

Abstract
The molecular basis for control of the cytoskeleton by the Arf GTPase-activating protein AGAP1 has not been characterized. AGAP1 is composed of G-protein-like (GLD), pleckstrin homology (PH), Arf GAP, and ankyrin repeat domains. Kif2A was identified in screens for proteins that bind to AGAP1. The GLD and PH domains of AGAP1 bound the motor domain of Kif2A. Kif2A increased GAP activity of AGAP1, and a protein composed of the GLD and PH domains of AGAP1 increased ATPase activity of Kif2A. Knockdown (KD) of Kif2A or AGAP1 slowed cell migration and accelerated cell spreading. The effect of Kif2A KD on spreading could be rescued by expression of Kif2A-GFP or FLAG-AGAP1, but not by Kif2C-GFP. The effect of AGAP1 KD could be rescued by FLAG-AGAP1, but not by an AGAP1 mutant that did not bind Kif2A efficiently, ArfGAP1-HA or Kif2A-GFP. Taken together, the results support the hypothesis that the Kif2A·AGAP1 complex contributes to control of cytoskeleton remodeling involved in cell movement.
Keywords: ADP ribosylation factor (ARF), cell biology, enzyme, GTPase activating protein (GAP), kinesin, protein complex, small GTPase, AGAP1, Kif2A, kinesin-13, Arf GTPase-activating protein, PH domain
Introduction
The ADP-ribosylation factor (Arf)3 GTPase-activating proteins (GAPs) are a family of structurally diverse proteins with the common function of catalyzing the hydrolysis of GTP bound to Arf, thereby converting Arf·GTP to Arf·GDP (1–3). The five Arf isoforms in humans regulate membrane traffic and actin remodeling (4). Arfs have no intrinsic GTPase activity; consequently, their function is dependent on the GAPs (1, 5). With 31 genes in human, the Arf GAPs outnumber the Arfs. In addition to functioning as isoform-specific regulators, the GAPs are thought to be site-specific regulators of Arfs, and there has been some speculation that they function as Arf effectors (2, 6, 7). However, site-specific targeting, regulation, and effector function of Arf GAPs remain relatively unexplored.
AGAPs are a subtype of Arf GAPs named for the domain structure of G-protein-like, also called mitochondria Rho-like (miro), split PH, Arf GAP, and ankyrin repeats (see Fig. 1A for schematic) (8). Like other Arf GAPs, the AGAPs studied to date affect membrane trafficking and actin cytoskeleton. AGAP1 binds directly to the clathrin adaptor protein AP-3, which together with clathrin can form a vesicular coat (9). AGAP1 also binds to muscarinic receptor and affects its trafficking (10). The related protein AGAP2 binds to β-arrestin, which affects Erk signaling (11), and to focal adhesion kinase, which controls focal adhesions and, presumably, cell migration (12), a plausible function for AGAP1 as well given its effect on both actin cytoskeleton and membrane traffic (8).
FIGURE 1.

Interaction between AGAP1 and Kif2A. A, schematic of the domain structure of AGAP1 and recombinant proteins. Ank, ankyrin repeat. B, coimmunoprecipitation of kinesins with FLAG-tagged AGAPs. HeLa cells were transfected with an empty pCI vector or pCI with cDNA for FLAG-AGAP1, FLAG-AGAP2, or myc-AGAP3. The cells were lysed after 24 h. Proteins from the lysates were immunoprecipitated with an antibody against the FLAG or myc epitope, and the precipitates were probed with an antibody against Kif2A, Kif2A, Kif3A, and Kif5B. IB, immunoblotting. C, association of GST-AGAP1 with Kif2A in a cell lysate. HeLa cell lysate was incubated with GST or GST-AGAP at 4 °C overnight. GST was precipitated with glutathione beads, and Kif2A was detected in the precipitates by immunoblotting using a polyclonal antibody against Kif2A. The total Hela cell lysate was included as a positive control. IP, immunoprecipitation. D, co-immunoprecipitation of Kif2A with four Arf GAP subtypes. HeLa cells were transfected with empty pCI vector or pCI with cDNA for FLAG-ASAP1, FLAG-ARAP1, FLAG-ACAP1, or FLAG-AGAP1. The cells were lysed after 24 h. Proteins from the lysates were immunoprecipitated with an antibody against the FLAG epitope. The precipitates were probed with an antibody against Kif2A. E, colocalization of FLAG-AGAP1 and endogenous Kif2A. Hela cells were transfected with FLAG-AGAP1 for 24 h. Cells were replated on fibronectin-coated coverslips in Opti-MEM for 6 h. The cells were fixed and stained using a monoclonal anti-FLAG antibody and a polyclonal rabbit anti-Kif2A serum. A 0.9-μm slice of the ventral surface is shown. F, quantification of colocalization of Kif2A with FLAG-AGAP1 and FLAG-ARAP1. Pearson's coefficients for Kif2A with either FLAG-AGAP1 or FLAG-ARAP1 in the periphery of the cells were determined for 30 cells (10 from each of 3 experiments). The data presented are the mean ± S.D. p < 0.0001 for FLAG-AGAP1-Kif2A. G, specificity of Kif2A antibody. Kif2A-GFP, Kif2B-GFP, and Kif2C-GFP were expressed in HeLa cells. The cell lysates were analyzed by immunoblotting using either the anti-Kif2A antibody and/or the anti-GFP antibody. Although all three proteins were expressed at similar levels, indicated by the signal with the anti-GFP antibody, signal was only observed with Kif2A when using the anti-Kif2A antibody.
Kinesins are microtubule-binding proteins (13, 14). Kinesins have a nucleotide-binding fold that binds and hydrolyzes ATP. For most kinesins, the energy of ATP hydrolysis is used to translocate along microtubules. Kif2A is a member of the kinesin-13 family, which are unconventional kinesins (15). The motor domain is located midway along the amino acid sequence of the protein with a targeting domain N-terminal to the motor and a dimerization domain C-terminal of the motor. The closely related Kif2C diffuses along the microtubule lattice independent of ATP hydrolysis. It binds the end of the microtubules where ATP hydrolysis drives the removal of tubulin dimers (16). Unlike Kif2C (17), Kif2A is expressed in terminally differentiated cells including neurons (18) and cardiomyocytes (19). A limited number of studies have examined kinesin-13s in interphase cells. Kif2A has been found to affect microtubule dynamics (20) and axonal branching (18). Kif2 has been reported to affect lysosomal positioning (21). Increased Kif2A expression levels have also been reported to be an indicator of poor prognosis and higher metastatic potential, and to contribute to cell migration and invasion in vitro in squamous cell carcinoma and breast cancer (22, 23). In transformed bronchial epithelial cells, Kif2A and Kif2C levels are regulated by K-Ras and contribute to the invasive behavior of the cells, but not to epithelial-to-mesenchymal transition (24).
Here, we identified kinesins in a screen for proteins that bind to and regulate AGAP1 activity. Subsequent examination revealed a specific and functional interaction with Kif2A, resulting in activation of AGAP1 GAP activity. Reciprocally, AGAP1 increased ATPase activity of Kif2A. Furthermore, in interphase cells, Kif2A and AGAP1 had a common effect on cell migration and cell spreading. The effect of knockdown of Kif2A on cell spreading could be rescued by expression of AGAP1, but the effect of knockdown of AGAP1 was not rescued by Kif2A overexpression, suggesting that Kif2A may function upstream of AGAP1. We propose that the Kif2A and AGAP1 complex controls cell movement and migration.
Results
In previous work, we found that the GLD and PH domains of AGAP1 are critical for regulated GAP activity (25). Rho family GTP-binding proteins are binding partners that stimulate catalytic activity of AGAP1; however, the affinity is relatively low (Kd greater than 10 μm), and the biological significance is not clear. As an initial test of the possibility that there are other binding partners that bind to the GLDPH domains of AGAP1 to regulate catalytic and biological activity, we screened for binding partners using a proteomic approach (column 3 of Table 1). In these experiments, a protein composed of the GLDPH domain with an N-terminal fusion to 10 histidines was adsorbed to large unilamellar vesicles (LUVs) and mixed with a lysate of HeLa cells. The LUVs were floated through a sucrose gradient, and the associated proteins were identified by mass spectrometry of trypsinized fragments. A Ras family GTP-binding protein, actin and actin-associated proteins, and a kinesin (Kif5) were identified. We also have access to a database of two-hybrid screens (Center for Cancer Research Database for Antibodies and Protein Interactions, CCRDAPI). We examined the results for the three AGAPs containing the GTP-binding protein-like domain (also called the miro domain). We include the original screening results in Table 1. Column 1 has the results for AGAPs as bait, and column 2 shows protein baits that bound to AGAPs as prey. Ras superfamily GTP-binding proteins, actin-associated proteins, and a kinesin (Kif5B using AGAP3 as bait) were again identified.
TABLE 1.
Proteins identified in screens for binding partners of GLDPH tandem
Two-hybrid experiments were performed by Myriad Genetics. In proteomic studies, HeLa cell lysates were mixed with LUVs to which the GLDPH fragment of AGAP1 had been adsorbed. The mixture was mixed with sucrose and layered in a discontinuous sucrose gradient and was subject to centrifugation. Proteins that associated with LUVs that floated through the sucrose gradient were analyzed by mass spectrometry. MPRIP, myosin phosphatase Rho-interacting protein.
| AGAP as bait in two-hybrid experiments | AGAP as prey in two-hybrid experiments | Proteomics | |
|---|---|---|---|
| AGAP1 | Rock1, Calcoco2, RhoA, Cdc42, Rac1 | PLEKHB1 | Dynactin subunit 1, Kif5, myosin 1c, Rab11, actin |
| AGAP2 | Rock1, AGAP1, α-actinin, filamin G, MPRIP | PLEKHB1, Rab5, Rab2 | |
| AGAP3 | Kif5B, clathrin heavy chain, PI-4kinaseA |
We focused on kinesins, a class of proteins that have roles in membrane and cytoskeleton remodeling (13, 14), for further examination as possible binding partners. FLAG-AGAP1 was expressed in HeLa cells and immunoprecipitated from the cell lysates using an antibody to the FLAG epitope. Immunoblotting of the precipitates was used to determine the presence of representative kinesins, including Kif5B (kinesin 1), Kif2A and Kif2C (kinesin 13s), and Kif3A. The most robust signal was observed for Kif2A (Fig. 1B). We tested for specificity of the antibody by expressing GFP-tagged Kif2A, Kif2B, and Kif2C and comparing immunoblotting signals using antibodies to GFP or the antibody to Kif2A. Signal was similar for all three kinesins when blotting for GFP, but signal was only observed with GFP-Kif2A when using the antibody for Kif2A (Fig. 1G). GST-AGAP1 was incubated with HeLa cell lysates and precipitated with glutathione-conjugated beads. Kif2A was coprecipitated (Fig. 1D).
The association with Kif2A was specific for AGAP1. AGAP1 was compared with other Arf GAP types (Fig. 1C). FLAG-AGAP1, FLAG-ASAP1, FLAG-ACAP1, and FLAG-ARAP1 were expressed in HeLa Cells and immunoprecipitated from cell lysates. The precipitates were analyzed by immunoblotting for Kif2A. The most robust signal for Kif2A was observed with AGAP1. A faint signal was observed with ASAP1. AGAP1 was also compared with AGAP2 and AGAP3 using the same experimental design (Fig. 1B). The signal was most robust for AGAP1, although all three associated with Kif2A to some extent.
FLAG-AGAP1 and Kif2A colocalized in HeLa cells (Fig. 1E). FLAG-AGAP1 was expressed in HeLa cells. The cells were incubated on fibronectin-coated coverslips in serum-free medium for 6 h and then fixed and prepared for immunofluorescence. FLAG-AGAP1 and Kif2A colocalized in one of two distinct distributions. In many cells, the proteins were in large, indistinct structures, possibly representing aggregated proteins (not shown). This phenotype was associated with higher expression levels of AGAP1. In cells with lower expression levels, the proteins sometimes colocalized in structures in the cell periphery (Fig. 1E is a representative cell, and Fig. 1F is a summary of the Pearson's coefficients determined in three experiments, 10 cells/experiment). A related Arf GAP, ARAP1, did not colocalize with Kif2A (Fig. 1F). A diffuse perinuclear signal with the Kif2A antibody was also observed. Unfortunately, antibodies for endogenous AGAP1 suitable for immunofluorescence are not available for further analysis.
We next mapped the domains of Kif2A and AGAP1 that mediated the association of the proteins. For Kif2A, the motor domain was necessary and sufficient for binding AGAP1. Recombinant His10-[1–124]Kif2A and His10-[193–531]Kif2A, expressed in and purified from bacteria, were incubated with GST-AGAP1. GST-AGAP1 was precipitated with glutathione-conjugated agarose beads. The presence of His-tagged protein in the precipitates was determined by immunoblotting (Fig. 2A). His10-[193–531]Kif2A was detected, which comprises the motor domain. His10-[1–124]Kif2A was not detected. The C terminus of Kif2A was also examined but precipitated with the glutathione beads alone (not shown).
FIGURE 2.
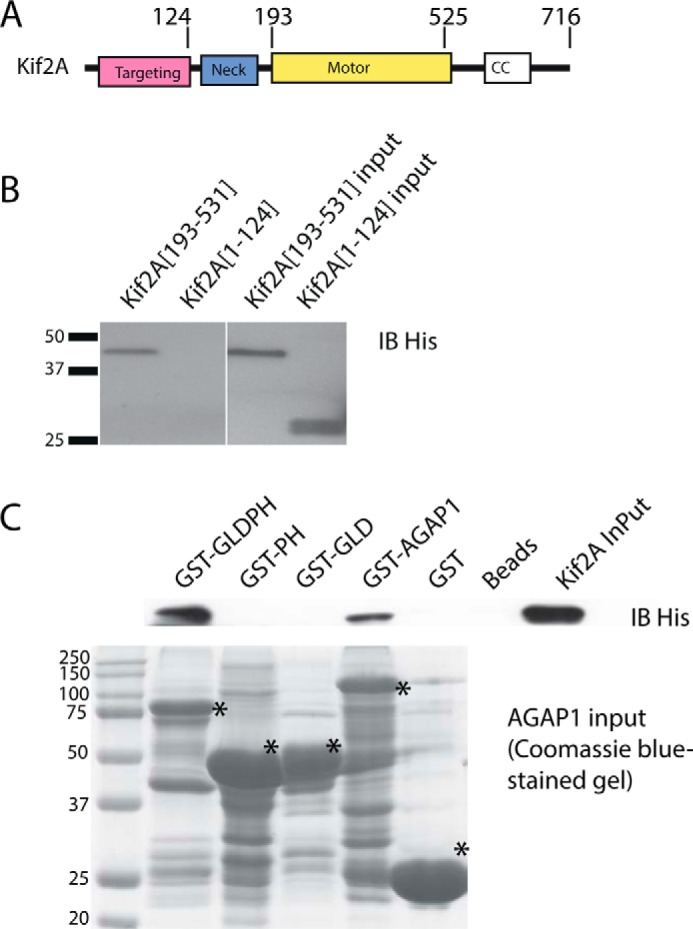
Identification of sites of Kif2A·AGAP1 association. A, schematic of Kif2A. B, domain of Kif2A that binds to AGAP1. 100 nm His10-[193–531]Kif2A or His10-[1–124]Kif2A was incubated with 30 μg of GST-AGAP1. After an incubation at 37 °C for 30 min, glutathione beads were added and precipitated. The beads were washed with PBS three times, and the samples were separated on SDS-PAGE and immunoblotted (IB) with polyclonal anti-His antibody. C, domains of AGAP1 that are necessary and required for binding to Kif2A motor domain. 100 nm His10-[193–531]Kif2A was incubated with 30 μg of GST-GLDPH, GST-PH, GST-GLD, and GST-AGAP1 (see schematic in Fig. 1 for domains). GST and no GST were used as controls. After an incubation at 37 °C for 30 min, the glutathione beads were washed with PBS three times, and the samples were separated on SDS-PAGE and immunoblotted with polyclonal anti-Kif2A antibody. CC, coiled-coil; *, indicates position of the GST fusion protein.
The PH and GLD domains both contribute to binding Kif2A. Our initial experiments designed to map the binding site involved expressing FLAG-tagged fragments of AGAP1 in HeLa cells, immunoprecipitating proteins from the cell lysates through the FLAG tag, and probing the precipitates for endogenous Kif2A by immunoblotting (not shown). We found that the PH domain of AGAP1 was necessary and sufficient to precipitate Kif2A. However, AGAPs may oligomerize through the PH domain; endogenous AGAP1 also coprecipitated with the PH domain.4 Therefore, it was possible that a domain in addition to the PH domain was necessary for binding. The experiment was repeated with purified GST-AGAP1 fragments and purified His10-[193–531]Kif2A, which contains the motor domain of Kif2A (Fig. 2B). In these experiments, GST-AGAP1 and GST-GLDPH domain precipitated Kif2A, but GST-PH or GST-GLD did not. These results support the idea that the GLD and PH domain together comprise the binding site.
We determined whether Kif2A binding to AGAP1 affected GAP activity. In initial experiments, we titrated AGAP1 into a reaction mixture containing myristoylated Arf-GTP (myrArf-GTP) and LUVs (Fig. 3A). In some reactions, the LUVs contained phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2). Reactions also contained Kif2A as indicated. Kif2A had no effect on activity when PtdIns(4,5)P2 was absent. PtdIns(4,5)P2 reduced the amount of AGAP1 needed for activity. In the presence of PtdIns(4,5)P2, Kif2A further reduced the amount of AGAP1 needed for half-maximal hydrolysis by ∼10-fold (note the logarithmic scale). We also titrated proteins composed of fragments of Kif2A into a reaction mixture containing AGAP1, myrArf1·GTP, and LUVs with PtdIns(4,5)P2 (Fig. 3B). Activation was most efficient with the motor domain present, with 30–60-fold differences in efficiency for activation between protein fragments containing the motor domain and those that do not.
FIGURE 3.
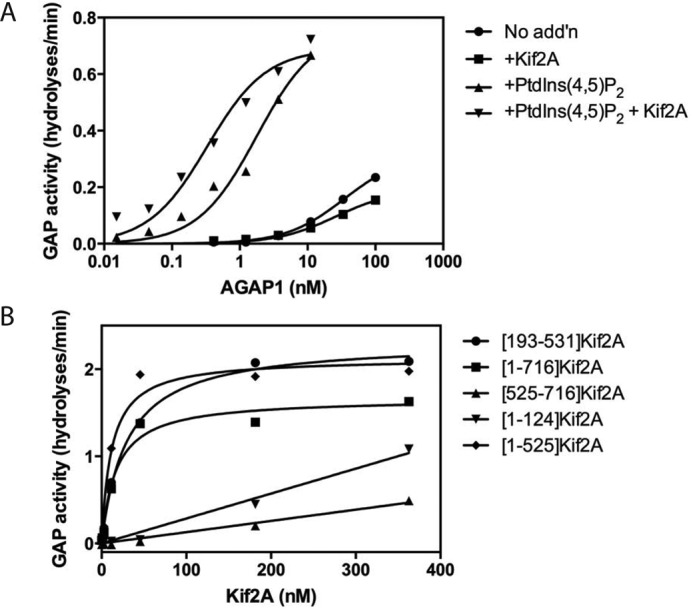
Effect of Kif2A on AGAP1 activity. A, PtdIns(4,5)P2 and Kif2A cooperatively stimulate AGAP1 activity. 100 nm [193–531]Kif2A and 5 μm PtdIns(4,5)P2 were used in the GAP assay, which contained 0.5 μm myrArf1·[α32P]GTP, LUVs with PtdIns(4,5)P2 as indicated, and the indicated concentration of His-AGAP1. The reaction was followed by the conversion of [α32P]GTP to [α32P]GDP. No add'n, no addition. B, determination of the domain of Kif2A that stimulates AGAP1. The indicated His10-tagged fragments of Kif2A were titrated into reactions containing LUVs (same composition as panel A + PtdIns(4,5)P2), 1.1 nm AGAP1, and 0.5 μm myrArf1·[α32P]GTP. The reaction was followed by the conversion of [α32P]GTP to [α32P]GDP.
We mapped residues within the GLD necessary for activation of AGAP1 (Fig. 4). We selected negatively charged residues on the surface including several within and at the edge of motifs equivalent to switch 1 and switch 2 in G-proteins (Fig. 4, A and B). A number of the mutants affected activity in the absence of Kif2A, particularly within the switch motifs, although the effects were less than 5-fold (Fig. 4, C and D). The most dramatic effects of the mutants were on Kif2A-stimulated activity (Fig. 4, F and G). Kif2A did not affect activity of [E125Q]AGAP1 at concentrations up to 180 nm, as compared with half-maximum activity at 20 nm for wild type AGAP1. In contrast, [D124N]AGAP1 was more sensitive to Kif2A than wild type protein.
FIGURE 4.

Effect of GLD of AGAP1 on GAP activity. A, sequence alignment of AGAP GLDs with H-Ras. Accession numbers are: AGAP1, gi:51338837; AGAP2, gi:6176569; AGAP3, gi:16799069; H-Ras, gi:231061. B, structure of AGAP3 GLD. The crystal structure of AGAP3 is shown with switch 1 colored red, switch 2 colored blue, and the residues aligning with the residues mutated in AGAP1 indicated. Protein Data Bank ID: 3IHW. C–E, activity of mutant AGAPs in the absence of Kif2A. The indicated AGAP1 mutants were titrated into a GAP reaction containing LUVs (same composition as in Fig. 3A, + PtdIns(4,5)P2), and 0.5 μm myrArf1·[α32P]GTP. F–H, effect of Kif2A on the activity of AGAP1 mutants. His10-[193–531]Kif2A was titrated in a reaction containing 0.5 μm myrArf1·[α32P]GTP, 500 μm LUVs (same composition as in Fig. 3A), and 1.0 nm of wild type AGAP1 or the indicated mutants. I, binding of mutant AGAP1 to Kif2A. FLAG-tagged wild type AGAP1 or the indicated mutants were expressed in HeLa cells. Cells were lysed, and the FLAG-tagged proteins were immunoprecipitated (IP) with an anti-FLAG antibody. Kif2A was detected in the precipitates by immunoblotting (IB) using an anti-Kif2A antibody. In the left panel, immunoprecipitated FLAG-tagged protein was detected by Ponceau S staining. In the right panel, immunoprecipitated FLAG-tagged protein was detected by immunoblotting for the FLAG epitope.
We also tested residues within the unique insert within the PH domain for effects on Kif2A-stimulated GAP activity. The insert has a stretch of positively charged amino acids that we considered to be potentially important for activity. Mutating two pairs of these residues to negatively charged amino acids (K474D,K475D, and K479D,K480D) reduced both basal and Kif2A-stimulated activity (Fig. 4, E and H).
We next determined the effect of the mutations in the GLD and PH domain on binding to Kif2A. For these experiments, AGAP1 proteins containing the indicated mutations were expressed in HeLa cells and immunoprecipitated, and Kif2A coprecipitation was detected by immunoblotting (Fig. 4I). None of the mutations in the GLD that we tested had an effect on the amount of Kif2A detected. However, Kif2A did not efficiently coprecipitate with either [K474D,K475D]AGAP1 or [K479D,K480D]AGAP1.
We examined the possibility that AGAP1 regulated the ATPase activity of Kif2A. In initial experiments, the motor domain of Kif2A was titrated into a reaction mixture containing microtubule fragments, ATP, and as indicated, the fragment of AGAP1 that efficiently bound Kif2A (GLDPH) (Fig. 5A). The reaction was initiated by the addition of ATP and terminated after 5 min. ATP hydrolysis, measured as the release of orthophosphate, was linearly dependent on Kif2A in this concentration range. The presence of GLDPH increased activity by ∼2-fold. GLDPH, the PH domain of AGAP1, and a recombinant AGAP1 lacking the GLD and PH domains (ZA) were titrated into reaction mixtures containing ATP and Kif2A as indicated (Fig. 5B). ATPase activity in reactions containing Kif2A was increased ∼75% by the addition of GLDPH. The PH domain increased activity by ∼30%. ZA had no effect. Neither GLDPH nor ZA had detectable ATPase activity by themselves.
FIGURE 5.
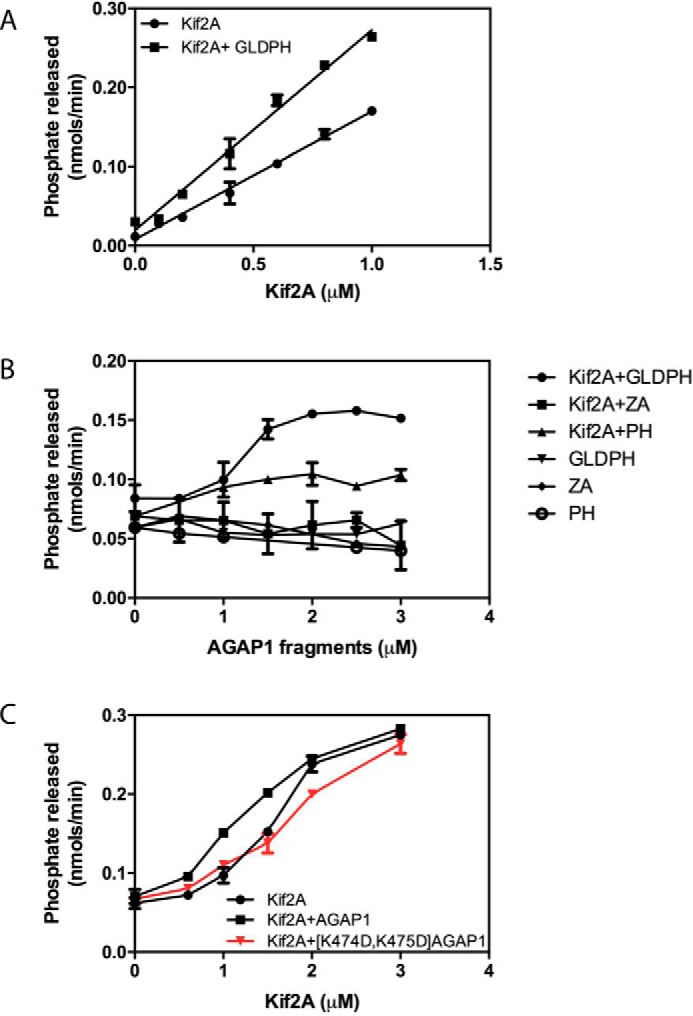
Effect of AGAP1 on Kif2A ATPase activity. A, titration of Kif2A with fixed GLDPH. Purified His10-[193–531]Kif2A was titrated into a 30-μl reaction containing 200 μm LUVs and 0.2 μg of microtubules with or without 1.0 μm of purified His10-GLDPH domain of AGAP1. The reactions were started by adding 0.3 mm ATP and stopped after 5 min. The data were analyzed by linear regression. The slope of Kif2A+GLDPH was greater than the slope of Kif2A alone, p < 0.001. B, titration of AGAP1 into a reaction with fixed Kif2A. The isolated His10-GLDPH of AGAP1 (the minimum binding domain), a recombinant protein composed of the Arf GAP and ankyrin repeats of AGAP1 fused to a 10-histidine tag (designated ZA in the figure, which does not bind to Kif2A), or the PH domain of AGAP1 was titrated into a reaction containing 200 μm LUVs, 0.2 μg of microtubules, and 0.7 μm His10-[191–531] Kif2A as indicated. Reactions were initiated by the addition of ATP and terminated after 5 min. Data were analyzed by two-way ANOVA. GLDPH increased Kif2A activity, p < 0.001; PH increased activity, p < 0.001; PH had less effect than GLDPH, p < 0.001; ZA had no significant effect. C, effect of mutation in the PH domain of AGAP1 on Kif2A ATPase activity. Purified His10-[193–531]Kif2A was titrated into a reaction mixture containing full-length AGAP1 or [K474D,K475D]AGAP1 as described for panel A. The data were analyzed by two-way ANOVA. AGAP1 plus Kif2A was greater than Kif2A alone, p < 0.001; [K474D,K475D]AGAP1 did not have a significant effect. Results shown are the mean ± S.E. from three independent experiments.
We examined the effect of GLDPH with mutations that reduced Kif2A-stimulated GAP activity. [E125Q]GLDPH stimulated Kif2A ATPase activity to the same extent as did wild type GLDPH (not shown). [K474D,K475D]AGAP1 (mutation in the context of full-length protein) was compared with wild type AGAP1 (Fig. 5C). Like GLDPH, full-length AGAP1 stimulated Kif2A ATPase activity, whereas [K474D,K475D]AGAP1, which does not bind Kif2A efficiently (Fig. 4I), did not.
AGAP1 cellular function has been examined exclusively in interphase cells. One effect of AGAP1 overexpression is to reduce actin stress fibers in cells. AGAP1 also regulates membrane traffic. Kif2A has been reported to affect migration and invasion of cancer cells, activities dependent on coordinated remodeling of membranes and actin. To explore the potential function of the complex of AGAP1 and Kif2A, we used siRNA to reduce expression (henceforth called “KD” for knockdown) of each and examined the effect on cell migration using a wound healing assay. We found that reduction of either AGAP1 or Kif2A reduced the rate at which cells moved into the wound (Fig. 6). The effect was seen with a pool of siRNA and individual siRNA for each protein.
FIGURE 6.

Effect of AGAP1 and Kif2A on cell migration. A, efficiency of Kif2A and AGAP1 KD. MDA-MB-231 cells were transfected with control siRNA (Control Si) or siRNA targeting either Kif2A or AGAP1 (SMARTpool siGENOME human siRNA from Dharmacon). Cell lysates were prepared 72 h later and analyzed by immunoblotting (IB) for Kif2A and AGAP1. B, wound healing assay. The effect of reduced expression of Kif2A or AGAP1, by siRNA treatment, on cell migration was examined using a wound healing assay using Ibidi wound healing assay chambers as described under “Materials and Methods.” A single time point from a representative experiment is presented. C, migration of cells with reduced expression of Kif2A and AGAP1. Migration of cells treated with SMARTpool siRNA targeting either Kif2A or AGAP1 was determined using the wound healing assay. Migration was quantified as described under “Materials and Methods.” D and E, effect of reduced AGAP1 or Kif2A expression on cell migration is independent of the specific siRNA. The experiment was conducted as described for Fig. 7C but using several different individual siRNA targeting AGAP1 (D) or Kif2A (E).
We also examined the effect of AGAP1 and Kif2A on cell spreading, another cell function dependent on actin and membrane remodeling (Fig. 7). KD of either AGAP1 or Kif2A accelerated the rate of cell spreading. The effect of KD of AGAP1 was reversed by expressing FLAG-AGAP1 from a plasmid but not by ArfGAP1, another Arf GAP that also used Arf1·GTP as a substrate (Fig. 7A). The KD of Kif2A was reversed by expression of Kif2A-GFP but not the related Kif2C-GFP (Fig. 7B). The contribution of each domain of AGAP1 to the effects on cell spreading was examined. Expression of FLAG-PZA (AGAP1 without GLD) was able to partially rescue the effect of knockdown, whereas AGAP1 lacking GAP activity, FLAG-[R599K]AGAP1, or AGAP1 that was not activated by Kif2A, FLAG-[E125Q]AGAP1, rescued as well as did wild type FLAG-AGAP1 (Fig. 7C). It is plausible that the mutants are not fully active but are able to rescue because of the degree of overexpression. Expression of FLAG-[K474D,K475D]AGAP1 did not rescue the effect of AGAP knockdown (Fig. 7D). Expression of GLDPH could not reverse the effect of knockdown of AGAP1 (Fig. 7C) but instead accelerated spreading in the presence of endogenous AGAP1, consistent with its function as a dominant negative (Fig. 7E).
FIGURE 7.

Effect of AGAP1 and Kif2A on cell spreading. A and B, reduced expression of endogenous AGAP1 or Kif2A and replacement with FLAG-AGAP1 and Kif2A-GFP. Hela cells treated with siRNA targeting noncoding regions on either AGAP1 or Kif2A and transfected with either empty plasmid or plasmids for expression of FLAG-AGAP1, Arf GAP1-HA, Kif2A-GFP, or Kif2C-GFP, as indicated, were plated on fibronectin-coated cover glass for 20 min. The area of rhodamine-phalloidin-stained cells was determined for at least 40 cells per condition using ImageJ version 1.46r. Results shown are the mean ± S.E. from three independent experiments. Differences were tested by ANOVA followed by multiple comparison post-tests using the program PrismTM. A, control Si versus AGAP1 Si + empty vector, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + FLAG-AGAP1, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + ArfGAP1-HA, p < 0.05. B, control Si + empty vector versus Kif2A Si + empty vector, p < 0.001; Kif2A Si + empty vector versus Kif2A Si + Kif2A GFP, p < 0.005; Kif2A Si + empty vector versus Kif2A Si + Kif2C-GFP, not significant. C, effect of expression of AGAP1 mutants on spreading of cells with reduced endogenous AGAP1. Hela cells treated with siRNA targeting the noncoding region of AGAP1 and transfected with plasmids for expression of the indicated fragments of AGAP1 or mutants of AGAP1 were plated on fibronectin-coated coverslips for 20 min, and cell surface area was determined as in A. Control Si + empty vector versus AGAP1 Si + empty vector, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + FLAG-GLDPH, not significant; AGAP1 Si + empty vector versus AGAP1 Si + FLAG-PZA, + [R599K]AGAP1, + [E125Q]AGAP1, p < 0.001. AGAP1Si + PZA versus AGAP1Si + FLAG-[R599K]AGAP1, p < 0.001. D, effect of expression of FLAG-[K474D,K475D]AGAP1 on spreading of cells with reduced endogenous AGAP1 expression. The experiment is similar to that described in C except that FLAG-[K474D,K475D] AGAP1 was the only mutant examined. Control Si + empty vector versus AGAP1 Si + empty vector, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + FLAG-AGAP1, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + FLAG-[K474D,K475D]AGAP1, not significant. E, effect of overexpression of AGAP1 and GLDPH on spreading of cells. Spreading of cells transfected with empty vector (pCI) or plasmids for expression of FLAG-AGAP1 or FLAG-GLDPH was determined as in A. Empty vector versus FLAG-GLDPH, p < 0.001; empty vector versus FLAG-AGAP1, p < 0.01.
To gain insight into which protein was mediating the effects on cell spreading, we determined whether overexpression of one member of the AGAP1·Kif2A complex could compensate for the loss of the other protein (Fig. 8). FLAG-AGAP1 overexpression rescued Kif2A KD. The unrelated ArfGAP1-HA did not. In contrast, Kif2A-GFP overexpression did not rescue AGAP1 KD. Kif2C-GFP did not rescue either, which was anticipated given that it could not rescue Kif2A KD. These results might indicate that AGAP1 functions downstream of Kif2A.
FIGURE 8.
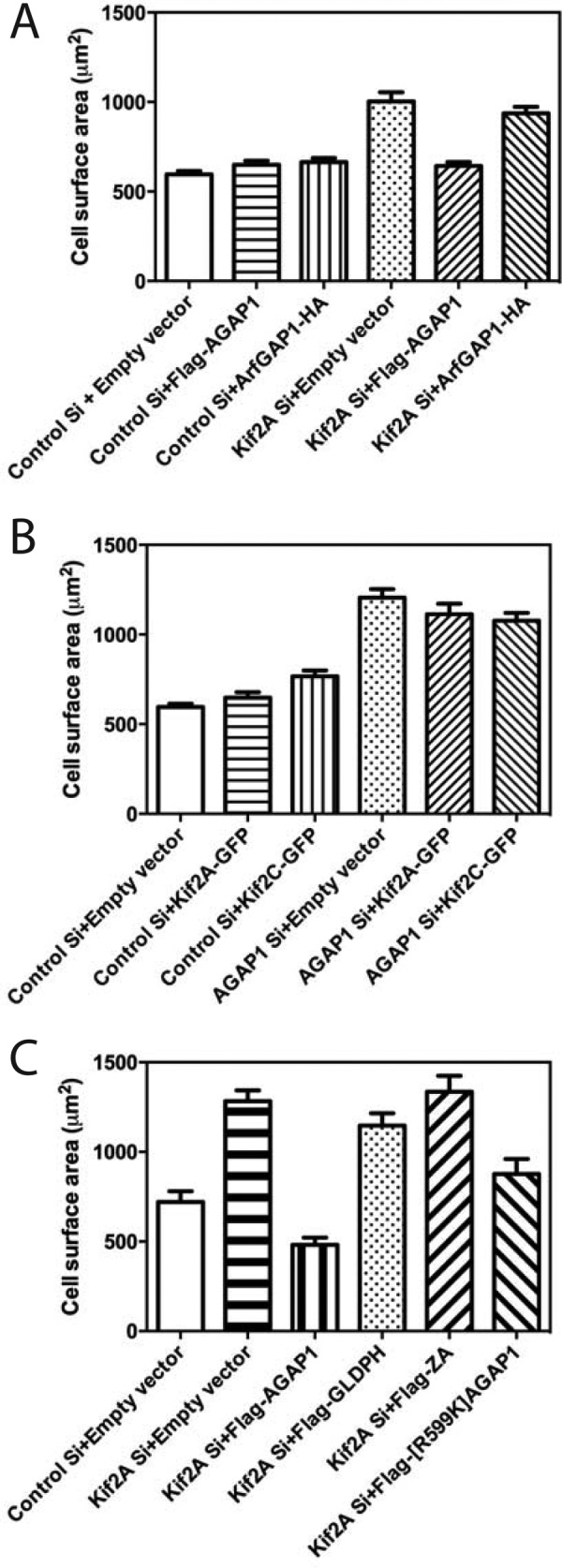
Effect of Kif2A-GFP overexpression on AGAP1 knockdown and effect of FLAG-AGAP1 overexpression on Kif2A knockdown. Spreading of HeLa cells treated with siRNA targeting the noncoding region of AGAP1 or Kif2A and transfected with plasmids for expression of either Kif2A-GFP, Kif2C-GFP, FLAG-AGAP1, or ArfGAP1-HA was determined, and data analysis was carried out as in Fig. 7. A, effect of AGAP1 overexpression on Kif2A knockdown. Control Si + empty vector versus Kif2A Si + empty vector, p < 0.001; Kif2A Si + empty vector versus Kif2A Si + FLAG-AGAP1, p < 0.001; Kif2A Si + empty vector + ArfGAP1-HA, not significant. B, effect of Kif2A-GFP or Kif2C-GFP overexpression of AGAP1 knockdown. Control Si + empty vector versus AGAP1 Si + empty vector, p < 0.001; AGAP1 Si + empty vector versus AGAP1 Si + Kif2A-GFP or AGAP1 Si + Kif2C-GFP, not significant. C, effect of AGAP1 and AGAP1 mutants on Kif2A knockdown. The results were analyzed by ANOVA with post-test multiple comparisons. Control Si + empty vector versus Kif2ASi + empty vector, p < 0.0001; control Si + empty vector versus Kif2ASi + FLAG-AGAP1, not significant; Kif2A Si +empty vector versus Kif2A Si + FLAG-AGAP1, p < 0.0001; Kif2A Si +empty vector versus Kif2A Si + FLAG-GLDPH, not significant; Kif2A Si +empty vector versus Kif2A Si + FLAG-ZA, not significant; Kif2A Si +empty vector versus Kif2A Si + FLAG-[R599K]AGAP, p < 0.01; Kif2A Si +FLAG-AGAP1 versus Kif2A Si + FLAG-[R599K]AGAP, p < 0.05. Results shown are the mean ± S.E. from three independent experiments.
The effect of AGAP1 fragments and mutants on accelerated spreading in Kif2A knockdown was determined (Fig. 8C). Neither FLAG-GLDPH nor FLAG-ZA reversed the effect of Kif2A knockdown. FLAG-[R599K]AGAP1 reversed the effect, but not to the same extent as did wild type FLAG-AGAP1.
Discussion
We set out to find binding partners of AGAP1 with two goals. AGAP1 is regulated by its GLD and PH domain, but the molecular basis is not understood. Identification of binding partners for the regulatory domains could provide insights into the mechanisms of regulation. The second goal was to gain insight into the biological function of AGAP1. We discovered that AGAP1 binds Kif2A, a kinesin-13 family member with interphase functions that include the control of cell migration and invasion of cancer cells. Kif2A, synergistically with PtdIns(4,5)P2, stimulated AGAP1 GAP activity. Conversely, AGAP1 stimulated Kif2A ATPase activity. KD of either Kif2A or AGAP1 similarly accelerated cell migration and cell spreading. Expression of wild type FLAG-AGAP1 reversed the effect of KD of endogenous AGAP1. Expression of a point mutant or a deletion of a mutant of AGAP1 that bound Kif2A poorly only partly reversed the effect of AGAP1 KD. A deletion mutant of AGAP1 containing only the Kif2A-binding domains(GLD-PH) functioned as a dominant negative, accelerating spreading. AGAP1 overexpression compensated for reduced Kif2A expression, but Kif2A overexpression did not compensate for reduced AGAP1 expression. In addition to identifying allosteric control of AGAP1 by PtdIns(4,5)P2 and Kif2A, these results support the idea that the AGAP1·Kif2A complex regulates cytoskeleton remodeling associated with cell movement.
We are considering two roles for the Kif2A·AGAP1 complex. First, it is possible that Kif2A-stimulated Arf GAP activity controls a membrane trafficking step, which must occur in parallel with microtubule changes. Reciprocal regulation of the GAP and Kif2A would ensure that the events are coordinated. However, GAP activity is not required for rescue of the spreading phenotype of AGAP1 knockdown. Another possibility is that cooperative binding of PtdIns(4,5)P2, kinesin, and Arf1·GTP functions as a coincidence detector, recruiting AGAP1 to a specific site where it might function as an adaptor. In this case, overexpressed AGAP1 might overcome a deficiency in Kif2A because efficient recruitment is not required when the AGAP1 levels are elevated, which is analogous to high concentrations of Kif2C not requiring interaction with EB1 for targeting and microtubule depolymerization (26, 27). Kif2A stimulation of GAP activity could serve as a feedback mechanism if Arf·GTP contributes to regulating Kif2A. These functions of regulated GAP activity are not mutually exclusive.
The regulation of AGAP1 by PtdIns(4,5)P2 and Kif2A may involve a number of interdomain dependences. The first is related to binding the ligands. The GLD domain is required but not sufficient for AGAP1 binding to Kif2A. The PH domain is also required. One plausible explanation for the dependence is that Kif2A and AGAP1, via its PH domain, bind to PtdIns(4,5)P2 to concentrate the two proteins on the same surface to promote binding of Kif2A to the GLD of AGAP1. However, other mechanisms should be considered. APPL1 is an example of a protein in which the binding surface for the GTP-binding protein Rab5 is contributed by both the BAR and the PH domain (28). Similarly, Kif2A may contact both the GLD and the PH domains.
The mechanism of allosteric regulation of GAP activity by Kif2A and PtdIns(4,5)P2 similarly involves interdomain dependences. Both the GLD and the PH domains contribute to regulation of AGAP1 GAP activity. The effect of Kif2A on GAP activity depends on PtdIns(4,5)P2. Similar to the possible role of the PH domain in concentrating AGAP1 on a surface that also contains Kif2A, binding of Kif2A and PtdIns(4,5)P2 could concentrate AGAP1 more specifically on a surface for site-specific activity against Arf·GTP. Alternatively, PtdIns(4,5)P2 may modify the conformation of the PH domain, promoting binding to Kif2A, or may affect the interaction of the PH domain with the GLD, possibly creating a binding site. However, the regulation of the GAP activity is likely to involve more than control of binding of the two ligands. Kif2A bound to [E125Q]AGAP1, but did not stimulate GAP activity, indicating that binding to PtdIns(4,5)P2 and Kif2A is not sufficient. We are further investigating the molecular basis of the cooperative interaction.
Kinesin-13s are regulated by both phosphorylation and direct protein interactions. During mitosis, phosphorylation of the N-terminal domain of Kif2A (29) and Kif2C (30) by aurora B inhibits activity, whereas Plk1 phosphorylation promotes Kif2B function (31). Phosphorylation of the motor domain of the Drosophila kinesin-13 KLP10 by casein kinase inhibits activity. Interphase function of kinesin-13s is also regulated. Kif2A controls axonal branching (18). The effects on axonal branching and microtubule-depolymerizing activity are regulated by phosphatidylinositol 4-phosphate 5-kinase α, which binds to the targeting and neck domains of Kif2A to accelerate depolymerization activity. Regulation by an Arf GAP and regulation through interaction with the motor domain, however, are unprecedented.
Kinesin-13 function in interphase has not been extensively explored. Function in positioning lysosomes has been described (21), although the molecular mechanism was not examined. More recently, a connection to cell migration has been established (22, 23). The effect could be mediated by control of focal adhesions (FAs). Microtubules regulate FAs (32). Stabilization of microtubules increases FA turnover, whereas depolymerization of microtubules stabilizes FAs. AGAP1-regulated Kif2A could affect FA dynamics by its activity to depolymerize microtubules. It is also plausible that cell migration is affected by control of organellar positioning, and with that, cell polarity (33), which are possible functions of the AGAP1 downstream of Kif2A.
In summary, we have discovered a complex of AGAP1·Kif2A. Further studies are necessary to understand the molecular basis of reciprocal regulation of these two enzymes and the role of the complex in cellular behaviors.
Materials and Methods
Plasmids and Antibody
Plasmids for FLAG- or GST-tagged AGAP1, GLD ([69–317]AGAP1), PH ([347–535]AGAP1 for GST and [347–540]AGAP1 for FLAG), PZA ([347–804]AGAP1), ZA ([545–804]AGAP1), and FLAG-tagged ASAP1, ARAP1, and ACAP1 have been described previously (34–36). His-tagged PH ([347–540]AGAP) and GLDPH ([64–540]AGAP1) were prepared by amplifying the open reading frame by PCR and subcloning the product into the NdeI and XhoI sites of pET19b. GST GLDPH ([64–540]AGAP1), PH ([347–535]AGAP1), and GLD ([69–317]AGAP1) were generated by subcloning the corresponding ORFs into BamHI and XhoI sites of pGEX4T-1. A plasmid for bacterial expression of human Kif2A fused to a histidine tag (ID 25548) was purchased from Addgene. A plasmid for expressing mouse Kif2A fused to EGFP in mammalian cells (pMX155) has been described (37). The ORFs for mouse [1–716]Kif2A, [525–716]Kif2A, [1–124]Kif2A, and [1–525]Kif2A were amplified by PCR and subcloned into the NdeI and XhoI sites of pET19b, resulting in cDNA encoding His10-Kif2A fusion proteins.
Anti-FLAG M2 affinity gel (catalog number A2220), anti-FLAG M5 (catalog number F4042) and anti-FLAG polyclonal Ab (catalog number F7425), and anti-Kif5B (catalog number AV33904) were purchased from Sigma-Aldrich, anti-Kinesin-like protein KIF2A (catalog number AB6050) was from Millipore (Temecula, CA), and anti-Kif3A (catalog number Ab95885) was from Abcam (Cambridge, MA). Anti-FLAG mouse monoclonal (9A3, catalog number 8146) was from Cell Signaling (Danvers, MA).
Protein Purification
Bovine myrArf1 and His-AGAP1 were expressed in and purified from bacteria as described (8, 38). His-tagged PH ([347–540]AGAP1) and GLDPH ([64–540]AGAP1) were expressed as described for His-AGAP1. Proteins were purified by gradient chromatography on a 5-ml HisTrap HPTM column (GE HealthCare Life Sciences), eluting with imidazole, followed by a size exclusion column (HiPrep 26/60 Sephacryl S-100 HR) developed in 20 mm Tris, pH 8.0, 200 mm NaCl, and 1 mm DTT. Full-length Kif2A and fragments of Kif2A were purified by a 5-ml HisTrap HPTM column followed by a HiLoad 16/60 Superdex 75 size exclusion column using the same buffers as for AGAP1.
Cell Culture, Immunoprecipitation, and GST Pulldown
HeLa cells were maintained in Dulbecco's modified enriched medium (DMEM) containing 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) at 37 °C with 5% CO2. Cells were transfected with plasmids directing expression of FLAG-tagged AGAP1, ASAP1, ARAP1, and ACAP1 using Lipofectamine 2000 (Invitrogen). Immunoprecipitation was carried out as described previously (29). Monoclonal anti-FLAG Ab or polyclonal anti-Kif2 Ab were used. GST pulldown experiments were performed by incubating 1 mg of Hela cell lysate with 30 μg of GST-AGAP1 or GST on GSH beads for 1 h at 4 °C. After washing the GSH beads with PBS three times, proteins were eluted from the beads in a 2-fold concentrate of SDS sample buffer and analyzed by SDS-PAGE followed by immunoblotting using an anti-Kif2A antibody. For in vitro GST pulldown, 30 μg of GST-fused fragments of AGAP1 were incubated with 100 nm His10-Kif2A[193–531] at 37 °C for 30 min. The beads were then washed with PBS three times, and proteins were eluted and analyzed as described for the cell lysates.
Immunofluorescence
To analyze the relative localization of proteins, HeLa cells were transfected, using Lipofectamine 2000, with plasmids directing expression of relevant proteins. 24 h later, the cells were replated on fibronectin-coated coverslips and maintained in serum-free Opti-MEM (Invitrogen) for 6 h prior to fixing and immunostaining. Images from fixed cells were collected with a Leica SP8 laser scanning confocal microscope, using a 63×, 1.4 NA objective (Leica Microsystems Inc., Buffalo Grove, IL). 2–3-mm Z stacks with a spacing of 0.3 mm were taken of the area contacting the coverslip.
Leica AF software was used to produce images. Single confocal slices were exported, and then Adobe Photoshop and Illustrator (Adobe Systems Inc., San Jose CA) were used to prepare composite figures. Scale bars were removed from the original images and replaced with a more visible version in the final composite image.
GAP Assay
Arf GAP assays were performed as described in Ref. 39. Lipid necessary for the reaction was provided as LUVs composed of 40% (mole percent) phosphatidylcholine (PtdCho), 25% phosphatidylethanolamine (PtdEt), 15% phosphatidylserine (PtdSer), 9% phosphatidylinositol (PtdIns), 1% PtdIns(4,5)P2, and 10% cholesterol prepared as described previously (38, 40).
ATPase Assay
ATPase assays were carried out using the HTS kinesin ATPase Endpoint Assay Biochem Kit (BK053) from Cytoskeleton, Inc. (Denver, CO) according to the manufacturer's manual using half-area 96-well plates and a SpectraMax M5.
Liposome Floatation Assays and Mass Spectrometry
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). 10 μm AGAP1 GLDPH protein was incubated with 1 mm LUVs (40% PtdCho, 19.8% PtdEt, 0.2% rhodamine-PtdEt, 5% 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] DGS-NTA (Ni2+), 15% PtdSer, 9% PtdIns, 1% PtdIns(4,5)P2, and 10% cholesterol) for 5 min at room temperature. 300 μg of HeLa cell lysates were added to the LUVs in a total of 200 μl and incubated for 15 min at room temperature. For the floatation assay, 200 μl of lysis buffer (50 mm HEPES, pH 7.4, 120 mm potassium acetate, 1 mm MgCl2, and 1 mm DTT) containing 60% (w/v) sucrose were mixed with 200 μl of the mixtures of LUVs and lysates to a final concentration of 30%, and then overlaid with 60 μl of 25% sucrose in lysis buffer. The bound proteins were floated with LUVs by centrifugation in an S55-S swinging bucket rotor (Thermo Scientific) at 53,000 rpm (240,000 × g) for 30 min at 4 °C. The bound proteins were dissolved in SDS sample buffer, separated by SDS-PAGE, and visualized with Coomassie Blue dye. The protein bands were reduced, alkylated with iodoacetamide, and in-gel trypsin-digested for 16 h at 37 °C, as described (41). For mass spectrometry analysis, the dried peptides were resuspended in water containing 2% acetonitrile, 0.5% acetic acid and injected onto a 0.2 × 50-mm Magic C18AQ reverse phase column (Michrom Bioresources, Inc.) using the Paradigm MS4 HPLC (Michrom Bioresources, Inc.). Peptides were separated at a flow rate of 2 μl/min followed by online analysis by tandem mass spectrometry using an LTQ ion trap mass spectrometer (Thermo Scientific) equipped with an ADVANCE CaptiveSpray ion source (Michrom Bioresources, Inc.). The raw mass spectrometry data were searched against the International Protein Index (IPI) database using TurboSEQUEST in BioWorks version 3.2 (Thermo Electron).
Cell Spreading and Migration
For cell spreading and migration assays, SMARTpool siGENOME human AGAP1 siRNA, SMARTpool siGENOME human Kif2A siRNA, the set of four individual siGENOME AGAP1 siRNA or siGENOME Kif2A siRNA, and control siRNA (siCONTROL non-targeting siRNA #4) were from GE Dharmacon. For rescue experiments, siRNA against AGAP1 (AGAP1 siRNA targeted region: 3′-UTR catalog number A-020452-13) and Kif2A (siGENOME Kif2A siRNA targeted region 33′-UTR, 5′-GGAUAUAGCUGCUGGACCAUU-3′) and control siRNA (siCONTROL non-targeting siRNA #4) were also from GE Dharmacon. Subconfluent HeLa cells were transfected with 100 nm siRNA using DharmaFECT transfection reagent 1 (GE Dharmacon, Lafayette, CO). 48 h after siRNA transfection, cells were transfected with plasmids expressing FLAG-AGAP1, Kif2-GFP, or an empty vector (pCI) using Lipofectamine 2000 reagent (Invitrogen). To measure cell spreading rates, 72 h after the initial siRNA transfection, cells were replated onto coverslips that had been coated with 10 μg/ml fibronectin. After 20 min, the cells were fixed with 4% paraformaldehyde for 20 min. Cells on coverslips were washed with PBS and quenched with 50 mm NH4Cl/PBS. The cell area was measured based on F-actin staining by rhodamine-phalloidin. Cell migration was measured in a wound healing assay using Ibidi wound assay chambers (Ibidi GmbH, Martinsried, Germany). 48 h after transfection with siRNA targeting AGAP1 or Kif2A or with an irrelevant control siRNA, MDA-MB-231 cells were trypsinized and resuspended in DMEM with 10% FBS. 70 μl of cell suspension (5 × 105 cells/ml) were seeded into each well of the insert. After 24 h, the culture inserts were removed, and the cells were incubated with fresh medium DMEM with 10% FBS for 8–10 h. The cell migration into the defined cell-free gap (500 μm) was observed by taking time-lapse phase contrast images for up to 10 h. Repair of the artificial “wound” (% of migration) was quantified as follows: migration (%) = ((1−(width of GAP at 0 h/width of GAP at a specific hour)) × 100.
Two-hybrid Screens
The two-hybrid screens were carried out by Myriad Genetics, Inc. Myriad's method is based on the nuclear yeast two-hybrid methodology originally developed by Fields and Song (42). Myriad used the following protocol for the screen.
The DNA-binding domain vector for constructing baits was pGBT.superB. It carries an Escherichia coli origin of replication (ori) and a kanamycin resistance gene (Kan) for maintenance and selection of the plasmid in E. coli. The Saccharomyces cerevisiae TRP1 gene is included for selection in yeast, and CEN and ARS sequences are included for maintenance of the plasmid at single copy in yeast. Baits were placed in-frame, C-terminal to the Gal4 DNA-binding domain (nucleotide 1–441 coding sequence in GAL4), followed by the ADH1 transcriptional terminator sequence. The ADH1 promoter drives expression of the fusion between the Gal4 DNA-binding domain and bait. This vector also contains a 1.6-kb CEN6 sequence inserted into the multiple cloning site between EcoRI and SalI sites. This prevents empty bait vector from propagating in yeast.
Prey libraries were constructed in pGAD.PN2. It carries an E. coli origin of replication (ori) and an ampicillin resistance gene (Amp) for maintenance. The S. cerevisiae LEU2 gene is included for selection in yeast, and CEN and ARS sequences are included for maintenance of the plasmid at single copy in yeast. Preys are placed C-terminal to the Gal4 activation domain (nucleotide 2301–2643 coding sequence in GAL4), followed by the PGK1 transcriptional terminator sequence. The ADH1 promoter drives expression of the fusion between the Gal4 activation domain and prey.
The yeast strain used to maintain the bait plasmids is PNY200 (MATα ura3-52 ade2-101 trp1-901 his3-Δ200 leu2-3,112 gal4Δ gal80Δ). The yeast strain used to maintain the prey constructs is BK100 (MATa ura3-52 trp1-901 his3-Δ200 leu2-3,112 gal4Δ gal80ΔGAL2-ADE2 LYS2::GAL1-HIS3 met2::GAL7-lacZ). This strain is a derivative of PJ69-4A (43).
Double poly(A) selected mRNA were used for construction of the prey plasmids. First strand cDNA synthesis is initiated by priming with random decamers that contain a common tag sequence and a biotin blocker. Second strand synthesis is performed according to the Gubler-Hoffman procedure, followed by blunt-ending with T4 DNA polymerase. The cDNA is purified and ligated with a DNA adaptor. The adaptor-ligated cDNA is subjected to gel filtration to remove free adaptors, followed by PCR amplification with a pair of primers that anneal to the 5′ and 3′ tags and contain tails for homologous recombination in yeast. The PCR-amplified DNA is gel-purified to remove fragments containing inserts shorter than ∼250 bp and co-transformed with linear pGAD.PN2 vector into BK100 for homologous recombination in vivo. The yeast transformants are harvested from the selection plates and dispensed into aliquots for −80 °C storage for future library screening experiments.
A mating-based method was used to screen for bait-prey interactions. Approximately 25–30 million MATα yeast cells containing single bait were mixed with 60 million MATa library yeast cells and allowed to mate on filters. Between 5 and 10 million diploid yeast cells are routinely obtained for each mating. After mating, the cells are plated onto selective media. Transcription of two auxotrophic reporter genes (HIS3 and ADE2) with dissimilar promoters (see BK100 genotype) occurs if the bait and prey protein interact. Colonies are picked from the selection plates, and the prey inserts are identified by sequence analysis.
To confirm the interactions, the bait and prey plasmids are isolated from yeast diploids and electroporated into E. coli. Both bait and prey plasmids are purified followed by sequencing to confirm their identities. The bait and prey plasmid DNAs are co-transformed into a naive yeast strain to recapitulate the interaction. The confirmation test takes advantage of a third reporter gene (lacZ) and is based on a chemiluminescent reporter gene assay system. The specificity of the prey is investigated in a separate false positive test where the prey is tested against a mixture of several heterologous baits.
Author Contributions
R. L. conceived of the project, designed and performed experiments, and wrote and edited the manuscript. P. W. C. guided cell migration and spreading work and designed and performed preliminary experiments. M. W. provided guidance on ATPase assays and designed and performed experiments. X. J. designed and performed experiments. L. J. performed mass spectrometry and analysis. L. W. defined project goals, designed experiments, and wrote and edited the manuscript. P. A. R. conceived and directed the project and wrote and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Roberto Weigert for critical review of the manuscript and Marielle E. Yohe for insightful discussions.
This work was supported by the intramural program of the National Cancer Institute (Project BC007365) and by the National Institute of General Medical Sciences (Grant GM069429) (to L. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
R. Luo and P. A. Randazzo, unpublished data.
- Arf
- ADP-ribosylation factor
- GAP
- GTPase-activating protein
- PH
- pleckstrin homology
- GLD
- G-protein-like domain
- KD
- knockdown
- LUV
- large unilamellar vesicle
- FA
- focal adhesion
- PtdIns
- phosphatidylinositol
- PtdIns(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PtdCho
- phosphatidylcholine
- PtdEt
- phosphatidylethanolamine
- PtdSer
- phosphatidylserine
- DMEM
- Dulbecco's modified enriched media
- CEN
- centromere
- ARS
- autonomously replicating sequences
- ANOVA
- analysis of variance
- myrArf
- myristoylated Arf.
References
- 1. Kahn R. A., Bruford E., Inoue H., Logsdon J. M. Jr., Nie Z., Premont R. T., Randazzo P. A., Satake M., Theibert A. B., Zapp M. L., and Cassel D. (2008) Consensus nomenclature for the human ArfGAP domain-containing proteins. J. Cell Biol. 182, 1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Randazzo P. A., Inoue H., and Bharti S. (2007) Arf GAPs as regulators of the actin cytoskeleton. Biol. Cell 99, 583–600 [DOI] [PubMed] [Google Scholar]
- 3. Donaldson J. G., and Jackson C. L. (2011) ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 12, 362–375; Correction (2011) Nat. Rev. Mol. Cell Biol.12, 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kahn R. A., Cherfils J., Elias M., Lovering R. C., Munro S., and Schurmann A. (2006) Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J. Cell Biol. 172, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Randazzo P. A., and Kahn R. A. (1994) GTP Hydrolysis by ADP-ribosylation factor is dependent on both an ADP-ribosylation factor GTPase-Activating protein and acid phospholipids. J. Biol. Chem. 269, 10758–10763 [PubMed] [Google Scholar]
- 6. East M. P., and Kahn R. A. (2011) Models for the functions of Arf GAPs. Semin. Cell Dev. Biol. 22, 3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Inoue H., and Randazzo P. A. (2007) Arf GAPs and their interacting proteins. Traffic 8, 1465–1475 [DOI] [PubMed] [Google Scholar]
- 8. Nie Z., Stanley K. T., Stauffer S., Jacques K. M., Hirsch D. S., Takei J., and Randazzo P. A. (2002) AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J. Biol. Chem. 277, 48965–48975 [DOI] [PubMed] [Google Scholar]
- 9. Nie Z., Boehm M., Boja E. S., Vass W. C., Bonifacino J. S., Fales H. M., and Randazzo P. A. (2003) Specific regulation of the adaptor protein complex AP-3 by the Arf GAP AGAP1. Dev. Cell 5, 513–521 [DOI] [PubMed] [Google Scholar]
- 10. Bendor J., Lizardi-Ortiz J. E., Westphalen R. I., Brandstetter M., Hemmings H. C. Jr., Sulzer D., Flajolet M., and Greengard P. (2010) AGAP1/AP-3-dependent endocytic recycling of M5 muscarinic receptors promotes dopamine release. EMBO J. 29, 2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Y., Zhao Y., Ma X., Zhu Y., Patel J., and Nie Z. (2013) The Arf GAP AGAP2 interacts with β-arrestin2 and regulates β2-adrenergic receptor recycling and ERK activation. Biochem. J. 452, 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Y., Wu Y., Kim J. I., Wang Z., Daaka Y., and Nie Z. (2009) Arf GTPase-activating protein AGAP2 regulates focal adhesion kinase activity and focal adhesion remodeling. J. Biol. Chem. 284, 13489–13496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirokawa N., Noda Y., Tanaka Y., and Niwa S. (2009) Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696 [DOI] [PubMed] [Google Scholar]
- 14. Hirokawa N., and Takemura R. (2004) Kinesin superfamily proteins and their various functions and dynamics. Exp. Cell Res. 301, 50–59 [DOI] [PubMed] [Google Scholar]
- 15. Walczak C. E., Gayek S., and Ohi R. (2013) Microtubule-depolymerizing kinesins. Annu. Rev. Cell Dev. Biol. 29, 417–441 [DOI] [PubMed] [Google Scholar]
- 16. Helenius J., Brouhard G., Kalaidzidis Y., Diez S., and Howard J. (2006) The depolymerizing kinesin MCAK uses lattice diffusion to rapidly target microtubule ends. Nature 441, 115–119 [DOI] [PubMed] [Google Scholar]
- 17. Ginkel L. M., and Wordeman L. (2000) Expression and partial characterization of kinesin-related proteins in differentiating and adult skeletal muscle. Mol. Biol. Cell 11, 4143–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Homma N., Takei Y., Tanaka Y., Nakata T., Terada S., Kikkawa M., Noda Y., and Hirokawa N. (2003) Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114, 229–239 [DOI] [PubMed] [Google Scholar]
- 19. Drum B. M., Yuan C., Li L., Liu Q., Wordeman L., and Santana L. F. (2016) Oxidative stress decreases microtubule growth and stability in ventricular myocytes. J. Mol. Cell. Cardiol. 93, 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mennella V., Rogers G. C., Rogers S. L., Buster D. W., Vale R. D., and Sharp D. J. (2005) Functionally distinct kinesin-13 family members cooperate to regulate microtubule dynamics during interphase. Nat. Cell Biol. 7, 235–245 [DOI] [PubMed] [Google Scholar]
- 21. Santama N., Krijnse-Locker J., Griffiths G., Noda Y., Hirokawa N., and Dotti C. G. (1998) KIF2β, a new kinesin superfamily protein in non-neuronal cells, is associated with lysosomes and may be implicated in their centrifugal translocation. EMBO J. 17, 5855–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang C.-Q., Qu X., Zhang X.-Y., Zhou C.-J., Liu G.-X., Dong Z.-Q., Wei F.-C., and Sun S.-Z.(2010) Overexpression of Kif2a promotes the progression and metastasis of squamous cell carcinoma of the oral tongue. Oral Oncol. 46, 65–69 [DOI] [PubMed] [Google Scholar]
- 23. Wang J., Ma S., Ma R., Qu X., Liu W., Lv C., Zhao S., and Gong Y. (2014) KIF2A silencing inhibits the proliferation and migration of breast cancer cells and correlates with unfavorable prognosis in breast cancer. BMC Cancer 14, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaganjor E., Osborne J. K., Weil L. M., Diaz-Martinez L. A., Gonzales J. X., Singel S. M., Larsen J. E., Girard L., Minna J. D., and Cobb M. H. (2014) Ras regulates kinesin 13 family members to control cell migration pathways in transformed human bronchial epithelial cells. Oncogene 33, 5457–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo R., Akpan I. O., Hayashi R., Sramko M., Barr V., Shiba Y., and Randazzo P. A. (2012) GTP-binding protein-like domain of AGAP1 is protein binding site that allosterically regulates ArfGAP protein catalytic activity. J. Biol. Chem. 287, 17176–17185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Domnitz S. B., Wagenbach M., Decarreau J., and Wordeman L. (2012) MCAK activity at microtubule tips regulates spindle microtubule length to promote robust kinetochore attachment. J. Cell Biol. 197, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montenegro Gouveia S., Leslie K., Kapitein L. C., Buey R. M., Grigoriev I., Wagenbach M., Smal I., Meijering E., Hoogenraad C. C., Wordeman L., Steinmetz M. O., and Akhmanova A. (2010) In vitro reconstitution of the functional interplay between MCAK and EB3 at microtubule plus ends. Curr. Biol. 20, 1717–1722 [DOI] [PubMed] [Google Scholar]
- 28. Miaczynska M., Christoforidis S., Giner A., Shevchenko A., Uttenweiler-Joseph S., Habermann B., Wilm M., Parton R. G., and Zerial M. (2004) APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 116, 445–456 [DOI] [PubMed] [Google Scholar]
- 29. Knowlton A. L., Vorozhko V. V., Lan W., Gorbsky G. J., and Stukenberg P. T. (2009) ICIS and Aurora B coregulate the microtubule depolymerase Kif2a. Curr. Biol. 19, 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andrews P. D., Ovechkina Y., Morrice N., Wagenbach M., Duncan K., Wordeman L., and Swedlow J. R. (2004) Aurora B regulates MCAK at the mitotic centromere. Dev. Cell 6, 253–268 [DOI] [PubMed] [Google Scholar]
- 31. Hood E. A., Kettenbach A. N., Gerber S. A., and Compton D. A. (2012) Plk1 regulates the kinesin-13 protein Kif2b to promote faithful chromosome segregation. Mol. Biol. Cell 23, 2264–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ezratty E. J., Partridge M. A., and Gundersen G. G. (2005) Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 7, 581–590 [DOI] [PubMed] [Google Scholar]
- 33. Bornens M. (2008) Organelle positioning and cell polarity. Nat. Rev. Mol. Cell Biol. 9, 874–886 [DOI] [PubMed] [Google Scholar]
- 34. Jackson T. R., Brown F. D., Nie Z., Miura K., Foroni L., Sun J., Hsu V. W., Donaldson J. G., and Randazzo P. A. (2000) ACAPs are Arf6 GTPase-activating proteins that function in the cell periphery. J. Cell Biol. 151, 627–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miura K., Jacques K. M., Stauffer S., Kubosaki A., Zhu K., Hirsch D. S., Resau J., Zheng Y., and Randazzo P. A. (2002) ARAP1: a point of convergence for Arf and Rho signaling. Mol. Cell 9, 109–119 [DOI] [PubMed] [Google Scholar]
- 36. Brown M. T., Andrade J., Radhakrishna H., Donaldson J. G., Cooper J. A., and Randazzo P. A. (1998) ASAP1, a phospholipid-dependent Arf GTPase-activating protein that associates with and is phosphorylated by Src. Mol. Cell. Biol. 18, 7038–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moore A. T., Rankin K. E., von Dassow G., Peris L., Wagenbach M., Ovechkina Y., Andrieux A., Job D., and Wordeman L. (2005) MCAK associates with the tips of polymerizing microtubules. J. Cell Biol. 169, 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jian X., Brown P., Schuck P., Gruschus J. M., Balbo A., Hinshaw J. E., and Randazzo P. A. (2009) Autoinhibition of Arf GTPase-activating Protein activity by the BAR domain in ASAP1. J. Biol. Chem. 284, 1652–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo R., Ahvazi B., Amariei D., Shroder D., Burrola B., Losert W., and Randazzo P. A. (2007) Kinetic analysis of GTP hydrolysis catalysed by the Arf1-GTP-ASAP1 complex. Biochem. J. 402, 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nie Z., Hirsch D. S., Luo R., Jian X., Stauffer S., Cremesti A., Andrade J., Lebowitz J., Marino M., Ahvazi B., Hinshaw J. E., and Randazzo P. A. (2006) A BAR domain in the N terminus of the Arf GAP ASAP1 affects membrane structure and trafficking of epidermal growth factor receptor. Curr. Biol. 16, 130–139 [DOI] [PubMed] [Google Scholar]
- 41. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 42. Fields S., and Song O. (1989) A novel genetic system to detect protein-protein interactions. Nature 340, 245–246 [DOI] [PubMed] [Google Scholar]
- 43. James P., Halladay J., Craig E. A. (1996) Genomic libraries and a host strain designed for highly efficient two hybrid selection in yeast Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]