

Abstract
Infection with Bacillus anthracis, the causative agent of anthrax, can lead to persistence of lethal secreted toxins in the bloodstream, even after antibiotic treatment. VHH single-domain antibodies have been demonstrated to neutralize diverse bacterial toxins both in vitro and in vivo, with protein properties such as small size and high stability that make them attractive therapeutic candidates. Recently, we reported on VHHs with in vivo activity against the protective antigen component of the anthrax toxins. Here, we characterized a new set of 15 VHHs against the anthrax toxins that act by binding to the edema factor (EF) and/or lethal factor (LF) components. Six of these VHHs are cross-reactive against both EF and LF and recognize the N-terminal domain (LFN, EFN) of their target(s) with subnanomolar affinity. The cross-reactive VHHs block binding of EF/LF to the protective antigen C-terminal binding interface, preventing toxin entry into the cell. Another VHH appears to recognize the LF C-terminal domain and exhibits a kinetic effect on substrate cleavage by LF. A subset of the VHHs neutralized against EF and/or LF in murine macrophage assays, and the neutralizing VHHs that were tested improved survival of mice in a spore model of anthrax infection. Finally, a bispecific VNA (VHH-based neutralizing agent) consisting of two linked toxin-neutralizing VHHs, JMN-D10 and JMO-G1, was fully protective against lethal anthrax spore infection in mice as a single dose. This set of VHHs should facilitate development of new therapeutic VNAs and/or diagnostic agents for anthrax.
Keywords: anthrax toxin, antibody, epitope mapping, microbial pathogenesis, phage display, toxin, EF, LF, VHH, VNA
Introduction
Anthrax is a frequently fatal disease caused by the Gram-positive, spore-forming bacterium Bacillus anthracis. Central to the development of anthrax signs and symptoms in the host is the bacterium's production of two toxins, edema toxin (ET)2 and lethal toxin (LT). ET comprises edema factor (EF) and protective antigen (PA), whereas LT comprises lethal factor (LF) and PA. In each case, PA binds to cell surface receptors and allows entry of the toxin to the cell via the formation of a pore. After binding to the receptor, PA is cleaved from the PA83 (83 kDa) to the PA63 (63 kDa) form by cell-surface proteases, leading to PA oligomerization and the formation of binding sites for LF/EF. Upon entry into the cell, EF is toxic via activity as an adenylate cyclase, whereas LF is toxic via activity as a zinc-dependent metalloprotease that cleaves mitogen-activated protein kinases (MEKs) (for review, see Ref. 1).
The development of antibody-based therapeutics against the anthrax toxins provides a means of neutralizing the toxicity and lethality of ET and LT. Many existing antibodies block the binding of PA to cellular receptors, thus preventing entry of the toxins into the cell (2). Recently, we reported on the identification and antitoxin application of VHH single-domain antibodies that neutralize PA (3). VHHs consist of the 15-kDa variable domain from heavy chain-only antibodies that occur naturally in camelid species (e.g. camels, llamas, alpacas) and include the complete binding site for an epitope without the need for a separate light chain. The small size and single-chain nature of VHHs allow efficient production from bacteria as well as access to epitopes and protein regions that may not be accessible to conventional antibodies. VHHs also have higher stability to pH and temperature extremes than conventional antibodies (4, 5). We previously reported development of neutralizing VHHs against a range of toxins, including several bioterror threat agents such as ricin (6) and botulinum neurotoxin (7). The anti-PA VHHs exhibited neutralization of PA in vitro and provided protection against B. anthracis in a mouse infection model (3). Linking two or more neutralizing VHHs recognizing different epitopes into heteromultimers (VHH-based neutralizing agents, VNAs) often dramatically improves the in vivo potency of these antitoxin antibodies (3, 6–9). In the case of PA, we showed that two linked VHHs targeting different neutralizing epitopes provided strong protection against anthrax infection in mice (3).
Here we report the characterization of a set of VHHs against the EF and/or LF components of the anthrax toxins. Toxin-neutralizing VHHs with nanomolar range IC50 values were identified that recognize various epitopes in either or both toxins. A subset of these VHHs cross-react against an epitope in the homologous N terminus of both toxins in a region responsible for association with PA. Other VHHs selectively neutralize LF or EF, including one VHH that appears to bind the C terminus of LF. We demonstrate therapeutic effectiveness of these agents in a spore model of anthrax in mice and discuss the potential for enhanced therapeutics that combine these EF/LF-neutralizing VHHs with anti-PA VHHs.
Results
EF and LF Binding and Neutralization Potencies of VHHs
To identify a pool of VHHs that bind to EF and/or LF, two alpacas were immunized with purified toxin proteins, and a phage display library prepared from the immune B cells was separately panned for phage binding to each protein. VHHs from clones of interest were re-expressed in Escherichia coli as fusions to thioredoxin, and the soluble products were purified for further in vitro characterization. VHH sequences are depicted in supplemental Fig. S1.
First, the series of purified VHHs was screened for binding to EF and LF by dilution ELISA. Based on the results of these assays, VHHs were divided into three categories: EF-specific, LF-specific, and cross-specific for both EF and LF (Table 1, EC50 columns). As EF and LF must bind to PA63 to gain entry to the cell, the proteins share sequence similarity in their N-terminal PA binding domains (supplemental Fig. S2; potential shared epitopes in the N terminus highlighted with dark blue bars). Therefore, cross-specific VHHs would be expected to recognize one of these shared sequence stretches.
TABLE 1.
Properties of anti-EF, anti-LF, and anti-EF/LF VHHs
>100 or >1000 indicates no detectable signal within the limits of the assay.
| VHH Name | Plasmid | Competition groupa | EC50b EF | IC50c EF | EC50b LF | IC50c LF |
|---|---|---|---|---|---|---|
| nm | nm | nm | nm | |||
| EF-specific | ||||||
| JMN-E2 | JMY-3 | EF2 | 4 | >1000 | >100 | >1000 |
| JMN-F3 | JMY-7 | EF1 | 2 | 1028 ± 317 | >100 | >1000 |
| LF-specific | ||||||
| JMO-A2 | JMY-9 | LF4 | >100 | >1000 | 0.8 | >1000 |
| JMO-B9 | JMY-15 | LF2 | >100 | >1000 | 0.8 | 9.4 ± 4.7 |
| JMO-C1 | JMY-17 | LF4 | >100 | >1000 | 20 | >1000 |
| JMO-C10 | JMY-21 | LF3 | >100 | >1000 | 20 | 29 ± 3 |
| JMO-F4 | JMY-23 | ND | >100 | >1000 | 125 | >1000 |
| JMO-F12 | JMY-27 | ND | >100 | >1000 | 20 | >1000 |
| JMO-C9 | JMY-19 | LF1 | ≈150 | >1000 | 0.4 | 2.4 ± 0.9 |
| EF/LF-specific | ||||||
| JMN-D10 | JMY-1 | EF1/LF1 | 0.3 | 2.7 ± 3.8 | 20 | >1000 |
| JMO-B3 | JMY-13 | EF1/LF1 | >100 | 103 ± 57 | 0.4 | 6.6 ± 1.5 |
| JMO-G1 | JMY-29 | EF1/LF1 | 0.2 | 13.7 ± 6.7 | 0.3 | 2.1 ± 1.0 |
| JMN-E12 | JMY-33 | EF1/LF1 | 0.4 | 30.5 ± 15.3 | 20 | 117 ± 16 |
| JMN-F1 | JMY-5 | EF1/LF1 | 0.3 | 52 ± 49 | 0.4 | 7.7 ± 2.2 |
| JMO-A4 | JMY-11 | EF1/LF1 | 0.3 | 409 ± 393 | 100 | 408 ± 99 |
a Group identification determined by competition ELISAs, as depicted in Fig. 2 and supplemental Fig. S4. ND, each of these VHHs exhibited poor competitive ability in the ELISA assays; because they also demonstrated poor neutralization, competition groups for these VHHs were not examined in more detail.
b EC50 assessed by dilution ELISAs.
c IC50 assessed by RAW cell neutralization (MTT and cAMP) assays.
To aid in the selection of therapeutic candidates, each VHH was further characterized for its potency in neutralizing LF in a standard macrophage toxicity assay and in neutralizing EF in a cAMP production assay (Table 1, IC50 columns; representative results in Fig. 1, A–D). The two EF-specific VHHs, JMN-E2 and JMN-F3, exhibited little to no EF or LF neutralizing potency. Three of the seven LF-specific VHHs, JMO-B9, JMO-C9, and JMO-C10, potently neutralized LF while, as expected from the ELISA results, exhibiting no activity on EF. Most of the EF/LF cross-specific VHHs neutralized both EF and LF, with varying potencies. EF and LF binding as assessed by ELISA (EC50 values) did not always correlate with toxin neutralizing potency, as evidenced by the differences in EF binding versus EF neutralization observed for JMO-A4 (Table 1).
FIGURE 1.

LF and EF neutralization by VHHs. Representative LF (A and B) and EF (C and D) neutralization experiments measuring toxicity of LT or ET for macrophages in the presence of various doses of antibody are shown. Assays were performed using 250 ng/ml concentrations of each toxin component. In addition to testing the new EF and LF binding VHHs (Table 1), 7F10 (12), JKH-C7 (3), and 14B7 (24) are previously characterized neutralizing mAbs and VHHs included here as positive controls. Percent survival of macrophages is calculated relative to medium-treated cells.
Epitope Binning of VHHs
Next, VHHs were placed into competition groups based on sandwich ELISA data (Table 1; ELISA schematic in supplemental Fig. S3). In these studies (Fig. 2), each of the VHHs was assessed as both a test agent and as a blocking agent in combination with each other VHHs recognizing the EF or LF targets. Based on the sandwich ELISA results, the two EF-specific VHHs recognize distinct, non-competing epitopes (EF1 and EF2). Although the EF/LF cross-specific VHHs vary significantly in their relative EC50 and IC50 values for EF and LF, all six are in the same competition group for both EF binding (Fig. 2A) and LF binding (Fig. 2B), which we named EF1/LF1. Presumably, the epitope recognized by the EF1/LF1 group is within a region of conserved sequence between the EF and LF proteins. Surprisingly, the EF-specific JMN-F3 (as well as the LF-specific JMO-C9, described below) is in the same competition group as the EF/LF cross-specific VHHs. We propose that this is due to partially overlapping epitopes within the same competition group.
FIGURE 2.

EF/LF epitope mapping of VHHs. Sandwich ELISAs, as shown graphically in supplemental Fig. S3, were performed with a subset of anti-EF VHHs (A) and anti-LF VHHs (B) to investigate epitope binding competition. Briefly, HRP-labeled EF (A) or LF (B) was preincubated with a blocking VHH or PBS control for 1 h followed by application to an ELISA plate coated with a test VHH. Test VHHs are identified below each cluster of bars, and blocking agents are identified under each individual bar. ELISA values are expressed as a fraction of the signal in the PBS control wells, with smaller bars indicating increased competition between the blocking and the test VHHs. Pairs in which the binding of EF/LF to the test VHH was reduced ∼3-fold or more by the blocking VHH are indicated by black bars. Bars represent the average of three technical replicates ± S.D. Results were effectively duplicated within the experiment, as each VHH was tested as both a blocking and test agent with each other VHH; additionally, a matching pattern of competition was observed for two independent experiments.
Within the EF1 group, JMN-D10, JMO-G1, and JMO-A4 exhibited the best ability to block EF binding by other members of the group when added as the blocking VHH. However, JMO-A4 is not a good therapeutic candidate due to its poor neutralizing ability (IC50 value, Table 1).
Within the LF1 group, JMN-F1, JMO-B3, and JMO-G1 were all excellent blocking antibodies in the sandwich ELISA (Fig. 2B). In Fig. 2 it is evident that some VHHs exhibited weak or no self-competition in the sandwich assay. The VHHs with the highest apparent target affinities (lowest EC50) were typically the most potent blocking agents within their competition group, especially in the case of LF binding, suggesting that weak competition was a result of weak binding affinity. Therefore, to complete the LF epitope mapping, we performed a modified version of the sandwich assay that was less sensitive to VHH affinity on a subset of the VHHs (supplemental Fig. S4). Based on the data in Fig. 2 and supplemental Fig. S4, at least three non-overlapping LF epitopes are recognized by the set of VHHs (LF1, LF2, and LF3). VHHs binding to each of these LF epitopes were toxin-neutralizing (Fig. 1). JMO-A2 and JMO-C1 bound well to LF coated on plastic (with a high affinity in the case of JMO-A2 in particular; EC50 = 0.8 nm). However, JMO-A2 bound LF poorly in the sandwich ELISAs, both as the blocking antibody and as a test antibody. When applied to a plate coated with LF, JMO-A2 (but not JMO-G1 or JMO-B9) inhibited the binding of subsequently added JMO-A2 or JMO-C1 ≈2-fold; therefore, we placed these two antibodies into a final, non-neutralizing competition group, LF4.
Biochemical Characterization of a Selected Group of VHHs
Based on our ELISA and toxin neutralization results, four VHHs were selected for further analysis. JMO-G1 was selected as the most potent cross-neutralizer in the EF1/LF1 group; JMN-D10 was selected for its excellent EF binding and neutralization capabilities; JMO-B3 was selected as being distinct in properties from the rest of the EF1/LF1 group, possibly suggesting a partially different epitope; JMO-B9 was selected as a good LF neutralizer within a different LF epitope group (LF2). All four of these selected VHHs were highly stable, as assessed by nanoscale differential scanning fluorimetry (ranging from 67–75 °C; Table 2). JMO-G1 exhibited the highest Tm (highest stability) of the set.
TABLE 2.
Biochemical properties of a selected set of VHHs
VHH properties were assessed via SPR to obtain kd, ka, and KD values and by nanoscale differential scanning fluorimetry to obtain Tm (protein melting temperature) values.
| VHH | Tm | Kd, EF | ka, EF | KD, EF | kd, LF | ka, LF | KD, LF |
|---|---|---|---|---|---|---|---|
| °C | m | ||||||
| JMN-D10 | 65.7 °C | (1.3 ± 0.5) × 10−5 | (3.9 ± 1.2) × 105 | (1.6 ± 0.2) × 10−11 | (5.4 ± 2.2) × 10−4 | (1.5 ± 0.3) × 106 | (1.6 ± 0.2) × 10−9 |
| JMO-B3 | 69.8 °C | No binding | No binding | No binding | (7.6 ± 0.5) × 10−6 | (7.7 ± 1.5) × 105 | (2.0 ± 0.5) × 10−11 |
| JMO-B9 | 66.6 °C | No binding | No binding | No binding | |||
| JMO-G1 | 75.3 °C | (1.5 ± 0.3) × 10−5 | (6.6 ± 0.3) × 105 | (2.2 ± 0.4) × 10−11 | (6.6 ± 3.0) × 10−6 | (5.5 ± 0.6) × 105 | (1.1 ± 0.5) × 10−11 |
In a screen of VHH KD values by surface plasmon resonance (SPR) at a VHH concentration of ≈100 nm, we observed that JMO-B3 and JMO-G1 had subnanomolar affinity for LF, and JMN-D10 and JMO-G1 had subnanomolar affinity for EF (Fig. 3; Table 2). JMO-B3 did not exhibit any appreciable binding to EF by SPR at 100 nm, consistent with its ELISA binding behavior. JMO-B9 did not exhibit binding to LF by SPR at 100 nm (data not shown) despite its tight binding indicated in ELISA binding and competition studies and its potent LF neutralization; we suggest that its binding mode to LF is not compatible with the orientation or conformation of the LF on the SPR chip. Based on the SPR results, the poor EC50 and IC50 values of JMN-D10 for LF may be related to its comparatively rapid dissociation rate from LF (kd column, Table 2).
FIGURE 3.
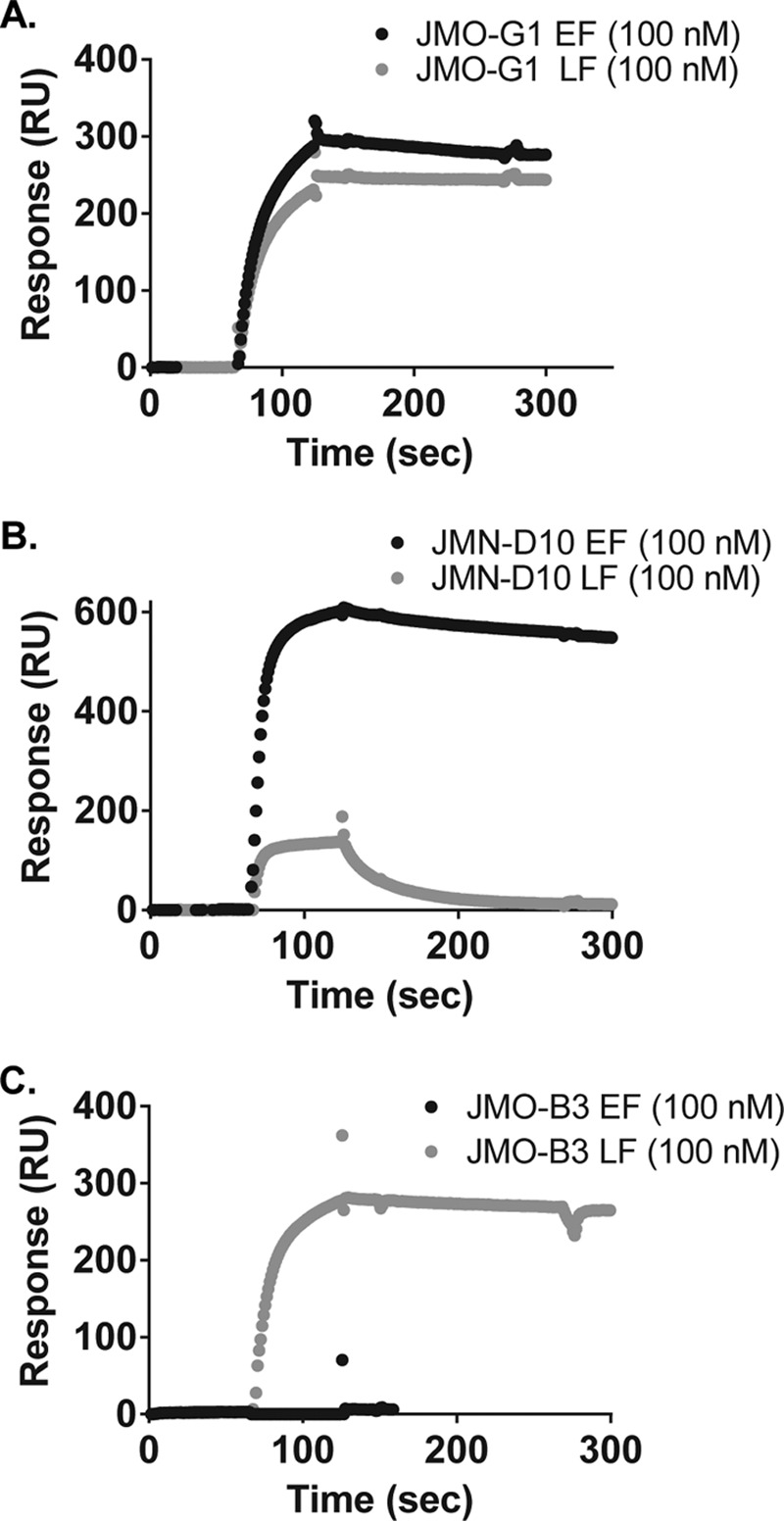
SPR binding curves for selected EF/LF binding VHHs. Binding of JMO-G1 (A), JMN-D10 (B), and JMO-B3 (C) to EF (black curves) and LF (gray curves) were assessed by SPR. Association and dissociation phases of curves are depicted for chips coupled to EF or LF and assessed with ∼100 nm VHH protein in the flow chamber. Representative curves are shown; triplicate curves were utilized to generate the mean binding parameters ± S.E. displayed in Table 2.
Epitope Characterization of the VHH Subset
Although three of the VHHs in the selected subset share the same competition group, they are distinct in their binding and/or neutralization properties.
This suggests that the antibodies recognize overlapping or contiguous, but not matching, epitopes, and/or that they differ in the kinetics of epitope binding to their paratopes. Therefore, we examined the locations and nature of each of the epitopes. We started by characterizing the binding of each of the VHHs in our experimental subset to the different domains of the LF protein (Fig. 4A). The LFN domain represents the N-terminal 263 amino acids of the 809 amino acid LF protein (Fig. 4A, red) and is the domain responsible for interaction with PA (Fig. 4B). The EF1/LF1 family binds to both LFN and LF by ELISA, confirming that these VHHs recognize an epitope(s) in the N terminus (Fig. 4D; supplemental Fig. S5A). In contrast, JMO-B9 binds to LF but not LFN (Fig. 4C; supplemental Fig. S5A). Therefore, JMO-B9 likely recognizes an epitope in the C terminus of LF. Alternatively, it may recognize a conformational epitope that is no longer present in recombinant LFN.
FIGURE 4.
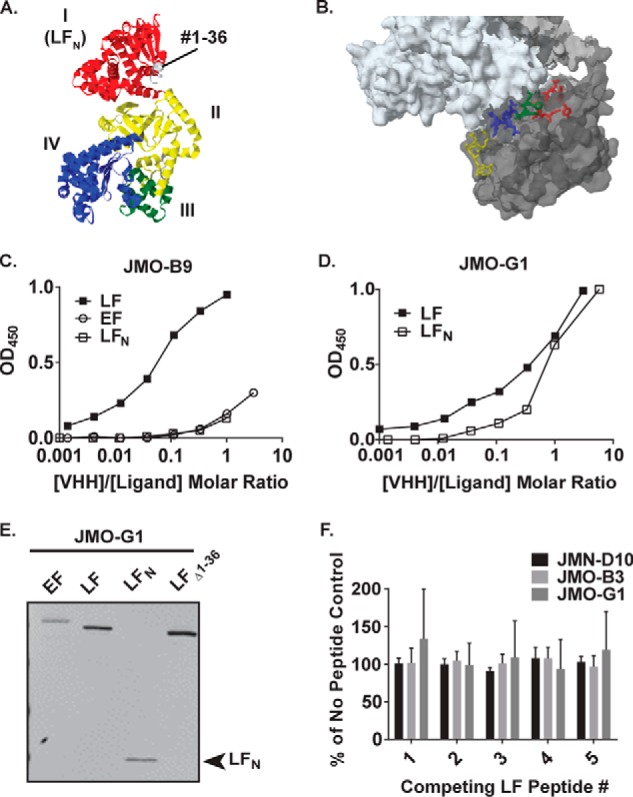
Additional epitope mapping of VHH binding to LF. A, crystal structure of LF (PDB 1J7N; Ref. 29) colored by domain. The LFN domain is colored in red, and residues 1–36 are colored in gray. B, location of LF residues represented by peptides in supplemental Table S1, as depicted on the co-crystal structure of PA (white) with LF (gray) (PDB 3KWV). Yellow, residues #179–184; blue, residues #231–236; green, residues #227–231; red, residues #136–143. C and D, ELISA curves assessing direct binding of HRP-labeled VHHs to plates coated with LF, EF, or LFN. Panel C depicts binding of JMO-B9, and panel D depicts binding of JMO-G1. Note that molar ratios of [VHH]/[antigen] are calculated as based on the total concentration of VHH in the reaction; however, only a fraction of the VHH was HRP labeled. As depicted here in a representative experiment, JMO-B9 failed to effectively bind to LFN in four independent experiments. E, boiled LF, EF, LFN, and LF(Δ1–36) proteins were blotted and probed with E-tagged JMO-G1 as indicated using an anti-E tag goat antibody and an IR dye-coupled anti-goat antibody for detection. The same pattern was observed for three other members of the EF1/LF1 family in three sets of replicate blots. In a control experiment (data not shown), no reactivity was observed in a parallel blot in which the VHH primary antibody incubation was omitted. F, results of peptide competition assay for binding of HRP-VHHs to LF. ELISA signals for JMN-D10, JMO-B3, and JMO-G1 in the presence of peptides 1–5 (supplemental Table S1) are expressed as a percentage of controls in which HRP-VHHs were incubated in the absence of competing peptides. Data are the mean ± S.D. of six replicates across two independent experiments. Continued increases in the concentration of competing peptides did not block binding in two additional experiments conducted with JMN-D10 and JMO-B3 (data not shown).
LFN docks to PA at two separate sites: at the α-clamp, involving an interaction between the first 36 amino acids of LFN and the N terminus of PA, and at the C-terminal PA subsite (10). We characterized the interaction between JMN-D10, JMO-B3, or JMO-G1 (EF1/LF1 family) and a Δ1–36 LF deletion construct. Binding was retained as assessed by ELISA (supplemental Fig. S5, B and C), indicating that the cross-neutralizing VHHs do not neutralize by blocking the interaction between LF and the PA α-clamp binding subsite and instead block binding to the PA C-terminal site.
For finer mapping, Thullier et al. (11) previously exploited the same observation of cross-reaction between EF and LF to map the epitopes of a different neutralizing antibody within LFN. The authors combined an LFN/EFN sequence alignment with a solvent exposure analysis to predict a series of potential cross-reactive epitopes: LF(97–103), LF(136–143), LF(178–184), LF(227–231), and LF(231–236). As a means of examining whether the epitopes for our VHH subset fall within any of these locations, as depicted on the structure of LF (Fig. 4B), we synthesized a set of linear peptides with sequences matching each of these five regions on LFN (supplemental Table S1). With the exception of peptide LF1, each of the peptides comprises residues that are located within the second LFN-PA binding cleft (as determined by the co-crystal structure; Fig. 4B).
Although in general VHHs are expected to recognize conformational epitopes, we did observe some reactivity of LF and EF in a Western blot probed with VHHs from the EF1/LF1 competition group (Fig. 4E), suggesting that these VHHs retain some recognition of the denatured forms of the epitopes. However, when used in direct binding ELISA (with coating of biotinylated peptides to a streptavidin plate, probed with the VHHs), none of the VHHs in our subset demonstrated evidence of peptide binding as compared with a biotinylated full-length LF control (data not shown). Additionally, pre-binding of the peptides to the VHHs did not reduce the level of binding to an ELISA plate coated with full-length LF (Fig. 4F). Other residues on the solvent-exposed surface of LFN remain as potential epitope binding sites, including LF residues in the range of 188–225 (another component of the second PA-LFN binding cleft; Ref. 10). Alternatively, the VHHs may not effectively recognize these short linear peptides.
Finally, we compared the epitope recognized by EF1/LF1 to the epitope(s) recognized by two anti-EF monoclonal antibodies that were previously described as binding to the N terminus of EF (12): 4A6 and 7F10. In the sandwich ELISA described above, no competition was observed (including between 4A6 and 7F10), indicating the presence of three distinct epitopes with neutralizing potential in the EF N terminus (data not shown).
Examination of the Neutralizing Mechanism of VHHs
The data from the epitope mapping experiments suggest that the mode of action for toxin neutralization by VHHs in the EF1/LF1 competition group is a hindrance of the binding of EF/LF to PA63, which prevents formation of the complete ET and LT toxins on the cell surface. ELISAs were performed to determine whether preincubation of LF and EF with the VHHs can inhibit toxin binding to PA63 oligomer (Fig. 5A). Consistent with the cell-based neutralization assays (Fig. 1; Table 1), JMN-D10 inhibited EF but not LF binding, JMO-B3 inhibited LF but not EF binding, and JMO-G1 inhibited both EF and LF binding to PA63 (Fig. 5B). JMO-B9, the LF-specific antibody in the distinct LF2 neutralization group, did not inhibit PA63 binding to either toxin, suggesting that it does not impact the interaction between LF/EF and PA63.
FIGURE 5.

VHH neutralization of LF and EF binding to PA63 oligomer. A, schematic diagram illustrating ELISA methods used in B. B, LF and EF were preincubated with JMN-D10, JMO-B3, and JMO-G1 at a molar ratio of 1:5 (LF or EF to VHH). The ability of LF and EF preincubated with VHH antibodies to bind to PA63 oligomer was assessed by ELISA and compared with LF and EF binding to PA63 oligomer in the absence of any VHH. Data are the mean ± S.E. of eight technical replicates from three independent experiments. The table (bottom) summarizes the ability of each VHH to neutralize LF or EF binding to PA63 oligomer. C, RAW264.7 cells were treated for 1 h with PA (1 μg/ml) and with either vehicle, VHHs, LF (1 μg/ml), or LF (1 μg/ml) preincubated for 1 h with VHHs (1.75 μg/ml) as in B, at a molar ratio of 1:5 (LF to VHH). Western blotting of cell lysates was performed to assess cleavage of MEK1, -2, and -3 by LF. The MEK1 and MEK2 antibodies recognize N-terminal epitopes that are degraded upon cleavage, and the MEK3 antibody recognizes an N-terminal epitope that is not degraded after cleavage. The larger MEK3 band is the full-length protein, and the smaller band is the N-terminal cleavage product. Actin is a control to demonstrate equal protein loading. Data are representative of three independent experiments.
Additional assays were performed to examine the inhibitory effects of the subset of VHHs on LF function under cellular conditions. Briefly, RAW264.7 cells were treated with LF that had been preincubated with VHHs and PA, and Western blotting was performed to assess MEK cleavage as a readout of toxin entry and activity (Fig. 5C; supplemental Fig. S6). Consistent with the ELISA data, JMO-B3 and JMO-G1 significantly inhibited LF entry and cleavage of MEK1, -2, and -3 (p < 0.05), whereas JMN-D10 did not (p > 0.05) (unpaired t test; Fig. 5C; supplemental Fig. S6). Surprisingly for an LF neutralizer, we found that JMO-B9 also did not prevent LF entry and MEK cleavage by the toxin, even at doses of toxin used in the neutralization assays and conditions where cells were protected from death (Fig. 5C; supplemental Fig. S6). Together, these data demonstrate that JMO-B3 and JMO-G1 are able to inhibit LF binding to PA63, toxin entry, and MEK cleavage, although JMO-B9, which binds to an epitope outside the PA-interacting N terminus of LF, does not prevent MEK cleavage.
Under two sets of assay conditions, JMO-B9 consistently led to a small but reproducible reduction in MEK cleavage (Fig. 5C; supplemental Fig. S6), suggesting the possibility of altered kinetics for interaction with this toxin substrate. The macrophage death utilized in the standard LF neutralization assay requires the toxin cleavage of the NLRP1b inflammasome sensor, which is expressed at undetectable levels in cells (data not shown). It is possible that a small impact on NLRP1b cleavage can result in protection from pyroptosis under our neutralization assay's “low toxin” conditions. Therefore, to test the hypothesis that the difference in impact of JMO-B9 on cleavage of MEK proteins relative to cell death is a kinetic effect, we compared JMO-B9, JMO-G1, and JMO-B3 to the potent PA-neutralizing VHH JKH-C7 under “high toxin” concentration neutralization assays (supplemental Fig. S7). JMO-B9 was no longer able to neutralize toxin when concentrations of 750 or 1500 ng/ml were used in neutralization assays, whereas JMO-B3, JMO-G1, and JKH-C7 were still neutralizing, albeit with higher IC50 values. These findings indicate that although JMO-B9 is identified as an LF-neutralizing VHH in the standard anthrax toxin neutralization assay, it is ineffective as a neutralizing agent over higher toxin concentrations.
In Vivo Therapeutic Efficacy of VHHs in Mice
To characterize the VHHs as therapeutic candidates, we first tested EF-neutralizing JMN-D10 for its ability to inhibit ET-induced edema in mouse footpads. BALB/cJ mice were IV injected with the PA-neutralizing VHH JKH-C7 or with JMN-D10 before footpad administration of ET, and edema was assessed at 21 h post-toxin administration. A single administration of 25 μg of either VHH was sufficient to significantly inhibit edema (p < 0.0001) almost to the levels of control footpads injected with PBS (Fig. 6A). The in vivo efficacy of JMN-D10 in this model suggests striking potency for this antibody acting in the first minutes after toxin administration and before VHH clearance.
FIGURE 6.
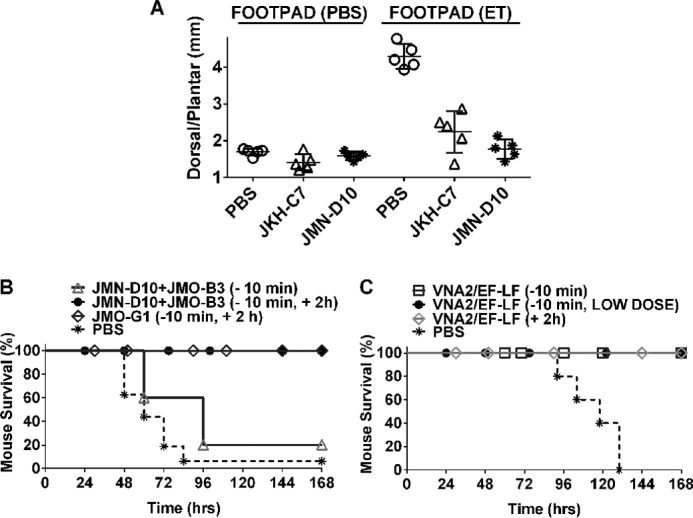
Therapeutic efficacy of VHHs in mice. A, footpad edema model. BALB/cJ mice (n = 5/group) were injected with PBS, JKH-C7, or JMN-D10 (25 μg/100 μl, i.v.) 10–20 min before footpad administration of ET (0.2 μg/20 μl) or PBS (20 μl). Each circle represents the average of three dorsal/plantar measurements for a single footpad/mouse at 21 h post-toxin administration. The p value comparing the ET footpad measurements of both antibody pretreated groups to the PBS group is <0.0001 using a standard unpaired t test. B and C, spore challenge model. C57BL/6J mice were challenged with a lethal dose of 5 × 107 spores (SC, 400 μl). In B, mice received PBS (n = 16) or VHH treatments, either as a single administration (16 μg/VHH, SC, 10 min pre-spore infection, n = 5) or as two administrations (16 μg, SC, 10 min pre-spore infection followed by 32 μg, SC, 2 h post-spore infection, n = 5). JMO-G1, targeting both LF and EF, was injected alone (n = 9), whereas JMN-D10 (targeting EF) and JMO-B3 (targeting LF) were administered to mice as a mixture (n = 10). Results shown are from three studies. In C, VNA2/EF-LF was administered at either 16 μg, SC, 10 min pre-spore infection, or 2 h post-spore infection. In one group (LOW DOSE), the VNA was administered at 4 μg, SC, only 10 min pre-spore infection. Control mice received PBS at 10 min pre-infection (n = 5/treatment group).
To establish infection and disseminate, B. anthracis requires LF and EF to disable innate immune cells early in infection (13). We next tested EF-neutralizing JMN-D10 in combination with the LF-neutralizing JMO-B3 for their ability to protect against lethal spore infection of C57BL/6J mice. Antibodies against EF alone are usually only partially protective against B. anthracis Sterne spore challenge in mice (12, 14), but in combination with LF antibodies (data not shown) or LF inhibitors (14) there is an additive protective effect. It is thought that both toxins impact the innate immune system, with LF playing the primary role in disabling immune cells to allow bacterial dissemination (13).
Although a single administration of these antibodies (16 μg, 10 min before infection) only delayed malaise and death, an administration of two doses (16 μg, 10-min pre-infection + 32 μg, 2-h post-infection) led to full protection against lethal spore challenge (Fig. 6B). As >90% of spores germinate within 5 min of subcutaneous administration in mice, the two injections likely allow for a more complete neutralization of the toxin produced in the first few hours, which is crucial for the dismantling of the innate immune response (13). The first injection allows a head start for antibody distribution in the body before spore germination and toxin production, whereas the second ensures an antibody supply in the most important window for bacterial dissemination, allowing protection. Similarly, administration of two injections of JMO- G1, which targets both LF and EF binding to PA63, was also fully protective (Fig. 6B).
Finally, we tested a bispecific VHH heterodimer called VNA2/EF-LF (a “VHH-based neutralizing agent”) containing two linked VHHs, JMN-D10 and JMO-G1, for its ability to protect mice in similar infection studies. This VNA also contains a short albumin binding peptide to improve serum persistence (15). Administration of a single dose of VNA2/EF-LF, even at a dose of 4 μg, protected all mice (Fig. 6C). Administration of VNA2/EF-LF as a post-treatment 2 h after spore administration was also protective. These results indicate that both the individual VHHs and the bispecific VNA are capable of neutralizing toxins produced in early stages of infection, preventing dissemination of the germinated bacteria, likely by enabling the innate immune response to destroy them.
Discussion
In our previous work (3) we reported a series of VHHs that neutralize the protective antigen of the anthrax toxins, thus preventing the entry of LF or EF into cells. Here, we expand our repertoire of anti-anthrax toxin VHHs, developing antibodies of this stable and easy-to-produce type against the LF and/or EF components of the toxins themselves. We identify multiple competition groups for neutralization, including an EF/LF cross-reactive group that neutralizes the toxins in a cell-based assay via steric occlusion of the PA-LF/EF interaction in the N terminus of LF and EF (i.e. LFN, EFN). Of particular interest in this EF1/LF1 cross-reactive group was the significant level of diversity in EF/LF binding affinity and neutralizing potential, suggesting paratope diversity and partial (as opposed to complete) epitope overlap within this competition group. This diversity is exemplified by VHHs like JMN-D10, with excellent anti-EF neutralizing activity but poor LF neutralization, and JMO-G1, which potently neutralizes both EF and LF.
The only LF neutralizing antibody tested that did not interact with LFN was JMO-B9. This VHH is thought to neutralize by a distinct mechanism involving the C terminus of LF. Most anti-LF monoclonal antibody therapeutics are similar to the other VHHs described in this work and neutralize via LFN binding (2). Exceptions include monoclonal antibodies 5B13B1 and 3C16C3, which have neutralizing activity via binding of domain III of LF (16). JMO-B9 neutralizing action did not involve hindrance of binding of LF to the PA63 pore, toxin translocation, or even inhibition of LF cleavage of its cellular substrates. Based on the difference between the cellular neutralization results and the MEK cleavage results for this antibody, we propose that the neutralization of LF by JMO-B9 represents a kinetic effect on the rate of cleavage, possibly due to impact on the rate of LF entry into the cytoplasm. We verified that under high toxin conditions, JMO-B9 was no longer able to neutralize LT and thus represents a poor therapeutic candidate (despite its interest from an epitope mapping perspective).
Combinatorial targeting of EF and LF has previously been shown to have added benefit in protection against anthrax infection (14). In this study we also show that combination of anti-EF and anti-LF targeting VHHs or a VNA made of such VHH antibodies is protective in a mouse model of anthrax. Our anti-PA, anti-EF, and anti-LF VHH candidates provide a set of new reagents with a range of affinities for potential use in diagnostic and/or biotechnology applications. The VHHs were readily produced in high yield as highly stable molecules that can be combined with other VHHs for enhanced therapeutic efficacy as well as fused to domains that provide properties such as improved serum stability, simplified purification, etc. From a diagnostic standpoint, the identification of EF- and LF-specific VHHs also could lead to assays that determine the specific levels of ET and LT pools in the patient, in contrast to overall toxin quantitation as measured using anti-PA antibodies.
The VHHs identified in this work were effective in protecting mice from footpad edema and lethality induced by B. anthracis Sterne infection. VNA2/EF-LF, a bispecific neutralizing agent composed of two of these antibodies, appeared to display more potent efficacy than monomers in the infection model. Multispecific VHH neutralizing agents, called VNAs, have routinely displayed greater potency when compared with the unlinked component VHH monomer pools (3, 6, 7, 9, 17). A similar assertion, though, cannot be confirmed for VNA2/EF-LF based only on the data presented here for two reasons. First, the VNA, but not the VHHs, contains an albumin-binding peptide that substantially increases its serum half-life (15), and this improved persistence may fully explain the VNA improved potency compared with VHH monomers. Secondly, we could not effectively test the relative per molar efficacy of the bispecific VNA against the VHHs because a combined LT/ET toxin challenge model does not exist in mice, primarily due to the time-dependent opposing effects that LT and ET toxins have on immune cells and the vasculature in mice.
VHHs and VNAs have a number of advantages over conventional polyclonal and monoclonal antibody antitoxin as well as some possible disadvantages. These protein agents generally have comparable affinities and antitoxin potencies, whereas the VHH-based agents can be produced at less cost using microbial hosts and are more stable than conventional antibodies (for review, see Refs. 4 and 5). VHH-based agents are also more versatile as they express well as multifunctional fusion proteins (6–8, 18–20) and can be delivered efficiently by gene therapy methods (15, 21). A possible disadvantage of VHH-based agents for some applications is their shorter serum half-lives compared with conventional antibodies. When longer serum stability is desired, the short half-life of VHHs and VNAs (<1.5 h) can be substantially extended by use of serum protein binding partners or gene therapy (15, 22). Some concerns have also been raised regarding the possible immunogenicity of camelid VHH-based agents in patients. Although these proteins are considered poorly immunogenic (4, 5), immunogenicity has been demonstrated with long term exposures (15), although it is unclear if this is more pronounced than the anti-idiotypic responses that can follow prolonged exposure to monoclonal antibodies (23).
In sum, these studies pave the way for development of a VNA that combines multiple anthrax toxin-neutralizing VHHs: anti-PA VHHs from our previous work (3) and anti-LFN + EFNVHHs from this work. This type of combined VNA, possibly delivered by a gene therapy vehicle, has the potential to be a superior, cross-neutralizing product of interest for further therapeutic development.
Experimental Procedures
Ethics Statement
All studies used protocols that were approved by the Animal Care and Use Committees of Tufts University and the National Institutes of Health.
Reagents
Horseradish peroxidase (HRP)-conjugated and non-conjugated anti-E-tag antibodies were purchased from Bethyl Labs (Montgomery, TX), and IR-tagged secondary antibodies were purchased from Rockland Immunochemicals (Boyertown, PA) or LI-COR Biosciences (Lincoln, NE). HRP-conjugated anti-rabbit antibody (sc-2054), anti-MEK3 NT (sc-959), and anti-MEK2 (sc-524) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-MEK1 NT antibody (M2865–04B) was purchased from United States Biological (Salem, MA). VHHs, LF, and EF were HRP-labeled for ELISA experiments with a conjugation kit from Abcam (Cambridge, MA). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma. Peptides for epitope mapping were ordered conjugated to biotin from the Peptide Technologies Branch of NIAID, National Institutes of Health. ELISA developer solutions (DY999 and DY994) were purchased from R&D Systems (Minneapolis, MN). Anti-EF monoclonal antibodies 4A6 and 7F10 (12), anti-PA VHH JKH-C7 (3), and anti-PA monoclonal 14B7 (24) have been previously described.
Toxins and Spores
Endotoxin-free preparations of PA, LF, and EF were purified from B. anthracis as previously described (25). The LF used here is a recombinant protein having an N-terminal sequence beginning with HMAGG. The EF used here is a recombinant protein having the original N-terminal sequence of EF. LFN (amino acids 1–255), with a C-terminal extension of 10 amino acids, was produced by cleavage of a fusion protein.
Spores were prepared from the toxigenic non-encapsulated Sterne-like A35 strain that has been previously described (26). Bacteria were grown on nutrient broth yeast extract (NBY) agar medium at 37 °C for 1 day followed by 5 days at 30 °C and inspected by microscopy to verify >95% sporulation. Spores were purified off plates by washing with sterile water (10–20 ml/Petri plate) followed by four additional rounds of centrifugation and water washes. Spores were treated for 45 min at 75 °C to kill any remaining vegetative bacteria, and viable spore counts were completed by dilution plating.
Preparation of VHH Phage Display Library from Immunized Alpacas
Two alpacas were immunized with both LF and EF (100 μg each) by five successive multisite subcutaneous (SC) injections at 3-week intervals. For the first immunization, the antigen was in alum/CpG adjuvant, and subsequent immunizations contained only alum. Both alpacas achieved anti-LF or anti-EF titers of >105. Blood was obtained for lymphocyte preparation 3 days after the fifth immunization. Frozen lymphocytes were treated with RNAlater-ICE as described by the manufacturer (Thermo Fisher Scientific, Waltham, MA), and RNA was prepared using the RNeasy Plus Mini kit (Qiagen, Valencia, CA). A VHH display phage library was prepared in TG1 electroporation-competent cells (Agilent, Santa Clara, CA) as described previously (3), yielding a library (JMG-1) having a complexity of ∼5 × 106 independent clones with >95% containing VHH inserts.
Identification and Purification of VHHs
Phage library panning and phage recovery have been previously described (7, 17, 27). The JMG-1 VHH display library (described above) was panned on full-length LF or EF coated onto Nunc Immunotubes (Thermo Fisher Scientific). Initial panning was performed on plastic coated with 10 μg/ml target followed by a second round of panning at high stringency coated at 1 μg/ml and employing a 10-fold lower titer of input phage, shorter binding times, and longer washes. Ninety-five random clones from the selected population were screened for expression of VHHs that bound to EF or LF, and more than half demonstrated significant binding to their panning target protein (>2× background), with many binding to both targets.
About 25 clones producing the strongest positive signal on one or both protein targets were characterized by DNA fingerprinting, and the sequence of the coding DNA for the best (highest ELISA signal) clone possessing each unique fingerprint was obtained. From this initial screen we identified two unique VHHs (having no evidence of a common B cell clonal origin) that were specific only to EF, seven unique VHHs specific only to LF, and six unique VHHs that recognized both LF and EF. All 15 of these unique EF, LF, or EF/LF VHHs were expressed and purified as His6-tagged recombinant E. coli thioredoxin fusions with a C-terminal E-tag, as previously described (17). Specifically, expression plasmids containing the VHH coding DNAs were transformed into Rosetta-gamiTM 2(DE3)pLacI cells, induced at 0.6 A600 with 1 mm IPTG, and cultured overnight at 15 °C. Cells were disrupted in BugBuster (Thermo Fisher Scientific), and the VHHs were purified on nickel-agarose (Thermo Fisher Scientific) as recommended by the manufacturer and then dialyzed against PBS. Purified proteins were characterized for purity and protein concentration by SDS-PAGE band intensity versus standards. An alignment of protein sequences of the VHHs selected for expression is displayed in supplemental Fig. S1, and Table 1 summarizes the binding properties of the VHHs as determined below.
Expression and Purification of VNA2/EF-LF
A synthetic gene encoding both JMO-G1 and JMN-D10 VHHs separated by a flexible spacer ((GGGGS)3) was ligated into pET32b (EMD Millipore, Darmstadt, Germany) in-frame with an N-terminal E. coli thioredoxin, flanked by two E-tag sequences and having a C-terminal albumin-binding peptide (15). Expression and purification of the VNA was performed as for the VHHs above.
ELISA Experiments
Dilution ELISAs for EC50 Measurement
VHHs were serially diluted in Immulon 4HBX polystyrene 96-well plates (Thermo Fisher Scientific) coated with 1 μg/ml LF or EF, incubated for 1 h, and detected with HRP-anti-E-tag antibody, as described in Moayeri et al. (3). EC50 values displayed in Table 1 reflect the VHH concentration that produced a signal equal to 50% of the peak binding signal.
ELISAs for Epitope Binning
VHHs were placed into competition groups, as displayed in Table 1, via sandwich ELISA (supplemental Fig. S3). Plates were coated with 1 μg/ml of the test VHH followed by blocking with 3% BSA. HRP-labeled EF or LF (1 μg/ml) was preincubated for 1 h with “blocking” VHH antibodies (1:5 molar ratio of HRP-LF/HRP-EF to VHH) before adding to the plate. After washing and developing, signals were expressed as a percentage of the signal from positive controls in which HRP-LF or HRP-EF was preincubated with vehicle (PBS), with the use of wells without HRP- LF/HRP-EF addition as a negative control. A parallel sandwich ELISA experiment was performed for HRP-EF with two previously described anti-EF mouse monoclonal antibodies: 4A6 and 7F10 (12). For a subset of VHHs, a modified sandwich ELISA with LF (supplemental Fig. S4) was utilized for additional epitope binning.
ELISAs to Detect VHH Blocking of PA63 Binding
ELISAs were performed to assess the ability of preincubation with various VHH antibodies to block the binding of LF or EF to PA63 oligomer. Briefly, Immulon plates were coated with PA63 (5 μg/ml, 2 h) and then incubated with LF or EF (1 μg/ml) that had been preincubated for 1 h with VHH antibodies (1:5 molar ratio of LF/EF to VHH) or vehicle. LF or EF binding to PA63 oligomer was detected with a polyclonal rabbit-anti-LF or -EF antibody, anti-rabbit IgG-HRP, and ELISA developer solutions.
Toxin Neutralization Experiments
RAW264.7 mouse macrophages were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 10 mm HEPES, and 50 μg/ml gentamicin (all purchased from Gibco). PA83 and either LF or EF (final concentration 250 ng/ml for each toxin component) were preincubated with various dilutions of antibody for 1 h. The LT-VHH or ET-VHH mixtures were transferred to macrophages and incubated at 37 °C. For LT neutralization assays, cells were monitored every 30 min by light microscopy, and viability staining was performed when >90% of toxin-treated controls were lysed. Viability was measured by MTT dye as previously described (28). For ET neutralization, cells were incubated for 1 h with toxin, and total cAMP levels were assessed using the BioTRAK cAMP immunoassay (GE Healthcare) according to manufacturer's protocols. EF-neutralizing monoclonal antibody 7F10 (12), PA-neutralizing monoclonal antibody 14B7 (24), and PA-neutralizing VHH JKH-C7 (3) were used as controls in these assays. In certain LF neutralization experiments (supplemental Fig. S7), toxin concentrations of 750 ng/ml or 1500 ng/ml were used.
Affinity Screening by SPR
Studies to assess the kinetic parameters of VHH binding to LF or EF were carried out on a Biacore 3000 instrument (GE Healthcare). Full-length EF or LF was immobilized to a CM5 chip by amine coupling chemistry, as described in Moayeri et al. (3). VHHs were passed over the chip surface at 100 nm and 100 μl/min for 60 s, and dissociation was recorded for 200+ seconds in the standard running buffer of 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% Tween 20. The surface was regenerated between runs with 10 mm glycine, pH 2 or 3. Dissociation and association phases of each curve were fit separately with BIAevaluation software (GE Healthcare, 1:1 Langmuir model) to determine ka, kd, and KD values.
Western Blotting
Western blots were performed on LF and EF using VHHs as probing antibodies. Purified proteins were boiled (in 1× Tris-glycine loading buffer; Thermo Fisher) and separated on 4–20% Tris-glycine gels (Thermo Fisher), transferred to nitrocellulose, and probed with purified (E-tagged) VHHs at ≈2 μg/ml final concentration in Odyssey Blocking Buffer + 0.05% Tween 20 overnight at room temperature. VHHs were detected with (goat) anti-E-tag antibody at a 1:2,000 dilution (1 h, room temperature) followed by an anti-goat antibody (IR800 conjugated; 1:10,000); blots were visualized on the LI-COR Odyssey Infrared Imaging system.
To detect VHH inhibition of LF-mediated MEK cleavage, RAW264.7 cells were treated with PA (1 μg/ml) and with either vehicle, VHHs, LF, or LF preincubated with VHHs at a molar ratio of 1:5 (LF:VHH). Cells were lysed with radioimmunoprecipitation assay buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS in PBS) containing protease inhibitors, and Western blotting was performed using primary antibodies against MEK2 and MEK3 followed by probing with IR dye-conjugated secondary antibodies and imaging as described above.
Tm determination by Nanoscale Differential Scanning Fluorimetry
Tms (melting temperatures) were determined for VHH- thioredoxin fusion proteins on the Prometheus NT.48 differential scanning fluorimetry instrument (NanoTemper Technology, Munchen, Germany). Protein samples at ≈1 mg/ml were melted at a rate of 0.8 °C/min from 25 to 90 °C. Absorbance at 350 nm was monitored as a readout of the chemical environment of tryptophans in the protein, and Tms were fit with a polynomial function. Two Tms were observed for each protein, with the lower Tm (values displayed in Table 2) reflecting the VHH domain, and the higher Tm (≈80 °C) reflecting the thioredoxin domain.
Epitope Mapping Experiments
In an attempt to identify shared LF/EF epitope(s) recognized by the VHHs, direct binding ELISA and peptide competition ELISA were performed using five synthesized, biotinylated peptides (supplemental Table S1). In the direct binding ELISA, peptides were bound overnight (at 5 μg/ml) to a streptavidin-coated plate followed by application of HRP-labeled VHHs in competition group EF1/LF1 for 1–2 h to probe for binding. Biotin-LF protein was used as a positive control for VHH binding. In the peptide competition ELISA, LF was coated to a plate at 5 μg/ml. HRP-labeled VHHs were individually preincubated with each of the peptides at a >100:1 peptide:VHH molar ratio before application to the plate, and signals were compared with those for HRP-VHH preincubated with vehicle (PBS).
Spore Challenge Studies
C57BL/6J mice (8–10 weeks old, female; The Jackson Laboratory, Bar Harbor, ME) were challenged with a lethal dose of 5 × 107 spores (SC, 400 μl). Mice also received either VHH or VNA (4 μg, 16 μg, or 32 μg, SC, distal site) or PBS (SC) at 10 min before or both 10 min before and 2 h post spore infection.
Footpad Edema Studies
BALB/cJ mice (8–10 weeks old, female; The Jackson Laboratory) (n = 5/group) were IV injected with PBS or VHH antibodies 10–20 min before injection of ET (0.2 μg/20 μl, right footpad) or PBS (20 μl, left footpad). Edema was assessed at 21 h by dorsal/plantar measurements using digital calipers.
Author Contributions
C. E. V., M. M., A. B. K., and A. J. G. designed and performed the experiments, analyzed the data, and drafted the manuscript. D. O. performed the experiments. J. T. designed and performed the experiments and analyzed the data. C. B. S. and S. H. L. drafted the manuscript, provided reagents, and analyzed/interpreted the data. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Rasem Fattah of the NIAID, National Institutes of Health (NIH) for protein purification, Sergey Tarasov and Marzena Dyba of the NCI (NIH) as well as NanoTemper Technologies for melting temperature determinations, and David Garboczi and Apostolos Gittis of NIAID for assistance with surface plasmon resonance. We also thank Dr. Daniela Bedenice and Dr. Jean Mukherjee and her group for their skilled efforts in acquiring the alpacas, performing the immunizations and bleeds, and preparation of the PBLs.
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) and by National Institutes of Health Grant U54 AI057159 (to C. B. S). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1 and Figs. S1–S7.
- ET
- edema toxin
- LT
- lethal toxin
- EF
- edema factor
- PA
- protective antigen
- LF
- lethal factor
- VHH
- variable domains of camelid heavy chain-only antibody
- VNA
- VHH-based neutralizing agent/heteromultimer of neutralizing VHHs
- Tm
- melting temperature
- SPR
- surface plasmon resonance
- MTT
- 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide
- SC
- subcutaneous.
References
- 1. Moayeri M., Leppla S. H., Vrentas C., Pomerantsev A. P., and Liu S. (2015) Anthrax pathogenesis. Annu. Rev. Microbiol. 69, 185–208 [DOI] [PubMed] [Google Scholar]
- 2. Chen Z., Moayeri M., and Purcell R. (2011) Monoclonal antibody therapies against anthrax. Toxins 3, 1004–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moayeri M., Leysath C. E., Tremblay J. M., Vrentas C., Crown D., Leppla S. H., and Shoemaker C. B. (2015) A heterodimer of a VHH (variable domains of camelid heavy chain-only) antibody that inhibits anthrax toxin cell binding linked to a VHH antibody that blocks oligomer formation is highly protective in an anthrax spore challenge model. J. Biol. Chem. 290, 6584–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 [DOI] [PubMed] [Google Scholar]
- 5. Hassanzadeh-Ghassabeh G., Devoogdt N., De Pauw P., Vincke C., and Muyldermans S. (2013) Nanobodies and their potential applications. Nanomedicine 8, 1013–1026 [DOI] [PubMed] [Google Scholar]
- 6. Vance D. J., Tremblay J. M., Mantis N. J., and Shoemaker C. B. (2013) Stepwise engineering of heterodimeric single domain camelid VHH antibodies that passively protect mice from ricin toxin. J. Biol. Chem. 288, 36538–36547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mukherjee J., Tremblay J. M., Leysath C. E., Ofori K., Baldwin K., Feng X., Bedenice D., Webb R. P., Wright P. M., Smith L. A., Tzipori S., and Shoemaker C. B. (2012) A novel strategy for development of recombinant antitoxin therapeutics tested in a mouse botulism model. PLoS ONE 7, e29941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tremblay J. M., Mukherjee J., Leysath C. E., Debatis M., Ofori K., Baldwin K., Boucher C., Peters R., Beamer G., Sheoran A., Bedenice D., Tzipori S., and Shoemaker C. B. (2013) A single VHH-based toxin-neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect. Immun. 81, 4592–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herrera C., Tremblay J. M., Shoemaker C. B., and Mantis N. J. (2015) Mechanisms of ricin toxin neutralization revealed through engineered homodimeric and heterodimeric camelid antibodies. J. Biol. Chem. 290, 27880–27889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feld G. K., Thoren K. L., Kintzer A. F., Sterling H. J., Tang I. I., Greenberg S. G., Williams E. R., and Krantz B. A. (2010) Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 17, 1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thullier P., Avril A., Mathieu J., Behrens C. K., Pellequer J. L., and Pelat T. (2013) Mapping the epitopes of a neutralizing antibody fragment directed against the lethal factor of Bacillus anthracis and cross-reacting with the homologous edema factor. PLoS ONE 8, e65855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leysath C. E., Chen K. H., Moayeri M., Crown D., Fattah R., Chen Z., Das S. R., Purcell R. H., and Leppla S. H. (2011) Mouse monoclonal antibodies to anthrax edema factor protect against infection. Infect. Immun. 79, 4609–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu S., Miller-Randolph S., Crown D., Moayeri M., Sastalla I., Okugawa S., and Leppla S. H. (2010) Anthrax toxin targeting of myeloid cells through the CMG2 receptor is essential for establishment of Bacillus anthracis infections in mice. Cell Host Microbe 8, 455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moayeri M., Crown D., Jiao G. S., Kim S., Johnson A., Leysath C., and Leppla S. H. (2013) Small-molecule inhibitors of lethal factor protease activity protect against anthrax infection. Antimicrob. Agents Chemother. 57, 4139–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukherjee J., Dmitriev I., Debatis M., Tremblay J. M., Beamer G., Kashentseva E. A., Curiel D. T., and Shoemaker C. B. (2014) Prolonged prophylactic protection from botulism with a single adenovirus treatment promoting serum expression of a VHH-based antitoxin protein. PLoS ONE 9, e106422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lim N. K., Kim J. H., Oh M. S., Lee S., Kim S. Y., Kim K. S., Kang H. J., Hong H. J., and Inn K. S. (2005) An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infect. Immun. 73, 6547–6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tremblay J. M., Kuo C. L., Abeijon C., Sepulveda J., Oyler G., Hu X., Jin M. M., and Shoemaker C. B. (2010) Camelid single domain antibodies (VHHs) as neuronal cell intrabody binding agents and inhibitors of Clostridium botulinum neurotoxin (BoNT) proteases. Toxicon. 56, 990–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pant N., Marcotte H., Hermans P., Bezemer S., Frenken L., Johansen K., and Hammarström L. (2011) Lactobacilli producing bispecific llama-derived anti-rotavirus proteins in vivo for rotavirus-induced diarrhea. Future Microbiol. 6, 583–593 [DOI] [PubMed] [Google Scholar]
- 19. Hultberg A., Temperton N. J., Rosseels V., Koenders M., Gonzalez-Pajuelo M., Schepens B., Ibañez L. I., Vanlandschoot P., Schillemans J., Saunders M., Weiss R. A., Saelens X., Melero J. A., Verrips C. T., Van Gucht S., and de Haard H. J. (2011) Llama-derived single domain antibodies to build multivalent, superpotent and broadened neutralizing anti-viral molecules. PLoS ONE 6, e17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaliberov S. A., Kaliberova L. N., Buggio M., Tremblay J. M., Shoemaker C. B., and Curiel D. T. (2014) Adenoviral targeting using genetically incorporated camelid single variable domains. Lab. Invest. 94, 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moayeri M., Tremblay J. M., Debatis M., Dmitriev I. P., Kashentseva E. A., Yeh A. J., Cheung G. Y., Curiel D. T., Leppla S., and Shoemaker C. B. (2016) Adenoviral expression of a bispecific VHH- based neutralizing agent that targets protective antigen provides prophylactic protection from anthrax in mice. Clin. Vaccine Immunol. 23, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adams R., Griffin L., Compson J. E., Jairaj M., Baker T., Ceska T., West S., Zaccheo O., Dave E., Lawson A. D., Humphreys D. P., and Heywood S. (2016) Extending the half-life of a fab fragment through generation of a humanized anti-human serum albumin fv domain: an investigation into the correlation between affinity and serum half-life. Mabs 10.1080/19420862.2016.1185581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. López-Requena A., Burrone O. R., and Cesco-Gaspere M. (2012) Idiotypes as immunogens: facing the challenge of inducing strong therapeutic immune responses against the variable region of immunoglobulins. Front. Oncol. 2, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Little S. F., Leppla S. H., and Cora E. (1988) Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect. Immun. 56, 1807–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park S., and Leppla S. H. (2000) Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr. Purif. 18, 293–302 [DOI] [PubMed] [Google Scholar]
- 26. Pomerantsev A. P., Sitaraman R., Galloway C. R., Kivovich V., and Leppla S. H. (2006) Genome engineering in Bacillus anthracis using Cre recombinase. Infect. Immun. 74, 682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maass D. R., Harrison G. B., Grant W. N., and Shoemaker C. B. (2007) Three surface antigens dominate the mucosal antibody response to gastrointestinal L3-stage strongylid nematodes in field immune sheep. Int. J. Parasitol. 37, 953–962 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z., Moayeri M., Crown D., Emerson S., Gorshkova I., Schuck P., Leppla S. H., and Purcell R. H. (2009) Novel chimpanzee/human monoclonal antibodies that neutralize anthrax lethal factor, and evidence for possible synergy with anti-protective antigen antibody. Infect. Immun. 77, 3902–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pannifer A. D., Wong T. Y., Schwarzenbacher R., Renatus M., Petosa C., Bienkowska J., Lacy D. B., Collier R. J., Park S., Leppla S. H., Hanna P., and Liddington R. C. (2001) Crystal structure of the anthrax lethal factor. Nature 414, 229–233 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.