

Abstract
Fairly recently, it was recognized that human ribosomopathies—developmental defects caused by mutations in ribosome biogenesis proteins—can exhibit tissue-specific defects rather than the expected global defects. This apparent anomaly—that seemingly ubiquitously expressed and required ribosomal proteins can have distinct functions in cell and tissue differentiation—has spurred new areas of research focused on better understanding translational mechanisms, biogenesis, and function in diverse cell types. This renewed appreciation for, and need to better understand, roles for ribosomal proteins in human development and disease has identified surprising similarities and differences in a variety of human ribosomopathies. Here, we discuss ribosomal protein functions in health and disease, focusing on the ribosome biogenesis protein Utp5/WDR43. New and exciting research in this field is anticipated to provide insight into a variety of previously understudied craniofacial dysostoses and result in significantly improved knowledge and understanding of roles for translational machinery in human craniofacial development and disease.
Keywords: craniofacial anomalies, craniofacial biology/genetics, developmental biology, zebrafish, cell biology, genetics
Ribosomopathies and Craniofacial Dysostoses
Craniofacial dysostosis is a prominent tissue-specific manifestation of ribosomopathies (Trainor and Merrill 2014). Ribosomopathies are genetic diseases associated with mutations in proteins present in ribosomes (i.e., the protein synthesis factories of cells) or in proteins required to make ribosomes (Freed et al. 2010; Hannan et al. 2013; McCann and Baserga 2013; Armistead and Triggs-Raine 2014; Sondalle and Baserga 2014; Trainor and Merrill 2014; De Keersmaecker et al. 2015). Although the requirement for ribosomes is ubiquitous in all cells, there is a surprisingly diverse array of disease manifestations associated with ribosomopathies. For example, ribosomopathies are associated with bone marrow failure (Burwick et al. 2012; Chirnomas and Kupfer 2013; Raiser et al. 2013), liver cirrhosis (Chagnon et al. 2002; Freed and Baserga 2010; Freed et al. 2012), exocrine pancreatic deficiency (Boocock et al. 2003; Finch et al. 2011; Provost et al. 2012), failure to develop a spleen (Bolze et al. 2013), and craniofacial defects (Trainor and Merrill 2014). Interestingly, craniofacial defects are characteristic of disparate ribosomopathies, including Diamond Blackfan anemia (Gazda et al. 2008) and Treacher Collins syndrome (Valdez et al. 2004; Dauwerse et al. 2011). As all cells require protein synthesis and, therefore, ribosomes, it is surprising that mutations that cause defective ribosome synthesis and function would be compatible with life.
Several ribosomopathies cause abnormal development of the face during embryogenesis. One ribosomopathy with marked craniofacial defects is Treacher Collins syndrome (Dixon et al. 2006; Jones et al. 2008; Dauwerse et al. 2011). Treacher Collins syndrome is a mandibulofacial dysostosis often associated with maxillary, mandibular, and zygomatic hypoplasia as well as conductive hearing loss (Trainor and Merrill 2014; Vincent et al. 2015). Patients with Treacher Collins syndrome have mutations in proteins required for the transcription of pre-ribosomal RNA (pre-rRNA) by RNA polymerase I (RNAPI). The act of transcription of pre-rRNA by RNAPI is fundamental to all of life, as it is the first step of making ribosomes. Proteins mutated in Treacher Collins syndrome patients include a transcription factor for RNAPI called TCOF1 (Treacher Collins Syndrome Collaborative Group 1996; Valdez et al. 2004) and 2 subunits of RNAPI: POLR1C and POLR1D (Dauwerse et al. 2011). Furthermore, another RNAPI subunit, POLR1A, is mutated in patients with a craniofacial disorder called acrofacial dysostosis, Cincinnati type (Weaver et al. 2015). Unlike Treacher Collins syndrome, acrofacial dysostoses are characterized by developmental defects of the limbs and craniofacial structures (Wieczorek 2013). Thus, mutations in multiple proteins physically involved in the transcription or pre-rRNA or in the initiation of pre-rRNA transcription cause a human disease associated with defects in craniofacial development.
The craniofacial phenotype observed in Treacher Collins syndrome patients has been replicated in animal models of the disease. In 2006, Dixon et al. generated a heterozygous Tcof1+/-mouse model, which exhibited many of the same features of Treacher Collins syndrome, including underdeveloped jaw structures. Interestingly, Tcof1+/- heterozygosity in some mouse strains was not compatible with postnatal survival, such as the C57BL/6 strain. However, in the DBA mouse strain background, the heterozygous Tcof1 loss-of-function mutation was compatible with postnatal survival. This suggests that there is some contribution of other genetic factors, in addition to Tcof1, to the severity of craniofacial developmental defects. In Tcof1+/- mice, neural crest cells (i.e., the progenitor cells responsible for the development of the jaw) demonstrated reduced proliferation and increased apoptosis, illustrating the mechanism by which craniofacial defects arise. Furthermore, the craniofacial defect in Tcof1+/- mice could be rescued by inhibition of p53, highlighting the significant role that this protein plays in the pathogenesis of Treacher Collins syndrome (Jones et al. 2008). The role of p53 in the development craniofacial defects due to defects in ribosome biogenesis has been illustrated in other animal models, including zebrafish and frogs (Zhao et al. 2014; Griffin et al. 2015).
In addition to known patient mutations in transcription factors or subunits of RNAPI, depletion or dysfunction of other proteins required for pre-rRNA transcription causes craniofacial developmental defects in animal models, including Nol11, DDX11, and WDR43 (Griffin et al. 2015; Sun et al. 2015; Zhao et al. 2014). Despite possible alternative functions for these proteins associated with craniofacial defects, all of them are required for efficient transcription of pre-rRNA, thus offering a strong argument that defects in ribosome synthesis—and the likely consequential disruptions in messenger RNA (mRNA) translation and protein synthesis—lie at the heart of the etiology of defective craniofacial development. For this review, we focus our discussion concerning the relationship between ribosome biogenesis and craniofacial development on the protein WDR43, also called Utp5, whose significance in craniofacial development has only recently been identified (Zhao et al. 2014). Many erudite reviews have explored the connection between other ribosome biogenesis proteins and craniofacial development (Sakai and Trainor 2009; Trainor and Andrews 2013; van Gijn et al. 2013; Ross and Zarbalis 2014; Trainor and Merrill 2014).
Overview of Eukaryotic Ribosome Biogenesis
The synthesis of ribosomes consists of many steps that occur in multiple cellular compartments. In short, ribosomes are first assembled in the nucleolus, which is located in the nucleus, and then transported to the cytoplasm, where they perform the vital function of translating mRNAs into proteins. The process of making ribosomes, aptly named ribosome biogenesis, initiates in the large non–membrane bound nuclear body called the nucleolus in eukaryotic cells (Henras et al. 2008; Woolford and Baserga 2013). Ribosomes are composed of 2 major constituents: ribosomal proteins and ribosomal RNA (rRNA). The mRNAs for ribosomal proteins are transcribed by RNA polymerase II, subsequently translated in the cytoplasm, and transported back to the nucleus and then nucleolus to be assembled into nascent ribosomal subunits: the large and small subunits.
Ribosome biogenesis initiates with the transcription of ribosomal DNA into rRNA. Of the 4 rRNAs, 3 (the 18S, 5.8S, and 28S [called 25S in yeast] rRNA) are not transcribed individually but are instead transcribed as one large pre-rRNA by RNAPI. This pre-rRNA contains the sequences for all 3 mature rRNAs (Fig. 1A). In addition to the sequences for the mature rRNAs, 2 external transcribed sequences and 2 internal transcribed spacers are removed through a series of cleavages to form the mature rRNAs (Fig. 1A). The fourth rRNA, called the 5S rRNA, is transcribed separately by RNA polymerase III and is incorporated later into assembling ribosomes (Woolford and Baserga 2013). The 18S rRNA is assembled into the small subunits, while the 5S, 5.8S, and 28S rRNAs are assembled into the large subunits (Fig. 1B). The final steps of the maturation of ribosomes occur with the transport of ribosomes to the cytoplasm, where they participate in translation—the synthesis of proteins from mRNAs. The complex and energy-intensive process of ribosome biogenesis requires the coordinated action of several small nucleolar RNAs (snoRNAs) and >200 proteins (Woolford and Baserga 2013). The ribosome biogenesis protein that is the subject of this review, named Utp5 in yeast and WDR43 in other eukaryotes, is essential for pre-rRNA transcription and processing (Fig. 1A; Gallagher et al. 2004; Prieto and McStay 2007).
Figure 1.
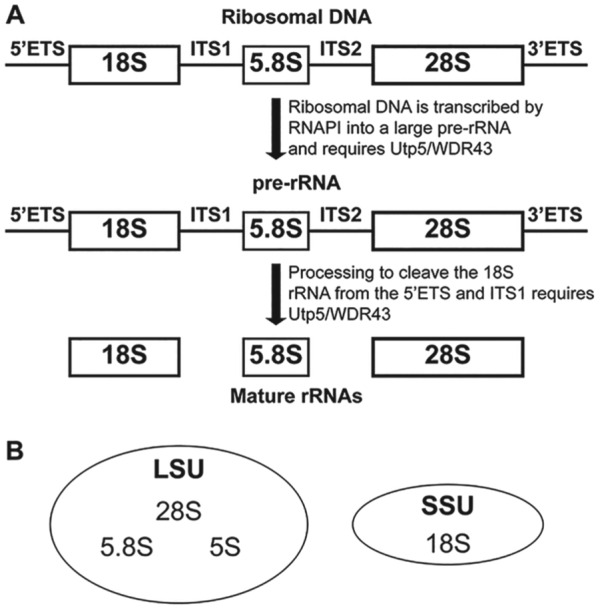
Overview of the requirements of Utp5/WDR43 for ribosome biogenesis. (A) Ribosomal DNA (rDNA) is transcribed by RNA polymerase I (RNAPI) in the nucleolus as a large precursor RNA encoding 3 of the 4 rRNAs and requires Utp5/WDR43. Multiple pre-rRNA processing steps remove the 5′ and 3′ external transcribed sequences (ETSs) and internal transcribed spacers (ITSs) 1 and 2 and results in the formation of 3 mature rRNAs from one large precursor. Formation of the mature small ribosomal subunit (SSU) specifically requires Utp5/WDR43. (B) Three rRNAs are found in the large subunit (LSU) of the ribosome: 5S, 5.8S, and 28S. One rRNA, 18S, is found in the small SSU of the ribosome.
Discovery and Role of Utp5 in the Yeast Saccharomyces cerevisiae
The role of Utp5 in ribosome biogenesis was first described with the purification of a large nucleolar ribonucleoprotein that contains the U3 snoRNA, called the small subunit processome (Dragon et al. 2002). The U3 snoRNA is a nucleolar-specific noncoding RNA required for ribosome biogenesis (Tyc and Steitz 1989; Kass et al. 1990). While the U3 snoRNA was known to be required for the maturation of the 18S rRNA, the panoply of proteins associated with U3 had remained elusive. To identify U3 interacting proteins, Dragon et al. (2002) tagged the Nop5/58 protein, which binds to the box C/D class of RNAs that includes the U3 snoRNA. A known U3-specific binding protein called Mpp10 was tagged like Nop5/58 but with a different epitope. Nop5/58 and its binding partners were purified from whole cell yeast extract with beads that would bind to the tag on Nop5/58. Subsequently, Nop5/58 and its binding partners were eluted from the beads. Then Mpp10 and its binding partners were purified from the Nop5/58 elution with beads that would bind to the tag on Mpp10. Mass spectrometry was used to analyze the resulting purified proteins that bind to the U3 snoRNA. This analysis identified 17 previously unknown components of the U3 ribonucleoprotein, one of which was Utp5, with Utp being used as an acronym for “U three protein.” The large ribonucleoprotein complex was named the small subunit processome because it is required for the processing and maturation of the ribosomal small subunit rRNA (the 18S rRNA). Depletion of Utp5 and other Utps decreases the levels of mature 18S rRNA in yeast (Dragon et al. 2002), causes pre-rRNA processing defects (Gallagher et al. 2004), and is required for cell cycle progression at G1 (Bernstein and Baserga 2004). Coinciding with the requirement of ribosomes for life, all but 1 Utp are encoded by essential genes (Dragon et al. 2002).
Utp5 was subsequently shown to be a member of the t-Utp/UTPA subcomplex (Gallagher et al. 2004; Krogan et al. 2004). This subcomplex contains Utps 4, 5, 8, 9, 10, 15, and 17 (Gallagher et al. 2004) and is largely conserved to humans (Prieto and McStay 2007). All members of the t-Utp/UTPA subcomplex are required for efficient RNAPI transcription of pre-rRNA, in addition to their requirement for pre-rRNA processing. Within the t-Utp/UTPA subcomplex, Utp5 interacts directly with Utp4 (Freed and Baserga 2010), Utp10 (Freed and Baserga 2010), and Utp15 (Uetz et al. 2000; Freed and Baserga 2010) as shown by yeast 2-hybrid (Fig. 2). By an alternative method of detecting protein-protein interactions called the protein fragment complementation assay, Utp5 directly interacts with all the t-Utps except Utp17 (Tarassov et al. 2008; Lim et al. 2011). Furthermore, in vitro reconstitution of the t-Utp/UTPA subcomplex demonstrates that Utp5 directly interacts with Utp4 and Utp15 (Poll et al. 2014). Since depletion or dysfunction of Utp5 or any of its interacting partners in the t-Utp/UTPA subcomplex results in defects in pre-rRNA transcription and processing (Gallagher et al. 2004) and because ribosome biogenesis defects result in craniofacial dysmorphology, all the protein-protein interactions of Utp5/WDR43 are likely to have significance in the development of craniofacial structures. This hypothesis is supported by a zebrafish mutant with a premature stop codon in WDR43, which results in a C-terminal truncation that abrogates the interaction between WDR43 and UTP15 and the interaction between WDR43 and UTP4 (Zhao et al. 2014). The same mutation in WDR43 that disrupts protein-protein interactions also causes severe craniofacial defects.
Figure 2.
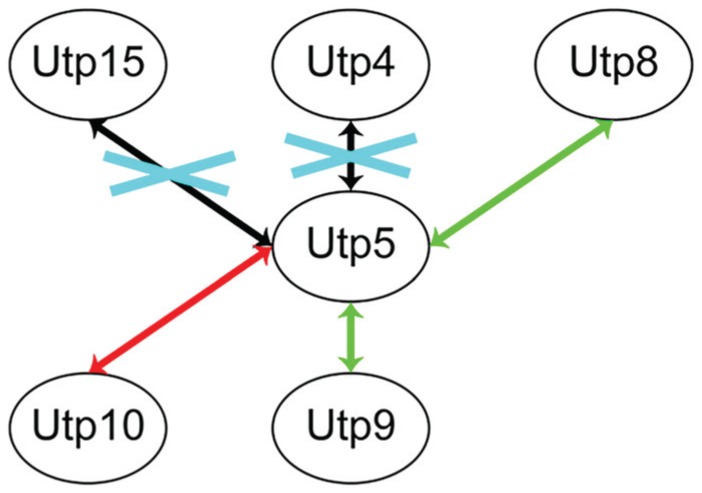
Binary interactions of Utp5 with its interacting partners. Green arrows indicate an interaction found only by protein fragment complementation (PCA; Tarassov et al. 2008). Red arrows indicate an interaction found by PCA (Tarassov et al. 2008) and yeast 2-hybrid (Uetz et al. 2000; Freed and Baserga 2010). Black arrows indicate an interaction found by PCA (Tarassov et al. 2008), yeast 2-hybrid (Uetz et al. 2000; Freed and Baserga 2010), in vitro reconstitution (Poll et al. 2014) and that the interaction is conserved for the human orthologs of the proteins (Sato et al. 2013). Blue crosses indicate protein-protein interactions that are disrupted in fantome mutant zebrafish and result in craniofacial defects (Zhao et al. 2014).
Role and Conservation of Utp5/WDR43 in Other Eukaryotes
Utp5/WDR43 is an evolutionarily conserved protein at the level of the amino acid sequence. The conservation of Utp5/WDR43 is supported by bioinformatics analyses demonstrating that Utp5/WDR43 is found in all 5 eukaryotic supergroups (Feng et al. 2013), including a parasitic protist (Srivastava et al. 2014), and can be traced to the last eukaryotic common ancestor (Feng et al. 2013). BLAST (http://blast.ncbi.nlm.nih.gov/) searches reveal that Utp5/WDR43 is well conserved at the amino acid level (Table). As its name suggests, WDR43 has a predicted conserved WD40 domain in the N-terminal region of the protein in addition to a Utp12 domain in the C-terminal region of the protein (Fig. 3A). A phylogenic tree of the evolution of Utp5/WDR43 reveals that vertebrate WDR43 and fungal Utp5 originated from a common ancestor (Fig. 3B). Altogether, Utp5/WDR43 has diverged little in amino acid sequence and predicted domain structure during evolution.
Table.
Comparison of Utp5/WDR43 in Various Eukaryotes.
| Percentage to Human |
Amino Acid of Predicted Domain: Start to End |
||||
|---|---|---|---|---|---|
| Species | Identity | Similarity | WD40 | Utp12 | Total Amino Acids |
| Homo sapiens | 100 | 100 | 19 to 272 | 473 to 579 | 677 |
| Mus musculus | 85 | 91 | 19 to 271 | 471 to 577 | 677 |
| Xenopus tropicalis | 62 | 76 | 15 to 251 | 466 to 572 | 665 |
| Danio rerio | 54 | 69 | 12 to 271 | 457 to 562 | 650 |
| Saccharomyces cerevisiae | 24 | 40 | 3 to 256 | 448 to 548 | 643 |
Sequence identity and similarity to human WDR43, amino acid positions of the predicted conserved WD40 (NCBI accession no. cd00200) and Utp12 (NCBI accession no. pfam04003) domains, and total amino acid length of Utp5/WDR43 for each indicated model organism. This figure is available in color online at http://jdr.sagepub.com.
Figure 3.
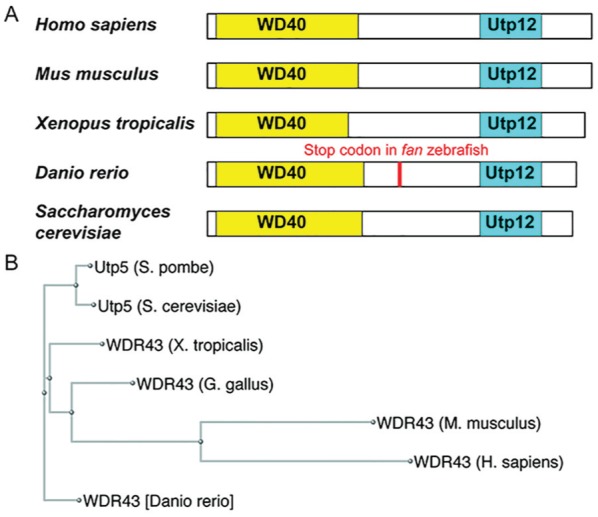
Evolutionary conservation of Utp5/WDR43. (A) Conserved WD40 and Utp12 domains predicted by the NCBI Conserved Domain database for Utp5/WDR43 in humans (Homo sapiens) and model organisms, including mice (Mus musculus), frogs (Xenopus tropicalis), zebrafish (Danio rerio), and yeast (Saccharomyces cerevisiae). White boxes indicate the relative lengths of the protein and are drawn to scale with the N-terminus to the left and C-terminus to the right. Yellow boxes indicate WD40 conserved domains (NCBI accession no. cd00200) and are drawn to scale. Cyan boxes indicate Utp12 conserved domains (NCBI accession no. pfam04003) and are drawn to scale. The location of the premature stop codon in fantome mutant zebrafish, which results in craniofacial developmental defects, is indicated in red. (B) Phylogeny tree for Utp5/WDR43 proteins. A ClustalW alignment of the indicated proteins was used to generate a phylogeny tree in the NCBI Genome Browser. This figure is available in color online at http://jdr.sagepub.com.
The function of Utp5/WDR43 is also conserved throughout eukaryotes. Like its ortholog in yeast, WDR43 is required for pre-rRNA transcription and processing in humans (Prieto and McStay 2007) and vertebrates, including zebrafish (Zhao et al. 2014). Furthermore, WDR43 copurifies with members of the human t-UTP/UTPA subcomplex (Freed et al. 2012), highlighting its conserved evolutionary role. In humans and in zebrafish, WDR43 is a nucleolar protein like Utp5 in yeast (Ahmad et al. 2009; Jarboui et al. 2011; Zhao et al. 2014). WDR43 localizes to all 3 subcompartments of nucleoli in HeLa cells by immunofluorescence (Wada et al. 2014) and directly interacts with human UTP15 (hUTP15) and human UTP4/Cirhin (hUTP4/Cirhin) when reconstituted in vitro (Sato et al. 2013). Phosphorylation of hUTP4/Cirhin by a mitotic cell extract abrogates its interaction with WDR43, suggesting a level of regulation for this interaction during the cell cycle (Sato et al. 2013). A study that systemically tested whether human proteins could complement their yeast orthologs did not test Utp5/WDR43 (Kachroo et al. 2015), although its functional conservation suggests that human WDR43 may complement yeast Utp5. Furthermore, injection of human Wdr43 mRNA partially rescues the zebrafish fantome (fan) / wdr43 mutant craniofacial phenotype (Zhao et al. 2014). Overall, there is ample evidence to support a functional conservation for the role of Utp5/WDR43 from yeast to humans.
Database mining reveals that WDR43 is modified posttranslationally. In an analysis of human protein SUMOylation, WDR43 was found to have SUMO3 modifications at lysines 75 and 384 and a phosphate at serine 77 (Lamoliatte et al. 2014). SUMOylation has been shown to allow for high-affinity binding of nucleolar proteins to snoRNAs (Westman et al. 2010); however, the functional role of these modifications of WDR43 has yet to be further explored.
Utp5/WDR43 and Disease
There is precedent for dysfunction of nucleolar factors required for ribosome biogenesis causing defects in craniofacial development. WDR43 is among this group of nucleolar factors, as disruption of WDR43 function causes craniofacial defects in zebrafish. In an ENU chemical mutagenesis screen of zebrafish embryos for mutants with defects in craniofacial development, the Yelick laboratory identified the fan mutant, which exhibited extremely reduced pharyngeal arch cartilage formation at 4 d postfertilization (Fig. 4; Zhao et al. 2014). Positional cloning revealed that fan mutants contained a nonsense mutation and, thus, a premature stop codon in the zebrafish WDR43 gene at amino acid 356 of 650 (Fig. 3). Analysis of fan mutants by whole mount in situ hybridization revealed a marked decrease of cranial neural crest (CNC) markers in fan mutant embryos (Zhao et al. 2014). As mentioned in the discussion of Treacher Collins syndrome, the CNC is a population of migratory cells that originate at the crest of the developing neural tube and significantly contribute to the development of craniofacial structures, including the pharyngeal arches (Mayor and Theveneau 2013). The fan mutant embryos exhibited increased apoptosis and reduced proliferation of the developing CNC. Furthermore, fan mutant embryos have decreased steady-state levels of pre-rRNAs and pre-rRNA processing defects (Zhao et al. 2014), as expected from previous studies of the molecular function Utp5/WDR43 (Gallagher et al. 2004; Prieto and McStay 2007).
Figure 4.
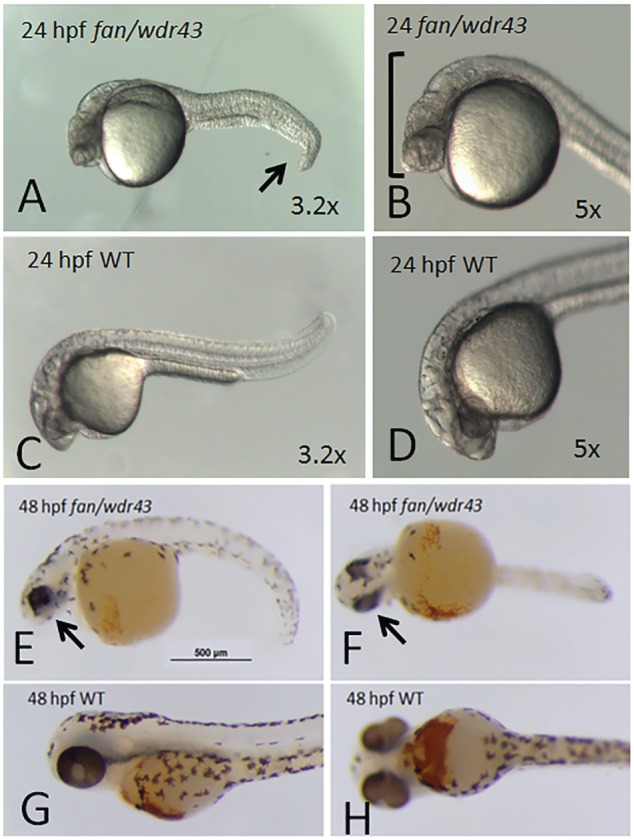
fantome (fan) / wdr43 mutant zebrafish phenotypes. At 24 h postfertilization (hpf), fan/wdr43 mutants exhibit distinct craniofacial defects, hydrocephaly, and tail phenotypes (A, B) as compared with age-matched wild-type (WT) siblings (C, D). At 48 hpf, fan/wdr43 mutants, stained with benzidine to reveal hemoglobin, exhibit even more dramatic craniofacial and tail defects as well as heart edema (E, F) as compared with age-matched WT siblings (G, H).
Apoptosis mediated by p53 due to defects in ribosome biogenesis is conserved across species. A hallmark of nucleolar disruption, such as defects in pre-rRNA processing, is p53 activation and apoptosis through a process known as nucleolar stress (Rubbi and Milner 2003; reviewed in Boulon et al. 2010). Indeed, fan mutant embryos exhibit increased p53 signaling, and the development of craniofacial structures could be rescued by depletion of p53 through a translation blocking morpholino (Zhao et al. 2014). This is in concordance with a study showing that inhibition of p53 in Tcof+/- mice, which model Treacher Collins syndrome, also rescues their craniofacial phenotype (Jones et al. 2008). A subsequent study found that depletion of another t-UTP, NOL11, in developing Xenopus tropicalis embryos causes a p53-mediated CNC defect associated with pre-rRNA processing defects and decreased pre-rRNA steady-state levels (Griffin et al. 2015). In contrast, in a zebrafish model for the ribosomopathy Shwachman-Diamond syndrome, inhibition of p53 was not able to rescue defects in organogenesis, including craniofacial cartilage defects (Provost et al. 2012). Unlike WDR43, TCOF, or NOL11, the protein affected in Shwachman-Diamond syndrome, SBDS, is required for late-stage cytoplasmic maturation of the large subunit, not pre-rRNA transcription (Finch et al. 2011). Taken together, these results demonstrate a conserved role for p53 in mediating CNC defects that are due to depletion or dysfunction of proteins required for pre-rRNA transcription. Importantly, the tissue specificity of these ribosome biogenesis protein mutations remains to be elucidated.
We can speculate about why the neural crest is uniquely sensitive to defects in pre-rRNA transcription. One possibility is that, as compared with other cell types in a developing embryo, the neural crest may require larger numbers of ribosomes due to the high proliferation exhibited by these cells during craniofacial development. Alternatively, neural crest cells may have unique mechanisms distinct from other cell types, to sense defects in ribosome biogenesis. This latter idea is supported by results from Griffin et al. (2015), which demonstrate that depletion of p53 rescues craniofacial defects in a frog model that are due to depletion of the t-UTP NOL11. Depletion of p53, however, does not rescue the decrease in pre-rRNA steady-state levels. Likewise, in a zebrafish model, p53 depletion alone was sufficient to rescue craniofacial defects caused by a premature stop codon in WDR43 (Zhao et al. 2014), and inhibition of p53-mediated cell death rescued craniofacial defects in Treacher Collins model (Tcof+/-) of mice (Jones et al. 2008). This is significant for the physiologic impact of disruptions of ribosome biogenesis and craniofacial dysmorphology, as it indicates that the cellular response to defects in ribosome biogenesis—and not to defects in ribosome biogenesis itself—results in craniofacial dysmorphology. However, the unique cellular mechanisms in neural crest cells that would be used to sense defects in ribosome biogenesis differently than other cell types and to activate apoptosis are, as of yet, unknown.
WDR43 is also linked to the primordial growth disorder 3-M syndrome (Hanson et al. 2014): a normocephalic primordial short-stature disorder with exclusively growth-related defects. Mutations in 3 proteins (CUL7, OBSL1, and CCDC8) are associated with the syndrome. These proteins are responsible for ubiquitination leading to targeted degradation by the proteasome; however, their specific role in the pathogenesis of 3-M syndrome has yet to be determined. In an effort to understand the molecular niche within which CUL7, OBSL1, and CCDC8 function, Hanson et al. (2014) performed immunoprecipitation, followed by mass spectrometry, to identify the interacting partners. WDR43 was coimmunoprecipitated by CUL7 and OBSL1. Furthermore, other members of the t-UTP/UTPA subcomplex were immunoprecipitated, including human UTP17/WDR75, NOL11, human UTP4/Cirhin, and human UTP10/HEATR1 (Hanson et al. 2014). As 3-M syndrome is a growth disorder, this leads to the intriguing hypothesis that defective ribosome biogenesis, which affects cellular growth, may at least partially contribute to the pathogenesis of the disease.
Conclusions and Perspectives
Our understanding of the function of Utp5/WDR43 in normal growth and development is far from complete. Although much is known about the role of Utp5/WDR43 in ribosome biogenesis at the molecular level, much remains to be understood about its role at the organismal level. However, any understanding of the physiologic impact of defects in Utp5/WDR43 must be built on a detailed molecular understanding of the protein and its functions. Therefore, a strong foundation for future studies on the physiology of defects in Utp5/WDR43 has been laid by the current progress in understanding the biochemical basis of ribosome biogenesis.
The fan/wdr43 mutant zebrafish may provide an important animal model for answering a variety of questions, including “Why are CNC cells more sensitive to defects in nucleolar function as compared with other, less affected tissues?” Zebrafish are particularly important for these investigations, as no WDR43 knockout mouse model exists in the International Knockout Mouse Consortium, Knockout Mouse Project, or International Mouse Phenotyping Consortium databases. This is not surprising, as Utp5 is an essential protein in yeast (Dragon et al. 2002) and fan/wdr43 homozygous zebrafish live only 5 to 6 d post-fertilization (Zhao et al. 2014). In contrast, heterozygous fan/wdr43 mutant zebrafish were phenotypically normal. Thus, mice homozygous null for WDR43 would likely be embryonic lethal, and heterozygous knockout of WDR43 would likely not produce craniofacial defects.
We have already speculated that neural crest cells may have unique mechanisms not present in other cell types by which they sense defects in ribosome biogenesis and activate apoptosis. One may also speculate that neural crest cells destined to develop into craniofacial structures may require more protein synthesis and, thus, more ribosomes. Perhaps neural crest cells proliferate faster than other cell types, which would require efficient ribosome biogenesis? As such, WDR43 is a viable model protein for addressing these questions and for studying how different tissues sense defects in pre-rRNA processing and transcription. Furthermore, since dysfunction of WDR43 causes craniofacial defects in an animal model, we begin to wonder: Is WDR43 mutated in patients with craniofacial dysostoses? Online Mendelian Inheritance in Man (omim.org) does not contain linkage between Utp5/WDR43 and any human disease as of yet. It is, however, tempting to predict that as DNA from increasing numbers of patients with craniofacial defects continues to be sequenced, mutations in WDR43 and/or related nucleolar factors required for the synthesis of ribosomes may be identified as the molecular etiology.
Author Contributions
S.B. Sondalle, contributed to conception, design, data analysis, and interpretation, drafted the manuscript; S.J. Baserga, P.C. Yelick, contributed to conception, design, and data interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
We thank the following funding sources that have supported these studies: National Institutes of Health (NIH) / Diabetes and Digestive and Kidney Diseases / General Medical Sciences / Child Health and Human Development (F30DK109582, T32GM07205, and T32HD007149 to S.B.S.); NIH / General Medical Sciences (R01GM115710 to S.J.B.); the Bohmfalk Scholar Award, Yale University (S.J.B.); and NIH / National Institute for Dental and Craniofacial Research (R01DE018043 to P.C.Y.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Ahmad Y, Boisvert FM, Gregor P, Cobley A, Lamond AI. 2009. NOPdb: Nucleolar Proteome Database—2008 update. Nucleic Acids Res. 37(Database issue):D181–D184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armistead J, Triggs-Raine B. 2014. Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett. 588(9):1491–1500. [DOI] [PubMed] [Google Scholar]
- Bernstein KA, Baserga SJ. 2004. The small subunit processome is required for cell cycle progression at G1. Mol Biol Cell. 15(11):5038–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, Abhyankar A, Itan Y, Patin E, Brebner S, et al. 2013. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 340(6135):976–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. 2003. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 33(1):97–101. [DOI] [PubMed] [Google Scholar]
- Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. 2010. The nucleolus under stress. Mol Cell. 40(2):216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwick N, Coats SA, Nakamura T, Shimamura A. 2012. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 120(26):5143–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagnon P, Michaud J, Mitchell G, Mercier J, Marion JF, Drouin E, Rasquin-Weber A, Hudson TJ, Richter A. 2002. A missense mutation (R565W) in cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am J Hum Genet. 71(6):1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirnomas SD, Kupfer GM. 2013. The inherited bone marrow failure syndromes. Pediatr Clin North Am. 60(6):1291–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer-Haas C, Maiwald R, et al. 2011. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 43(1):20–22. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker K, Sulima SO, Dinman JD. 2015. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood. 125(9):1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. 2006. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 103(36):13403–13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, et al. 2002. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 417(6892):967–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JM, Tian HF, Wen JF. 2013. Origin and evolution of the eukaryotic SSU processome revealed by a comprehensive genomic analysis and implications for the origin of the nucleolus. Genome Biol Evol. 5(12):2255–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, Gonzalez Fernandez A, Simpson P, D’Santos CS, Arends MJ, et al. 2011. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 25(9):917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EF, Baserga SJ. 2010. The C-terminus of Utp4, mutated in childhood cirrhosis, is essential for ribosome biogenesis. Nucleic Acids Res. 38(14):4798–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EF, Bleichert F, Dutca LM, Baserga SJ. 2010. When ribosomes go bad: diseases of ribosome biogenesis. Mol Biosyst. 6(3):481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EF, Prieto JL, McCann KL, McStay B, Baserga SJ. 2012. NOL11, implicated in the pathogenesis of North American Indian childhood cirrhosis, is required for pre-rRNA transcription and processing. PLoS Genet. 8(8):e1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ. 2004. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 18(20):2506–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, et al. 2008. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 83(6):769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JN, Sondalle SB, Del Viso F, Baserga SJ, Khokha MK. 2015. The ribosome biogenesis factor Nol11 is required for optimal rDNA transcription and craniofacial development in Xenopus. PLoS Genet. 11(3):e1005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan KM, Sanij E, Rothblum LI, Hannan RD, Pearson RB. 2013. Dysregulation of RNA polymerase I transcription during disease. Biochim Biophys Acta. 1829(3–4): 342–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson D, Stevens A, Murray PG, Black GC, Clayton PE. 2014. Identifying biological pathways that underlie primordial short stature using network analysis. J Mol Endocrinol. 52(3):333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci. 65(15):2334–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarboui MA, Wynne K, Elia G, Hall WW, Gautier VW. 2011. Proteomic profiling of the human T-cell nucleolus. Mol Immunol. 49(3):441–452. [DOI] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, et al. 2008. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 14(2):125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM. 2015. Evolution: systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science. 348(6237):921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass S, Tyc K, Steitz JA, Sollner-Webb B. 1990. The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell. 60(6):897–908. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, et al. 2004. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell. 13(2):225–239. [DOI] [PubMed] [Google Scholar]
- Lamoliatte F, Caron D, Durette C, Mahrouche L, Maroui MA, Caron-Lizotte O, Bonneil E, Chelbi-Alix MK, Thibault P. 2014. Large-scale analysis of lysine SUMOylation by SUMO remnant immunoaffinity profiling. Nat Commun. 5:5409. [DOI] [PubMed] [Google Scholar]
- Lim YH, Charette JM, Baserga SJ. 2011. Assembling a protein-protein interaction map of the SSU processome from existing datasets. PLoS One. 6(3):e17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor R, Theveneau E. 2013. The neural crest. Development. 140(11):2247–2251. [DOI] [PubMed] [Google Scholar]
- McCann KL, Baserga SJ. 2013. Genetics: mysterious ribosomopathies. Science. 341(6148):849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poll G, Li S, Ohmayer U, Hierlmeier T, Milkereit P, Perez-Fernandez J. 2014. In vitro reconstitution of yeast tUTP/UTP A and UTP B subcomplexes provides new insights into their modular architecture. PLoS One. 9(12):e114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto JL, McStay B. 2007. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev. 21(16):2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E, Wehner KA, Zhong X, Ashar F, Nguyen E, Green R, Parsons MJ, Leach SD. 2012. Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development. 139(17):3232–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiser DM, Narla A, Ebert BL. 2013. The emerging importance of ribosomal dysfunction in the pathogenesis of hematologic disorders. Leuk Lymphoma. 55(3):491–500. [DOI] [PubMed] [Google Scholar]
- Ross AP, Zarbalis KS. 2014. The emerging roles of ribosome biogenesis in craniofacial development. Front Physiol. 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi CP, Milner J. 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22(22):6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai D, Trainor PA. 2009. Treacher Collins syndrome: unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 41(6):1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Araki N, Kumeta M, Takeyasu K, Taguchi Y, Asai T, Furukawa K, Horigome T. 2013. Interaction, mobility, and phosphorylation of human orthologues of WD repeat-containing components of the yeast SSU processome t-UTP sub-complex. Biochem Cell Biol. 91(6):466–475. [DOI] [PubMed] [Google Scholar]
- Sondalle SB, Baserga SJ. 2014. Human diseases of the SSU processome. Biochim Biophys Acta. 1842(6):758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Ahamad J, Ray AK, Kaur D, Bhattacharya A, Bhattacharya S. 2014. Analysis of U3 snoRNA and small subunit processome components in the parasitic protist Entamoeba histolytica. Mol Biochem Parasitol. 193(2):82–92. [DOI] [PubMed] [Google Scholar]
- Sun X, Chen H, Deng Z, Hu B, Luo H, Zeng X, Han L, Cai G, Ma L. 2015. The Warsaw breakage syndrome-related protein DDX11 is required for ribosomal RNA synthesis and embryonic development. Hum Mol Genet. 24(17):4901–4915. [DOI] [PubMed] [Google Scholar]
- Tarassov K, Messier V, Landry CR, Radinovic S, Serna Molina MM, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick SW. 2008. An in vivo map of the yeast protein interactome. Science. 320(5882):1465–1470. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Andrews BT. 2013. Facial dysostoses: etiology, pathogenesis and management. Am J Med Genet C Semin Med Genet. 163C(4):283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor PA, Merrill AE. 2014. Ribosome biogenesis in skeletal development and the pathogenesis of skeletal disorders. Biochim Biophys Acta. 1842(6):769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treacher Collins Syndrome Collaborative Group. 1996. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 12(2):130–136. [DOI] [PubMed] [Google Scholar]
- Tyc K, Steitz JA. 1989. U3, U8 and U13 comprise a new class of mammalian snRNPs localized in the cell nucleolus. EMBO J. 8(10):3113–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 403(6770):623–627. [DOI] [PubMed] [Google Scholar]
- Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. 2004. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci U S A. 101(29):10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gijn DR, Tucker AS, Cobourne MT. 2013. Craniofacial development: current concepts in the molecular basis of Treacher Collins syndrome. Br J Oral Maxillofac Surg. 51(5):384–388. [DOI] [PubMed] [Google Scholar]
- Vincent M, Genevieve D, Ostertag A, Marlin S, Lacombe D, Martin-Coignard D, Coubes C, David A, Lyonnet S, Vilain C, et al. 2015. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med. 18(1):49–56. [DOI] [PubMed] [Google Scholar]
- Wada K, Sato M, Araki N, Kumeta M, Hirai Y, Takeyasu K, Furukawa K, Horigome T. 2014. Dynamics of WD-repeat containing proteins in SSU processome components. Biochem Cell Biol. 92(3):191–199. [DOI] [PubMed] [Google Scholar]
- Weaver KN, Watt KE, Hufnagel RB, Navajas Acedo J, Linscott LL, Sund KL, Bender PL, Konig R, Lourenco CM, Hehr U, et al. 2015. Acrofacial dysostosis, Cincinnati type, a mandibulofacial dysostosis syndrome with limb anomalies, is caused by POLR1A dysfunction. Am J Hum Genet. 96(5):765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westman BJ, Verheggen C, Hutten S, Lam YW, Bertrand E, Lamond AI. 2010. A proteomic screen for nucleolar SUMO targets shows SUMOylation modulates the function of Nop5/Nop58. Mol Cell. 39(4):618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek D. 2013. Human facial dysostoses. Clin Genet. 83(6):499–510. [DOI] [PubMed] [Google Scholar]
- Woolford JL, Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 195(3):643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Andreeva V, Gibert Y, LaBonty M, Lattanzi V, Prabhudesai S, Zhou Y, Zon L, McCann KL, Baserga S, et al. 2014. Tissue specific roles for the ribosome biogenesis factor Wdr43 in zebrafish development. PLoS Genet. 10(1):e1004074. [DOI] [PMC free article] [PubMed] [Google Scholar]