

Abstract
Rifampicin monooxygenase (RIFMO) catalyzes the N-hydroxylation of the natural product antibiotic rifampicin (RIF) to 2′-N-hydroxy-4-oxo-rifampicin, a metabolite with much lower antimicrobial activity. RIFMO shares moderate sequence similarity with well characterized flavoprotein monooxygenases, but the protein has not been isolated and characterized at the molecular level. Herein, we report crystal structures of RIFMO from Nocardia farcinica, the determination of the oligomeric state in solution with small angle x-ray scattering, and the spectrophotometric characterization of substrate binding. The structure identifies RIFMO as a class A flavoprotein monooxygenase and is similar in fold and quaternary structure to MtmOIV and OxyS, which are enzymes in the mithramycin and oxytetracycline biosynthetic pathways, respectively. RIFMO is distinguished from other class A flavoprotein monooxygenases by its unique middle domain, which is involved in binding RIF. Small angle x-ray scattering analysis shows that RIFMO dimerizes via the FAD-binding domain to form a bell-shaped homodimer in solution with a maximal dimension of 110 Å. RIF binding was monitored using absorbance at 525 nm to determine a dissociation constant of 13 μm. Steady-state oxygen consumption assays show that NADPH efficiently reduces the FAD only when RIF is present, implying that RIF binds before NADPH in the catalytic scheme. The 1.8 Å resolution structure of RIFMO complexed with RIF represents the precatalytic conformation that occurs before formation of the ternary E-RIF-NADPH complex. The RIF naphthoquinone blocks access to the FAD N5 atom, implying that large conformational changes are required for NADPH to reduce the FAD. A model for these conformational changes is proposed.
Keywords: enzyme kinetics, enzyme structure, flavoprotein, small-angle X-ray scattering (SAXS), x-ray crystallography
Introduction
Rifampicin (RIF)3 (Fig. 1) is a potent frontline antibiotic against tuberculosis and other mycobacterial infections, but extensive usage of RIF and its derivatives has contributed to bacterial resistance, which neutralizes antibiotic activity (1, 2). In addition to the point mutations in RNA polymerase that are responsible for resistance in mycobacteria (3, 4), some bacterial species, such as soil actinomycetes and parasitic bacteria, employ secondary enzyme-mediated inactivation mechanisms that chemically modify RIF to less active forms or degradation products (5).
FIGURE 1.

The reaction catalyzed by RIFMO.
At least four RIF-deactivating enzymes have been described: ADP-ribosyltransferase (Arr) (6), glycosyltransferase (Rgt) (7, 8), phosphotransferase (Rph) (8–11), and RIF monooxygenase (12, 13). Arr and Rgt act on a critical hydroxyl group (C23) located on the ansa aliphatic chain of RIF, whereas Rph adds a phosphate group to the C21 hydroxyl. These hydroxyls are important for antibiotic action because they hydrogen-bond to a conserved region of the β-subunit in RNA polymerase, which is the target of RIF (14). Covalent modification of RIF hydroxyls with ADP-ribose or phosphate results in high level resistance in Escherichia coli, such as a 64-fold increase in the RIF minimal inhibition concentration (6, 10). Modification of RIF by Rgt results in a 4-fold increase of Streptomyces speibonae cultures (7) and markedly reduces the activity against Gram-positive bacteria (8).
Little is known about the functions of RIF monooxygenase (RIFMO, also known as Rox). The enzyme was discovered from pathogenic Nocardia farcinica (15). Phylogenetic analysis suggests that RIFMO is distinguishable from other known flavoprotein monooxygenases (13). A highly similar gene (iri) that confers moderate RIF resistance was also found in Rhodococcus equi (12). RIFMO decreases the potency of RIF by converting it to 2′-N-hydroxy-4-oxo-rifampicin (RIF-O) (Fig. 1), which has a markedly higher minimal inhibition concentration against a spectrum of Gram-negative and Gram-positive bacteria (13). The addition of a hydroxyl group on the N2′ atom of the piperazine moiety, which is absent in other rifamycins, leads to decolorization of RIF (i.e. decomposition) in N. farcinica-containing media. Understanding the molecular basis of RIFMO and its mechanisms for substrate binding and catalysis is a key to overcoming RIFMO-mediated drug resistance. However, the three-dimensional structure of RIFMO has not been determined, and kinetic characterization of the purified enzyme has not been reported.
Herein we report two crystal structures of RIFMO along with characterization of the oligomeric state in solution and an assessment of RIF binding using spectroscopic methods. The structures identify RIFMO as a class A flavoprotein monooxygenase and provide a basis for studying the chemical mechanism of the enzyme.
Results
RIFMO Is a Flavoenzyme
Full-length RIFMO was expressed in E. coli and purified to homogeneity. The purified protein exhibited the intense yellow color that is characteristic of proteins containing an oxidized flavin cofactor. The UV-visible spectrum of the purified enzyme has absorbance maxima at 366 and 449 nm, with the latter peak having an associated shoulder near 470 nm (Fig. 2A). These features are indicative of a well folded flavoenzyme. These results confirm that RIFMO is a flavoenzyme, which is consistent with the prediction based on sequence analysis (13).
FIGURE 2.

UV-visible spectra and RIF binding data. A, UV-visible spectra of RIFMO-bound FAD (black), free FAD (blue), and RIFMO (30 μm) in the presence of 15 μm RIF (orange). B, UV-visible spectrum of 15 μm RIF (in 100 mm sodium phosphate buffer, pH 7.5) showing peak maxima at 236, 254, 334, and 475 nm. C, flavin spectra monitored as a function of increasing concentration of RIF (0–30 μm). The inset shows spectral differences after subtracting the spectrum of RIFMO with 0 μm RIF. D, determination of the KD value of RIF. The absorbance at 525 nm was plotted as a function of RIF concentration to determine a KD value of 13 ± 2 μm.
The addition of RIF to the enzyme alters the UV-visible spectrum, which suggests a binding event. The spectrum of RIF alone has maxima at 236, 254, 334, and 475 nm (Fig. 2B). The titration of the enzyme with increasing concentration of RIF results in increases in absorbance at several wavelengths (320, 360, 400, 443, and 525 nm) and an isosbestic point at ∼479 nm (Fig. 2C). The change in absorbance at 525 nm was monitored to estimate a dissociation constant for RIF binding to RIFMO of KD = 13 ± 2 μm (Fig. 2D).
Structure of the RIFMO Protomer
Crystal structures of apo-RIFMO and RIFMO complexed with RIF were determined at resolutions of 2.10 and 1.80 Å, respectively. The structures were solved by molecular replacement using a search model based on OxyS (47% sequence identity to RIFMO) (Table 1).
TABLE 1.
Crystallographic data processing and refinement statistics
Values for the outer resolution shell of data are given in parenthesis. NA, not applicable.
| Apo-RIFMO | RIF complex | |
|---|---|---|
| PDB code | 5KOW | 5KOX |
| Diffraction source | ALS 4.2.2 | ALS 4.2.2 |
| Space group | P6522 | P6522 |
| Unit cell parameters (Å) | a = 81.5, c = 282.2 | a = 81.4, c = 287.4 |
| Resolution range (Å) | 63.12–2.10 (2.16–2.10) | 57.47–1.80 (1.84–1.80) |
| Total no. of reflections | 662,524 (45,101) | 988,942 (33,161) |
| No. of unique reflections | 33,607 (2,666) | 52,691 (2,595) |
| Completeness (%) | 100.0 (100.0) | 98.4 (83.9) |
| Redundancy | 19.7 (16.9) | 18.8 (12.8) |
| 〈I/σ(I)〉 | 25.6 (5.8) | 36.3 (4.6) |
| Rmerge | 0.101 (0.514) | 0.064 (0.439) |
| Rmeas | 0.106 (0.546) | 0.067 (0.474) |
| Rpim | 0.032 (0.182) | 0.021 (0.173) |
| Mean CC½ | 0.999 (0.946) | 1.000 (0.884) |
| Wilson B factor (Å2) | 21.8 | 17.1 |
| Rcryst | 0.1867 | 0.1796 |
| Rfreea | 0.2351 | 0.2151 |
| No. of non-hydrogen atoms | ||
| All | 3,744 | 3,888 |
| Protein | 3,611 | 3,614 |
| FAD | 53 | 53 |
| RIF | NA | 52 |
| Water | 76 | 162 |
| Root mean square deviation | ||
| Bonds (Å) | 0.006 | 0.015 |
| Angles (degrees) | 0.829 | 1.510 |
| Average B factors (Å2) | ||
| All | 27.6 | 21.5 |
| Protein | 27.9 | 21.6 |
| FAD | 18.9 | 13.2 |
| RIF | NA | 23.9 |
| Water | 21.4 | 21.6 |
| Ramachandran plotb | ||
| Favored (%) | 97.0 | 97.9 |
| Allowed (%) | 3.0 | 2.1 |
| Outliers | 0.0 | 0.0 |
| Clashscoreb (percentile) | 2.06 (100) | 2.98 (99) |
| Molprobity scoreb (percentile) | 1.15 (100) | 1.12 (100) |
| Coordinate error (Å)c | 0.25 | 0.18 |
a Random 5% test set.
b Generated with MolProbity (44).
c Maximum likelihood-based coordinate error estimate from phenix.refine.
The RIFMO protomer comprises three domains: FAD-binding (residues 1–170 and 259–377), middle (residues 171–258), and C-terminal (residues 378–473) (Fig. 3A). The FAD-binding domain features a central five-stranded parallel β-sheet (β1, β2, β5, β9, β14) that resembles the sheet of Rossmann dinucleotide-binding domains. The ADP of FAD binds at the C termini of the central sheet, as expected. The Rossmann β-sheet is stacked between a three-stranded antiparallel β-sheet (β6–β8) and a three-helix bundle (α1, α4, α8) to make a three-layer ββα substructure. This domain also includes three α-helices (α9–α11) that form a protruding triangular substructure that is involved in dimerization (see below). Overall, the FAD-binding domain recapitulates conserved themes of FAD-binding motifs and residues in other flavoprotein monooxygenases (16).
FIGURE 3.

Crystal structure of RIFMO. A, schematic diagram with the FAD-binding domain in blue, middle domain in gray, and C-terminal domain in red. FAD is shown in yellow sticks. RIF is colored pink. B, a view into the RIF pocket. C, a view into the active site opening on the opposite side of the FAD from RIF. Arg41 is noted in green. As described under “Results,” Arg41 interacts with the FAD pyrophosphate and ribityl chain (Fig. 4) and is proposed to change conformation during the catalytic cycle (Fig. 9).
The middle domain is inserted between residues 170 and 259 of the FAD-binding domain (Fig. 3A). It consists of two interlaced βαβ folds that form a curved β-sheet. The β-sheet is structurally similar to that of OxyS (17), except that strand β14 in OxyS is replaced with helix α6 in RIFMO.
The middle domain forms a cap over the FAD-binding domain, resulting in two large openings that allow access to the FAD (Fig. 3, B and C). The opening near α6–β11 of the cap provides access to the N5 edge and pyrimidine ring of the FAD isoalloxazine (Fig. 3B). As described below, RIF binds in this opening. The other opening, which is near β13 of the cap, exposes the dimethylbenzene edge of the isoalloxazine as well as the FAD ribityl and ADP groups (Fig. 3C).
The C-terminal domain has a conserved thioredoxin-like α/β-fold, which packs against the FAD-binding domain (Fig. 3A). A similar C-terminal domain is also present in some aromatic polyketide hydrolases and is probably involved in oligomerization in phenol hydrolase (18) and meta-hydroxybenzoate hydrolase (19). However, the C-terminal domain of RIFMO is remote from the active site and dimer interface, so apparently it is not directly involved in catalysis or oligomerization. The presence of the C-terminal domain in many related enzymes suggests that it is probably needed for proper folding and stability. Domain deletion mutagenesis could be used to address this issue.
A survey of the Protein Data Bank (PDB) with PDBeFold (20) shows the closest structural homologs of RIFMO to be MtmOIV (21) and OxyS (17), which are flavoprotein monooxygenases in the mithramycin and oxytetracycline biosynthetic pathways, respectively. RIFMO superimposes with these enzymes with a root mean square deviation of 1.6 Å covering an alignment region of >440 residues.
The FAD-binding Site of RIFMO
Electron density maps clearly define the conformation of FAD and its interactions with the enzyme (Fig. 4). The isoalloxazine is planar, which is consistent with the oxidized state of the FAD. Most of the FAD-enzyme interactions are similar to those in MtmOIV (21) and OxyS (17). A unique aspect of RIFMO involves Arg41. This residue reaches across one of the active site openings to interact with the FAD pyrophosphate and ribityl chain (Figs. 3C and 4). Although OxyS has a Lys residue in place of Arg41, it does not interact with the FAD.
FIGURE 4.
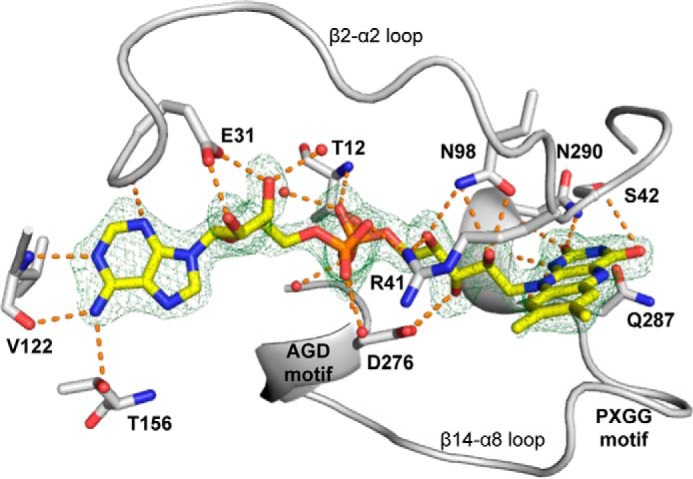
FAD-binding site of apo-RIFMO. FAD is colored in yellow, and the hydrogen bonds are in orange. The cage represents a simulated annealing FO − FC map contoured at 3.0σ.
RIFMO Is a Functional Homodimer
The oligomeric state and quaternary structure of RIFMO were determined using small angle x-ray scattering (SAXS). Three samples differing in protein concentration were analyzed to account for the possibility of concentration-dependent effects, such as nonspecific aggregation, oligomerization, and interparticle repulsion (22). The qualitative shape of the scattering curve is constant with increasing concentration, consistently displaying a small bump near q = 0.10–0.15 Å−1 (Fig. 5A). The Guinier plots exhibit good linearity and yield a radius of gyration (Rg) in the range of 34–36 Å (Fig. 5A and Table 2). Calculations of the pair distribution function suggest a maximum particle dimension (Dmax) of 107–108 Å and Rg of 34–35 Å (Fig. 5B). For reference, the Rg of a RIFMO monomer calculated from the crystal structure is 23 Å, which indicates that RIFMO forms an oligomer in solution.
FIGURE 5.

SAXS analysis. A, SAXS curves from three RIFMO samples at different concentrations (from the top: 7.2, 6.0, and 3.9 mg/ml). Black curves, experimental data. The highest concentration curve has been offset for clarity. The red curves are theoretical SAXS profiles calculated from the crystallographic homodimer with FoXS (23). Goodness of fit values (χ) are listed on the right. The inset shows Guinier plots. B, distance distribution functions, P(r). C, shape reconstruction calculated with assumed 2-fold symmetry.
TABLE 2.
SAXS parameters of RIFMO
The RIFMO oligomer was characterized by comparing the experimental SAXS curve with theoretical ones calculated from oligomer models using FoXS (23). Analysis of the crystal lattice with PDBePISA (24) suggests a stable dimer with Rg of 34 Å (The crystallographic asymmetric unit contains one protomer of this dimer). The SAXS curve calculated from the dimer has excellent agreement with the experimental profiles (Fig. 5A). The envelope from shape reconstruction calculations performed with GASBOR, assuming a 2-fold symmetric dimer, is consistent with the crystallographic dimer in terms of overall shape and dimensions (Fig. 5C).
The RIFMO homodimer has dimensions of 110 × 80 × 70 Å (Fig. 5C) The interface is formed primarily by α-helices 9, 10, and 11, which form a triangular structure that protrudes from the FAD-binding domain. Analysis with PDBePISA shows that the dimer interface includes contributions from 26 residues and has a buried surface area of 1000 Å2. The interface includes eight hydrogen bonds and 14 ion pairs. The RIFMO dimer is similar to those formed by OxyS (PDB code 4K2X), MtmOIV (PDB 4K5S), p-hydroxybenzoate hydroxylase (PDB 1PBE), BexE (PDB 4X4J), PgaE (PDB 2QA1), and CabE (PDB 2QA2).
Kinetic Measurements
Oxygen consumption was used as a proxy for N-hydroxylation activity. Incubation of RIFMO with NADPH in the absence of RIF resulted in a very slow oxygen consumption rate of 0.13 ± 0.01 s−1 (Fig. 6A). The inclusion of RIF in addition to NADPH in the assay accelerated oxygen consumption ∼20-fold to a rate of 2.3 ± 0.2 s−1.
FIGURE 6.
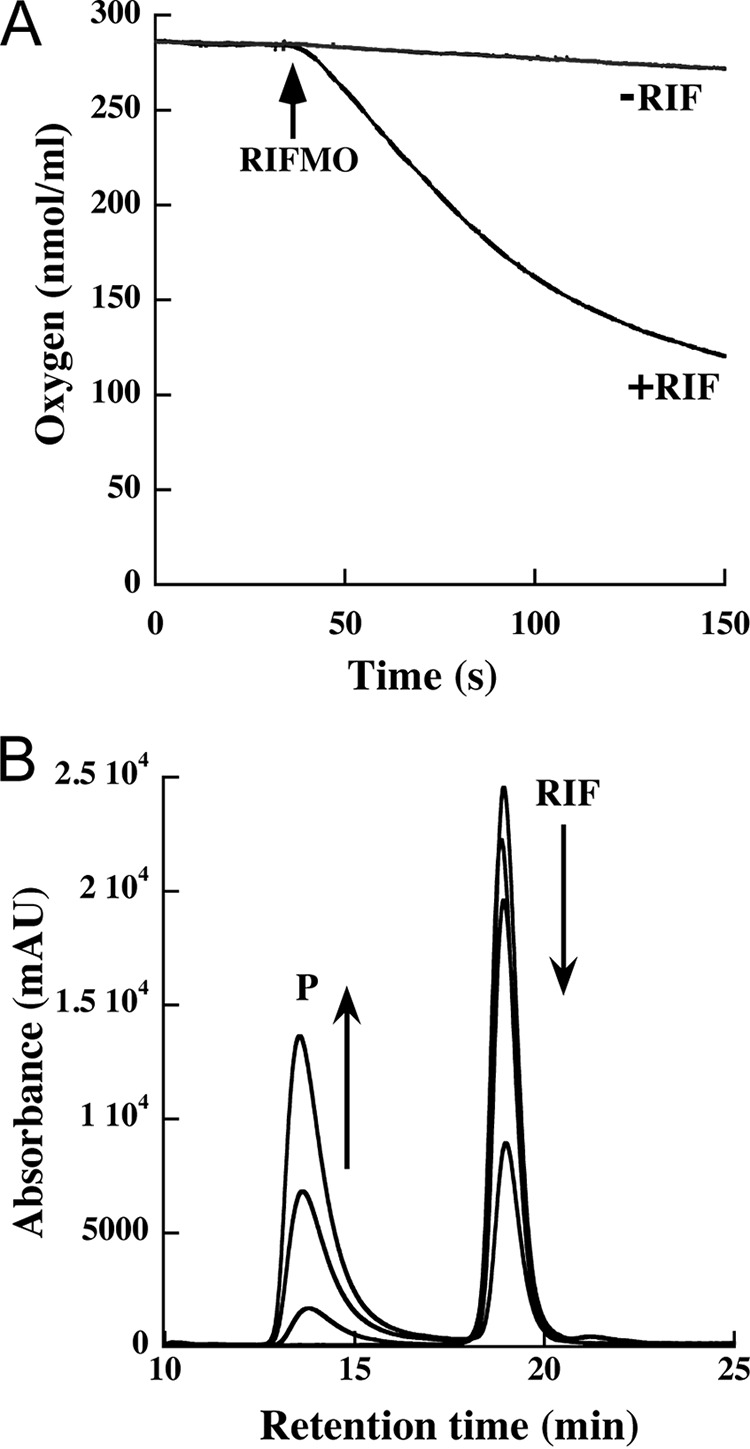
Steady-state kinetics and HPLC analysis of RIF oxidation. A, oxygen consumption monitored with 0.5 mm NADPH in the absence (rate = 0.13 ± 0.01 s−1) and presence of 50 μm RIF substrate (rate = 2.3 ± 0.2 s−1). The arrow indicates the reaction initiation by the addition of 1 μm RIFMO. The oxygen consumption assays were done in a 1-ml mixture of 100 mm sodium phosphate, pH 7.5, at 25 °C. The rates were obtained from the slope of the initial linear portions of the curves, just after the addition of RIFMO. B, production of hydroxylated RIF by RIFMO. An HPLC chromatogram (340 nm) shows the elution of RIF and hydroxylated RIF (P). The arrows show the RIF peak decreasing and the P peak increasing at 0, 10, 30, and 60 s. mAU, milliabsorbance units.
The full reaction for RIF monooxygenation was also monitored on a shorter time scale by HPLC (Fig. 6B). Samples of the reaction mixture were analyzed over the course of 1 min. The chromatogram peak for RIF was observed to decrease, whereas a new peak with a shorter retention time appeared. This peak was isolated and characterized by 1H NMR as RIF-2′-N-oxide, a resonance form of RIF-O, as reported previously (13). This result demonstrates that recombinant RIFMO is catalytically active.
Structure of the RIFMO-RIF Complex
The electron density map clearly defines the conformation of the entire substrate except for the methylpiperazine group (Fig. 7A). The region between the 70s loop and the ansa chain of RIF is the only available space for the methylpiperazine (Fig. 7B). We modeled the methylpiperazine into this space to guide the eye but have deleted these atoms in the deposited PDB file to reflect the lack of observable electron density. The lack of electron density is consistent with conformational disorder of the methylpiperazine.
FIGURE 7.
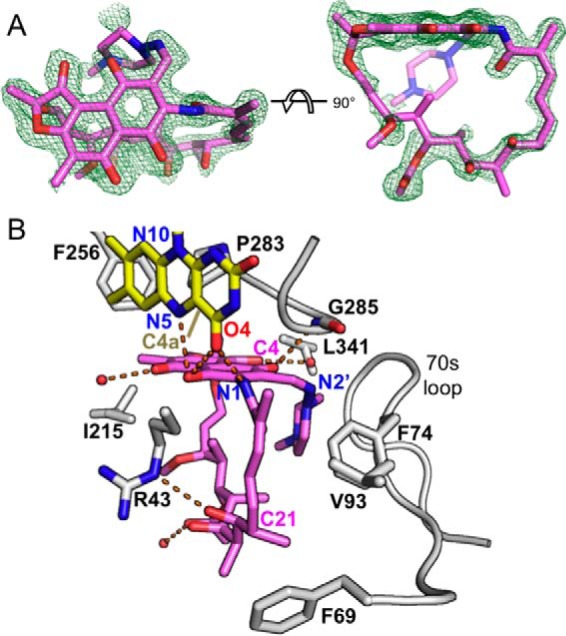
Electron density and interactions for RIF bound to RIFMO. A, two views of the electron density for RIF. The cage represents a simulated annealing FO − FC map contoured at 3.0 σ. Note that the methylpiperazine atoms have been deleted in the deposited PDB file to reflect the lack of observable electron density. B, interactions between RIF and RIFMO. The residues involved with interactions are in white. RIF (pink), FAD (yellow), and residues contacting RIF are shown as sticks. Water molecules are shown as red spheres, and hydrogen bonds are colored as orange dashed lines.
RIF binds in the large active site pocket below the α6–β11 region of the middle domain (Fig. 3, A and B). We note that premithramycin B occupies the analogous pocket in MtmOIV, a close structural homolog of RIFMO (PDB code 4K5S). The RIF naphthoquinone binds deep in the pocket, near the N5 edge of the isoalloxazine, whereas the ansa bridge spans the outer rim of the pocket (Fig. 3B). The naphthoquinone of RIF sits atop the Pro283–Gly286 loop and is oriented perpendicular to the isoalloxazine (Fig. 7B).
The binding site is expansive and primarily nonpolar. RIF contacts 30 carbon atoms from 14 residues (3.9 Å cut-off). Nonpolar residues in the active site include Phe69, Phe74, Val93, Ile215, Phe256, and Leu341 (Fig. 7B). Just a few electrostatic interactions are present. The N1 of RIF forms a hydrogen bond with the FAD O4. The C21 hydroxyl hydrogen bonds with Arg43. The C4 hydroxyl of the naphthoquinone accepts a hydrogen bond from Gly285.
Conformational Changes Associated with the Binding of RIF
Comparison of the two RIFMO structures provides information about the conformational changes attendant to RIF binding (Fig. 8). Large structural differences are evident in the FAD and protein. The isoalloxazine rotates 17° around the N5–N10 axis (Fig. 8A). This rotation appears to be coordinated with rearrangement of the 280s loop, upon which the RIF naphthoquinone sits. The center of this loop moves by over 4 Å upon RIF binding. The conformational changes of the 280s loop enable Gly285 to hydrogen-bond with the C4 hydroxyl and several carbon atoms of the loop to contact the RIF naphthoquinone.
FIGURE 8.
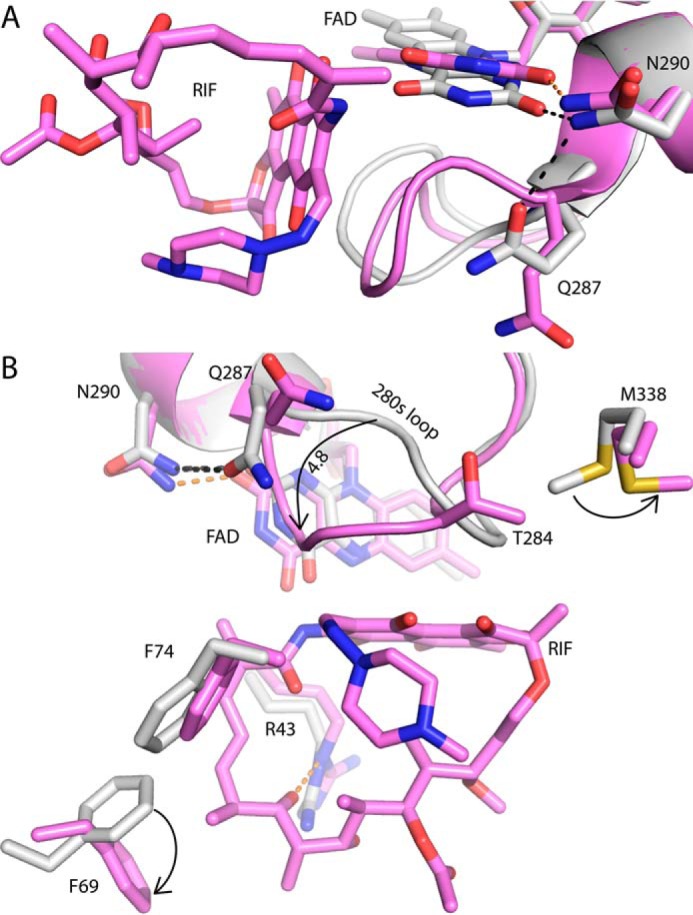
Superposition of apo-RIFMO (white) and the RIFMO-RIF complex (pink). A, close-up view emphasizing the rotation of the FAD and rupture of the Asn290–Gln287 hydrogen bond. B, a view emphasizing conformational changes in the 280s loop and side chain rotations. The arrows indicate the direction of conformational changes associated with RIF binding.
Side chain rotations are also observed (Fig. 8B). Phe74 rotates toward the substrate, whereas Phe69 flips away to make room for the C31 methyl. The guanidinium of Arg43 moves 1.8 Å to achieve optimal hydrogen bonding with the C21 hydroxyl. The methyl group of Met338 rotates 180° to avoid Thr284, whereas Gln287 flips 180° to avoid Gly286. The latter rotation breaks the hydrogen bond between Gln287 and Asn290. Rupture of this interaction is potentially significant, because Asn290 stabilizes the FAD by forming a hydrogen bond to the isoalloxazine O2 carbonyl.
Discussion
We described the first structure of a member of the RIFMO group of flavoprotein monooxygenases. RIFMO has the three-domain architecture found in other flavoprotein monooxygenases, yet it also exhibits structural differences in the outer end of the middle domain, which forms one edge of the RIF binding site. RIFMO has only one Rossmann dinucleotide-binding domain, which binds FAD. The absence of a second Rossmann domain (for NAD(P)H) identifies RIFMO as a class A flavoprotein monooxygenase (25).
Our kinetic data suggest the order of substrate binding. Slow oxygen consumption by RIFMO was observed in the presence of NADPH alone, but the addition of RIF increased the rate 20-fold (Fig. 6A). A possible explanation is that RIF binds to RIFMO first, followed by NADPH (Scheme 1). Based on other class A flavoprotein monooxygenases (16), additional proposed steps would include reduction of the FAD, release of NADP+, binding of O2, formation of the reactive RIFMO-C4a-hydroperoxyflavin, hydroxylation of RIF, and regeneration of the oxidized FAD (Scheme 1).
SCHEME 1.
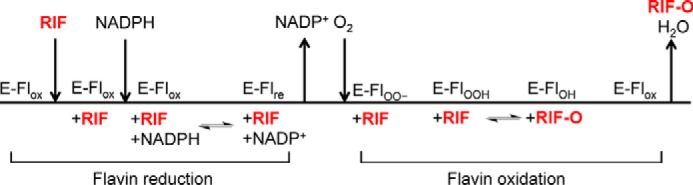
Proposed catalytic scheme for RIFMO. The left-hand side of the scheme depicts the steps involved in the binding of RIF and the reduction of the FAD by NADPH (reductive half-reaction). The right-hand side shows the steps involved in the hydroxylation of RIF and regeneration of the oxidized FAD (oxidative half-reaction). Downward arrows denote substrate binding, and upward arrows denote product dissociation. For each step, the redox state of the FAD is listed above the horizontal line. Enzyme-bound substrates and products are indicated below the horizontal line. E-Flox, RIFMO with oxidized FAD; E-Flre, RIFMO with reduced FAD; E-FlOO(H), RIFMO C4a-hydroperoxyflavin intermediates; E-FlOH, RIFMO C4a-hydroxyflavin. Adapted from Ref. 16.
Apparently, NADPH is unable to associate productively with RIFMO in the absence of RIF. One possibility is that RIF binding promotes a new protein/FAD conformation that unveils the NADPH site. Another possibility is that RIF binding hides a secondary, unproductive NADPH binding site.
The RIFMO-RIF structure potentially represents the initial binary E-Flox-RIF complex of the proposed catalytic scheme (Scheme 1). Examination of this structure suggests a major role for conformational change during the catalytic cycle. For example, the naphthoquinone of RIF blocks access to the N5 edge of the FAD (Fig. 7B), and thus NADPH could not reduce the flavin with RIF and FAD in the observed poses. Furthermore, the target of hydroxylation (N2′) is 8 Å away from the C4a atom of the FAD and appears to be poorly positioned to react with the putative C4a-OOH intermediate (Fig. 7B).
Other flavoprotein monooxygenases provide clues about the conformational changes associated with the reductive half of the reaction (Scheme 1). The NADPH binding site of MtmOIV, a very close structural homolog of RIFMO, has been proposed from low resolution electron density maps that indicate a fragment of NADPH (21). The site contains four Arg residues from the FAD-binding domain that are predicted to contact NADPH. RIFMO has all four of these Arg residues (positions 154, 158, 159, and 261) (Fig. 9A). The predicted NADPH site also includes the loop equivalent to RIFMO residues 218–226 (Fig. 9A). Assuming that NADPH binds at the analogous site in RIFMO, the isoalloxazine would have to move from its location in the current structures for reaction.
FIGURE 9.
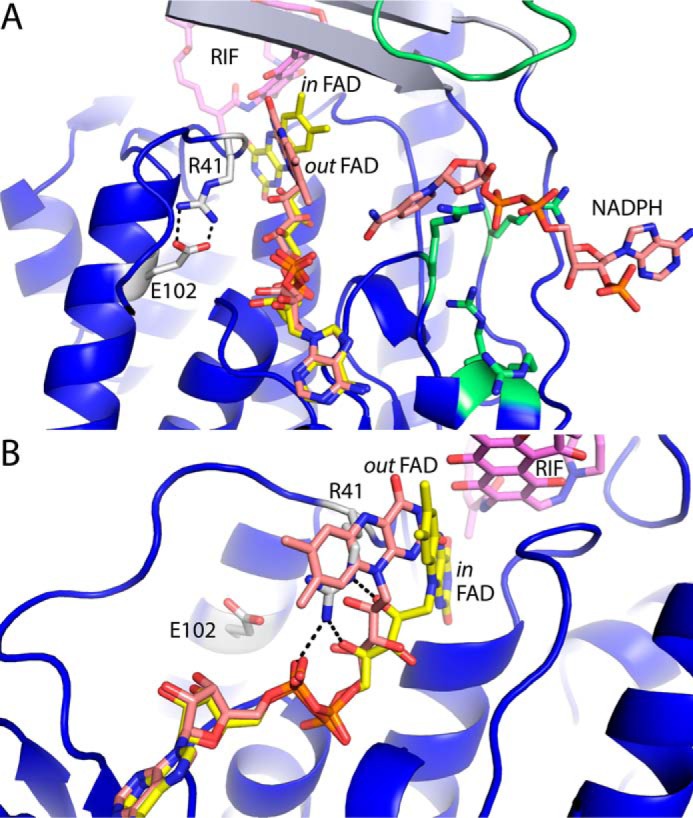
A model for conformational changes in RIFMO. A, a model of RIFMO with the FAD in the out conformation and NADPH bound. The out FAD conformation (salmon) was obtained by docking the FAD from RdmE (PDB code 3IHG (27)) onto RIFMO. The conformation of Arg41 and the ion pair with Glu102 are also modeled after RdmE. The in conformation FAD of RIFMO is shown in yellow. The coordinates of NADPH were obtained from a model of MtmOIV (21). Arg residues and a loop in RIFMO that have been shown to be important for NADPH binding in MtmOIV are colored green. B, steric clash between the out conformation FAD and Arg41 as it appears in the RIFMO structures. Black dashes represent hydrogen bonds formed by Arg41 in the RIFMO-RIF complex. The clash necessitates a conformational change in Arg41 when the FAD swings to the out conformation.
Ample precedent exists for dramatic movement of the isoalloxazine in class A flavin monooxygenases. para-Hydroxybenzoate hydroxylase (PHBH) is the best studied example (26). Flavin conformations designated as “in” and “out” have been described for PHBH. The FAD in the RIFMO structures resembles the in conformation. The out conformation occurs in PHBH during the reductive half-reaction, when close interaction between NADPH and FAD is required. The FAD moves between these states by a rotation about the ribityl chain in the plane of the isoalloxazine.
A structure of aklavinone-11-hydroxylase (RdmE) provides a good example of the out conformation (27) (PDB code 3IHG). We docked the FAD from this structure onto ours to model the out conformation of RIFMO (Fig. 9A). Movement of the FAD from the in to the out conformation in RIFMO brings the isoalloxazine toward the proposed NADPH site, which may allow sufficient room for RIF to adjust in preparation for reaction with the C4a-hydroperoxyflavin (Fig. 9).
Swinging of the FAD is probably concerted with protein motion. Only one residue of RIFMO has steric clash with the modeled out conformation FAD: Arg41 (Fig. 9B). This residue interacts with the pyrophosphate and ribityl of the in conformation FAD. Rupture of these interactions seems necessary for FAD to swing out. We again look to RdmE for hints about conformational changes in RIFMO. Arg41 is structurally analogous to RdmE Arg45. In RdmE, Arg45 ion-pairs with Glu123, which corresponds to Glu102 of RIFMO. We therefore propose that the transition of the FAD from the in to the out conformation is accompanied by a conformation change of Arg41 that breaks its interactions with the FAD and forms a new ion pair with Glu102 (Fig. 9B).
Experimental Procedures
Protein Expression and Purification
The N. farcinica IFM 10152 rox gene was codon-optimized for expression in E. coli and cloned into pET15b for expression as an N terminus His6-tagged protein (GenScript USA Inc.). The protein was expressed in E. coli and purified using immobilized metal ion affinity chromatography. The His tag was removed with thrombin.
Spectroscopic Measurements
Binding of RIF to oxidized RIFMO was monitored by recording the change in the flavin spectrum on an Agilent 8453 diode array spectrophotometer (Agilent Technologies, Santa Clara, CA) as a function of RIF concentration. Each solution consisted of 200 μl of 100 mm sodium phosphate buffer (pH 7.5), RIFMO (30 μm), and various RIF concentrations (0–30 μm). The solutions were incubated on ice for 10 min before data acquisition. The spectrum of each RIF concentration alone was subtracted from the spectrum of its corresponding RIFMO-RIF complex. The spectral changes showed a distinct absorbance pattern at 525 nm. Additionally, absorbance values of each RIFMO-RIF complex spectrum were subtracted from the spectrum of the free enzyme; absorbance differences at 525 nm were plotted as a function of RIF concentration to calculate the KD value.
Oxygen Consumption Assay
A Hansatech Oxygraph (Norfolk, UK) was used to monitor the oxygen consumption activity of RIFMO (1 μm) with 0.5 mm NADPH in the absence of RIF or with 50 μm RIF. A total reaction volume of 1 ml of 100 mm sodium phosphate buffer, pH 7.5, was used. The reactions proceeded for 2 min at 25 °C with constant stirring.
High Performance Liquid Chromatography Analysis
The RIFMO reaction was monitored using a Shimadzu HPLC system with photodiode array detector. Separation was achieved with a C18 reversed-phase column (Luna®, 5 μm, 100 Å, 250 × 4.6 mm) using an isocratic mobile phase consisting of 65% 10 mm potassium phosphate buffer (pH 7.5) and 35% acetonitrile. Samples (50 μl) were injected at flow rate of 1 ml/min, and each run proceeded for 25 min at 25 °C. For a total reaction volume of 0.2 ml in 100 mm sodium phosphate buffer, pH 7.5, final concentrations of 500 μm NADPH, 40 μm RIF, 0.5 μm RIFMO were used. The reaction was initiated by the addition of the enzyme and allowed to incubate for 10, 30, and 60 s at 25 °C. Reactions were then quenched with acetonitrile (35% final concentration), and protein was removed by centrifugation at 13,000 rpm for 2 min. The eluted substrate and product were monitored using a diode array detector at 340 nm. Product preparation and NMR analysis of the RIFMO reaction were done as described previously (13).
SAXS
Shutterless SAXS data collection was performed with a Pilatus detector at beamline 12.3.1 of the Advanced Light Source through the SIBYLS mail-in high throughput SAXS program (28). The RIFMO sample was passed through a 13/30 Superdex 200 prep grade size exclusion chromatography column that had been equilibrated with 25 mm Hepes (pH 7.5), 100 mm NaCl, 2.5% glycerol, and 0.5 mm Tris(3-hydroxypropyl)phosphine. Proteins were concentrated to about 10 mg/ml after the sizing column using an Amicon Ultra centrifugal filter (10 kDa). Protein concentration was estimated with the BCA assay. Twenty-five images were collected in 5 s for the RIFMO protein at three different concentrations (3.9, 6.0, and 7.2 mg/ml). Images for background subtraction were collected similarly on the effluent from the size exclusion column. PRIMUS (29) was used to analyze the data for radiation damage and to identify the best buffer replicate for subtraction. The first 12 images were found to be appropriate for averaging for each sample. The maximum particle dimension was estimated from calculations of the pair distribution function using GNOM (30) via PRIMUS, and the output GNOM.out files were used for shape reconstruction by GASBOR (31). Forty-eight shape reconstructed envelopes from independent calculations were averaged by DAMAVER (32) to give a representative model, which was converted into a volumetric map using pdb2vol (33).
Crystallization of RIFMO
The protein was exchanged into a buffer containing 25 mm Hepes and 100 mm NaCl at pH 7.5 and concentrated to 10.8 mg/ml (210.8 μm; Bradford assay). The initial hit condition was discovered from a broad screening with Hampton crystallization screens at room temperature. Hexagonal crystals of RIFMO obtained by vapor diffusion arise from conditions using sodium acetate, PEG 3350, or PEG 4000 as precipitant. Whereas these crystals appear to be isomorphous, some of the crystals grown in PEGs suffered from varying degrees of merohedral twinning. Therefore, the initial structure of RIFMO was obtained from crystals grown in sodium acetate.
Crystals of apo-RIFMO (RIF-free) were grown in sodium acetate as follows. The protein was mixed in a 1:1 ratio with a well solution containing 2.4 m sodium acetate trihydrate at pH 7.0 and suspended over the well solution in Linbro hanging drop trays. Yellow crystals with symmetry consistent with space group P6522 grew to 40 × 40 × 200 μm3 over a period of a week at room temperature. Crystal growth could be accelerated by introducing 0.3 μl of the well solution containing micro-crystals from previous experiments into the crystallization drops. Crystals were cryo-protected by adding 10% glycerol before flash-freezing in liquid nitrogen.
Crystallization of a RIFMO-RIF Complex
Crystals of RIFMO complexed with RIF were obtained by co-crystallization. To make the complex, RIF was first added to the protein sample at a final concentration of 5 mm (2.5% dimethyl sulfoxide), mixed well, and allowed to equilibrate for 30 min. The inclusion of RIF apparently inhibited growth of the sodium acetate crystal form, so a PEG condition was used. The co-crystals were prepared by mixing the RIFMO-RIF complex sample in a 1:1 ratio with a well solution comprising 17% (w/v) PEG 3350, 200 mm magnesium chloride, and 2.5% glycerol. Microseeds made from the apo-crystals (0.3 μl) were added to the drop to encourage crystal formation and to mitigate the twinning issue.
Diffraction Data Collection and Structure Determination
Diffraction data were collected in shutterless mode from single crystals at the MBC beamline 4.2.2 at the Advanced Light Source (Berkeley, CA). Each data set consisted of a wedge of 180° of data collected with a Taurus-1 detector over a period of 180 s. Images were written to disk every 0.2 s, so that the data set consisted of 900 images with an effective oscillation width of 0.2°. The data were indexed, integrated, and scaled with the XDS package (34). The space group is P6522 with unit cell parameters of a = b = 81.5 Å and c = 282.2 Å. The asymmetric unit contains one RIFMO protomer, which implies a solvent content of 53.6% (35). Intensities were converted to amplitudes with Truncate (36). Data processing statistics are provided in Table 1.
Initial phases were determined by molecular replacement with the program PHASER (37). The search model was generated with CHAINSAW (38) using the sequence of RIFMO and monomer A of OxyS (47% sequence identity, PDB entry 4K2X (17)). These calculations showed that the space group is P6522 with one protein molecule in an asymmetric unit. The initial solution from molecular replacement was further modified (in COOT) by deleting parts of the model that had poor fit to the electron density map. In all, 117 of 473 residues were deleted, and side chains for the remaining residues were trimmed to Cγ if not identical to OxyS.
Iterative rounds of model building and restrained refinement were carried out with COOT (39, 40), AutoBuild (41), and phenix.refine (42). The apo-structure was used to solve the co-complex structure with RIF bound. No alternate conformations were modeled because a single conformation of all residues could satisfactorily explain the electron density. The restraint files for FAD and RIF were generated in eLBOW (43). Refined structures were validated with MolProbity (44). In the apo-structure, the side chains of five residues (His0, Asp224, Arg225, Arg265, and Thr284) are disordered and therefore omitted beyond Cβ. In the RIF complex, only the side chains of Asp224 and Arg225 are disordered and trimmed to Cβ. The C terminus of the polypeptide is convincingly shown in both structures by the presence of electron density for the carboxylate of Thr473.
Author Contributions
L.-K. L. designed and performed crystallography experiments, analyzed data, and wrote the paper. H. A. and J. S. M. D. C. generated enzyme, performed biochemical experiments, and analyzed data. R. M.-C. grew crystals. P. S. designed experiments, analyzed data, and wrote the paper. J. J. T. designed experiments, analyzed data, and wrote the paper.
Acknowledgments
We thank Dr. Jay Nix for help with x-ray diffraction data collection and Kathryn Burnett for collecting the SAXS data through the SIBYLS mail-in program at the Advanced Light Source. We thank Dr. Nicholas Noinaj for providing the model of MtmOIV complexed with NADPH. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231. Additional support for the SYBILS beamline comes from the National Institute of Health project MINOS (R01GM105404).
This work was supported by National Science Foundation Grants CHE-1506206 and MCB 1021384. This work was also supported by a grant from the Douglas D. Randall Young Scientists Development Fund (to L.-K. L.) from the University of Missouri and a Joint Supervision Fellowship (to H. A.) funded by the Cultural Affairs and Missions sector of the Egyptian Ministry of Higher Education. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5KOW and 5KOX) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RIF
- rifampicin
- RIFMO
- rifampicin monooxygenase
- RIF-O
- 2′-N-hydroxy-4-oxo-rifampicin
- SAXS
- small-angle x-ray scattering
- PDB
- Protein Data Bank
- MtmOIV
- Baeyer-Villiger monooxygenase from the mithramycin biosynthetic pathway in Streptomyces argillaceus
- OxyS
- anhydrotetracycline hydroxylase from Streptomyces rimosus
- RdmE
- aklavinone-11 hydroxylase from Streptomyces purpurascens
- PHBH
- para-hydroxybenzoate hydroxylase.
References
- 1. Aristoff P. A., Garcia G. A., Kirchhoff P. D., and Hollis Showalter H. D. (2010) Rifamycins: obstacles and opportunities. Tuberculosis 90, 94–118 [DOI] [PubMed] [Google Scholar]
- 2. Goldstein B. P. (2014) Resistance to rifampicin: a review. J. Antibiot. 67, 625–630 [DOI] [PubMed] [Google Scholar]
- 3. Severinov K., Soushko M., Goldfarb A., and Nikiforov V. (1993) Rifampicin region revisited: new rifampicin-resistant and streptolydigin-resistant mutants in the β subunit of Escherichia coli RNA polymerase. J. Biol. Chem. 268, 14820–14825 [PubMed] [Google Scholar]
- 4. Telenti A., Imboden P., Marchesi F., Matter L., Schopfer K., Bodmer T., Lowrie D., Colston M. J., and Cole S. (1993) Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 341, 647–650 [DOI] [PubMed] [Google Scholar]
- 5. Tupin A., Gualtieri M., Roquet-Banères F., Morichaud Z., Brodolin K., and Leonetti J.-P.(2010) Resistance to rifampicin: at the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 35, 519–523 [DOI] [PubMed] [Google Scholar]
- 6. Baysarowich J., Koteva K., Hughes D. W., Ejim L., Griffiths E., Zhang K., Junop M., and Wright G. D. (2008) Rifamycin antibiotic resistance by ADP-ribosylation: structure and diversity of Arr. Proc. Natl. Acad. Sci. U.S.A. 105, 4886–4891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spanogiannopoulos P., Thaker M., Koteva K., Waglechner N., and Wright G. D. (2012) Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental actinomycetes. Antimicrob. Agents Chemother. 56, 5061–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yazawa K., Mikami Y., Maeda A., Akao M., Morisaki N., and Iwasaki S. (1993) Inactivation of rifampin by Nocardia brasiliensis. Antimicrob. Agents Chemother. 37, 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qi X., Lin W., Ma M., Wang C., He Y., He N., Gao J., Zhou H., Xiao Y., Wang Y., and Zhang P. (2016) Structural basis of rifampin inactivation by rifampin phosphotransferase. Proc. Natl. Acad. Sci. U.S.A. 113, 3803–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spanogiannopoulos P., Waglechner N., Koteva K., and Wright G. D. (2014) A rifamycin inactivating phosphotransferase family shared by environmental and pathogenic bacteria. Proc. Natl. Acad. Sci. U.S.A. 111, 7102–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stogios P. J., Cox G., Spanogiannopoulos P., Pillon M. C., Waglechner N., Skarina T., Koteva K., Guarné A., Savchenko A., and Wright G. D. (2016) Rifampin phosphotransferase is an unusual antibiotic resistance kinase. Nat. Commun. 7, 11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andersen S. J., Quan S., Gowan B., and Dabbs E. R. (1997) Monooxygenase-like sequence of a Rhodococcus equi gene conferring increased resistance to rifampin by inactivating this antibiotic. Antimicrob. Agents Chemother. 41, 218–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoshino Y., Fujii S., Shinonaga H., Arai K., Saito F., Fukai T., Satoh H., Miyazaki Y., and Ishikawa J. (2010) Monooxygenation of rifampicin catalyzed by the rox gene product of Nocardia farcinica: structure elucidation, gene identification and role in drug resistance. J. Antibiot. 63, 23–28 [DOI] [PubMed] [Google Scholar]
- 14. Floss H. G., and Yu T.-W.(2005) Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 105, 621–632 [DOI] [PubMed] [Google Scholar]
- 15. Ishikawa J., Chiba K., Kurita H., and Satoh H. (2006) Contribution of rpoB2 RNA polymerase beta subunit gene to rifampin resistance in Nocardia species. Antimicrob. Agents Chemother. 50, 1342–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crozier-Reabe K., and Moran G. (2012) Form follows function: structural and catalytic variation in the class A flavoprotein monooxygenases. Int. J. Mol. Sci. 13, 15601–15639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang P., Bashiri G., Gao X., Sawaya M. R., and Tang Y. (2013) Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc. 135, 7138–7141 [DOI] [PubMed] [Google Scholar]
- 18. Enroth C., Neujahr H., Schneider G., and Lindqvist Y. (1998) The crystal structure of phenol hydroxylase in complex with FAD and phenol provides evidence for a concerted conformational change in the enzyme and its cofactor during catalysis. Structure 6, 605–617 [DOI] [PubMed] [Google Scholar]
- 19. Hiromoto T., Fujiwara S., Hosokawa K., and Yamaguchi H. (2006) Crystal structure of 3-hydroxybenzoate hydroxylase from Comamonas testosteroni has a large tunnel for substrate and oxygen access to the active site. J. Mol. Biol. 364, 878–896 [DOI] [PubMed] [Google Scholar]
- 20. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 21. Bosserman M. A., Downey T., Noinaj N., Buchanan S. K., and Rohr J. (2013) Molecular insight into substrate recognition and catalysis of Baeyer-Villiger monooxygenase MtmOIV, the key frame-modifying enzyme in the biosynthesis of anticancer agent mithramycin. ACS Chem. Biol. 8, 2466–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jacques D. A., and Trewhella J. (2010) Small-angle scattering for structural biology: expanding the frontier while avoiding the pitfalls. Protein Sci. 19, 642–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneidman-Duhovny D., Hammel M., and Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 25. van Berkel W. J. H., Kamerbeek N. M., and Fraaije M. W. (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124, 670–689 [DOI] [PubMed] [Google Scholar]
- 26. Entsch B., Cole L. J., and Ballou D. P. (2005) Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch. Biochem. Biophys. 433, 297–311 [DOI] [PubMed] [Google Scholar]
- 27. Lindqvist Y., Koskiniemi H., Jansson A., Sandalova T., Schnell R., Liu Z., Mäntsälä P., Niemi J., and Schneider G. (2009) Structural basis for substrate recognition and specificity in aklavinone-11-hydroxylase from rhodomycin biosynthesis. J. Mol. Biol. 393, 966–977 [DOI] [PubMed] [Google Scholar]
- 28. Dyer K. N., Hammel M., Rambo R. P., Tsutakawa S. E., Rodic I., Classen S., Tainer J. A., and Hura G. L. (2014) High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol. Biol. 1091, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 30. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 31. Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wriggers W. (2010) Using Situs for the integration of multi-resolution structures. Biophys. Rev. 2, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 36. French G. S., and Wilson K. S. (1978) On the treatment of negative intensity observations. Acta Crystallogr. A34, 517–525 [Google Scholar]
- 37. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stein N. (2008) CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 41, 641–643 [Google Scholar]
- 39. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moriarty N. W., Grosse-Kunstleve R. W., and Adams P. D. (2009) electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]