

Abstract
Macrophages are the predominant innate immune cells recruited to tissues following injury or infection. These early-responding, pro-inflammatory macrophages play an essential role in the amplification of inflammation. However, macrophage pro-inflammatory gene expression should be tightly regulated to avert host tissue damage. In this study, we identify the Kruppel-like transcription factor 6 (KLF6)-B cell leukemia/lymphoma 6 (BCL6) signaling axis as a novel regulator of macrophage inflammatory gene expression and function. Utilizing complementary gain- and loss-of-function studies, we observed that KLF6 is essential for macrophage motility under ex vivo and in vivo conditions. Concordant with these observations, myeloid-specific deficiency of KLF6 significantly attenuates macrophage pro-inflammatory gene expression, recruitment, and progression of inflammation. At the molecular level, KLF6 suppresses BCL6 mRNA and protein expression by elevating PR domain-containing 1 with ZNF domain (PRDM1) levels in macrophages. Interestingly, pharmacological or genetic inhibition of BCL6 in KLF6-deficient macrophages completely abrogated the attenuation of pro-inflammatory cytokine/chemokine expression and cellular motility. Collectively, our observations reveal that KLF6 repress BCL6 to enhance macrophage inflammatory gene expression and function.
Keywords: host defense, inflammation, Kruppel-like factor (KLF), macrophage, vascular biology
Introduction
Inflammation is a pathophysiological immune response that protects the host from infection or injury (1, 2). Monocyte-derived macrophages are the major innate immune cells that are recruited to the sites of inflammation (3). These recruited macrophages play a critical role in initiation, amplification, and resolution of inflammation. During the onset of inflammation, recruited macrophages are predominantly pro-inflammatory in nature (4). These macrophages express elevated levels of cytokines, chemokines, matrix metalloproteinases, and reactive oxygen/nitrogen species (5). Excessive production of chemotactic agents by these pro-inflammatory macrophages helps to recruit additional inflammatory cells to the sites of infection/injury, which results in amplification of inflammation (6). However, newly recruited pro-inflammatory macrophages may be able to eliminate the sources of inflammation with significant tissue/organ damage (1). Thus, unbridled activation of these pro-inflammatory macrophages is acknowledged to contribute to the pathogenesis of various disorders, including, but not limited to, sepsis, inflammatory bowel diseases (IBDs),2 metabolic syndrome, atherosclerosis, arthritis, and autoimmune diseases (7). However, during the resolution phase of inflammation, these originally recruited macrophages can transcriptionally reprogram to produce anti-inflammatory cytokines and mediators that contribute to tissue repair through matrix remodeling, recruitment of fibroblasts, and elimination of dead cells and debris (8–10). Macrophages achieve these diverse functional phenotypes by actively modulating gene expression (11). Therefore, dynamic changes in macrophage gene expression during different phases of inflammation must be precisely governed and orchestrated by transcription factors (12).
Activation of macrophages by cytokines or inflammatory agent such as LPS alter the expression of a large number of genes (13). Classical pro-inflammatory macrophage activation induces expression of inflammatory cytokines and chemokines such as IL1α, IL1β, IL6, IL12, TNF, CCL2, and CCL3. These pro-inflammatory mediators provide a chemotactic gradient for the recruitment of additional immune cells to sites of inflammation (14). This establishes a positive feedforward loop that leads to amplification of inflammation. However, inducible negative feedback regulators such as IκBα and B cell lymphoma 6 (BCL6) restrain inflammatory gene expression and then bridle the process of inflammation (15). The Bcl6 gene, originally identified as a proto-oncogene, encodes a zinc finger-containing, sequence-specific transcriptional repressor protein (16). Studies utilizing experimental murine models indicated that a systemic deficiency of BCL6 significantly attenuated growth. Further, loss of BCL6 resulted in multiple immunological deformities, including lack of germinal center formation and spontaneous development of lethal pulmonary vasculitis as well as myocarditis (15). Large collections of studies indicated that BCL6 represses inflammatory gene expression at the transcriptional level through interactions with other co-repressor proteins, including histone deacetylases, nuclear receptor co-repressor 2, NCOR1, BCL6 corepressor, and C-terminal binding protein (17). Interestingly, elevated PR domain zinc finger protein 1 (PRDM1 or Blimp1) repressed BCL6 expression in a broad spectrum of cellular systems (18–20). Recent genomics studies revealed that deficiency of BCL6 significantly induced a large number of inflammatory genes following LPS exposure (21). At the molecular level, BCL6 can bind to specific DNA sequences through the carboxyl terminus of Kruppel-type zinc finger motifs and recruit transcriptional co-repressors through amino-terminal interactions with proteins containing the broad-complex, tramtrack and bric a brac/poxvirus and zinc finger and second repression domain (17, 22). However, whether Kruppel-like factor 6 (KLF6) enhances pro-inflammatory gene expression and function by regulating the BCL6 signaling pathway has not been investigated.
The Klf6 gene is a member of the zinc finger family of transcription factors that mediates various cellular processes, including proliferation, differentiation, development, and programmed cell death (23). Alterations in its expression or function has been associated with the pathogenesis of numerous human diseases, including IBD, cancer, hepatic steatosis, and hepatic fibrosis (23). Our previous studies indicated that KLF6 is most abundantly expressed in macrophages and significantly induced by pro-inflammatory agents such as LPS or interferon γ (24). At the molecular level, KLF6 cooperates with NF-κB to promote pro-inflammatory gene expression while inhibiting PPARγ or STAT3 function to restrain anti-inflammatory gene expression in macrophages (24, 25). However, whether KLF6 promotes pro-inflammatory gene expression by curbing sequence-specific inducible negative feedback regulators of inflammation has not been investigated. In this study, we provide evidence that KLF6 suppresses BCL6 to enhance pro-inflammatory gene expression in macrophages. KLF6 deficiency significantly enhances basal or LPS-induced BCL6 expression at the mRNA and protein levels. Elevated BCL6 levels in KLF6-deficient macrophages results in the repression of BCL6 pro-inflammatory cytokines, and chemokines target gene expression. This results in diminished macrophage motility and inflammation under ex vivo and in vivo conditions. Based on our observations, we propose that KLF6 is a novel transcriptional repressor of inducible negative feedback regulators of inflammation in macrophages.
Results
KLF6 Enhances Macrophage Motility ex Vivo and in Vivo
Macrophages are the major innate immune cells recruited to sites of infection/injury that orchestrate progression of inflammation. Given the role of activated macrophages in a wide variety of chronic and acute human inflammatory disease conditions, we sought to investigate whether KLF6 regulates macrophage motility under in vivo and ex vivo conditions. Accordingly, Lyz2cre and Lyz2cre:KLF6fl/fl mice were subjected to thioglycollate-induced peritonitis, and the total number of macrophages that accumulated at the site of inflammation was counted as described under “Experimental Procedures.” The result indicates that deficiency of KLF6 significantly attenuated recruitment of macrophages to sites of inflammation (Fig. 1A). Several factors, including macrophage migration and invasion properties, could significantly affect their recruitment to sites of inflammation. Therefore, we examined whether altering KLF6 levels would affect inflammatory stimulus-induced cellular motility of macrophages. Accordingly, BMDMs derived from Lyz2cre, Lyz2cre:KLF6fl/fl mice and RAW264.7 cells overexpressing KLF6 (pCI-neo-KLF6) or empty vector (pCI-neo) were stimulated with LPS and evaluated for altered cellular migration and invasion functions (Fig. 1, B–E). The results illustrate that deficiency of KLF6 attenuates and overexpression of KLF6 augments LPS-induced migration (Fig. 1, B and C) and invasion (Fig. 1, D and E) of macrophages. Taken together, our results indicate that KLF6 is required for inflammatory stimulus-induced macrophage motility.
FIGURE 1.

KLF6 is required for macrophage motility. A, Lyz2cre and Lyz2cre:KLF6fl/fl mice were subjected to thioglycollate-induced peritonitis. Responding inflammatory cells from the peritoneal cavity were harvested using sterile 1× PBS, and the number of macrophages (CD11b+/SiglecF−/Ly6G−) was quantified. This experiment was performed three times with similar results. B–E, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice (B and D) and RAW264.7 cells transfected with the pCI-neo-KLF6 or pCI-neo plasmids (C and E) were stimulated with 100 ng/ml LPS for 4 h. These cells were added to the upper chamber of migration or invasion tissue culture inserts and incubated for 18 h. The number of cells migrated (B and C) or invaded (D and E) across the membrane in control group (untreated Lyz2cre BMDMs or RAW264.7 cells transfected with pCI-neo plasmids) was assigned as 100%, and -fold changes over this are indicated. Each experiment was performed three times with four replicates. The box plot represents the median with first and third quartiles, and whiskers represent minimum/maximum. Graphs represent mean ± S.D. *, p ≤ 0.05; N.S., not significant. p < 0.05 between indicated groups is considered significant.
KLF6 Augments Macrophage Recruitment to Sites of Inflammation
Solid tissue architecture and microenvironments could significantly affect macrophage motility and the progression of inflammation. The TPA-induced skin inflammation model is characterized by massive myeloid cell infiltration (26). Therefore, to investigate whether KLF6 regulates macrophage recruitment and progression of inflammation, Lyz2cre and Lyz2cre:KLF6fl/fl mice were subjected to the TPA-induced cutaneous model of inflammation. Our results demonstrate that TPA exposure significantly enhanced leukocyte infiltration, epidermal hyperplasia, interstitial edema, and tissue thickening in Lyz2cre mice (Fig. 2, A–C). Interestingly, all of these hallmark features of inflammation, including leukocyte infiltration, were significantly attenuated in Lyz2cre:KLF6fl/fl mice (Fig. 2, A–C). Concurrently, we examined whether KLF6 deficiency affected macrophage recruitment to sites of inflammation by staining tissue sections with anti-F4/80 antibody (a macrophage-specific marker). Our results indicate that KLF6 deficiency significantly attenuated TPA-induced macrophage recruitment to sites of inflammation in Lyz2cre:KLF6fl/fl mice compared with Lyz2cre mice (Fig. 2, D and E). Next we examined tissue levels of pro-inflammatory cytokines and chemokines by ELISA analysis. Our results indicate that myeloid-KLF6 deficiency significantly attenuated TPA-induced CCL2, IL1α, IL1β, and IL6 levels (Fig. 2, F–I). Collectively, our studies indicate that KLF6 is quintessential for inflammatory stimulus-induced macrophage motility, macrophage recruitment to sites of inflammation, and the progression of inflammation.
FIGURE 2.

KLF6 enhances macrophage recruitment to sites of inflammation. A–C, Lyz2cre and Lyz2cre:KLF6fl/fl mice were subjected to TPA-induced cutaneous inflammation. Control and TPA-treated tissues from these mice were stained with hematoxylin and eosin (A). The CellProfiler Analyst software was utilized to quantify epithelial cell thickness (B) and total tissue thickness (C) of control and TPA-treated mice. D and E, control and TPA-treated mouse tissues were subjected to immunohistochemical analysis using anti-F4/80 antibody to detect macrophage infiltration (D). These immunohistochemical images were utilized for quantification of macrophage number by NIS-Elements imaging software (E). n = 5 mice/group. F–I, tissue levels of CCL2, IL1α, IL1β, and IL6 were analyzed by Quantikine ELISA kit and normalized to tissue weight. This experiment was repeated three times. Scale bars = 100 μm. Graphs represent mean ± S.D. Box plots represent the median with first and third quartiles, and whiskers represent minimum or maximum. *, p ≤ 0.05. p < 0.05 between indicated groups is considered significant.
KLF6 Enhances Pro-inflammatory Chemokine and Cytokine Expression in Macrophages
Macrophage-derived pro-inflammatory chemokines and cytokines play an essential role in the elaboration of inflammation. Our studies so far indicated that KLF6 is required for macrophage recruitment to sites of inflammation as well as progression of inflammation (Figs. 1 and 2). Therefore, we examined whether amending KLF6 expression would affect LPS-induced pro-inflammatory chemokine and cytokine expression in macrophages. Accordingly, RAW264.7 cells overexpressing KLF6 or empty vector were stimulated with LPS and evaluated for pro-inflammatory chemokine and cytokine expression by quantitative PCR analysis (Fig. 3A). The results reveal that overexpression of KLF6 significantly enhanced LPS-induced Ccl7, Il6, Il12a, and Thbs4 expression in the RAW264.7 macrophage cell line (Fig. 3A). To evaluate the effects of KLF6 deficiency on inducible pro-inflammatory chemokine/cytokine expression, we utilized BMDMs derived from myeloid-specific KLF6-null (Lyz2cre:KLF6fl/fl) and control (Lyz2cre) mice. Concordant with our overexpression studies (Fig. 3A), deficiency of KLF6 significantly attenuated LPS-induced Ccl7, Il6, Il12a, and Thbs4 expression in primary macrophages (Fig. 3B). Taken together, these results implicate a critical role for KLF6 in macrophage pro-inflammatory chemokine/cytokine gene expression, cellular migration, and macrophage recruitment to sites of inflammation (Figs. 1–3).
FIGURE 3.

KLF6 enhances pro-inflammatory chemokine and cytokine expression. A and B, RAW264.7 cells transfected with pCI-neo-KLF6 or pCI-neo plasmids (A) and BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice (B) were stimulated with 100 ng/ml LPS for 4 h. Total RNA from these experiments was collected, and cDNA was analyzed for expression of Ccl7, Il6, Il12a, and Thbs4 by quantitative PCR and normalized to 36B4. Each experiment was performed four times with three replicates in each group. Graphs represent mean ± S.D. *, p ≤ 0.05. p < 0.05 between indicated groups is considered significant.
KLF6 Represses BCL6 Expression in Macrophages by Elevating PRDM1
Previous studies have demonstrated that BCL6 is required for macrophage quiescence and repression of the pro-inflammatory response (21). Interestingly, deficiency of BCL6 significantly elevated LPS induced pro-inflammatory chemokines and cytokines, including gene targets that are attenuated in KLF6-deficient macrophages (21, 24). Therefore, we hypothesized that KLF6 enhances pro-inflammatory gene expression by repressing BCL6 expression in macrophages. To test this hypothesis, RAW264.7 cells overexpressing KLF6 (pCI-neo-KLF6) or empty vector and BMDMs derived from Lyz2creand Lyz2cre:KLF6fl/fl mice were stimulated with LPS. Total RNA from these experiments was analyzed for Bcl6 expression (Fig. 4, A and B). Our results indicate that overexpression of KLF6 significantly attenuated LPS-induced Bcl6 expression in the RAW264.7 macrophage cell line (Fig. 4A). Concordant with this observation, deficiency of KLF6 significantly elevated LPS-induced Bcl6 expression in Lyz2cre:KLF6fl/fl BMDMs (Fig. 4B). Concurrently, we examined whether these observations were recapitulated at the protein level (Fig. 4C). Our results indicate that LPS stimulation significantly elevated BCL6 protein expression in Lyz2cre:KLF6fl/fl BMDMs (Fig. 4C, fourth lane) compared with Lyz2cre BMDMs (Fig. 4C, third lane). Subsequently, we evaluated whether KLF6 regulates LPS-induced BCL6 expression by directly occupying KLF6 binding sites on the BCL6 promoter. However, our KLF6-ChIP analysis results indicated that LPS stimulation did not enrich KLF6 on any putative binding sites on the BCL6 promoter (data not shown). Recent studies have indicated that PRDM1 (Blimp1) represses BCL6 expression in numerous cell types (18–20). Therefore, we examined whether KLF6 regulates PRDM1 by enriching on its promoter following LPS stimulation. Interestingly, the KLF6-ChIP analysis results revealed that LPS stimulation of wild-type BMDMs significantly induced KLF6 occupancy on the PRDM1 promoter (−1616 to −1463) compared with the vehicle control group (Fig. 4D). Based on these observations, we hypothesized that KLF6 represses BCL6 expression by up-regulating PRDM1 expression in macrophages. To evaluate this hypothesis, BMDMs derived from Lyz2cre, Lyz2cre:KLF6fl/fl mice and RAW264.7 cells overexpressing KLF6 or empty vector were stimulated with LPS. The total RNA derived from these experiments was examined for Prdm1 expression (Fig. 4, E and F). The results revealed that overexpression of KLF6 significantly enhanced LPS induced Prdm1 expression in RAW264.7 cells (Fig. 4E). Consistent with this observation, loss of KLF6 completely abolished LPS-induced Prdm1 expression in Lyz2cre:KLF6fl/fl BMDMs (Fig. 4F). Simultaneously, we investigated whether these observations were emulated at the protein level (Fig. 4G). The results revealed that LPS-induced PRDM1 protein expression was completely attenuated in Lyz2cre:KLF6fl/fl mice BMDMs (Fig. 4G, fourth lane) compared with Lyz2cre BMDMs (Fig. 4G, third lane).
FIGURE 4.

KLF6 represses inducible BCL6 expression in a PRDM1-dependent manner. A–C, RAW264.7 cells transfected with pCI-neo-KLF6 or pCI-neo plasmids (A) and BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice (B) were stimulated with 100 ng/ml LPS for 4 h. Total RNA and protein samples were extracted. Expression of BCL6 at the mRNA (A and B) and protein (C) levels was analyzed by quantitative PCR and Western blotting, respectively. D, BMDMs derived from wild-type mice were stimulated with 100 ng/ml LPS for 4 h. The ChIP analysis was performed on the PRDM1 promoter (−1616 to −1464) utilizing anti-KLF6 antibody or isotype-specific IgG. E–G, RAW264.7 cells transfected with pCI-neo-KLF6 or pCI-neo plasmids (E) and BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice (F) were stimulated with 100 ng/ml LPS for 4 h. Expression of PRDM1 at the mRNA (E and F) and protein (G) level were analyzed by quantitative PCR and Western blotting, respectively. H–K, RAW264.7 cells transfected with pCI-neo-KLF6 or pCI-neo plasmids were co-transfected with siControl or siPrdm1 siRNA. These cells were stimulated with 100 ng/ml LPS for 5 h and evaluated for Bcl6, Il1α, Il1β, and Il6 expression by quantitative PCR analysis. L, expression of KLF6 levels in Lyz2cre and Lyz2cre:KLF6fl/fl mouse BMDMs was analyzed by Western blotting. M, overexpression of KLF6 in RAW264.7 cells was analyzed by Western blotting. N, PRDM1 protein expression levels in RAW264.7 cells following siPrdm1 siRNA transfection were analyzed by Western blotting. 36B4 and β-Actin were used as the housekeeping genes for quantitative PCR and Western blotting analysis, respectively. Each experiment was performed four times. Quantitative PCR and ChIP analyses contained three replicates in each experiment. Western blots were subjected to densitometry analysis utilizing Quantity One 1-D analysis software and normalized to β-Actin. Graphs represent mean ± S.D. Box plots represent the median with first and third quartiles, and whiskers represent minimum/maximum. *, p ≤ 0.05; N.S., not significant. p < 0.05 between indicated groups is considered significant.
Next we examined whether KLF6 enhanced pro-inflammatory gene expression is PRDM1-dependent. Accordingly, RAW264.7 cells were overexpressed with KLF6 in the presence or absence of siRNA specifically targeting PRDM1 expression (Fig. 4, H–K). As anticipated, overexpression of KLF6 significantly attenuated LPS-induced Bcl6 expression in RAW264.7 cells (Fig. 4H, fourth box). Importantly, silencing of Prdm1 significantly enhanced LPS-induced Bcl6 mRNA expression (Fig. 4H, sixth box). Compellingly, overexpression of KLF6 did not diminished Bcl6 elevation in attenuation of Prdm1 expression (Fig. 4H, eighth box). These results conclusively demonstrate that KLF6 suppress Bcl6 expression in a PRDM1-dependent manner. Further, analysis of pro-inflammatory target genes of KLF6 indicates that overexpression of KLF6 significantly elevated LPS-induced Il1α, Il1β, and Il6 expression (Fig. 4, I–K, fourth box). Attenuation of Prdm1 expression significantly diminished LPS-induced Il1α, Il1β, and Il6 expression (Fig. 4, I–K, sixth box). Interestingly, deficiency of Prdm1 in KLF6-overexpressing cells completely abrogated the inductive effects of KLF6 on Il1α, Il1β, and Il6 expression following LPS stimulation (Fig. 4, I–K, eighth box). We confirmed silencing of KLF6 (Fig. 4L, in Lyz2cre:KLF6fl/fl mouse BMDMs) overexpression of KLF6 (Fig. 4M, in RAW264.7 cells) and silencing of PRDM1 (Fig. 4N, in RAW264.7 cells) at the protein level by Western blotting analysis. Taken together, our results are the first to reveal that pro-inflammatory transcription factor KLF6 represses LPS-induced BCL6 expression and induce pro-inflammatory gene expression by elevating PRDM1 expression in macrophages.
Diminished Chemokine/Cytokine Expression in KLF6-null Macrophages Are BCL6-dependent
Our results so far indicated that KLF6 suppresses BCL6 expression in macrophages. BCL6 is known to repress pro-inflammatory chemokine and cytokine expression in macrophages (21). Therefore, we investigated whether attenuation of pro-inflammatory chemokine and cytokine expression in KLF6-deficient macrophages is BCL6-dependent. Accordingly, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were nucleofected with BCL6-specific siRNA or control siRNA. Cells with a more than 80% reduction in Bcl6 mRNA levels were utilized for these studies. These cells were stimulated with LPS, and the expression of pro-inflammatory chemokines/cytokines that are the direct targets of KLF6 was evaluated by quantitative PCR analysis. We confirmed a reduction in BCL6 (Fig. 5G) at the protein level by Western blotting analysis. Our results indicate that deficiency of KLF6 significantly attenuated LPS-induced pro-inflammatory chemokine Ccl2 and Ccl7 expression in macrophages (Fig. 5, A and B). Interestingly, loss of BCL6 in KLF6-null macrophages strongly reversed attenuated Ccl2 and Ccl7 expression following LPS exposure (Fig. 5, A and B). Next, we examined whether attenuation of pro-inflammatory cytokine expression in KLF6-null macrophages is BCL6-dependent (Fig. 5, C–F). The results illustrate that deficiency of KLF6 significantly attenuated LPS-induced Il1α, Il1β, Il6, and Il12a expression in macrophages (Fig. 5, C–F). Intriguingly, loss of BCL6 in KLF6-null macrophages completely reversed attenuated Il1α, Il1β, Il6, and Il12a expression following LPS stimulation (Fig. 5, C–F).
FIGURE 5.

Inhibition of BCL6 reverses suppression of pro-inflammatory chemokine/cytokine expression in KLF6-deficient macrophages. A–F, BMDMs derived from Lyz2cre, Lyz2cre:KLF6fl/fl mice were nucleofected with BCL6-specific siRNA or control siRNA. These cells were stimulated with 100 ng/ml LPS for 4 h. Total RNA from these experiments was collected, and cDNA was analyzed for expression of Ccl2 (A), Ccl7 (B), Il1α (C), Il1β (D), Il6 (E), and Il12a (F) by quantitative PCR and normalized to 36B4. G, BCL6 protein expression levels in BMDMs following siBcl6 siRNA transfection were analyzed by Western blotting. Western blots were subjected to densitometry analysis utilizing Quantity One 1-D analysis software and normalized to β-Actin. Each experiment was performed four times with six replicates. Graphs represent mean ± S.D. Box plots represent the median with first and third quartiles, and whiskers represent minimum/maximum. *, p ≤ 0.05; N.S., not significant. p < 0.05 between indicated groups is considered significant.
To further corroborate these observations, we utilized a pharmacological approach to block BCL6 function in macrophages. Accordingly, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were treated with a BCL6 inhibitor, 79-6 (27) and subsequently stimulated with LPS (Fig. 6, A–E). The total RNA derived from these experiments was assessed for expression of pro-inflammatory chemokine and cytokine expression by quantitative PCR analysis. As anticipated, our results revealed that a deficiency of KLF6 significantly attenuated LPS-induced Ccl2, Il1α, Il1β, Il6, and Il12a expression in Lyz2cre:KLF6fl/fl BMDMs (Fig. 6, A–E). Concordant with previous observations (Fig. 5), pharmacological inhibition of BCL6 in KLF6-deficient macrophages completely reversed attenuated Ccl2, Il1α, Il1β, Il6, and Il12a expression following LPS stimulation (Fig. 6, A–E). Taken together, our results reveal that attenuation of pro-inflammatory chemokine/cytokine expression in KLF6-deficient macrophages is BCL6-dependent.
FIGURE 6.

Pharmacological inhibition of BCL6 reverses suppression of pro-inflammatory chemokine/cytokine expression in KLF6-deficient macrophages. A–E, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were treated with the BCL6-specific inhibitor 79-6 (250 μm). These cells were stimulated with 100 ng/ml LPS for 4 h. Total RNA from these experiments was collected, and cDNA was analyzed for expression of Ccl2 (A), Il1α (B), Il1β (C), Il6 (D), and Il12a (E) by quantitative PCR and normalized to 36B4. Each experiment was performed four times with six replicates. Graphs represent mean ± S.D. *, p ≤ 0.05; N.S., not significant. p < 0.05 between indicated groups is considered significant.
KLF6-BCL6 Signaling Regulates Macrophage Motility
Our studies so far indicated that a deficiency of KLF6 significantly attenuated pro-inflammatory chemokine/cytokine expression and cellular motility in macrophages. Further, we established that the elevated BCL6 expression in KLF6-null macrophages suppresses chemokine/cytokine expression following LPS stimulation. Therefore, we investigated whether the diminished cellular migration and invasion observed in KLF6-null macrophages are BCL6-dependent. Accordingly, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were nucleofected with BCL6-specific siRNA or control siRNA separately. These cells were stimulated with LPS and subjected to cellular migration and invasion function analysis (Fig. 7, A and B). As demonstrated above (Fig. 1, B and D), deficiency of KLF6 significantly curtailed LPS-induced macrophage migration and invasion (Fig. 7, A and B). Compellingly, loss of BCL6 expression in KLF6-null macrophages completely reversed attenuated cellular migration and invasion (Fig. 7, A and B). To further confirm these observations, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were treated with a BCL6-specific inhibitor, 79-6, and subjected to LPS-induced cellular migration and invasion analysis (Fig. 7, C and D). Consistent with genetic inhibition studies, pharmacological blockade of BCL6 in KLF6-deficient macrophages completely reversed diminished cellular migration and invasion (Fig. 7, C and D). Taken together, our results reveal that the attenuation of cellular motility observed in KLF6-deficient macrophages is BCL6-dependent.
FIGURE 7.

The KLF6-BCL6 signaling axis regulates macrophage motility. A and B, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were nucleofected with BCL6-specific siRNA or control siRNA. These cells were stimulated with 100 ng/ml LPS for 4 h and added to the upper chamber of migration or invasion tissue culture inserts. C and D, BMDMs derived from Lyz2cre and Lyz2cre:KLF6fl/fl mice were treated with the BCL6-specific inhibitor 79-6 (250 μm). These cells were stimulated with 100 ng/ml LPS for 4 h and subjected to migration or invasion analysis. The number of cells that migrated (A and C) or invaded (B and D) across the membrane in the control group (untreated Lyz2cre BMDMs or RAW264.7 cells transfected with pCI-neo plasmids) was assigned as 100%, and -fold changes over this are indicated. Each experiment was performed three times with four replicates. Graphs represent mean ± S.D. *, p ≤ 0.05; N.S., not significant. p < 0.05 between indicated groups is considered significant.
Discussion
Our discoveries are the first to identify that KLF6 enhances macrophage-mediated inflammation by suppressing the expression of an inducible, sequence-specific negative feedback regulator such as BCL6. Inhibition of BCL6 completely reversed attenuated pro-inflammatory chemokine/cytokine expression and cellular motility in KLF6-deficient macrophages. The key findings of this study are as follows: KLF6 enhances macrophage mobility under ex vivo and in vivo conditions; KLF6 promotes macrophage recruitment to sites of inflammation and progression of inflammation; KLF6 heightens LPS induced pro-inflammatory chemokine/cytokine expression in macrophages; KLF6 suppresses LPS-induced BCL6 expression in macrophages; KLF6 induces PRDM1 expression by directly enriching its promoter; KLF6 elevates pro-inflammatory gene expression in a PRDM1-dependent manner; pharmacological or genetic inhibition of BCL6 in KLF6-deficient macrophages reverses diminished pro-inflammatory cytokine and chemokine expression; and blockage of BCL6 completely abrogates the attenuation of cellular motility observed in KLF6-null macrophages. Collectively, these findings indicate that KLF6 boosts inflammation by suppressing BCL6 expression in macrophages (Fig. 8).
FIGURE 8.
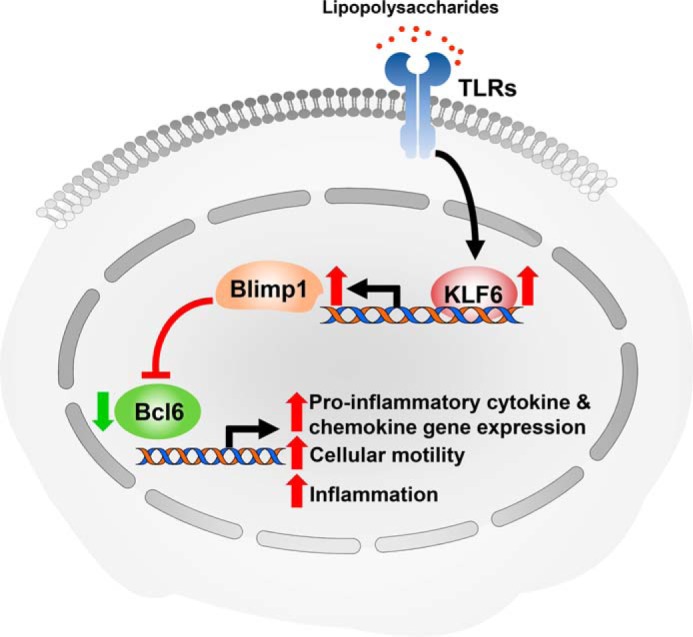
KLF6 enhances macrophage-mediated inflammation by repressing BCL6 expression through PRDM1.
Monocyte-derived macrophages are essential components of the innate immune system and play a central role in the pathogenesis of inflammation. A number of transcription factors are implicated in the regulation of inflammation in a signal-specific manner (28). Our studies are the first to define a role for KLF6 in macrophage inflammatory gene expression and polarization function (24). Originally, KLF6 was discovered and cloned by screening human placental DNA-binding proteins that interact with a core promoter element on a solid support (29). Further, studies from several laboratories indicated that KLF6 is expressed in multiple tissues, including lung, liver, skeletal muscle, kidney, placenta, and heart. Similarly, KLF6 dysfunction has been associated with a broad spectrum of disease conditions (23). Further, recent human studies have revealed elevated KLF6 expression in several chronic inflammatory disease conditions, including psoriasis and IBD (25, 30). Our studies have conclusively documented that KLF6 promotes pro-inflammatory gene programming in macrophages (24, 25). Myeloid-specific deficiency of KLF6 attenuated pro-inflammatory gene expression in experimental murine models of inflammation (25). Further, other independent studies have recapitulated our observations (31). This study revealed that KLF6 is essential for macrophage migration and invasion functions. Deficiency of KLF6 significantly attenuated LPS-induced macrophage motility under ex vivo and in vivo conditions. Evaluation of protective and pathogenic macrophage populations indicated that macrophage recruitment is fundamental in amplification to the resolution of inflammation. To this end, our investigation conclusively documents that KLF6 deficiency significantly attenuates macrophage recruitment to sites of inflammation.
The transcriptional control of inducible inflammatory gene expression requires the coordinated regulation of multiple transcription factors, co-activators, and repressors. Mononuclear phagocytes are transcriptionally dynamic throughout the processes of inflammation (13). During initiation of inflammation, transcriptionally quiescent monocytes are recruited to the site of inflammation and differentiate into activated inflammatory macrophages (32). Macrophage activation is a pathophysiological event that requires de novo expression of numerous signaling molecules, cytokines, and growth factors (12). Our studies revealed that a deficiency of KLF6 significantly attenuated pro-inflammatory chemokine/cytokines, including CCL2, CCL7, IL1α, IL1β, IL6, and IL12a. Further, diminished expression of these inflammatory agents in myeloid-KLF6-null mice may also contribute to reduced macrophage recruitment and significantly diminished adverse effects of inflammation. Concordant with these observations, studies from our group and others have indicated that elevated levels of myeloid-KLF6 are associated with adverse outcomes in human and experimental models of inflammatory disease conditions (25, 30). Studies from numerous laboratories implicated that transcription factors such as STATs, NF-κB, interferon regulatory factors, HIF-1α, AP-1, and PPARs play a critical role in the regulation of inflammatory gene expression in macrophages (7). Investigations from our group and others have indicated that KLF6 actively cooperates with NF-κB to induce pro-inflammatory gene expression and antagonize PPARγ or STAT3 to suppress anti-inflammatory gene expression in macrophages (24, 25, 31). Taken together, our studies, for the first time, reveal that KLF6 is essential for macrophage motility, recruitment to sites of inflammation, pro-inflammatory chemokine/cytokine expression, and progression of inflammation.
It is well established that the induction of inflammatory gene expression is essential for host defense during infection or injury (2). Our studies reveal that KLF6 is one such transcription activator of pro-inflammatory gene expression in macrophages. Importantly, KLF6 sustains pro-inflammatory gene expression in macrophages by inducing PRDM1. However, it is important to curb pro-inflammatory gene expression in a timely manner to limit the detrimental effects of inflammation. Generally, inhibition of inflammatory gene expression in macrophages is achieved through basal or inducible repressors (12). BCL6 is one such inducible, sequence-specific repressor of inflammatory gene expression in macrophages (15, 21). Studies from multiple laboratories indicated that a deficiency of BCL6 dramatically elevated basal and inducible pro-inflammatory chemokine and cytokine expression in macrophages (17, 21, 24). Specifically, studies from Barish et al. (21, 33) revealed that BCL6 represses one-third of the NF-κB target genes induced by LPS in a nuclear receptor co-repressor 2/nuclear receptor co-repressor 1-dependent manner. Our recent studies revealed that KLF6 enhances basal as well as LPS/IFNγ-induced NF-κB transcriptional activity in macrophages (24, 25). Consistent with these observations, overexpression of KLF6 enhanced and deficiency of KLF6 attenuated LPS-induced NF-κB target gene expression in macrophages. Taken together, the KLF6-BCL6 signaling axis works upstream of NF-κB signaling to regulate pro-inflammatory gene expression in macrophages. In parallel with these observations, our studies reveal that deficiency of KLF6 enhances BCL6 expression in macrophages. This resulted in attenuation of pro-inflammatory cytokine and chemokine expression in KLF6-null macrophages. Interestingly, across different cellular systems, PRDM1 is known to suppress BCL6 expression (18–20). Indeed, our studies illustrate that KLF6 induces PRDM1 expression by directly enriching its promoter. Further, diminished expression of PRDM1 in KLF6-null macrophages resulted in elevated BCL6 expression. A study from Barish et al. (33) indicated that myeloid-Bcl6 deficiency significantly accelerated the progression of inflammatory disease conditions such as atherosclerosis. Consistent with these observations, our study demonstrated that elevated BCL6 levels in KLF6-null macrophages significantly attenuated pro-inflammatory gene expression and cellular motility. We discovered that blockade of BCL6 in KLF6-null macrophages reversed pro-inflammatory gene expression as well as cellular migration and invasion functions. Collectively, our study reveals that KLF6 enhances inflammation by suppressing BCL6 expression in macrophages.
In summary, the in vitro, ex vivo, and in vivo observations presented here emphasize the importance of KLF6 in macrophage cytokine/chemokine gene expression and amplification of inflammation. This study is the first to identify that KLF6 elevates pro-inflammatory gene expression by suppressing BCL6 expression in macrophages. Further, loss of BCL6 completely reversed attenuated inflammatory phenotypes in KLF6-null macrophages. Taken together, our studies uncover a novel KLF6-BCL6 signaling pathway that regulates inflammatory gene expression and progression of inflammation, which can be implicated in a broad spectrum of human chronic and acute inflammatory disease conditions.
Experimental Procedures
Materials
LPS, 12-O-tetradecanoylphorbol-13-acetate (TPA), hematoxylin, eosin, and thioglycollate broth were purchased from Sigma-Aldrich (St. Louis, MO). The mouse monoclonal anti-β-Actin antibody was obtained from Santa Cruz Biotechnology, Inc. (SC-69879, lot DO111, Santa Cruz, CA). Rabbit monoclonal anti-BCL6 (4242S, lot 1, clone D665C10) and anti-PRDM1 (9115S, lot 6) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Phycoerythrin-Texas Red-labeled anti-integrin subunit alpha M (CD11b) antibody (RM2817, lot 1363544A) was obtained from Invitrogen. Phycoerythrin-labeled anti-SIGLEC5 (SiglecF) antibody (552126, lot 2244617) was obtained from BD Biosciences. FITC-conjugated anti-mouse LY-6G (Gr-1) antibody (catalog no. 11-5931-82, clone RB6–8C5, lot E013253) was obtained from eBioscience (San Diego, CA). Rat anti-mouse F4/80 antibody (MCA497R, batch no. 10108) was obtained from ABD Serotec. Goat anti-rabbit IgG conjugated with HRP (A16096, lot 27-108-04113), goat anti-mouse IgG conjugated with HRP (A16066, lot 31-118-043013), Lipofectamine® transfection reagents, and control and Prdm1-specific siRNAs were obtained from Thermo Fisher Scientific (Grand Island, NY). The BCL6 inhibitor 79-6 was obtained from EMD Millipore (Billerica, MA). Mouse MCP1 (MJE00), IL1α (MLA00), IL1β (MLB00C), IL6 (M6000B), the Quantikine ELISA kit, and cell migration and invasion chambers were obtained from R&D Systems (Minneapolis, MN). Control and BCL6-specific siRNAs were obtained from Qiagen. The Amaxa® mouse macrophage Nucleofector® kit was obtained from Lonza. The RAW264.7 cell line was obtained from the American Type Culture Collection (Manassas, VA). All tissue culture supplies were obtained from Corning Inc. (Lowell, MA). All other chemicals and reagents used were of analytical grade and were obtained from commercial sources.
Cell Culture
BMDMs were generated as described previously (34). Briefly, bone marrow cells from 8-week-old wild-type, Lyz2cre, and Lyz2cre:KLF6fl/fl mice were harvested from the femur and tibia. These bone marrow cells were cultured in cell culture medium supplemented with mouse recombinant macrophage colony stimulating factor 1 for 7 days. These BMDMs were collected and utilized for the indicated experiments. Similarly, peritoneal macrophages were obtained from the peritoneal cavity by inducing peritonitis with 3% thioglycollate broth in 8-week-old mice as described earlier (24). RAW264.7 cells were utilized for all in vitro experiments. All cells were cultured in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm glutamine in a humidified atmosphere of 5% CO2 and 95% air at 37 °C.
Experimental Mouse Models
All animal procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and conformed to guidelines established by the American Association for Accreditation of Laboratory Animal Care. The investigators performing all animal experiments were blinded to mouse genotype by non-continuous ear tag numbering. All mice were bred and maintained under pathogen-free conditions, fed standard laboratory chow (Harlan Teklad, Indianapolis, IN), and kept on a 12-h light/dark cycle. The mouse line expressing lysozyme-M promoter-driven Cre recombinase (Lyz2cre) was obtained from The Jackson Laboratory (Bar Harbor, ME). Myeloid KLF6-specific null mice were generated as described previously (24). Briefly, Klf6 floxed (KLF6fl/fl) mice were crossed with Lyz2cre mice to generate a mouse line harboring the Klf6 floxed allele and Lyz2cre allele. These mice were further cross-bred to generate male and female offspring expressing two Lyz2cre and Klf6 floxed alleles. The mice with two Klf6 floxed and Lyz2cre alleles were used as the KLF6 myeloid-specific null group (C57BL/6 background). Mice with only two Lyz2cre alleles were used as the control group. Myeloid KLF6-specific null mouse-derived primary macrophages displayed an ∼85–90% reduction in KLF6 expression, as assessed by quantitative PCR and Western blotting analyses (24).
Thioglycollate-induced Peritonitis and TPA-induced Inflammation Models
Lyz2cre and Lyz2cre:KLF6fl/fl mice were subjected to thioglycollate-induced peritonitis as described previously (35). Briefly, 10-week-old mice were injected intraperitoneally with 2 ml of sterile 3% thioglycollate broth per mouse. Mice were euthanized 72 h after thioglycollate injection, and the peritoneal cavity was flushed with 5 ml of sterile 1× PBS and 1 ml of air to harvest cells. This procedure was repeated two more times to collect all responding inflammatory cells. Equal volumes of peritoneal lavage from each mouse were stained with fluorescently labeled anti-CD11b (a macrophage marker), anti-LY-6G (a neutrophil marker), and anti-SiglecF (an eosinophil marker) antibodies. The total number of macrophages (CD11b high, LY-6G, and SiglecF low) that responded to the site of inflammation was enumerated by FACS analysis. TPA-induced cutaneous inflammation experiments were performed as described previously (35). Briefly, ear fur from 8-week-old control (Lyz2cre) and myeloid KLF6-null (Lyz2cre:KLF6fl/fl) mice was removed 2 days before the experiment. The right ear was treated with TPA twice at an interval of 24 h (2.5 μg of TPA in 20 μl of acetone). The left ear was treated similarly with acetone alone and served as a vehicle control. The mice were euthanized 24 h after the second TPA application. The ear tissues were collected and fixed with 4% paraformaldehyde to perform hematoxylin and eosin staining to examine histological changes. Further, ear tissues were subjected to immunohistochemical analysis using F4/80 antibody to detect macrophage infiltration and quantification. Images were acquired at room temperature using a Leica-DM2000LED microscope (×20 objective with 0.4 numerical aperture) equipped with a QImaging camera (model 01-RGB-HM-S) supported with QCapture Pro image and analysis software. Immunohistochemical images were quantified using the CellProfiler Analyst software (BROAD Institute) and NIS-Elements imaging software (Nikon, Melville, NY). The total number of macrophages per field was expressed as the average of total macrophage cell counts in five fields of 100 μm2. No experimental animal or data from the experiment were excluded from analyses.
Macrophage Migration and Invasion Assay
Bone marrow-derived macrophages from control (Lyz2cre) and myeloid KLF6-null (Lyz2cre:KLF6fl/fl) mice or RAW264.7 cells transfected with pCI-neo or pCI-neoKLF6 (Addgene, Cambridge, MA) were exposed to 100 ng/ml LPS for 4 h on cell culture plates. These cells were detached and added to the upper wells of a Boyden-type migration chamber. To examine macrophage invasive and migratory properties, cells with the indicated treatment were added to the upper wells of Boyden chamber coated with or without basement membrane extracellular matrix extract, respectively. The lower chambers were filled with DMEM consisting of 5% FBS to provide a chemotactic gradient. These chambers were placed in a humidified incubator for 18 h at 37 °C supplemented with 5% CO2. Following incubation, cells in the upper wells were removed by cotton swab, and the migrated cells on the lower side of the filter were stained with trypan blue dye to test cell viability. Further, these cells were fixed and stained with Giemsa. The migrated cells on the filter were counted under an inverted microscope (Leica). The number of cells migrated or invaded in the control group (unstimulated Lyz2Cre BMDMs or RAW264.7 cells transfected with the pCI-neo plasmid) were assigned as 100%, and the relative percentages of -fold changes are indicated.
RNA Extraction, Reverse Transcription, and Real-time Quantitative PCR Assay
Total RNA was isolated from the indicated cell types after completion of the specified treatment using the High Pure RNA Isolation Kit (Roche Life Science, Indianapolis, IN). One microgram of total RNA was used for reverse transcription using Moloney Murine Leukemia Virus reverse transcriptase (New England Biolabs, Ipswich, MA) in the presence of random hexamers and oligo(dT) primers (Thermo Fisher Scientific). The resulting cDNA was subjected to real-time quantitative PCR using gene-specific primers and universal SYBR Green PCR Master Mix on a Step One Plus real-time PCR system (Applied Biosystems, Foster City, CA).
Transient Transfection, Western Blotting, and Chromatin Immunoprecipitation
RAW264.7 cells were transiently transfected with the indicated nucleotide with Lipofectamine transfection reagents as specified by the instructions of the manufacturer. Similarly, BMDMs were transiently transfected with the indicated siRNA utilizing the Amaxa mouse macrophage Nucleofector kit according to the instructions of the manufacturer. Western blotting for the indicated proteins was performed as described before (24). Briefly, the indicated cell types were lysed using radioimmunoprecipitation lysis buffer (Sigma-Aldrich) supplemented with a protease and phosphatase inhibitor mixture tablet (Roche Applied Science) following the specified treatments. Equal quantities of total protein were separated by SDS-PAGE and transferred to nitrocellulose membrane (EMD Millipore). These nitrocellulose membranes were incubated with primary antibody against β-Actin, KLF6, BCL6, or PRDM1 (1:1000 dilution), and HRP-conjugated goat anti-rabbit IgG was used as a secondary antibody (1:5000 dilution). Western blotting x-ray films were aligned on a nitrocellulose membrane to highlight the position of molecular weight markers. Further, Western blots were subjected to densitometry analysis utilizing Quantity One 1-D analysis software (Bio-Rad) and normalized to β-Actin expression.
Chromatin immunoprecipitation analyses were performed using the EZ-Magna ChIP G kit (Millipore Corp.) according to the instructions of the manufacturer. Briefly, BMDMs derived from wild-type mice were stimulated with LPS. Chromatin immunoprecipitations were performed using anti-KLF6 antibody (Sc-7158, lot C1710). Chromatin samples from these experiments were analyzed by real-time quantitative RT-PCR. Chromatin immunoprecipitation performed using isotype IgG was used as a negative control.
Statistical Analysis
Data are presented as mean ± S.D. in bar diagrams and minimum to maximum in box-whisker plots. The statistical significance of differences between two groups with normal sample distributions was analyzed using Student's t test, and without normal sample, distributions were analyzed using Mann-Whitney U test. p < 0.05 was considered statistically significant.
Author Contributions
G. H. M., L. D. H., and G. K. conceived and designed the study. G. K., G. H. M., R. D., S. M., and L. G. performed the experiments. G. K., G. H. M., L. D. H., R. D., L. G., and S. M. analyzed and interpreted the data. G. H. M. and G. K. wrote and edited the manuscript, and it was approved by all authors.
This work was supported by National Institutes of Health Grant HL126626, Cleveland Digestive Diseases Research Core Center Pilot/Feasibility Award P30DK097948, and Crohn's and Colitis Foundation of America Senior Research Award 421904 (to G. H. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IBD
- inflammatory bowel disease
- PPAR
- peroxisome proliferator-activated receptor
- BMDM
- bone marrow-derived macrophage
- TPA
- 12-O-tetradecanoylphorbol-13-acetate.
References
- 1. Laskin D. L., Sunil V. R., Gardner C. R., and Laskin J. D. (2011) Macrophages and tissue injury: agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Serbina N. V., Jia T., Hohl T. M., and Pamer E. G. (2008) Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26, 421–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murray P. J., and Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stout R. D., and Suttles J. (2004) Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukocyte Biol. 76, 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stout R. D., Jiang C., Matta B., Tietzel I., Watkins S. K., and Suttles J. (2005) Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 175, 342–349 [DOI] [PubMed] [Google Scholar]
- 6. Taylor P. R., Martinez-Pomares L., Stacey M., Lin H. H., Brown G. D., and Gordon S. (2005) Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23, 901–944 [DOI] [PubMed] [Google Scholar]
- 7. Sica A., and Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mantovani A., Biswas S. K., Galdiero M. R., Sica A., and Locati M. (2013) Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229, 176–185 [DOI] [PubMed] [Google Scholar]
- 9. Arnold L., Henry A., Poron F., Baba-Amer Y., van Rooijen N., Plonquet A., Gherardi R. K., and Chazaud B. (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yona S., Kim K. W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., and Jung S. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smale S. T., and Natoli G. (2014) Transcriptional control of inflammatory responses. Cold Spring Harb. Perspect. Biol. 6, a016261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Medzhitov R., and Horng T. (2009) Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 9, 692–703 [DOI] [PubMed] [Google Scholar]
- 13. Nykter M., Price N. D., Aldana M., Ramsey S. A., Kauffman S. A., Hood L. E., Yli-Harja O., and Shmulevich I. (2008) Gene expression dynamics in the macrophage exhibit criticality. Proc. Natl. Acad. Sci. U.S.A. 105, 1897–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schall T. J., and Bacon K. B. (1994) Chemokines, leukocyte trafficking, and inflammation. Curr. Opin. Immunol. 6, 865–873 [DOI] [PubMed] [Google Scholar]
- 15. Dent A. L., Shaffer A. L., Yu X., Allman D., and Staudt L. M. (1997) Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 276, 589–592 [DOI] [PubMed] [Google Scholar]
- 16. Ye B. H., Rao P. H., Chaganti R. S., and Dalla-Favera R. (1993) Cloning of bcl-6, the locus involved in chromosome translocations affecting band 3q27 in B-cell lymphoma. Cancer Res. 53, 2732–2735 [PubMed] [Google Scholar]
- 17. Basso K., and Dalla-Favera R. (2010) BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 105, 193–210 [DOI] [PubMed] [Google Scholar]
- 18. Miyauchi Y., Ninomiya K., Miyamoto H., Sakamoto A., Iwasaki R., Hoshi H., Miyamoto K., Hao W., Yoshida S., Morioka H., Chiba K., Kato S., Tokuhisa T., Saitou M., Toyama Y., et al. (2010) The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J. Exp. Med. 207, 751–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diehl S. A., Schmidlin H., Nagasawa M., van Haren S. D., Kwakkenbos M. J., Yasuda E., Beaumont T., Scheeren F. A., and Spits H. (2008) STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J. Immunol. 180, 4805–4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaffer A. L., Lin K. I., Kuo T. C., Yu X., Hurt E. M., Rosenwald A., Giltnane J. M., Yang L., Zhao H., Calame K., and Staudt L. M. (2002) Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 17, 51–62 [DOI] [PubMed] [Google Scholar]
- 21. Barish G. D., Yu R. T., Karunasiri M., Ocampo C. B., Dixon J., Benner C., Dent A. L., Tangirala R. K., and Evans R. M. (2010) Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24, 2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pasqualucci L., Migliazza A., Basso K., Houldsworth J., Chaganti R. S., and Dalla-Favera R. (2003) Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood 101, 2914–2923 [DOI] [PubMed] [Google Scholar]
- 23. McConnell B. B., and Yang V. W. (2010) Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 90, 1337–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Date D., Das R., Narla G., Simon D. I., Jain M. K., and Mahabeleshwar G. H. (2014) Kruppel-like transcription factor 6 regulates inflammatory macrophage polarization. J. Biol. Chem. 289, 10318–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goodman W. A., Omenetti S., Date D., Di Martino L., De Salvo C., Kim G. D., Chowdhry S., Bamias G., Cominelli F., Pizarro T. T., and Mahabeleshwar G. H. (2016) KLF6 contributes to myeloid cell plasticity in the pathogenesis of intestinal inflammation. Mucosal Immunol. 9, 1250–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cramer T., Yamanishi Y., Clausen B. E., Förster I., Pawlinski R., Mackman N., Haase V. H., Jaenisch R., Corr M., Nizet V., Firestein G. S., Gerber H. P., Ferrara N., and Johnson R. S. (2003) HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cerchietti L. C., Ghetu A. F., Zhu X., Da Silva G. F., Zhong S., Matthews M., Bunting K. L., Polo J. M., Farès C., Arrowsmith C. H., Yang S. N., Garcia M., Coop A., Mackerell A. D. Jr., Privé G. G., and Melnick A. (2010) A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 17, 400–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lawrence T., and Natoli G. (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 [DOI] [PubMed] [Google Scholar]
- 29. Koritschoner N. P., Bocco J. L., Panzetta-Dutari G. M., Dumur C. I., Flury A., and Patrito L. C. (1997) A novel human zinc finger protein that interacts with the core promoter element of a TATA box-less gene. J. Biol. Chem. 272, 9573–9580 [DOI] [PubMed] [Google Scholar]
- 30. Palau N., Julià A., Ferrándiz C., Puig L., Fonseca E., Fernández E., López-Lasanta M., Tortosa R., and Marsal S. (2013) Genome-wide transcriptional analysis of T cell activation reveals differential gene expression associated with psoriasis. BMC Genomics 14, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y., Lei C. Q., Hu Y. H., Xia T., Li M., Zhong B., and Shu H. B. (2014) Kruppel-like factor 6 is a co-activator of NF-κB that mediates p65-dependent transcription of selected downstream genes. J. Biol. Chem. 289, 12876–12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nilsson R., Bajic V. B., Suzuki H., di Bernardo D., Björkegren J., Katayama S., Reid J. F., Sweet M. J., Gariboldi M., Carninci P., Hayashizaki Y., Hume D. A., Tegner J., and Ravasi T. (2006) Transcriptional network dynamics in macrophage activation. Genomics 88, 133–142 [DOI] [PubMed] [Google Scholar]
- 33. Barish G. D., Yu R. T., Karunasiri M. S., Becerra D., Kim J., Tseng T. W., Tai L. J., Leblanc M., Diehl C., Cerchietti L., Miller Y. I., Witztum J. L., Melnick A. M., Dent A. L., Tangirala R. K., and Evans R. M. (2012) The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 15, 554–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davis B. K. (2013) Isolation, culture, and functional evaluation of bone marrow-derived macrophages. Methods Mol. Biol. 1031, 27–35 [DOI] [PubMed] [Google Scholar]
- 35. Nayak L., Goduni L., Takami Y., Sharma N., Kapil P., Jain M. K., and Mahabeleshwar G. H. (2013) Kruppel-like factor 2 is a transcriptional regulator of chronic and acute inflammation. Am. J. Pathol. 182, 1696–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]