

Abstract
Congenital insensitivity to pain with anhidrosis (CIPA) is a rare autosomal recessive disorder characterized by insensitivity to noxious stimuli and variable intellectual disability (ID) due to mutations in the NTRK1 gene encoding the NGF receptor TrkA. To get an insight in the effect of NTRK1 mutations in the cognitive phenotype we biochemically characterized three TrkA mutations identified in children diagnosed of CIPA with variable ID. These mutations are located in different domains of the protein; L213P in the extracellular domain, Δ736 in the kinase domain, and C300stop in the extracellular domain, a new mutation causing CIPA diagnosed in a Spanish teenager. We found that TrkA mutations induce misfolding, retention in the endoplasmic reticulum (ER), and aggregation in a mutation-dependent manner. The distinct mutations are degraded with a different kinetics by different ER quality control mechanisms; although C300stop is rapidly disposed by autophagy, Δ736 degradation is sensitive to the proteasome and to autophagy inhibitors, and L213P is a long-lived protein refractory to degradation. In addition L213P enhances the formation of autophagic vesicles triggering an increase in the autophagic flux with deleterious consequences. Mouse cortical neurons expressing L213P showed the accumulation of LC3-GFP positive puncta and dystrophic neurites. Our data suggest that TrkA misfolding and aggregation induced by some CIPA mutations disrupt the autophagy homeostasis causing neurodegeneration. We propose that distinct disease-causing mutations of TrkA generate different levels of cell toxicity, which may provide an explanation of the variable intellectual disability observed in CIPA patients.
Keywords: autophagy, ER quality control, neurodegeneration, protein aggregation, TRK1-transforming tyrosine kinase protein (Trk-A), autophagic flux, congenital insensitivity to pain with anhidrosis
Introduction
Congenital insensitivity to pain with anhidrosis (CIPA)5 (MIM number 256800), also known as hereditary sensory and autonomic neuropathy type IV (HSAN-IV), is a rare autosomal recessive disorder (1, 2). It is characterized by insensitivity to noxious stimuli, recurrent hyperpyrexia related to high ambient temperature, inability to sweat (anhidrosis), self-mutilation in the first months of life, and increased risk to bone fractures, multiple scars, osteomyelitis, joint deformities, and limb amputation as children grow older (1, 3). Death from hyperpyrexia occurs within the first 3 years of life in almost 20% of the patients and most patients show intellectual disability (ID) of varying severity.
Inactivating mutations in the NTRK1 gene encoding TrkA, a receptor tyrosine kinase for nerve growth factor (NGF), are responsible of CIPA development (4). The survival of nociceptive neurons and sympathetic ganglia derived from the neural crest depends on NGF stimulation (5–7). NGF/TrkA signaling is required not only for survival, but also for the sprouting and differentiation of nociceptor neurons during development (6, 8). The altered perception of pain and temperature and anhidrosis in CIPA are due to the absence of unmyelinated C-fibers, small-diameter myelinated Aδ-fibers, and lack of sweat glands innervations (1). In addition, CIPA patients present variable ID, although the causes for this are still unknown, its variability may indicate a mutation-specific phenotype.
To date, more than 50 TrkA mutations that cause CIPA have been described (9). Mutations are distributed all along the protein sequence of TrkA, from the extracellular to the kinase domain. Missense mutations usually affect the kinase domain where they disturb the kinase activity of TrkA by inducing conformational changes or misfolding. There are, as well, several nonsense mutations changing the open reading frame or introducing a stop codon. Although these mutations are clearly detrimental to the TrkA function, they generate truncated forms that may impinge the protein homeostasis, proteostasis, of the cell where they are expressed.
The folding of the secretory and the membrane proteins takes place in the endoplasmic reticulum (ER). The quality of the proteins folded in the ER is carefully monitored by an ER-quality control mechanism, which allows only correctly folded proteins to be transported to their final destinations (10–14). Misfolded and aggregated proteins are subsequently degraded by different clearance pathways like “ER-associated degradation” (ERAD) (11, 13–15) or by autophagy (16, 17) or a combination of both.
Autophagy allows recycling of cytoplasmic constituents or aged proteins for the maintenance of cellular homeostasis (18). Autophagy dysfunction can lead to cellular toxicity and diseases (19–21). Accumulation of abnormal protein aggregates, a common cause of neurodegenerative diseases, can be reduced through autophagic degradation. Defects in autophagy are implicated in neurodegenerative disorders (22, 23). Not only autophagy defects are detrimental, an excessive increase in autophagy can also cause neurodegeneration.
TrkA activity has been linked to autophagy-induced cell death. Several reports described the activation of autophagy upon TrkA overexpression in cancer cell lines (24–29), however, as the activation of autophagy is in some cases ligand-independent (30, 31) the physiological role of TrkA-induced autophagy is still questionable. Another possibility is that TrkA overexpression may saturate the lysosome pathway with toxic outcomes. Lysosomes constitute the degradation destiny of TrkA. Activation by NGF induces TrkA ubiquitination necessary for internalization and targeting to the lysosomal pathway for degradation (32, 33), however, how non-functional TrkA mutants are degraded has not been studied.
In addition, no study has addressed if TrkA mutations causing CIPA induce protein misfolding, aggregation, and cell toxicity. In this context, we biochemically characterized three mutations in NTRK1 causing CIPA that were identified in children with mild or absence of ID. One mutation is described in this work for the first time. We found that distinct TrkA mutations behaved differently in terms of misfolding, subcellular localization, degradation pathways, and cell toxicity.
Results
TrkA-Cys300stop a New Mutation in NTRK1 Causing CIPA
In this paper we report the clinical history of a family (Fig. 1A) with two members affected of CIPA: a 17-year-old male (index case) and his 8-year-old sister. This family also includes a non-affected sister and both parents. The index case was diagnosed at the age of 6 with HSAN-IV, which was characterized due to multiple fractures, hemarthrosis of lower limbs, especially of the left knee, and non-infectious fever related to high ambient temperature. At that time, he was using a wheelchair but was able to stand alone. Strength decrease in the lower limb (knees and ankles) was detected and the rest of the neurologic exploration was normal (coordination, balance, cranial pairs, tendon, and skin reflexes), except for the absence of nociceptive sensitivity (tactile and proprioceptive sensitivities were conserved). ID was not present at the time of the clinical history.
FIGURE 1.
Mutations in NTRK1 used in this study. A, pedigree of the family and results of the NTRK1 genetic analysis in CIPA patients. As indicated by the symbols in the pedigree, individuals II-1 and II-2 are affected by CIPA, whereas the parents and the sister are all carriers. The NTRK1 mutation detected in each individual is indicated under the corresponding symbol. Electropherograms demonstrating the presence of the NTRK1 point mutations. The position of each mutation is indicated by an asterisk. B, the mutations described in this report are named following the recommendations from the Human Genome Variation Society. C, location of the different mutations in the TrkA protein is shown in a scheme of the NTRK1 gene. Exons are numbered and shown as boxes.
Blood samples were collected from all the family members available: the two affected siblings, their healthy sister, the father, and the mother. Genomic DNA was purified from the blood of the 17-year-old affected male. All of the 17 exons and intron-exon boundaries of the longest NTRK1 isoform (NM_002529.3) were analyzed using PCR amplification of genomic DNA from the patient followed by direct DNA sequencing. The presence of the identified NTRK1 mutations was subsequently investigated in the other relatives using the same methods. Mutation screening in the index case (17-year-old male) revealed the presence of a new mutation in homozygosis in the NTRK1 gene, c.899_900delinsAA (p.Cys300*). The presence of this mutation was subsequently investigated in the other relatives using the same methods, confirming the homozygous status of the affected sister and the carrier status of the rest of the family members (Fig. 1A).
Together with C300stop, in this study we also investigated two other mutations in NTRK1 causing CIPA, which cover different domains of TrkA (Fig. 1B); the mutation TrkA-Δ736(c.2206–2A>G), which is a short in-frame deletion that eliminates 7 residues in the kinase domain of TrkA (Fig. 1B), which we previously described in a female child in Northern Spain with no evidences of ID (34); and the mutation L213P (c.637T>C), a missense mutation in the extracellular domain of TrkA originally characterized by Mardy et al. (35–38) with mild ID with speech problems. For practical reasons, we will refer to them as C300stop, Δ736, and L123P. These mutations causing variable ID are located in different regions of the TrkA protein (Fig. 1C) and are representative of the different types of mutations found in CIPA, missense, deletion, and truncated forms.
TrkA Signaling Pathways Are Impaired in All TrkA Mutants
To study how the new mutations influence TrkA activity, PC12nnr5 cells were transfected with plasmids encoding the TrkA-wt and all TrkA mutations. PC12 cells derived from a pheochromocytoma recapitulate neurite differentiation and survival in the presence of NGF and constitutes a good model of signaling and differentiation in the TrkA/NGF pathway (39). The PC12nnr5 cell line was generated by the Greene laboratory (40) and does not express TrkA, so it cannot differentiate in the presence of NGF being a good model for testing TrkA mutants. Two days after transfection cells were serum starved and treated with NGF for different time points and then, subjected to protein extraction and Western blot analysis (Fig. 2A). Upon transfection two TrkA bands are visible after Western blot analysis; a lower band that reflects the TrkA that has not completed Golgi-mediated processing of high-mannose N-glycans (41) (immature form of TrkA) and an upper mature band expressed in the plasma membrane (mature form of TrkA). It is established that the lowest band of TrkA is intracellular and its activation corresponds to spontaneous dimerization due to defective glycosylation (42). However, exposure to NGF substantially increases the phosphorylation of the upper TrkA band, consistent with that band protein being in the plasma membrane that has access to NGF. Upon NGF binding TrkA is activated by trans-phosphorylation in different tyrosine residues (43). We used two phospho-specific antibodies: one against phospho-Tyr-490, a site for Shc binding, and another against phospho-Tyr-674/675 in the kinase activation loop. The TrkA wild type is activated in a NGF-dependent manner, as we can see from the phosphorylation of the 140-kDa band after 5 and 15 min. The mutants Δ736, L213P, and the C300stop, however, are not activated by NGF and are not phosphorylated (Fig. 2A). In the L213P mutation, the lower band of TrkA corresponding to intracellular TrkA is recognized by the phospho-specific antibodies, indicating that the kinase domain is actually functional, although TrkA-L213P is causing CIPA due to a defective maturation process (35, 37). In the case of TrkA-Δ736, no phospho-reactive bands are observed, indicating that this mutation probably induces a global misfolding in the kinase domain (see below), making the kinase activity non-functional. C300stop has no kinase domain, and then no kinase activity was expected but it was used as an internal control (Fig. 2A). Western blotting against total TrkA indicates that TrkA-wt shows the bands corresponding to the mature form of TrkA, which is able to reach the plasma membrane (presence of the upper protein band), however, TrkA-Δ736 and TrkA-L213P are not able to mature and traffic correctly to the plasma membrane (absence of the upper protein band).
FIGURE 2.

Inactivation of the signaling and neurite differentiation mediated by TrkA mutations. A, Western blotting of PC12nnr5 cells transfected with the indicated constructs after stimulation with NGF (50 ng/ml) during different time points. B, differentiation of PC12nnr5 cells co-transfected with GFP and the indicated TrkA constructs upon stimulation with NGF (50 ng/ml) during 72 h. Presence of neurites is observed in TrkA-wt (white arrows). C, percentage of PC12nnr5 transfected with the indicated construct cells having neurites longer than the cell body.
To further analyze the effect of TrkA mutations in neurite development, PC12nnr5 cells were transfected and stimulated by NGF and the formation of neuronal-like dendrites was examined and quantified (Fig. 2, B and C). PC12nnr5 cells expressing TrkA-wt are able to form dendrites but not PC12nnr5 cells transfected with Δ736, L213P, or C300stop (Fig. 2, B and C). Altogether this analysis showed that all three TrkA mutations identified in CIPA patients induce inactivation of the TrkA receptor.
TrkA Mutants Are Retained in the ER and Do Not Traffic to the Plasma Membrane
As we noted above, Western immunoblotting analysis shows that TrkA-Δ736 and TrkA-L213P migrate with a unique protein band around 110 kDa, suggesting an improper processing or traffic to the plasma membrane (see Fig. 2A). We carried out different approaches to analyze the traffic of these mutants. First we used a surface-exposed biotinylation protocol to determine whether TrkA proteins are located at the cell surface. When expressed in HeLa cells the TrkA-wt is labeled with biotin but not the TrkA mutants (Fig. 3A), confirming that TrkA mutants are not exposed at the cell surface and suggesting that they have a defective traffic to the plasma membrane. Next we analyzed if the TrkA mutants are retained in the ER. We treated a total lysate of HeLa cells transfected with TrkA-wt and the mutants with endoglycosidase-H (Endo-H) (Fig. 3B), an enzyme that removes the glycans added to asparagine residues during transit from the ER to Golgi apparatus. As shown in Fig. 3B, the TrkA mutants are totally sensitive to the Endo-H treatment as well as some pools of TrkA-wt. As a control of deglycosylation the samples were incubated with N-glycosidase (N in Fig. 3B), an enzyme that removes all N-linked sugars. After treatment with N-glycosidase, a new expected band around 80 kDa appears, corresponding to the fully deglycosylated form of TrkA. This result suggests that all TrkA mutants tested, but not TrkA-wt, are retained during maturation through the ER, and do not reach the plasma membrane.
FIGURE 3.

TrkA CIPA mutants have impaired traffic to the plasma membrane. A, biotinylation protocol. Avidin-HRP immunodetection of TrkA immunoprecipitates from HeLa cells transfected with TrkA-wt and TrkA mutants. Mock indicates HeLa cells transfected with empty control plasmid. B, Western blotting of lysates from cells transfected with TrkA-wt and TrkA mutants treated with buffer (c), End-H (H), and N-glycosidase (N). C, immunolocalization of HeLa cells co-transfected with TrkA mutants (red) and GFP-Ras (green) in the absence and presence of 0.1% Triton X-100. DAPI nuclear staining and merge of three channels are shown. WB, Western blot.
We also analyzed the cellular localization of TrkA-wt and the three mutants by immunofluorescence (Fig. 3C). An N-terminal HA tag was introduced in TrkA constructs. We performed the immunofluorescence using an anti-HA antibody without permeating the cells. Only proteins in the plasma membrane would be able to be labeled with HA antibody. A construct encoding the fusion protein GFP-ras was used as a marker of the plasma membrane (green fluorescence in Fig. 3C). Our results showed that only TrkA-wt is localized in the plasma membrane but all the TrkA mutants tested remained intracellular (Fig. 3C). A control experiment in the presence of 0.1% Triton X-100 to permeate the cells before the immunofluorescence showed that all constructs are equally expressed (Fig. 3C).
We next studied the intracellular localization of TrkA-wt and TrkA mutants. We used two protein markers: calnexin, an ER resident transmembrane protein, and giantin a Golgi marker (Fig. 4). We used the Pearson's coefficient to assess co-localization of TrkA with calnexin (Fig. 4B) or giantin (Fig. 4D). Using this analysis we found that the three TrkA mutants are significantly co-localized with calnexin in comparison to TrkA-wt (Fig. 4, A and B). In addition, all the mutant constructs showed a poor co-localization with giantin, the Golgi marker (Fig. 4, C and D). These results indicate that the TrkA mutants are retained in the ER, although some pools of C300stop may reach some regions of the Golgi (Fig. 4D).
FIGURE 4.

TrkA CIPA mutants are retained in the ER. A, TrkA mutants (red) co-localization with calnexin (green) in transfected HeLa cells. DAPI nuclear staining and merge of three channels are shown. B, Pearson's coefficient to assess the co-localization of TrkA-wt and mutants with calnexin. C, TrkA mutants (red) co-localization with giantin (green) in transfected HeLa cells. DAPI nuclear staining and merge of three channels are shown. D, Pearson's coefficient to assess the co-localization of TrkA-wt and mutants with giantin. Error bars are S.E.; *, p < 0.05; **, p < 0.01; ***, p < 0.001. t test compared with wt.
For the case of TrkA-wt we observed that TrkA-wt overexpression induces Golgi fragmentation (Fig. 4C), supporting earlier reports (41). In this case the co-localization analysis is difficult. Although analyzed in the whole cell most of TrkA-wt is not co-localized with giantin (Fig. 4D), some TrkA-wt pools showed evidence of co-localization with giantin (yellow dots in Fig. 4C) promoting the Golgi fragmentation by autoactivation of TrkA, as previously reported (41).
CIPA Mutations Induce TrkA Misfolding and Protein Aggregation
ER retention is usually caused by protein misfolding. To prove misfolding we incubated a Triton X-100-soluble fraction of TrkA-wt and the mutants with an increasing concentrations of trypsin. The sensitivity to trypsin digestion is indicative of local or global misfolding. TrkA-wt digestion with trypsin is shown in Fig. 5A. We observed that all mutations induced a different protein band pattern in SDS-PAGE in comparison to the TrkA-wt. This indicates a different accessibility to the protease digestion suggesting a local or global misfolding (Fig. 5A).
FIGURE 5.
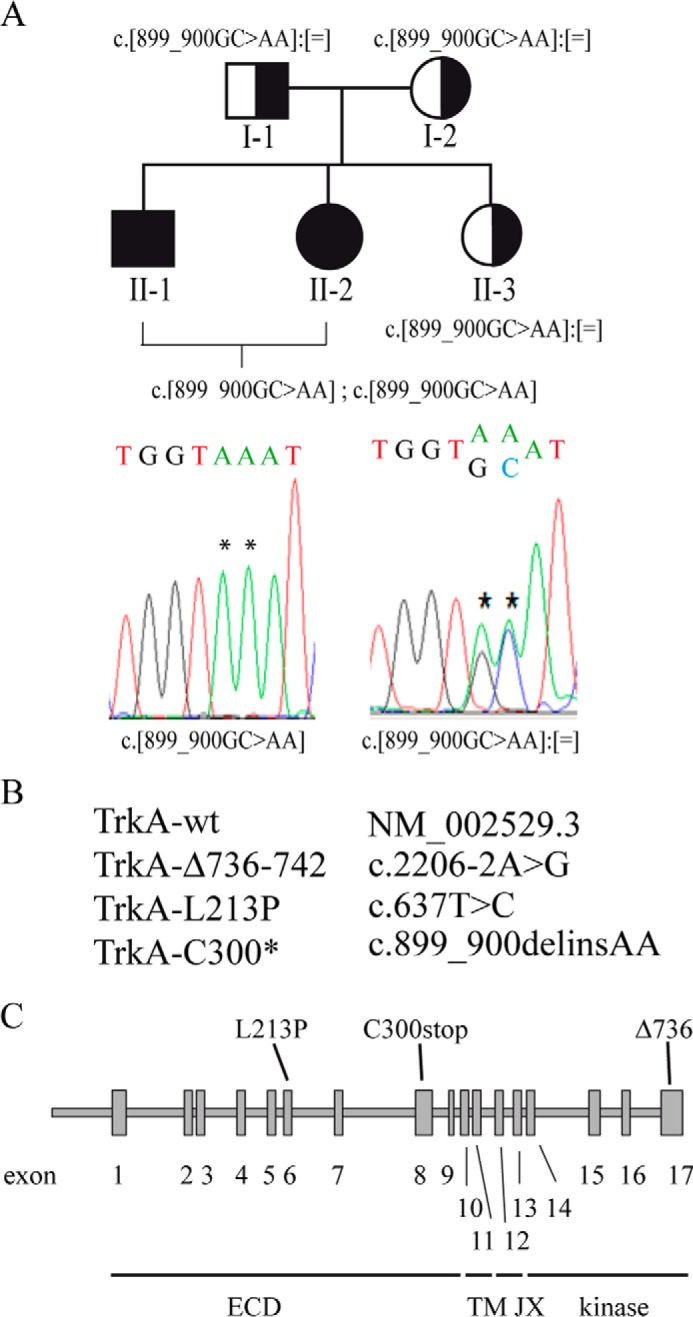
TrkA CIPA mutations induce protein misfolding and aggregation. A, trypsin digestion of Triton X-100-soluble fractions of TrkA mutants. Samples were incubated with the indicated concentrations of trypsin during 10 min on ice, quenched by adding SDS-PAGE sample buffer (2 times), boiled, and analyzed by SDS-PAGE/immunoblot with a specific antibody against the extracellular region of TrkA. B, analysis of disease protein solubility in the non-ionic detergent Triton X-100. Cell extracts were prepared in lysis buffer supplemented with 1% Triton X-100. Total, pellet, and supernatant were analyzed by SDS-PAGE and immunoblotting. C, the ratio of pellet to total (pellet/total) was quantified for two independent experiments. Error bars are S.E. WB, Western blot.
Protein misfolding may induce the exposition of hydrophobic residues to the protein surface promoting protein aggregation. We evaluated if the mutations on TrkA promote the formation of insoluble aggregates (44). Transfected HeLa cells with the different TrkA constructs were lysed with buffer containing the non-ionic detergent Triton X-100, and after centrifugation the pellet was treated with SDS-PAGE buffer containing 1% SDS and β-mercaptoethanol (Fig. 5B). TrkA-Δ736 and TrkA-C300stop are recovered in the Triton X-100-soluble fraction (Fig. 5B). However, TrkA-L213P is mainly found in the pellet, suggesting an aggregated state (Fig. 5, B and C). Due to Leu-213 residue localization in the middle of a β-sheet in the TrkA extracellular domain, the L213P mutation probably induces a structural change in that region and a subsequent aberrant aggregation of TrkA. The L213P mutation will probably induce a structural change in that region, potentially inducing an aberrant aggregation of TrkA. Some minor forms of TrkA-wt were also found in the aggregated pellet, suggesting the existence of off-pathways in the folding of TrkA when overexpressed.
TrkA Mutants Are Differentially Sensitive to Proteasome and Autophagy Inhibitors
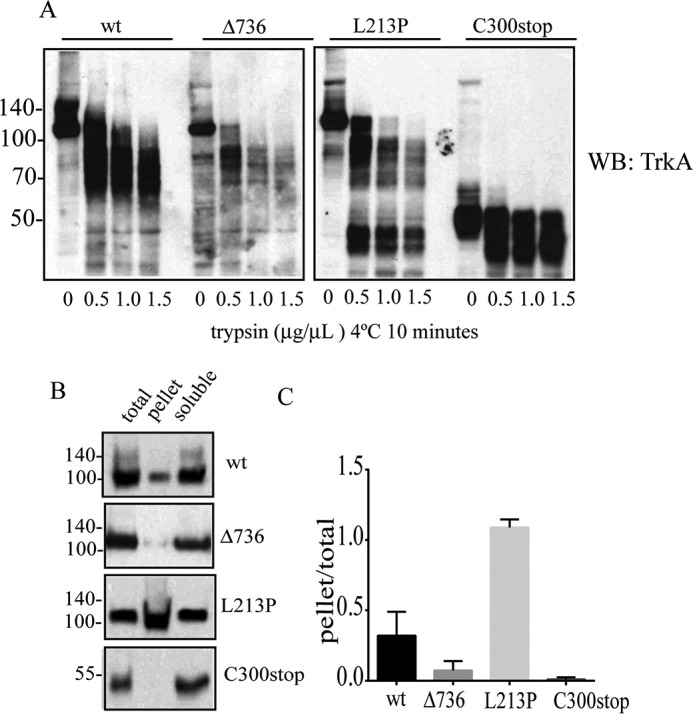
Misfolded proteins retained in the ER are targeted to degradation by an ER quality control (11). We determined the half-life times of the mutant proteins by a cycloheximide chase experiment (Fig. 6A). We found that TrkA-L213P (t½ = 26 h) is degraded to a slower rate than TrkA-wt (t½ = 4.5 h) suggesting a long-lived protein, probably due to the formation of aggregates. TrkA-C300stop (t½ = 0.7 h) is degraded faster than TrkA-wt, indicating a very unstable conformer (Fig. 6B) and TrkA-Δ736 (t½ = 6.5 h) is degraded to a similar rate as TrkA-wt (Fig. 6B). We then tested the degradation pathways of TrkA mutants using inhibitors of the proteasome or the autophagy (Fig. 6C). HeLa cells were transfected with the constructs that encode the different mutant TrkA, and treated with: epoxomycin, a proteasome inhibitor; wortmannin, an inhibitor of autophagosome formation; rapamycin, an inducer of autophagy; or compound E, an inhibitor of the γ-secretase complex (Fig. 6, C and D). All TrkA constructs showed resistance to degradation in the presence of wortmannin, indicating they are degraded mainly by the autophagy/lysosome pathway (Fig. 6, C and D). However, epoxomycin had a significant effect in the accumulation of the mutant TrkA-Δ736, suggesting that some pools of this mutant are also degraded by the proteasome. TrkA-L213P is a long-lived protein and even after 9 h some pools were still present implying a resistance of this construct to be degraded or a dysfunction of the degradation pathways in these cells.
FIGURE 6.
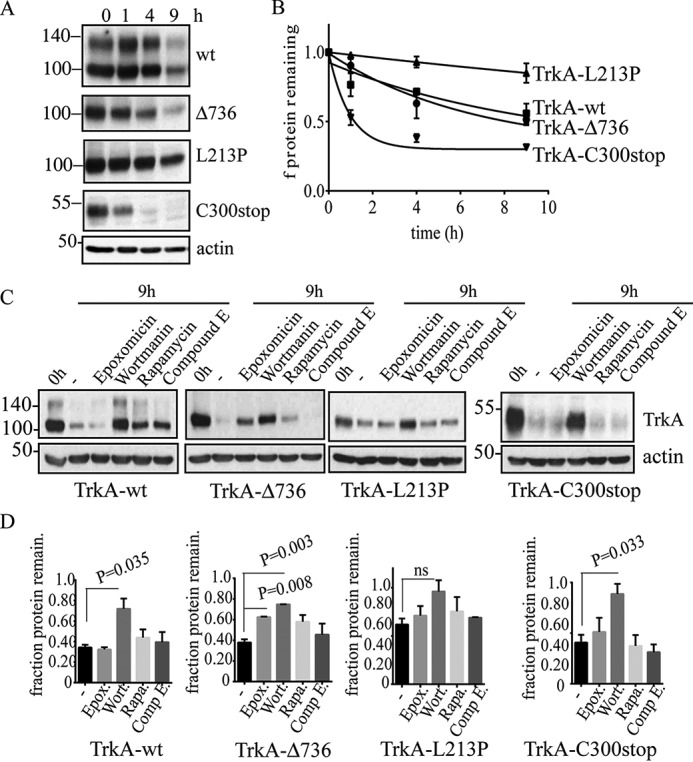
TrkA constructs have different degradation kinetics and differential sensitivity to proteasome and lysosome inhibitors. A, Western immunoblots of a representative experiment showing the levels of TrkA after protein translation inhibition with cycloheximide during the indicated time points. B, protein turnover half-lives of TrkA mutants were determined by quantifying the remaining levels of TrkA protein normalized to the actin blot. Three independent experiments consisting of independent transfections were performed. Data points were adjusted to a one exponential decay curve (black line) using GraphPad Prism software to calculate the half-lives times of each TrkA construct. C, Western blot analysis of cycloheximide treatment for 9 h and the sensitivity of the different TrkA constructs to the indicated inhibitors. D, fraction of the total protein remaining after 9 h. The values were normalized to time 0 h (1.0). Quantification of at least three independent experiments was quantified. Errors bars are S.E. p values are shown on the graph. t test compared with the control (−). ns, not significant.
TrkA-L213P Enhance the Formation of Autophagic Vesicles
HeLa cells were transfected with the TrkA mutants and with a plasmid encoding the fusion between microtubule-associated protein light chain 3 (LC3) and GFP, LC3-GFP, to evaluate the formation of autophagosome vesicles (AVs). Green fluorescent AVs were observed in the cells from all constructs (Fig. 7A). We did not observe a significant difference in the number of autophagosomes between TrkA-wt and Δ736 and C300stop constructs (Fig. 7B). However, we observed a significant increase in the number of GFP-positive AVs into the cells transfected with L213P (Fig. 7, A and B). The presence of large and abundant autophagosomes could be caused by a disruption of the autophagy flux, due to enhanced autophagosome synthesis or reduced autophagosome turnover (45). To test if L213P is influencing the autophagic flux we co-transfected HeLa cells with L213P and LC3-GFP and stimulated them with wortmannin, which prevents autophagosome formation, and with Bafilomycin A1 (BafA1), which inhibits the fusion between lysosomes and the autophagosome (Fig. 7C). Wortmannin treatment significantly reduced the number of AVs (Fig. 7, C and D), suggesting an efficient degradation of the AVs in L213P-transfected cells. This together with the observation that BafA1 induces the accumulation of more AVs (Fig. 7, C and D) suggests that L213P is not disrupting the degradation of the autolysosomes per se but that L213P enhances the formation of AVs disrupting the normal autophagy flux saturating their degradation and leading to an aberrant accumulation of AVs in the L213P cells.
FIGURE 7.

TrkA CIPA mutants induce the autophagic flux. A, HeLa cells were co-transfected with mock vector and the indicated TrkA constructs with LC3-GFP. A composite of confocal microscopy images for the channel green (LC3-GFP) and red (TrkA) is shown. B, the presence of green puncta indicative of AVs was quantified and plotted as the number of AVs per cell. C, HeLa cells co-transfected with TrkA-L213P and LC3-GFP were mock, wortmannin, and bafilomycin stimulated for 6 h, fixed, and analyzed by confocal microscopy. The number of green puncta of at least 20 cells was plotted in D. Error bars are S.E. *, p < 0.05; ** p < 0.01; ***, p < 0.001. One-way analysis of variance test with Tukey's multiple comparisons test (B) and t test (D) was compared with control.
TrkA Mutants Causing CIPA Induce Different Levels of Cell Toxicity
Given that a delayed degradation, an accumulation of misfolded proteins in the ER, and disruption of the autophagic flux can trigger cell toxicity, we examined whether the expression of TrkA mutants induced cell toxicity (Fig. 8). To evaluate this possibility we first transfected PC12nnr5 cells with all the TrkA constructs and quantified cell death. To increase ER stress and further challenge the cells, we added 2 μg/ml of tunicamycin. Tunicamycin induces ER stress by blocking glycosylation and ER retention. The addition of tunicamycin induces a faster migration in the SDS-PAGE of all TrkA constructs, indicating impairment in the glycosylation pathway (Fig. 8A). Cell death assays analyzed by flow cytometry revealed that TrkA-L213P and, to a lower extent, TrkA-Δ736, mutations increased basal cell toxicity, compared with cells transfected with constructs encoding TrkA-wt and TrkA-C300stop (Fig. 8B). The presence of tunicamycin significantly increased cell death caused by the expression of TrkA-L213P (p = 0.015, n = 3) and TrkA-Δ736 (p = 0.003, n = 3) (Fig. 8B). These results suggest that TrkA mutations causing CIPA differently increase the toxicity and vulnerability of cells to ER stress-induced cell death, pointing to mutation-specific cell toxicity.
FIGURE 8.
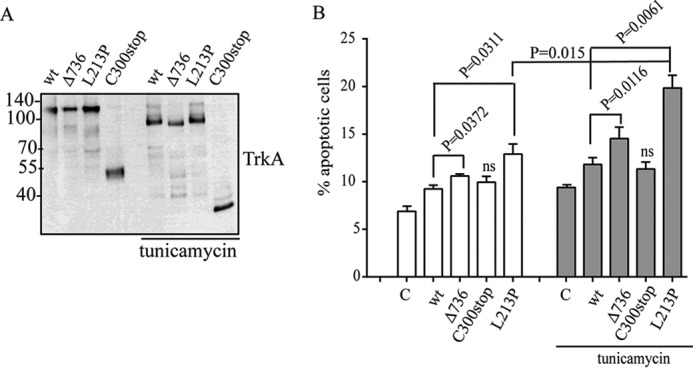
Cell toxicity of TrkA CIPA mutations in PC12nnr5 cells. Tunicamycin induces inhibition of protein glycosylation of all TrkA mutants. A, Western immunoblotting showing a decrease in the molecular weight due to inhibition of N-glycosylation of all the TrkA constructs upon tunicamycin treatment. B, percentage of PC12nnr5-transfected cells experimenting cell death (apoptotic cells) quantified by annexin V/propidium iodide. 2 μg/ml of tunicamycin were added to increase the basal ER stress. Three independent experiments were quantified. Error bars are S.E. p values are shown on the graph. t test compared with the control (C) is shown. ns, not significant.
TrkA CIPA Mutations Induce Dystrophic Neurites in Cortical Neurons
Autophagy has been reported to protect the neurons by eliminating dysfunctional organelles and aged aggregated proteins. We co-transfected TrkA-wt and L213P with LC3-GFP in mouse embryonic cortical neurons (E16–E17) and assessed for the presence of AVs (Fig. 9A). We found the presence of AVs in the dendrites and in the soma of neurons transfected with L213P mutant (Fig. 9A) but not in neurons expressing TrkA-wt (Fig. 9A). This supports the role of TrkA-L213P in enhancing the autophagy inducing an accumulation of AVs that may underlay neuronal toxicity.
FIGURE 9.
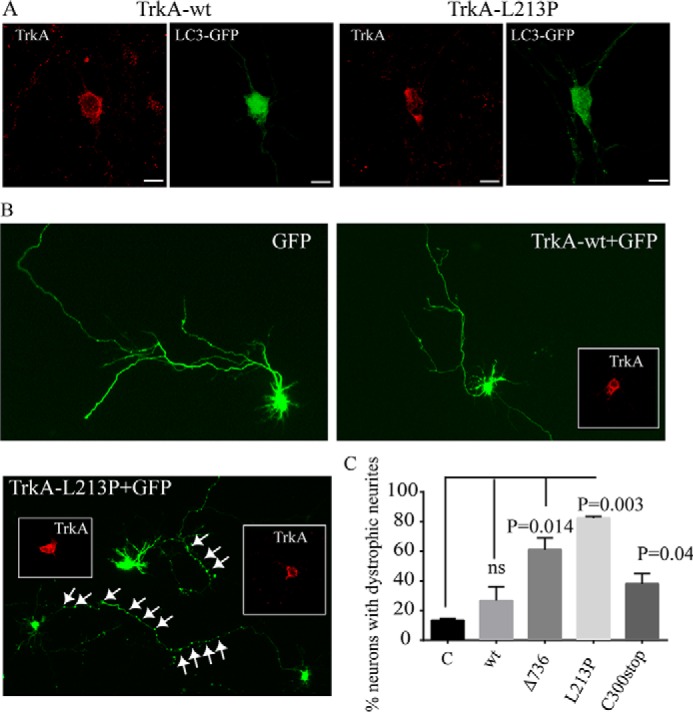
Presence of AVs and dystrophic neurites in L213P-transfected embryonic cortical neurons. A, embryonic cortical neurons were co-transfected with LC3-GFP and TrkA-wt or TrkA-L213P. 24 h post-transfection neurons were fixed and stained with TrkA antibody immunofluorescence (red). Several green positive AVs are observed in the soma and in the neurites of the neurons transfected with TrkA-L213P. B, E17 embryonic mouse cortical neurons were co-transfected with TrkA-wt and the CIPA mutants and GFP. Representative transfected neurons are shown. The presence of swollen regions in the neurites is indicated by white arrows. Inserts showed the TrkA staining. C, quantification of the percentage of transfected neurons bearing swollen regions. A positive neuron is counted if has more than 10 swollen regions in the axon or in the neurites. Error bars are S.E. p values are shown on the graph. t test compared with the control (C) is shown. ns, not significant.
We then co-transfected mouse embryonic cortical neurons with TrkA-wt and TrkA-L213P together with a vector encoding GFP. 48 h after transfection we noticed the presence of swelling neurites in the neurons expressing L213P and not in TrkA-wt (Fig. 9, B and C). Swollen neurites have been described as a sign of neurodegeneration and synaptic loss is some neurodegenerative diseases (46). Altogether this indicates that TrkA-L213P induce an increase in the formation of autophagosomes that causes cell toxicity and dystrophofic neurites in neurons.
Discussion
In this work we characterize three mutations in TrkA causing CIPA. We analyzed the subcellular localization, degradation half-live times, misfolding, aggregation, and cellular toxicity in cells and in primary culture of neurons. Our data suggest that distinct TrkA mutations causing CIPA induce different conformers with different fates and toxicities. The new CIPA mutation TrkA-C300stop forms soluble misfolded conformers that are rapidly cleared by autophagy. When overexpressed in neurons TrkA-C300stop induces mild toxic effects. TrkA-L213P mutation induces TrkA misfolding, and retention in the ER. Some pools of TrkA-L213P form Triton X-100-insoluble aggregates and a delayed degradation. When expressed in cells this mutant significantly increased the formation of autophagic vesicles inducing swollen regions in neurons and sensitized PC12nnr5 cells to cell death induced by tunicamycin. The mutant TrkA-Δ736 shows an intermediate phenotype between C300stop and L213P.
Misfolding could potentially affect not only the specific function of the protein but it could also lead to aggregation and toxicity. Accumulation of misfolded proteins in the ER is known to induce ER stress, which has also been implicated in human disease, including several neurodegenerative conditions (47–49). Our data suggest that some TrkA mutations increase the autopahgic flux and this promotes toxicity and neurodegeneration. Disruption of the autophagy homeostasis can lead to cellular dysfunction and diseases. Moreover, excessively deregulated autophagy can also cause neurodegeneration. In CIPA the absence of pain sensation is due to the lack of nociceptors neurons by disruption of TrkA/NGF signaling during embryonic development. However, the molecular mechanism by which CIPA patients present intellectual disability, a problem that must be localized in the central nervous system, is still unknown. TrkA is expressed in the cholinergic neurons from the basal forebrain during brain development and in adulthood and probably in other neuron populations of the CNS. As TrkA mutations induce different levels of cell toxicity, we suggest they may play a role in exacerbating some of the symptoms of CIPA patients in the central nervous system like mental retardation.
We think that depending of the type of mutation in TrkA, a higher or lower toxicity is induced in the population of neurons that expressed TrkA in the CNS and this may provide an explanation to the variable mental retardation or ID seen in CIPA patients. The patient with the mutation C300stop described here has CIPA but according to his clinical history is mentally healthy. The mutation Δ736 was described by our group (Dr. Garcia-Barcina) in a girl that at that time (2011) was mentally healthy, but because our study suggest that Δ736 induce some toxicity in neurons a follow-up of her cognitive abilities are needed to validate if some neurodegeneration will appear in the future. TrkA-L213P was first described in 2001 in a boy but no clinical data about his mental status was reported (35, 36, 50). In 2003 another boy with CIPA carrying the same mutation, L213P, was diagnosed with speech problems indicative of mild ID (38), which agrees with a stronger toxicity in the neurons expressing L213P.
In addition, our data suggest that the expression of toxic aggregates of TrkA in the neurons could be detrimental to its survival or could sensitize them to a neurodegenerative process promoted by normal aging. Recently a study in Arab-Bedouins children addressed the ID issue in CIPA (51). A cohort of 22 children (2–21 years old) with CIPA, carrying the same TrkA mutation (1926-ins-T) and 19 age-matched healthy children were assessed and documented for cognitive and adaptive behaviors (51). The results showed an inverse correlation between intelligent quotient (IQ) and age among children with CIPA; the older the child with CIPA, the lower the IQ score deterioration. We think this may reflect a neurodegenerative process in the CNS caused by the TrkA mutation during brain maturation. However, an analysis with more mutations and a standardized value of ID is needed to fully validate this hypothesis.
Finally, several works have linked TrkA activity to enhanced autophagy and cell death (24–29). Although we used small amounts of TrkA in our transfections, we observed that overexpression of TrkA-wt is able to increase the autophagic flux to levels that are toxic to HeLa cells (data not shown). This finding is supported by early work demonstrating that TrkA-induced cell death is dose-dependent (52) and that TrkA overexpression is able to trigger autophagy and apoptosis in glioblastoma (26) and in neuroblastoma cells (31), cancer cell lines where TrkA overexpression is of good prognosis. Furthermore, in medulloblastoma Daoy cells overexpression of TrkA induced cell death by micropinocytosis (28). Because in some of these systems the autophagy is activated in a ligand independent manner (30, 31, 53) or by TrkA inactivating mutations (this study) no kinase activity is necessary. One possibility is that TrkA protein by itself would be a specific target for autophagy receptors and able to nucleate the autophagosome formation. In this sense p62/sequestosome 1, one of the best characterized autophagy receptors (54), binds to TrkA and participates in TrkA degradation (55–57).
In summary our analysis indicates that different mutations in TrkA have different outcomes and this may provide an explanation for the variable phenotype seen in the CNS of CIPA patients. Although we do see significant differences between TrkA-wt and TrkA-L21P phenotypes, our studies are carried out by overexpressing the different constructs. We consider that generation of a mouse model of CIPA expressing endogenous levels of TrkA mutants is necessary to validate our hypothesis.
Experimental Procedures
Cell Lines
HeLa cells, a cell line derived from humans, were cultured in DMEM (Gibco) containing 10% fetal calf serum (ThermoFisher). PC12 and PC12nnr5 cells were cultured in DMEM with 10% FBS and 5% horse serum. All cell lines were cultured at 37 °C in a humidified atmosphere with 5% of CO2.
Identification of the New NTRK1 Mutation pCys300stop
Written informed consent was obtained from the patient and all family members participating in this study. This research work was approved by the Ethical Committee of Basurto University Hospital, conforming to Helsinki Declaration. Blood samples were collected from all the family members available. Genomic DNA was purified using a standard “salting-out” purification protocol and its quality was assessed using a spectrophotometer. Mutation screening was performed in the index case. Primers for PCR amplification were designed for all 17 exons and intron-exon boundaries of the longest NTRK1 isoform (NM_002529.3) using the Primer3 on-line application. Primer sequences and optimal annealing temperature for each primer pair are available upon request. All PCR were carried out using TaqGold DNA polymerase (Applied Biosystems) with 5% DMSO (Sigma). Direct sequencing of both strands of the amplified DNA fragments was performed using BigDye Terminator version 3.1 Sequencing kit (Applied Biosystems). Sequencing reactions were resolved on ABI PRISM 3130 Genetic Analyzer and analyzed using Sequence Scanner software (Applied Biosystems). The mutations described in this report are named following the recommendations from the Human Genome Variation Society.
In Vitro Site-directed Mutagenesis
To generate the constructs of the TrkA mutants a pair of selected oligonucleotides for each mutation was purchased from Sigma and the QuikChange mutagenesis kit (Agilent) was used following the commercial guidelines. Sequences of the oligonucleotides are as follows. For TrkA L213P, TrkA-L213Pfw, 5′-GGGGGATGACGTTTTTCCGCAGTGCCAGGTGGAGG-3′ and TrkA-L213P-rev, 5′-CCTCCACCTGGCACTGCGGAAAAACGTCATCCCCC-3′. For TrkA-C300STOP, a stop codon was introduced after the position Cys-300 of TrkA using the following pair of oligonucleotides; TrkA-C300fw, 5′-GGTGGAGATGCACCACTGGTAAATCCCCTTCTCTGTGG-3′ and TrkA-c300-rev, 5′-CCACAGAGAAGGGGATTTACCAGTGGTGCATCTCCACC-3′. For TrkA-Δ736, a deletion of seven residues in-frame was created using the following pair of oligonucleotides: TrkA-del736fw, 5′-TGGTACCAGCTCTCCAACACTGAGGGCCGGGAGCTGGAGCGG-3′ and TrkA-del736-rev, 5′-CCGCTCCAGCTCCCGGCCCTCAGTGTTGGAGAGCTGGTACCA-3′. The plasmid DNA was sequenced using the local genomic facilities of ISCIII.
Stimulation of Cells with NGF
The mutant or the wild-type TrkA-transfected cells and mock-transfected cells as well as non-transfected cells were treated under the same conditions in a six-well tissue culture plate. The cells were washed three times with serum-free medium and incubated for 2 h in a serum-free medium. After the medium was aspirated, 1 ml of fresh serum-free medium containing 50 ng of NGF (Alomone) was followed by incubation at 37 °C at the different time points. The cells were then immediately washed three times with phosphate-buffered saline and lysed using TNE buffer (Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA) supplemented with Triton X-100 (Sigma) and a mixture of protease inhibitors (Roche Applied Science) and phosphatase-like sodium orthovanadate (Na3VO4, Sigma) and sodium fluoride (NaF) (Sigma). Cells were harvested by scraping, transferred into a 1.5-ml tube, and collected by centrifuging at 12,000 × g for 5 min in a microcentrifuge. Supernatant was analyzed by SDS-PAGE or processed as indicated.
Immunoblotting
Primary antibodies used were as follows: a rabbit polyclonal antibody against TrkA (dilution 1:3000, 06-574 from Millipore) and two phospho-specific antibodies (TrkA(Tyr-490) 4619, dilution 1:1000, and TrkA(Tyr-674/675) 4621 dilution 1:1000 from Cell Signaling). Polyclonal anti-TrkA was used to directly detect total TrkA protein. The latter two phospho-specific antibodies were prepared to detect phosphorylated Tyr-490 and Tyr-674/675 residues. The membrane was incubated with the primary antibody diluted in TBS + 0.1% Tween 20 (Sigma) overnight at 4 °C. For detection we used ECL Western blotting detection reagents (Amersham Biosciences) and exposure to x-ray film.
Trypsin Digestion and Triton X-100 Solubility Assay
Cell pellets were lysed for 1 h at 4 °C in TNE buffer supplemented with 1% Triton X-100, pH 7.5. Protein concentrations of lysate were determined using the Bradford assay (Bio-Rad) and normalized across all samples. An aliquot of lysate was removed for the trypsin digestion assay. The remaining lysate was subjected to centrifugation at 20,000 × g for 15 min at 4 °C. Supernatant was removed and added to 67 μl of 4× SDS-sample buffer with 1.25% β-mercaptoethanol. Pellet was solubilized with the same volume of 2× SDS-sample buffer with 1.25% β-mercaptoethanol.
Trypsin digestion assays were performed by incubating the soluble lysate with three increasing concentrations of trypsin in PBS and on ice for 5 min. The reaction was stopped by adding 2× SDS-PAGE sample buffer and boiled for 10 min.
Cell Surface Biotinylation Assay
Transfected cells with the indicated TrkA constructs were washed sequentially using room temperature PBS and cold PBS, chilled on ice, and incubated in 0.5 μg/ml of Sulfo-NHS-SS-biotin (Pierce) dissolved in biotinylation buffer (PBS, 1 mm CaCl2, 0.5 mm MgCl2) for 20 min at 4 °C to label the membrane proteins. Free biotin was quenched with 0.1 m glycine for 15 min at 4 °C. Cells were then washed twice with cold PBS and lysed as indicated above. Biotinylated proteins were isolated from the total cell lysate by immobilization on NeutroAvidin beads (Pierce) overnight at 4 °C. Beads were washed three times with lysis buffer and 20 μl of 2× SDS sample buffer was added before boiling for 7 min. Proteins were subjected to SDS-PAGE and immunoblotted with HA antibodies to detect TrkA.
Cycloheximide Treatment and Half-life Calculations
HeLa cells were transfected with 1 μg of empty vector (control) or the indicated TrkA mutants. 48 h post-transfection, cells in a 6-well plate were incubated with 5 μg/ml of cycloheximide in the presence and absence of 15 μm chloroquine or 10 μm epoxomycin. Cells were harvested in TNE lysis buffer t = 0, 1, 4, and 9 h post-cycloheximide. The half-lives were calculated using the values obtained from densitometry of Western blots using ImageQuant (Molecular Dynamics) software. Values were fit to the half-life decay equation using GraphPad Prism software to an exponential regression of the form: N(t) = N(0 h) × e−λt. λ is the decay constant. Half-lives (t½) were calculated using the equation t½ = ln(2)/λ.
Confocal Microscopy
HeLa cells were grown and transfected on sterile coverslips. Thirty-six h post-transfection, coverslips were removed from media and fixed with 4% paraformaldehyde. Coverslips were washed 3 times in PBS, and 100 μl of blocking buffer (PBS with 1% FBS) were added on coverslips for 1 h. Blocking buffer was aspirated and replaced with primary antibody (rabbit anti-calnexin C5C9, number 2679 from Cell Signaling, dilution 1:500; rabbit anti-giantin number ab80864 from Abcam, dilution 1:1000; and mouse anti-HA antibody number H3663 from Sigma, dilution 1:2000) diluted in blocking buffer for 1 h. Primary antibody was then aspirated, coverslips were washed three times, and secondary antibody (anti-mouse Ig Alexa 555, number A31570 Invitrogen, anti-rabbit Ig Alexa 488 number A21206, Invitrogen) diluted in blocking buffer with DAPI stain was added. After 1 h, the solution was aspirated and coverslips were washed three times with PBS, and mounted onto slides using mounting media. Slides were allowed to dry in the dark at room temperature for at least 12 h before imaging. Cells were imaged using a Leica SP5 spectral confocal microscope. Pearson's coefficient of co-localization was calculated using Leica software with at least five different cells from each experiment.
Cell Viability and Apoptosis Assay
PC12nnr5 were transfected with the indicated constructs. 48 h after transfection, cells were lifted in PBS and analyzed by annexin V/propidium iodide apoptosis assay by flow cytometry. Propidium iodide and annexin V-FITC were used to discriminate between apoptotic and necrotic cells. According to the manufacturer's instructions, cells were incubated with the reagent for 20 min at room temperature in the dark and then analyzed via flow cytometry using a MACSQuant Analyzer cytometer (Miltenyi Biotec).
Embryonic Cortical Neurons Culture Methods
E16–E17 mice cortex was dissected. Briefly whole cortices from three embryos were collected in a Petri dish containing Hanks' balanced salt solution. After carefully removing the meninges, the tissue was divided into cortical hemispheres, dissected, and the non-cortical structures were removed. Then cortical tissue was digested in 1 ml of 2.5 mg/ml of trypsin and 0.5 ml of 200 units/ml of DNase I, incubated for 10 min at 37 °C and turned twice. The supernatant was removed and after that, the pellet was suspended in 0.5 ml of 4% BSA, 1 ml of NB/B-27. The solutions must be cold all the time. The tissue was disaggregated with a pipette until tissue was completely dispersed. 2.5 ml of 4% BSA was added in the bottom of the tube and the dissociated cell suspension was centrifuged at 200 × g for 5 min. The supernatant was replaced with 5 ml of 0.2% BSA in PBS. The cell suspension was passed through a 40-μm nylon filter. Viable cells were counted using trypan blue. After cells were counted, cells suspension was centrifuged again at 200 × g for 5 min and the supernatant replaced with plating medium (NB medium with B-27 supplement and 2 mm l-glutamine and 0.5% penicillin-streptomycin). Isolated cells were seeded onto cover slides of 24-well plates pre-coated with poly-d-lysine (100 μg/ml) and laminin (5 μg/ml) at a density of 250,000 cells/well (2 × 105 cell/cm3). Neurons were allowed to adhere and recover. On the second day, half of the medium was replaced with fresh NB, B27, and 2 μm AraC. 4 days after, the cells were transfected.
Author Contributions
M-L. F., C. M., P. A., I. C-B., A. L., E. S., M. G-B., and M. V. designed and performed the experiments. E. S. and M. G-B. sequenced and identified the new mutation Cys300stop in a CIPA patient. M. V. designed and supervised the entire project and wrote the manuscript with the supervision of all the authors.
Acknowledgments
We acknowledge Dr. J. M. Rojas for the GFP-ras construct, Dr. F. Gonzalez the help in the confocal microscopy, Dr. M. Llovera for PC12nnr5 cells, and Dr. Y. Campos for the bafilomycin A1 reagent.
This work was supported in part by the Spanish Minister of Economy and Competitiveness Grants BFU2013/42746-P (to M. V.) and TPY-M-1068/13 (to A. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- CIPA
- congenital insensitivity to pain with anhidrosis
- ID
- intellectual disability
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- ENDO-H
- endoglycosidase-H
- LC-3
- light chain 3
- AV
- autophagosome vesicle.
References
- 1. Indo Y. (2010) Nerve growth factor, pain, itch and inflammation: lessons from congenital insensitivity to pain with anhidrosis. Expert Rev. Neurother. 10, 1707–1724 [DOI] [PubMed] [Google Scholar]
- 2. Indo Y. (2002) Genetics of congenital insensitivity to pain with anhidrosis (CIPA) or hereditary sensory and autonomic neuropathy type IV. Clinical, biological and molecular aspects of mutations in TRKA(NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Clin. Auton. Res. 12, I20–32 [DOI] [PubMed] [Google Scholar]
- 3. Sztriha L., Lestringant G. G., Hertecant J., Frossard P. M., and Masouyé I. (2001) Congenital insensitivity to pain with anhidrosis. Pediatr. Neurol. 25, 63–66 [DOI] [PubMed] [Google Scholar]
- 4. Indo Y., Tsuruta M., Hayashida Y., Karim M. A., Ohta K., Kawano T., Mitsubuchi H., Tonoki H., Awaya Y., and Matsuda I. (1996) Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 13, 485–488 [DOI] [PubMed] [Google Scholar]
- 5. Lallemend F., and Ernfors P. (2012) Molecular interactions underlying the specification of sensory neurons. Trends Neurosci. 35, 373–381 [DOI] [PubMed] [Google Scholar]
- 6. Marmigère F., Montelius A., Wegner M., Groner Y., Reichardt L. F., and Ernfors P. (2006) The Runx1/AML1 transcription factor selectively regulates development and survival of TrkA nociceptive sensory neurons. Nat. Neurosci. 9, 180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Y., and Ma Q. (2011) Generation of somatic sensory neuron diversity and implications on sensory coding. Curr. Opin. Neurobiol. 21, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petruska J. C., and Mendell L. M. (2004) The many functions of nerve growth factor: multiple actions on nociceptors. Neurosci. Lett. 361, 168–171 [DOI] [PubMed] [Google Scholar]
- 9. Indo Y. (2001) Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Hum. Mutat. 18, 462–471 [DOI] [PubMed] [Google Scholar]
- 10. Merulla J., Fasana E., Soldà T., and Molinari M. (2013) Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic 14, 767–777 [DOI] [PubMed] [Google Scholar]
- 11. Brodsky J. L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., and Ye Y. (2012) Roles of p97-associated deubiquitinases in protein quality control at the endoplasmic reticulum. Curr. Protein Pept. Sci. 13, 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Araki K., and Nagata K. (2011) Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 3, a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Määttänen P., Gehring K., Bergeron J. J., and Thomas D. Y. (2010) Protein quality control in the ER: the recognition of misfolded proteins. Semin. Cell Dev. Biol. 21, 500–511 [DOI] [PubMed] [Google Scholar]
- 15. Vembar S. S., and Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Senft D., and Ronai Z. A. (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 40, 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ogata M., Hino S., Saito A., Morikawa K., Kondo S., Kanemoto S., Murakami T., Taniguchi M., Tanii I., Yoshinaga K., Shiosaka S., Hammarback J. A., Urano F., and Imaizumi K. (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuervo A. M., Bergamini E., Brunk U. T., Dröge W., Ffrench M., and Terman A. (2005) Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 1, 131–140 [DOI] [PubMed] [Google Scholar]
- 19. Schneider J. L., and Cuervo A. M. (2014) Autophagy and human disease: emerging themes. Curr. Opin. Genet. Dev. 26, 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morimoto R. I., and Cuervo A. M. (2014) Proteostasis and the aging proteome in health and disease. J. Gerontol. A Biol. Sci. Med. Sci. 69, S33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuervo A. M., and Wong E. (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frake R. A., Ricketts T., Menzies F. M., and Rubinsztein D. C. (2015) Autophagy and neurodegeneration. J. Clin. Invest. 125, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka K., and Matsuda N. (2014) Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 1843, 197–204 [DOI] [PubMed] [Google Scholar]
- 24. Bodnarchuk T. W., Napper S., Rapin N., and Misra V. (2012) Mechanism for the induction of cell death in ONS-76 medulloblastoma cells by Zhangfei/CREB-ZF. J. Neurooncol. 109, 485–501 [DOI] [PubMed] [Google Scholar]
- 25. Dadakhujaev S., Jung E. J., Noh H. S., Hah Y. S., Kim C. J., and Kim D. R. (2009) Interplay between autophagy and apoptosis in TrkA-induced cell death. Autophagy 5, 103–105 [DOI] [PubMed] [Google Scholar]
- 26. Hansen K., Wagner B., Hamel W., Schweizer M., Haag F., Westphal M., and Lamszus K. (2007) Autophagic cell death induced by TrkA receptor activation in human glioblastoma cells. J. Neurochem. 103, 259–275 [DOI] [PubMed] [Google Scholar]
- 27. Kaasinen S. K., Harvey L., Reynolds A. J., and Hendry I. A. (2008) Autophagy generates retrogradely transported organelles: a hypothesis. Int. J. Dev. Neurosci. 26, 625–634 [DOI] [PubMed] [Google Scholar]
- 28. Li C., Macdonald J. I., Hryciw T., and Meakin S. O. (2010) Nerve growth factor activation of the TrkA receptor induces cell death, by macropinocytosis, in medulloblastoma Daoy cells. J. Neurochem. 112, 882–899 [DOI] [PubMed] [Google Scholar]
- 29. Mnich K., Carleton L. A., Kavanagh E. T., Doyle K. M., Samali A., and Gorman A. M. (2014) Nerve growth factor-mediated inhibition of apoptosis post-caspase activation is due to removal of active caspase-3 in a lysosome-dependent manner. Cell Death Dis. 5, e1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jung E. J., and Kim D. R. (2011) Ectopic expression of H2AX protein promotes TrkA-induced cell death via modulation of TrkA tyrosine-490 phosphorylation and JNK activity upon DNA damage. Biochem. Biophys. Res. Commun. 404, 841–847 [DOI] [PubMed] [Google Scholar]
- 31. Jung E. J., and Kim D. R. (2008) Apoptotic cell death in TrkA-overexpressing cells: kinetic regulation of ERK phosphorylation and caspase-7 activation. Mol. Cells 26, 12–17 [PubMed] [Google Scholar]
- 32. Geetha T., Jiang J., and Wooten M. W. (2005) Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 20, 301–312 [DOI] [PubMed] [Google Scholar]
- 33. Arévalo J. C., Waite J., Rajagopal R., Beyna M., Chen Z. Y., Lee F. S., and Chao M. V. (2006) Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50, 549–559 [DOI] [PubMed] [Google Scholar]
- 34. Sarasola E., Rodríguez J. A., Garrote E., Arístegui J., and García-Barcina M. J. (2011) A short in-frame deletion in NTRK1 tyrosine kinase domain caused by a novel splice site mutation in a patient with congenital insensitivity to pain with anhidrosis. BMC Med. Genet. 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mardy S., Miura Y., Endo F., Matsuda I., and Indo Y. (2001) Congenital insensitivity to pain with anhidrosis (CIPA): effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum. Mol. Genet. 10, 179–188 [DOI] [PubMed] [Google Scholar]
- 36. Mardy S., Miura Y., Endo F., Matsuda I., Sztriha L., Frossard P., Moosa A., Ismail E. A., Macaya A., Andria G., Toscano E., Gibson W., Graham G. E., and Indo Y. (1999) Congenital insensitivity to pain with anhidrosis: novel mutations in the TRKA (NTRK1) gene encoding a high-affinity receptor for nerve growth factor. Am. J. Hum. Genet. 64, 1570–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miranda C., Di Virgilio M., Selleri S., Zanotti G., Pagliardini S., Pierotti M. A., and Greco A. (2002) Novel pathogenic mechanisms of congenital insensitivity to pain with anhidrosis genetic disorder unveiled by functional analysis of neurotrophic tyrosine receptor kinase type 1/nerve growth factor receptor mutations. J. Biol. Chem. 277, 6455–6462 [DOI] [PubMed] [Google Scholar]
- 38. Bonkowsky J. L., Johnson J., Carey J. C., Smith A. G., and Swoboda K. J. (2003) An infant with primary tooth loss and palmar hyperkeratosis: a novel mutation in the NTRK1 gene causing congenital insensitivity to pain with anhidrosis. Pediatrics 112, e237–241 [DOI] [PubMed] [Google Scholar]
- 39. Greene L. A., and Tischler A. S. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 73, 2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loeb D. M., Maragos J., Martin-Zanca D., Chao M. V., Parada L. F., and Greene L. A. (1991) The trk proto-oncogene rescues NGF responsiveness in mutant NGF-nonresponsive PC12 cell lines. Cell 66, 961–966 [DOI] [PubMed] [Google Scholar]
- 41. Schecterson L. C., Hudson M. P., Ko M., Philippidou P., Akmentin W., Wiley J., Rosenblum E., Chao M. V., Halegoua S., and Bothwell M. (2010) Trk activation in the secretory pathway promotes Golgi fragmentation. Mol. Cell. Neurosci. 43, 403–413 [DOI] [PubMed] [Google Scholar]
- 42. Watson F. L., Porcionatto M. A., Bhattacharyya A., Stiles C. D., and Segal R. A. (1999) TrkA glycosylation regulates receptor localization and activity. J. Neurobiol. 39, 323–336 [DOI] [PubMed] [Google Scholar]
- 43. Huang E. J., and Reichardt L. F. (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 44. Houck S. A., Ren H. Y., Madden V. J., Bonner J. N., Conlin M. P., Janovick J. A., Conn P. M., and Cyr D. M. (2014) Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol. Cell 54, 166–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barth S., Glick D., and Macleod K. F. (2010) Autophagy: assays and artifacts. J. Pathol. 221, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nixon R. A. (2014) Alzheimer neurodegeneration, autophagy, and Aβ secretion: the ins and outs (comment on DOI 10.1002/bies.201400002). Bioessays 36, 547. [DOI] [PubMed] [Google Scholar]
- 47. Matus S., Lisbona F., Torres M., León C., Thielen P., and Hetz C. (2008) The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr. Mol. Med. 8, 157–172 [DOI] [PubMed] [Google Scholar]
- 48. Doyle K. M., Kennedy D., Gorman A. M., Gupta S., Healy S. J., and Samali A. (2011) Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J. Cell. Mol. Med. 15, 2025–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gestwicki J. E., and Garza D. (2012) Protein quality control in neurodegenerative disease. Prog. Mol. Biol. Transl. Sci. 107, 327–353 [DOI] [PubMed] [Google Scholar]
- 50. Indo Y., Mardy S., Miura Y., Moosa A., Ismail E. A., Toscano E., Andria G., Pavone V., Brown D. L., Brooks A., Endo F., and Matsuda I. (2001) Congenital insensitivity to pain with anhidrosis (CIPA): novel mutations of the TRKA (NTRK1) gene, a putative uniparental disomy, and a linkage of the mutant TRKA and PKLR genes in a family with CIPA and pyruvate kinase deficiency. Hum. Mutat. 18, 308–318 [DOI] [PubMed] [Google Scholar]
- 51. Levy Erez D., Levy J., Friger M., Aharoni-Mayer Y., Cohen-Iluz M., and Goldstein E. (2010) Assessment of cognitive and adaptive behaviour among individuals with congenital insensitivity to pain and anhidrosis. Dev. Med. Child Neurol. 52, 559–562 [DOI] [PubMed] [Google Scholar]
- 52. Yan C., Liang Y., Nylander K. D., and Schor N. F. (2002) TrkA as a life and death receptor: receptor dose as a mediator of function. Cancer Res. 62, 4867–4875 [PubMed] [Google Scholar]
- 53. Jung E. J., and Kim D. R. (2010) Control of TrkA-induced cell death by JNK activation and differential expression of TrkA upon DNA damage. Mol. Cells 30, 121–125 [DOI] [PubMed] [Google Scholar]
- 54. Shaid S., Brandts C. H., Serve H., and Dikic I. (2013) Ubiquitination and selective autophagy. Cell Death Differ. 20, 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wooten M. W., Seibenhener M. L., Mamidipudi V., Diaz-Meco M. T., Barker P. A., and Moscat J. (2001) The atypical protein kinase C-interacting protein p62 is a scaffold for NF-κB activation by nerve growth factor. J. Biol. Chem. 276, 7709–7712 [DOI] [PubMed] [Google Scholar]
- 56. Geetha T., Seibenhener M. L., Chen L., Madura K., and Wooten M. W. (2008) p62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem. Biophys. Res. Commun. 374, 33–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wooten M. W., Geetha T., Babu J. R., Seibenhener M. L., Peng J., Cox N., Diaz-Meco M. T., and Moscat J. (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 283, 6783–6789 [DOI] [PubMed] [Google Scholar]