase (34), and histone deacetylase, HDAC2 (35). REST ubiquitylation and inactivation mediated by E3 ligase β-TRCP is essential for the differentiation of neurons (36). Proteasomal regulation of β-catenin in neural development and synaptogenesis involves β-TRCP. The regulation of transcription factor expression for the expansion of neural progenitors has been demonstrated in the cerebellum (37), and indeed, Huwe1 regulates the expansion of cerebellar neural progenitors through its action on Myc and ligands of Notch (38). Huwe1 promotes interaction of c-Myc with P300 and transcriptional activation via K63 ubiquitylation (39) and inhibits multimerization of the cytoplasmic scaffolding protein Dvl-3, resulting in down-regulation of Wnt signaling (40).
Huwe1 was recently shown to play a role in Atoh1 stability in the cerebellum through which it influenced the differentiation and distribution of cerebellar neural progenitors (15). That study identified Huwe1 as an E3 ligase that acted on Atoh1 but did not identify the linkage or the kinase targeting Atoh1 for ubiquitylation. Here we show that Atoh1 is ubiquitylated with the Lys-48 linkage that serves as a proteasomal degradation signal and that CK1 targets it for ubiquitylation. Identification of CK1 aided in the analysis of key phosphorylation sites for Atoh1 degradation by mass spectrometry. We suspect that CK1 overexpression made detection of phosphoserine 334 possible despite the rapid Atoh1 degradation by increasing phosphorylation at serine 334. Truncation of the C terminus abolished phosphorylation-mediated ubiquitylation, thereby indicating a site(s) in this region that targeted the protein for E3 ligase activity. Interestingly, the CK1 binding motif, (pS/T)XX(S/T), where pS/T indicates a primed serine/threonine residue, is repeated several times in Atoh1, spanning the serine-rich region from 325 to 334. Phosphorylation at three of the serine residues in this span was detected when CK1 was overexpressed. Of CK1 isoforms CK1α, CK1∂, CK1ϵ, and CK1γ, CK1∂ and CK1ϵ showed the highest activity. Overexpression of CK1∂ decreased, whereas blocking CK1∂ activity increased, Atoh1 abundance.
Several phosphorylation sites were reported to be relevant to Atoh1 stability, including C-terminal serines 328 and 339 (15) and 52 and 56 within the N terminus (20). Mutation of serine to alanine led to identification of serine 334, which was conserved across multiple species, as a key part of the Huwe1 degron stimulated by phosphorylation by CK1. Although we did see an effect of mutating serine 328 to alanine on Atoh1 half-life, a greater prolongation was observed after mutation of serine 334. Little effect was seen after mutating serine 339. Interestingly, serines 52 and 56, which constitute GSK-3β phosphorylation motifs (SXXXS), were reported to affect Atoh1 stability when mutated to alanine (20). However, mutation of these sites did not appear to alter the stability significantly in our hands. CK1α and GSK-3β are both involved in the phosphorylation of β-catenin, and priming by one kinase can convert a protein into a substrate for another kinase (42). Atoh1 is important for development and turnover of the intestinal epithelium (3, 43), and we expect that its regulation by the proteasome will be similar in that tissue. The report on tumor cell stability of Atoh1 (20) suggested coordinated regulation of Atoh1 and β-catenin through GSK-3β activity on both proteins. Although we cannot rule out the involvement of other kinases in degron formation, the effect of CK1 inhibition is quantitatively similar to that of proteasome inhibition on Atoh1.
Forcing expression of Atoh1 by manipulation of various upstream signaling pathways is one strategy for hair cell regeneration, but up-regulation of Atoh1 by drugs and by gene transfer suggest that the window in which Atoh1 is effective is limited (26, 44). Rapid degradation could account for some of these limitations. The data presented here provide new insight into Atoh1 regulation; CK1-mediated phosphorylation of Atoh1 facilitates Huwe1-Atoh1 interaction and subsequent degradation (Fig. 9). The role of both CK1 phosphorylation and Huwe1 ubiquitylation could provide a new approach to regeneration. The discovery that proteasome inhibition stabilized Atoh1 is valuable for this purpose, but proteasome inhibitors provide little specificity. Inhibition of the E3 ligase or its degron could be more selective for up-regulation of Atoh1, an option that can now be tested with the identification of the kinase and E3 ligase targeting Atoh1.
FIGURE 9.
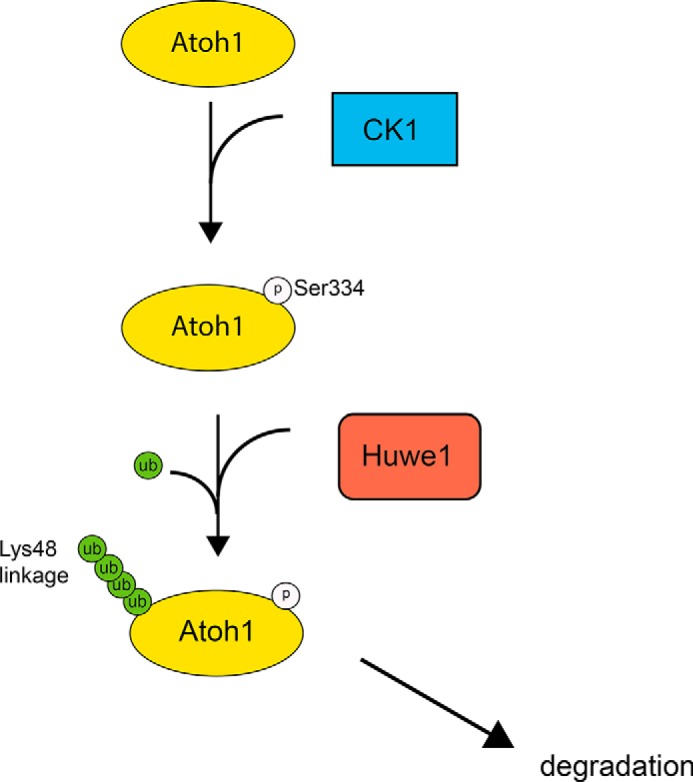
Atoh1 targeting by CK1 and Huwe1 is illustrated. CK1 phosphorylates Atoh1 at serine 334, facilitating Huwe1 interaction and initiating the polyubiquitylation and proteasomal degradation of Atoh1. Inactivation of either CK1 or Huwe1 results in accumulation of Atoh1.
Experimental Procedures
Cell Culture
HEK cells, FLAG-HA-Atoh1 293T cells, and HeLa cells were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mm Glutamax, and penicillin (100 units/ml)/streptomycin (100 μg/ml) (all from Invitrogen). All cultures were maintained in a 5% CO2/20% humidified incubator (Forma Scientific).
OC-1 cells (a gift from Dr. Federico Kalinec, House Ear Institute) (45) were cultured under permissive conditions (33 °C) in DMEM supplemented with 10% FBS and 50 units/ml γ-interferon (Genzyme) without antibiotics and moved to non-permissive conditions (39 °C in DMEM supplemented with 10% FBS) before quantitative PCR and Western blots.
Generation of Atoh1 Plasmids and Stably Expressing Cell Line
A construct consisting of Atoh1 cDNA modified to include two consecutive FLAG-tag sequences (GATTACAAGGATGACGA) preceding the start codon was subcloned into pcDNA3.1(+) to generate FLAG-Atoh1 plasmids (46). To generate FLAG-HA-Atoh1 plasmids, sequence-verified Atoh1 clones in pDONR223 were recombined into the Gateway destination vector pHAGE-N-FLAG-HA (Invitrogen) using λ-recombinase (14). To generate lentivirus for the 293T cell line stably expressing Atoh1, 1 μg of pHAGE-N-FLAG-HA-Atoh1 cDNA was co-transfected with 4 helper plasmids (2 μg of VSVG, 1 μg each of Tat1b, Mgpm2, and CMV-Rev) using Lipofectamine 2000 (Invitrogen) in 10-cm dishes of 293T cells. Virus particles were harvested 48 h post-transfection and used to infect 293T cells. Puromycin (Sigma, 1 μg/ml) was used for the selection of infected cells (14).
Western Blotting
Proteins extracted with radioimmune precipitation assay buffer from whole cells were separated on 4–12% NuPAGE Bis-Tris gels (Invitrogen) and electrotransferred to 0.2 μm nitrocellulose membranes (Bio-Rad). The membranes were probed with mouse anti-FLAG (Sigma), mouse anti-HA (Sigma), mouse anti-ubiquitin (Santa Cruz), rabbit anti-Atoh1 (1:1,000, Affinity BioReagent Antibody), myosin VIIa antibody (1:200, Proteus Biomedical), mouse anti-β-actin (Sigma), or mouse anti-HSC70 (1:10,000, Santa Cruz Biotechnology) antibodies followed by HRP-conjugated, anti-rabbit, anti-mouse IgG, or anti-mouse light chain antibody (Jackson ImmunoResearch Laboratories). The blots were processed with ECL or ECL-Plus Western blot Substrates (Thermo). Band intensity was quantified by densitometry using Quantity One software (Bio-Rad). Each band was normalized to β-actin or HSC70 and expressed as a ratio to the control.
RNA Preparation for Quantitative RT-PCR
Total RNA was extracted with the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. 1 μg of RNA was reverse-transcribed to cDNA using the Improm-II Reverse Transcription System (Promega). The reverse transcription conditions were 25 °C for 10 min followed by 37 °C for 60 min; the reaction was terminated at 95 °C for 5 min. The cDNA products were mixed with LightCycler Taqman Master Mix (Roche Applied Science) and Taqman primers (Invitrogen) in a 96-well plate according to the manufacturer's instructions. The quantitative PCR was run in triplicate on an ABI 7700 Real-Time PCR machine (Applied Biosystems, Inc.) with the initial denaturation at 95 °C for 2 min, denaturation at 95 °C for 15 s, and annealing/extension at 60 °C for 1 min for 45 cycles. Huwe1 gene expression was calculated relative to 18S RNA, and the amount of cDNA applied was adjusted to bring the Ct value for 18S RNA to within one half-cycle.
Cycloheximide Chase Assays for Stability
HEK cells were transfected with either FLAG-Atoh1 (wild type) or indicated mutant FLAG-Atoh1 plasmids (1 μg/ml) using Lipofectamine 2000 (3 μl per 1 μg of cDNA, Invitrogen). Forty-eight hours after transfection, 100 μg/ml cycloheximide (Sigma) was added to block protein synthesis. Cells were harvested at 0, 30, 60, 120, and 240 min. Equal amounts of protein from each treatment were taken for Western blotting. Protein bands were quantified by densitometry. The half-lives of the indicated proteins were calculated using GraphPad Prism 6 software and a one-phase exponential-decay model. Each experiment was repeated at least two times.
Co-immunoprecipitation
To determine if the ubiquitin-proteasome pathway was involved in Atoh1 degradation, HEK cells were transfected with FLAG-Atoh1 (1 μg/ml) using Lipofectamine 2000 (Invitrogen) for 48 h and either DMSO or MG132 (10 μm) for 6 h. Transfected cells were lysed in Pierce IP Lysis Buffer (Thermo) containing 1× complete protease inhibitors and 1× PhosSTOP phosphatase inhibitors (Roche Applied Science). Lysates were immunoprecipitated with anti-FLAG M2 Affinity Gel (Sigma) and immunoblotted by the procedures mentioned above.
To determine Lys-48 polyubiquitylation, HEK cells were co-transfected with FLAG-Atoh1 (1 μg/ml) and wild type, Lys-48 ubiquitin plasmids, or empty vector (0.5 μg/ml, from Addgene) using Lipofectamine 2000 (Invitrogen). At 48 h post-transfection, cells were lysed and immunoprecipitated with anti-FLAG M2 affinity gel and immunoblotted.
To prove the interaction between Atoh1 and Huwe1, FLAG-HA-Atoh1 293T were lysed, with IP buffer and the lysates were immunoprecipitated with HA-resin (Sigma), IgG-conjugated A/G agarose, or empty A/G-agarose (Santa Cruz Biotechnology) and immunoblotted with antibodies against HA and Huwe1. For reciprocal immunoprecipitation, similar lysates were immunoprecipitated with A/G-agarose conjugated with rabbit anti-Huwe1 antibody (Novus Biological) and immunoblotted with antibodies against HA or Huwe1.
To explore the role of Huwe1 in ubiquitylation of Atoh1, we generated Myc-Huwe1 with cysteine 4341 mutated to alanine (Myc-Huwe1-C4341) by recombining Huwe1 pENTR plasmids (Addgene) into pDEST-CMV-N-Myc using Gateway cloning. FLAG-HA-Atoh1 293T cells were transfected with wild-type Huwe1, Huwe1 shRNA (Sigma), or empty vector for 48 h and treated with MG132 (10 μm) or vehicle for 6 h. Cell lysates were immunoprecipitated with anti-FLAG-resin (Sigma) and immunoblotted with mouse anti-ubiquitin antibody (Santa Cruz Biotechnology) or reblotted with mouse anti-FLAG or rabbit anti-Huwe1 antibodies.
Wild-type or mutant Myc-Huwe1, wild-type or Lys-48 HA-ubiquitin, and FLAG-Atoh1 plasmids were co-transfected into HeLa cells. After 48 h, cell lysates were immunoprecipitated with anti-FLAG resin and immunoblotted with mouse anti-HA, mouse anti-FLAG, and rabbit anti-Huwe1 antibodies. Input was blotted with mouse anti-Myc, mouse anti-FLAG, and mouse anti-β-actin antibodies as loading controls.
Protein Purification
HEK293T cells with stable expression of Atoh1 from 15-cm tissue culture dishes at ∼80% confluence were lysed in a total volume of 4 ml of lysis buffer (50 mm Tris, pH 7.8, 150 mm sodium chloride, 0.5% Nonidet P-40 plus EDTA-free protease inhibitor mixture (Roche Applied Science) and incubated at 4 °C for 45 min. Lysates were centrifuged at 13,000 rpm for 10 min at 4 °C and filtered through 0.45-μm spin filters (Millipore) to remove cell debris. 60 μl of immobilized anti-HA resin (Sigma; 50% slurry) were used to immunoprecipitate the cleared lysates by gentle inversion overnight. Once the binding was complete, resin containing immunocomplex was washed with lysis buffer 4 times followed by PBS 4 times. Atoh1 was eluted with HA peptide (250 μg/ml) in PBS for 30 min (3 × 50 μl) at room temperature. Ten percent of the eluate was electrophoresed on a NuPAGE Novex 4–12% Bis-Tris gel and silver-stained to confirm immunoprecipitation of Atoh1, and the remaining eluate was subjected to trichloroacetic acid (TCA) precipitation for subsequent IP-MS/MS analysis. In other experiments, specific bands stained with Coomassie Blue after elution were excised for peptide mass spectrometric sequencing at the core facility of Harvard Medical School.
Mass Spectrometry
For identification of Atoh1 interacting proteins, the proteins precipitated by TCA were resuspended in 25 μg/μl sequencing grade trypsin (25 μg/μl in 30 μl of 100 mm ammonium bicarbonate, pH 8.0, with 10% acetonitrile) and incubated at 37 °C for 4 h. Digested samples were loaded onto stage tips and washed. Peptides were eluted with 50% formic acid, 5% acetonitrile to neutralize the trypsin followed by drying and resuspension in 10 μl of 5% formic acid 5% acetonitrile. The resulting spectra were analyzed by SEQUEST, a tandem mass spectrometry data analysis program for peptide sequencing and protein identification against a human database (47). The list of proteins was loaded into CompPASS for further processing and analysis (14).
In Situ Proximity Ligation Assay
HEK cells transfected with FLAG-Atoh1 and/or V5-Huwe1 (48) (a gift from Dr. Wei Gu, Columbia University) were fixed with 4% paraformaldehyde for 10 min and blocked with 15% donkey serum for 1 h. The fixed cells were incubated with rabbit anti-FLAG (Sigma) and mouse anti-V5 (Invitrogen) antibodies overnight. Proximity ligation assay probes were added after washing with PBS three times followed by ligation and amplification procedures. Images were obtained by confocal microscopy (Leica SP5).
Cell-free Ubiquitylation Assay
Recombinant GST-Atoh1 or GST-N-Myc (100 nm, both from Abnova) was incubated with 10 ng of human E1 (UBE1), 100 ng of human E2 (Ubc5), 2 μm ubiquitin (all from Boston Biochem), and 100 nm full-length recombinant Huwe1 (35) purified from baculovirus-infected insect cells (a gift from Dr. Qing Zhong, UT Southwestern) in an ATP-regenerating buffer for 1 h at 37 °C. The reaction was terminated by SDS sample buffer, fractionated by SDS-PAGE (4–12%), and analyzed by Western blotting with mouse anti-ubiquitin antibody.
Cochlear Explant Culture
Cochlear tissues were dissected from 1-day postnatal CD-1 mice (Charles River Laboratories). Spiral ganglion, Reissner's membrane, and the hook region of the organ of Corti were removed to obtain a flat cochlear surface preparation. Explants were plated onto 4-well plates (Greiner Bio-One) coated with Matrigel (BD Biosciences) diluted 1:10 in DMEM supplemented with 10% fetal bovine serum (FBS) overnight. The organs were incubated with Huwe1 or non-targeting, scrambled siRNA (100 nm) (Integrated DNA Technologies) for 72 h.
Gene Knockdown
ShRNA used for knockdown of Huwe1 was obtained from Sigma, whereas siRNA was purchased from IDT. shRNA (1 μg/ml) or siRNA (100 nm) was transfected into cells using Lipofectamine or RNAiMAX transfection reagents (both from Invitrogen), and tissues or cells were harvested 48–72 h later. We tested two shRNAs and two siRNAs and used the more efficient one based on quantitative PCR and Western blot for gene silencing. Non-targeting shRNA or siRNA was transfected in parallel.
Conditional Knock-out of Huwe1
The Sox2-Cre-ER mouse was described previously (49, 50). The Atoh1-Cre mouse was obtained from the Jackson Laboratory (stock no. 011104) (51). The Huwe1-flox mouse was a gift of Dr. Tak W. Mak (University of Toronto) (52). Male Sox2-Cre-ER mice were mated with Huwe1-flox female mice to obtain Huwe1 conditional knock-out in the supporting cells; tamoxifen was injected to mothers of the transgenic mice (Huwe1 is located on the X-chromosome, and Sox2-Cre-ER;Huwe1flox/y male offspring are, therefore, knockouts when the gene is deleted on the X-chromosome). Huwe1 conditional knock-out in the hair cells (Atoh1-Cre;Huwe1flox/y) were obtained from Huwe1-flox females mated with male Atoh1-Cre mice. Littermates without the Huwe1-flox transgene were used as controls.
The Sox2-Cre-ER and Atoh1-Cre mice were genotyped with Cre primers: forward (5′-TGG GCG GCA TGG TGC AAG TT-3′) and reverse (5′-CGG TGC TAA CCA GCG TTT TC-3′). The Huwe1-flox mouse was genotyped with the following primers: forward (5′-GTA TGG TCA TGA TTG AGT GCT TGG AAC T-3′) and reverse (5′-TAT ACC TGA ACA CAT GGG CAT ATA CAT-3′). All mouse experiments were approved by the Massachusetts Eye and Ear Infirmary Institutional Animal Care and Use Committees.
Immunohistochemistry
For embryonic cochlea, tamoxifen (250 mg/kg, Sigma) was given to the pregnant mice (intraperitoneal administration, once a day, for 2 consecutive days) at E15 to obtain conditional knock-out of Huwe1, and they were killed at the indicated time points. For newborn tissues, cochleae from embryos were dissected and processed as whole mount or section preparations.
For whole-mount staining of embryonic cochlea, tissue was harvested by removal of the cochlear capsule, lateral wall, and spiral ganglion. Organ of Corti was fixed for 10 min in 4% paraformaldehyde followed by 3 washes with PBS and blocking with 1% BSA and 5% goat serum in 0.1% Triton X-100 PBS (PBT1) for 1 h. Tissues were incubated overnight with PBT1 containing rabbit anti-myosin VIIa antibody (1:500, Proteus Biomedical) and rabbit anti-Sox2 (1: 500, Santa Cruz Biotechnology). Samples were washed with PBS for 10 min and incubated with secondary antibodies conjugated with Alexa 488, 594, or 647 (Invitrogen).
Adult mouse cochleae were decalcified (0.1 m EDTA) after perfusion with 4% paraformaldehyde for 2 h at room temperature through the round window and oval window (41). They were dissected into half-turns for whole mounts. The primary antibodies were mouse (IgG1) anti-CtBP2 (1:200, BD Biosciences), chicken anti-high molecular weight neurofilament (1:200, Millipore), and mouse (IgG2a) anti-GluR2 (1:200, Millipore). After overnight incubation, cochleae were incubated for 2× for 60 min at 37 °C in species-appropriate secondary antibodies with 1% Triton X-100. Staining was analyzed with a confocal microscope (Leica, SP5).
Cochlear Function Tests
DPOAEs and ABRs were recorded as described previously (41). The ABR stimuli were 5-ms tone pips with a 0.5-ms rise-fall time delivered at 30/s. Sound level was incremented in 5-db steps, from 25 db below threshold to 80 db sound pressure level. DPOAEs were recorded for primary tones with a frequency ratio of 1.2 and with the level of the f2 primary 10 db less than f1 level, incremented together in 5-db steps. The 2f1- f2 DPOAE amplitude and surrounding noise floor were extracted. DPOAE threshold is defined as the f1 level required to produce a response amplitude of 0 db sound pressure level.
Statistical Analysis
The mean values and S.E. were calculated and analyzed for significance by an unpaired two-tailed Student's t test with indicated alpha (0.05, 0.01, or 0.001) with Prism 6 software.
Author Contributions
Y. C., M. T., and A. S. B. E. designed the experiments, Y. C. and M. T. performed the experiments, Y. C. and A. S. B. E. wrote the paper, and all authors reviewed the data and approved the submission.
Acknowledgments
We thank John Lydeard and Wade Harper for assistance with identification of the E3 ligase by mass spectrometry. We thank Dan Finley, Bill Sewell, and Charlie Liberman for advice on experiments and Vasiliki Zoto for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 DC007174 and P30 DC005209 (NIDCD). This work was also supported by the American Hearing Research Foundation, the Yen Tjing Ling Medical Foundation (Taiwan), the Amelia-Peabody Foundation, the Shulsky Foundation, David H. Koch, and Robert Boucai. The authors declare that they have no conflicts of interest with the contents of this paper. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CK1
- casein kinase 1
- ABR
- auditory brainstem response
- DPOAE
- distortion product otoacoustic emission
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- bHLH
- basic helix-loop-helix.
References
- 1. Forge A., Li L., Corwin J. T., and Nevill G. (1993) Ultrastructural evidence for hair cell regeneration in the mammalian inner ear. Science 259, 1616–1619 [DOI] [PubMed] [Google Scholar]
- 2. Warchol M. E., Lambert P. R., Goldstein B. J., Forge A., and Corwin J. T. (1993) Regenerative proliferation in inner ear sensory epithelia from adult guinea pigs and humans. Science 259, 1619–1622 [DOI] [PubMed] [Google Scholar]
- 3. Bermingham N. A., Hassan B. A., Price S. D., Vollrath M. A., Ben-Arie N., Eatock R. A., Bellen H. J., Lysakowski A., and Zoghbi H. Y. (1999) Math1: an essential gene for the generation of inner ear hair cells. Science 284, 1837–1841 [DOI] [PubMed] [Google Scholar]
- 4. Chen P., Johnson J. E., Zoghbi H. Y., and Segil N. (2002) The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 129, 2495–2505 [DOI] [PubMed] [Google Scholar]
- 5. Zheng J. L., and Gao W. Q. (2000) Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 3, 580–586 [DOI] [PubMed] [Google Scholar]
- 6. Jeon S. J., Fujioka M., Kim S. C., and Edge A. S. (2011) Notch signaling alters sensory or neuronal cell fate specification of inner ear stem cells. J. Neurosci. 31, 8351–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kelley M. W. (2006) Regulation of cell fate in the sensory epithelia of the inner ear. Nat Rev. Neurosci 7, 837–849 [DOI] [PubMed] [Google Scholar]
- 8. Millimaki B. B., Sweet E. M., Dhason M. S., and Riley B. B. (2007) Zebrafish atoh1 genes: classic proneural activity in the inner ear and regulation by Fgf and Notch. Development 134, 295–305 [DOI] [PubMed] [Google Scholar]
- 9. Shi F., Cheng Y. F., Wang X. L., and Edge A. S. (2010) β-Catenin up-regulates Atoh1 expression in neural progenitor cells by interaction with an Atoh1 3′ enhancer. J. Biol. Chem. 285, 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang X., Peterson K. A., Liu X. S., McMahon A. P., and Ohba S. (2013) Gene regulatory networks mediating canonical Wnt signal directed control of pluripotency and differentiation in embryo stem cells. Stem Cells 31, 2667–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tai H.-C., and Schuman E. M. (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838 [DOI] [PubMed] [Google Scholar]
- 12. Naujokat C., and Sarić T.(2007) Concise review: role and function of the ubiquitin-proteasome system in mammalian stem and progenitor cells. Stem Cells 25, 2408–2418 [DOI] [PubMed] [Google Scholar]
- 13. Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 14. Sowa M. E., Bennett E. J., Gygi S. P., and Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forget A., Bihannic L., Cigna S. M., Lefevre C., Remke M., Barnat M., Dodier S., Shirvani H., Mercier A., Mensah A., Garcia M., Humbert S., Taylor M. D., Lasorella A., and Ayrault O. (2014) Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Dev. Cell 29, 649–661 [DOI] [PubMed] [Google Scholar]
- 16. Tan M. K., Lim H. J., Bennett E. J., Shi Y., and Harper J. W. (2013) Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol. Cell 52, 9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 18. Herold S., Hock A., Herkert B., Berns K., Mullenders J., Beijersbergen R., Bernards R., and Eilers M. (2008) Miz1 and HectH9 regulate the stability of the checkpoint protein, TopBP1. EMBO J. 27, 2851–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao X., Heng J. I., Guardavaccaro D., Jiang R., Pagano M., Guillemot F., Iavarone A., and Lasorella A. (2008) The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 10, 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsuchiya K., Nakamura T., Okamoto R., Kanai T., and Watanabe M. (2007) Reciprocal targeting of Hath1 and β-catenin by Wnt glycogen synthase kinase 3β in human colon cancer. Gastroenterology 132, 208–220 [DOI] [PubMed] [Google Scholar]
- 21. Shi F., Hu L., and Edge A. S. (2013) Generation of hair cells in neonatal mice by β-catenin overexpression in Lgr5-positive cochlear progenitors. Proc. Natl. Acad. Sci. U.S.A. 110, 13851–13856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cai T., Seymour M. L., Zhang H., Pereira F. A., and Groves A. K. (2013) Conditional deletion of Atoh1 reveals distinct critical periods for survival and function of hair cells in the organ of Corti. J. Neurosci. 33, 10110–10122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fritzsch B., Matei V. A., Nichols D. H., Bermingham N., Jones K., Beisel K. W., and Wang V. Y. (2005) Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev. Dyn. 233, 570–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Basch M. L., Ohyama T., Segil N., and Groves A. K. (2011) Canonical Notch signaling is not necessary for prosensory induction in the mouse cochlea: insights from a conditional mutant of RBPjκ. J. Neurosci. 31, 8046–8058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamamoto N., Chang W., and Kelley M. W. (2011) Rbpj regulates development of prosensory cells in the mammalian inner ear. Dev. Biol. 353, 367–379 [DOI] [PubMed] [Google Scholar]
- 26. Liu Z., Dearman J. A., Cox B. C., Walters B. J., Zhang L., Ayrault O., Zindy F., Gan L., Roussel M. F., and Zuo J. (2012) Age-dependent in vivo conversion of mouse cochlear pillar and Deiters' cells to immature hair cells by Atoh1 ectopic expression. J. Neurosci. 32, 6600–6610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Froyen G., Belet S., Martinez F., Santos-Rebouças C. B., Declercq M., Verbeeck J., Donckers L., Berland S., Mayo S., Rosello M., Pimentel M. M., Fintelman-Rodrigues N., Hovland R., Rodrigues dos Santos S., Raymond F. L., et al. (2012) Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am. J. Hum. Genet. 91, 252–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inoue S., Hao Z., Elia A. J., Cescon D., Zhou L., Silvester J., Snow B., Harris I. S., Sasaki M., Li W. Y., Itsumi M., Yamamoto K., Ueda T., Dominguez-Brauer C., Gorrini C., et al. (2013) Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 27, 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen D., Kon N., Li M., Zhang W., Qin J., and Gu W. (2005) ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 30. Zhong Q., Gao W., Du F., and Wang X. (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 31. Hall J. R., Kow E., Nevis K. R., Lu C. K., Luce K. S., Zhong Q., and Cook J. G. (2007) Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol. Biol. Cell 18, 3340–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noy T., Suad O., Taglicht D., and Ciechanover A. (2012) HUWE1 ubiquitinates MyoD and targets it for proteasomal degradation. Biochem. Biophys. Res. Commun. 418, 408–413 [DOI] [PubMed] [Google Scholar]
- 33. Wang X., Lu G., Li L., Yi J., Yan K., Wang Y., Zhu B., Kuang J., Lin M., Zhang S., and Shao G. (2014) HUWE1 interacts with BRCA1 and promotes its degradation in the ubiquitin-proteasome pathway. Biochem. Biophys. Res. Commun. 444, 549–554 [DOI] [PubMed] [Google Scholar]
- 34. Parsons J. L., Tait P. S., Finch D., Dianova I. I., Edelmann M. J., Khoronenkova S. V., Kessler B. M., Sharma R. A., McKenna W. G., and Dianov G. L. (2009) Ubiquitin ligase ARF-BP1/mule modulates base excision repair. EMBO J. 28, 3207–3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J., Kan S., Huang B., Hao Z., Mak T. W., and Zhong Q. (2011) Mule determines the apoptotic response to HDAC inhibitors by targeted ubiquitination and destruction of HDAC2. Genes Dev. 25, 2610–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Westbrook T. F., Hu G., Ang X. L., Mulligan P., Pavlova N. N., Liang A., Leng Y., Maehr R., Shi Y., Harper J. W., and Elledge S. J. (2008) SCFβ-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 452, 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao X., D' Arca D., Lim W. K., Brahmachary M., Carro M. S., Ludwig T., Cardo C. C., Guillemot F., Aldape K., Califano A., Iavarone A., and Lasorella A. (2009) The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev. Cell 17, 210–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stegmüller J., and Bonni A. (2010) Destroy to create: E3 ubiquitin ligases in neurogenesis. F1000 Biol. Rep. 2, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adhikary S., Marinoni F., Hock A., Hulleman E., Popov N., Beier R., Bernard S., Quarto M., Capra M., Goettig S., Kogel U., Scheffner M., Helin K., and Eilers M. (2005) The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123, 409–421 [DOI] [PubMed] [Google Scholar]
- 40. de Groot R. E., Ganji R. S., Bernatik O., Lloyd-Lewis B., Seipel K., Šedová K., Zdráhal Z., Dhople V. M., Dale T. C., Korswagen H. C., and Bryja V. (2014) Huwe1-mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci. Signal. 7, ra26. [DOI] [PubMed] [Google Scholar]
- 41. Yuan Y., Shi F., Yin Y., Tong M., Lang H., Polley D. B., Liberman M. C., and Edge A. S. (2014) Ouabain-induced cochlear nerve degeneration: synaptic loss and plasticity in a mouse model of auditory neuropathy. J. Assoc. Res. Otolaryngol. 15, 31–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Noort M., Weerkamp F., Clevers H. C., and Staal F. J. (2007) Wnt signaling and phosphorylation status of β-catenin: importance of the correct antibody tools. Blood 110, 2778–2779 [DOI] [PubMed] [Google Scholar]
- 43. Bossuyt W., De Geest N., Aerts S., Leenaerts I., Marynen P., and Hassan B. A. (2009) The atonal proneural transcription factor links differentiation and tumor formation in Drosophila. PLos Biol. 7, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kelly M. C., Chang Q., Pan A., Lin X., and Chen P. (2012) Atoh1 directs the formation of sensory mosaics and induces cell proliferation in the postnatal mammalian cochlea in vivo. J. Neurosci. 32, 6699–6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kalinec G. M., Webster P., Lim D. J., and Kalinec F. (2003) A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol. 8, 177–189 [DOI] [PubMed] [Google Scholar]
- 46. Parker M. A., Cheng Y. F., Kinouchi H., Bieber R., and Edge A. S. (2014) An independent construct for conditional expression of atonal homolog-1. Hum Gene. Ther. Methods 25, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 48. Chen D., Shan J., Zhu W. G., Qin J., and Gu W. (2010) Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 464, 624–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N., and Hochedlinger K. (2011) Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bramhall N. F., Shi F., Arnold K., Hochedlinger K., and Edge A. S. (2014) Lgr5-positive supporting cells generate new hair cells in the postnatal cochlea. Stem Cell Reports 2, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matei V., Pauley S., Kaing S., Rowitch D., Beisel K. W., Morris K., Feng F., Jones K., Lee J., and Fritzsch B. (2005) Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 234, 633–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hao Z., Duncan G. S., Su Y. W., Li W. Y., Silvester J., Hong C., You H., Brenner D., Gorrini C., Haight J., Wakeham A., You-Ten A., McCracken S., Elia A., Li Q., et al. (2012) The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 209, 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]