

Abstract
Photochemical control over irreversible inhibition was shown using Ru(II)-caged inhibitors of cathepsin L. Levels of control were dependent on where the Ru(II) complex was attached to the organic inhibitor, reaching >10:1 with optimal placement. A new strategy for photoreleasing Ru(II) fragments from inhibitor-enzyme conjugates is also reported.
Photolabile protecting groups allow researchers to gain spatial and temporal control over the release of biologically active molecules.1 Photocaging disrupts equilibrium binding between the active species and its target, such that inhibition can only take place once the protecting group is released with light. Metal complexes are an important class of highly tunable photocaging groups that release biologically active ligands with visible light.2 In this communication, we demonstrate for the first time the successful application of Ru(II) photocaging towards irreversible inhibitors, which are a proven class of pharmacologically active and clinically approved drugs.3 This method has potential for garnering spatiotemporal control over irreversible inhibition in biological systems. As part of this study, we show the effective substitution of phenyl for a pyridyl group as an approach for caging “uncageable” organic molecules that are devoid of an appropriate functional group, where pyridyl serves as a handle for coordination to photolabile metal complexes. Finally, we also report a new strategy where a caged inhibitor first binds to a target enzyme, followed by photorelease of the Ru(II) fragment, providing the first example of a homing strategy and the photoinitiated uncaging of a Ru(II) compound that can itself be biologically active.4
Through our work with photocaging nitrile-based inhibitors of cysteine cathepsins,5 we became interested in developing photocaged versions of epoxysuccinyl inhibitors, as chemical tools for gaining spatial control over irreversible inhibition. Epoxysuccinyl-based compounds are a class of cysteine protease inhibitors that rely on irreversible nucleophilic attack of active site thiolates on epoxide “warheads” to create a covalent bond between inhibitors and their enzyme targets (Figure 1A).6 Through major efforts, potent and selective epoxysuccinyl-based inhibitors of cysteine cathepsins were identified. The CLIK inhibitors were first described almost two decades ago (Figure 1 B), showing efficacy for reducing in vitro and in vivo activity of cathepsin L.7 This enzyme, a lysosomal cysteine protease whose dysregulation is linked with cancer,8 atherosclerosis9 and other inflammatory disease states,10 has well characterized inhibitor binding sites, including the S and S' subsites exploited herein.6 However, we recognized that photocaging epoxysuccinyl inhibitors would be inherently more challenging than caging nitrile-based inhibitors, which are covalent reversible binders.11 Unlike nitriles, which are released from their target over time, this new strategy could provide permanent inactivation of the cysteine protease. Given that epoxide ring opening is an irreversible process, kinetic rather than thermodynamic control was expected, because given enough time, even a caged irreversible inhibitor would be able to adopt the correct orientation to react within the active site.6
Figure 1.

A) CLIK inhibitors bearing epoxysuccinyl warheads show irreversible binding to cysteine cathepsins. B) Structures of known CLIK inhibitors and new analogs designed for this study.
Epoxysuccinyl inhibitors do not contain a nitrile for coordination to ruthenium, so another functional group was required. The compound CLIK-148 (Figure 1B) was deemed as an attractive lead because it contains a pyridyl substituent that could integrate well with a series of Ru(II)-based photocaging groups developed for binding to aromatic N-heterocycles.4, 12 Analysis of X-ray crystallographic data of CLIK-148 bound to papain,13 a close model of cathepsin L, suggested that coordination of a bulky metal complex to the pyridine ring, which binds in the S′ subsite, would deter inhibitor binding. However, the steric crowding around the 2-pyridyl substituent of CLIK-148 would likely not be a good candidate for Ru(II) binding, so a new analog 1 (Figure 1B) was designed to place the nitrogen donor at the less crowded 4 position. In addition, we sought to disrupt binding to the S subsites of cathepsin L. CLIK-181 was deemed as an attractive lead,7b even though it does not contain a pyridyl group for ruthenium coordination. We surmised that replacement of the P2 phenyl group of CLIK-181 with a pyridyl group 2 (Figure 1B) would maintain binding to cathepsin L, and would also provide a handle for caging with Ru(II) complexes. Pyridyl groups are not protonated at physiological pH, and would be only partially protonated under our assay conditions at pH 5.5.
Syntheses of compounds 1 and 2 were accomplished starting from advanced intermediates reported in the literature;14 full synthetic schemes and characterization data are included in the Supporting Information. Ru(II) caging was carried out by treating the known precursor [Ru(tpy)(Me2bpy)Cl]Cl12 with inhibitors 1 or 2 (4 equiv) and AgPF6 (2.2 equiv) in acetone:H2O (3:5) at 50 °C, followed by chloride exchange using Bu4NCl in acetone, providing the caged complexes [Ru(tpy)(Me2bpy)(1)]Cl2 (3) and [Ru(tpy)(Me2bpy)(2)]Cl2 (4) (Scheme 1).
Scheme 1.
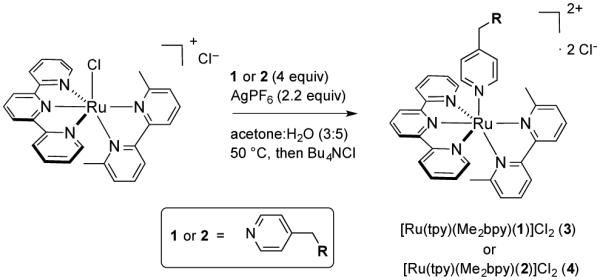
Synthesis of the ruthenium-caged epoxysuccinyl inhibitors 3 and 4.
Complexes 3 and 4 were characterized by 1H NMR, IR, electronic absorption spectroscopies, ESMS and elemental analysis. The 1H NMR spectrum of 3 exhibits a triplet at 4.9 ppm assigned to the α-proton of the phenylalanine residue and 2 singlets that are nearly coincidental at 3.02 ppm, consistent with the methine protons of the epoxide. As expected, the electronic absorption spectra of 3 and 4 are in close agreement with that of [Ru(tpy)(Me2bpy)(py)](PF6)2,12 with maxima at 474 nm (ε = 9,700 M−1cm−1) and 475 nm (ε = 10,400 M−1cm−1), respectively, consistent with the pyridyl group of 1 and 2 binding to the ruthenium center to generate complexes 3 or 4, respectively. The mass spectra of 3 and 4 show prominent ion clusters with major peaks at m/z 464.65 and 493.16, along with suitable isotopic distributions, consistent with the dications [Ru(tpy)(Me2bpy)(1)]2+ and [Ru(tpy)(Me2bpy)(2)]2+, respectively.
Cathepsin L inhibition was evaluated using a continuous fluorescence assay. This assay, where inhibitor and substrate are treated with enzyme, then substrate cleavage is monitored over time, is superior to IC50 determination for evaluating time-dependent inhibition observed by irreversible binders.6 Inhibition experiments were carried out with constant enzyme (4 nM) and Z-Phe-Arg-AMC substrate (10 μM) concentrations and inhibitor concentration was varied. Progress curves were fit satisfactorily to a model for competitive, irreversible inhibition model using Dynafit15 (Figures S23–28). Thermodynamics of inhibitor binding (Ki) and kinetics of inactivation (kinact) were determined, along with kinact/Ki, the second-order rate constant for irreversible inhibition, and the resulting data are tabulated in (Table 1). The second-order rate constant for inhibition of cathepsin L by 1 was within error for that of CLIK-148, indicating that shifting the N-atom from the 2- to the 4-position had no effect. Surprisingly, complex 3, the Ru(II)-caged version of 1, also showed potent inhibition of cathepsin L in the dark, with data for 1 and 3 within error. Irradiating solutions of 3 with visible light before adding enzyme had no effect on inhibition (data not shown). Monitoring the stability of 3 over time in the assay buffer by electronic absorption spectroscopy indicated that 3 is stable and does not release 1 during the inhibition experiment. The control complex [Ru(tpy)(Me2bpy)(py)](PF6)2 did not inhibit cathepsin L at the highest concentration tested (100 μM) under light (λirr ≥ 395 nm, 10 min) or dark conditions, consistent with the inactivity of the caging group alone. Taken together, these data show that Ru(II)-caged 3 is able to inhibit cathepsin L, with no discernible change from data for inhibitor 1, suggesting that binding a Ru(II) complex on the P′ side of 1 was not disrupting binding to cathepsin L.
Table 1.
Thermodynamic and kinetic parameters for inhibition of cathepsin L by CLIK-148, CLIK-181 and 1-4a
| Compound | Ki (⍰M) | kinact (s−1) | kinact/Ki (M−1 s−1) |
|---|---|---|---|
| CLIK-148 | 1.4 ± 0.7 | 0.015 ± 0.005 | 11000 ± 3000 |
| CLIK-181 | 1.3 ± 0.1 | 0.020 ± 0.003 | 15000 ± 2000 |
| 1 | 0.77 ± 0.27 | 0.011 ± 0.005 | 13000 ± 2800 |
| 2 | 1.6 ± 0.2 | 0.015 ± 0.001 | 9400 ± 1000 |
| 3 (dark) | 1.0 ± 0.2 | 0.016 ± 0.002 | 15000 ± 2000 |
| 4 (dark) | 14 ± 4 | 0.008 ± 0.002 | 570 ± 60 |
Inhibition data were acquired using cathepsin L (4 nM), Z-Phe-Arg-AMC (10 μM), and inhibitor (0.1–2.5 μM) in 0.4 M acetate buffer, pH 5.5, <1% DMSO, 4 mM EDTA, 0.01% Triton X-100, DTT = 8 at 25 °C. Data are averages of 3 independent experiments with errors equal to standard deviations. Data for 3 and 4 were collected under dark conditions. See SI for more details.
Data for inhibition of cathepsin L by CLIK-181, 2 and 4 were in marked contrast with the findings for 1 and 3. Replacing the Phe P2 residue of CLIK-181 with 4-pyridylalanine had a measurable, but not dramatic effect on inhibition by 2, with kinact/Ki within a factor of ~1.6. Gratifyingly, the Ru(II)-caged inhibitor 4 in the dark showed much slower inhibition than 2, with a second-order rate constant for inactivation of cathepsin L equal to 570 M−1 s−1, >16-fold slower than 2. Data indicated that binding of 4 to cathepsin L (Ki) was ~9 times weaker than that of 2, and the irreversible inactivation of cathepsin L (kinact) was ~2 times slower with 4 than with 2.
To gain further insight into the effectiveness of caging, compounds 2 and 4 (250 nM) were evaluated in an assay where enzyme and inhibitor were incubated under visible light (λirr ≥ 395 nm, 10 min irradiation prior to incubation) and dark conditions, and the enzyme activity was measured as a function of time of incubation. Cathepsin L (4 nM) was added every 10 min between 0–60 min, and enzyme activity was measured with the fluorgenic substrate Z-Phe-Arg-AMC (10 μM). Results are shown in (Figure 2). The free inhibitor 2 under light and dark conditions showed rapid inactivation of cathepsin L within ~20 min of incubation, but caged 4 was only active when it was irradiated prior to incubation, consistent with 4 releasing 2 upon irradiation with visible light. Conversely, 4 in the dark showed much slower inactivation of cathepsin L, with 70% of enzyme activity remaining after 60 min of incubation. Fitting the loss of enzyme activity over time to a single exponential gave krel = 10 for 4 under dark vs. light conditions. The same experiment carried out with 1 and 3 under dark and light conditions showed all data within error (krel = 1, Figure S29), consistent with results shown in (Table 1) where the Ru(II)-caged inhibitor 3 was a potent inactivator of cathepsin L. Taken together, these data indicate that fine tuning the position of the ruthenium-based caging group can provide >10-fold control over inhibition with light.
Figure 2.

Time-dependent inactivation of cathepsin L by 2 (blue with irradiation, red without) and 4 (green with irradiation and black without). Solutions of 2 or 4 (250 nM) were irradiated with visible light (λirr ≥ 395 nm, 10 min prior to incubation) or left in the dark and then were incubated with cathepsin L (4 nM) at 25 °C for 0–60 min. See Table 1 for conditions and SI for more details.
Due to its enhanced cathepsin L inactivation under irradiation, the photochemistry of 4 was investigated. Irradiation of 4 with visible light results in the exchange of 2 with a solvent molecule (S) to provide the photoproduct [Ru(tpy)(Me2bpy)(S)]2+ and one equivalent of free 2. To monitor this reaction, a solution of 4 in D2O was irradiated with λirr ≥ 455 nm for 5 h and the 1H NMR spectrum was monitored at various time points (Figure S30). Structures of the resulting photoproducts, a mixture of [Ru(tpy)(Me2bpy)(OD2)]2+ and 2, were elucidated by comparison to the products of the photolysis of [Ru(tpy)(Me2bpy)(py)]2+ in D2O (Figure S31), which produces [Ru(tpy)(Me2bpy)(OD2)]2+ and pyridine. For 4, the two singlets corresponding to the methyl groups on Me2bpy shift downfield from 1.41 and 1.95 ppm to 1.52 and 2.87 ppm upon irradiation, respectively, in agreement with the photoproduct from [Ru(tpy)(Me2bpy)(py)]2+. Additionally, the acetyl singlet at 1.90 ppm shifts downfield to 2.10 ppm, the two dimethylamide singlets shift downfield from 2.68 and 2.70 ppm to 2.84 and 2.91 ppm, and the phenylalanine α-proton triplet shifts downfield from 4.82 to 5.09 ppm; the new signals are consistent with the 1H NMR spectrum of 2 in D2O.
The efficiency of the photoinduced exchange of 2 with a solvent molecule upon irradiation of 4 was probed by electronic absorption spectroscopy. The 1MLCT transition of 4 blue shifts from 471 nm to 453 nm upon irradiation in CH3CN (λirr ≥ 455 nm), which is consistent with the release of 2 and formation of [Ru(tpy)(Me2bpy)(CH3CN)]2+ (Figure 3) This process occurs with a quantum yield (Φ500) of 0.079(4) with λirr = 500 nm. This lower value compared to that of [Ru(tpy)(Me2bpy)(py)]2+ (Φ500 = 0.16) can be attributed to the lower solubility of 2 in CH3CN as compared to that of pyridine, lowering the ability of the ligand to escape the solvent cage.
Figure 3.
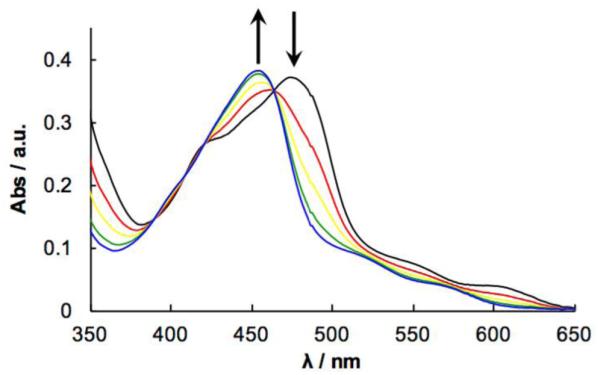
Electronic absorption spectra of 4 in CH3CN irradiated with λirr ≥ 455 nm for 0-4 min.
Given the stark differences between protein-binding behavior for 3 and 4, we probed the binding of ruthenium-inhibitor conjugates further using papain, a cysteine protease and model of cathepsin L (Figure 4). Following the conditions used to prepare papain bound to CLIK-148 for X-ray crystallographic analysis,13 the enzyme was treated with excess inhibitors 1-3 (dark) and cysteine in water with 20% DMSO and incubated for 18 h at rt. Samples were analyzed by LCMS analysis. Major peaks were observed consistent with modification of papain with 1-3 (Figures 4, S33–36). Importantly, when a solution of papain modified with 3 in the dark was irradiated (λirr ≥ 395 nm, 20 min), the major ion observed was lower by 517 units consistent with photochemical release of a [Ru(tpy)(Me2bpy)]2+ fragment. In solution, the exchange of 1 by a solvent molecule in 3 occurs with an efficiency similar to that of 4, with Φ500 = 0.097(6) in CH3CN and 0.043(3) in H2O. These data not only support the binding of 3 to a cysteine protease, but also show that photochemical release of a ruthenium complex can be achieved from the inhibitor-enzyme conjugate.
Figure 4.

ESMS characterization data of papain bound to 1-3. Data show the [Ru(tpy)(Me2bpy)]2+ fragment can be released by irradiation from papain modified with 3.
In conclusion, we demonstrated that kinetic control over irreversible inhibition can be achieved with ruthenium caging. We disclosed a method for photocaging bioactive compounds with metal complexes, using pyridyl groups in place of phenyl as a handle for metal coordination. This method is expected to be broadly applicable for caging organic molecules that are devoid of appropriate functional groups, whenever the potency of target binding is maintained upon pyridyl for phenyl substitution. An additional strategy for binding to proteases, followed by uncaging of the Ru(II) fragment represents a potential new manner for targeting cancer and other disease states where cysteine proteases are overexpressed. This approach may allow for targeted protease inhibition to be used in combination with cell death mediated by Ru(II) photosensitizers.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health (Grant EB 016062), National Science Foundation (CHE-1465067) and Wayne State University (grant to VGL) for their generous support of this research. We thank the Lumigen Instrument Facility for assistance with LCMS analysis
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Klan P, Solomek T, Bochet CG, Blanc A, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem. Rev. 2013;113119 doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deiters A. ChemBioChem. 2010;11:47. doi: 10.1002/cbic.200900529. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee H-M, Larson DR, Lawrence DS. ACS Chem. Biol. 2009;4:409. doi: 10.1021/cb900036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Zayat L, Calero C, Albores P, Baraldo L, Etchenique R. J. Am. Chem. Soc. 2003;125:882. doi: 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]; (b) Sgambellone MA, David A, Garner RN, Dunbar KR, Turro C. J. Am. Chem. Soc. 2013;135:11274. doi: 10.1021/ja4045604. [DOI] [PubMed] [Google Scholar]; (c) Farrer NJ, Salassa L, Sadler PJ. Dalton Trans. 2009:10690. doi: 10.1039/b917753a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pierri AE, Pallaoro A, Wu G, Ford PC. J. Am. Chem. Soc. 2012;134:18197. doi: 10.1021/ja3084434. [DOI] [PubMed] [Google Scholar]; (e) Chakraborty I, Carrington SJ, Mascharak PK. Acc. Chem. Res. 2014;47:2603. doi: 10.1021/ar500172f. [DOI] [PubMed] [Google Scholar]; (f) Karaoun N, Renfrew AK. Chem. Commun. 2015;51:14038. doi: 10.1039/c5cc05172j. [DOI] [PubMed] [Google Scholar]
- 3.Singh J, Petter RC, Baillie TA, Whitty A. Nat. Rev. Drug Discovery. 2011;10:307. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 4.Knoll JD, Albani BA, Turro C. Acc. Chem. Res. 2015;48:2280. doi: 10.1021/acs.accounts.5b00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Respondek T, Garner RN, Herroon MK, Podgorski I, Turro C, Kodanko JJ. J. Am. Chem. Soc. 2011;133:17164. doi: 10.1021/ja208084s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Herroon Mackenzie K, Sharma R, Rajagurubandara E, Turro C, Kodanko Jeremy J, Podgorski I. Biol. Chem. 2016;397:571. doi: 10.1515/hsz-2015-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers JC, Asgian JL, Ekici OD, James KE. Chem. Rev. 2002;102:4639. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Katunuma N, Murata E, Kakegawa H, Matsui A, Tsuzuki H, Tsuge H, Turk D, Turk V, Fukushima M, Tada Y, Asao T. FEBS Lett. 1999;458:6. doi: 10.1016/s0014-5793(99)01107-2. [DOI] [PubMed] [Google Scholar]; (b) Katunuma N. Proc. Jpn. Acad., Ser. B. 2011;87:29. doi: 10.2183/pjab.87.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sudhan DR, Siemann DW. Pharmacol. Ther. 2015;155:105. doi: 10.1016/j.pharmthera.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Kornmark L, Jonasson L, Forssell C, Yuan X-M. Atherosclerosis. 2009;202:92. doi: 10.1016/j.atherosclerosis.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 10.Conus S, Simon H-U. Swiss Med. Wkly. 2010;140:4. doi: 10.4414/smw.2010.13042. [DOI] [PubMed] [Google Scholar]
- 11.Frizler M, Stirnberg M, Sisay MT, Guetschow M. Curr. Top. Med. Chem. 2010;10:294. doi: 10.2174/156802610790725452. [DOI] [PubMed] [Google Scholar]
- 12.Knoll JD, Albani BA, Durr CB, Turro C. J. Phys. Chem. A. 2014;118:10603. doi: 10.1021/jp5057732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuge H, Nishimura T, Tada Y, Asao T, Turk D, Turk V, Katunuma N. Biochem. Biophys. Res. Commun. 1999;266:411. doi: 10.1006/bbrc.1999.1830. [DOI] [PubMed] [Google Scholar]
- 14.Chehade KAH, Baruch A, Verhelst SHL, Bogyo M. Synthesis. 2005:240. DOI: 10.1055/s-2004-837295. [Google Scholar]
- 15.Kuzmic P. Anal. Biochem. 1996;237:260. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.