

Abstract
The lipid bilayer is a dynamic environment that consists of a mixture of lipids with different properties that regulate the function of membrane proteins; these lipids are either annular, masking the protein hydrophobic surface, or specific lipids, essential for protein function. In this study, using tandem mass spectrometry, we have identified specific lipids associated with the Escherichia coli ABC transporter McjD, which translocates the antibacterial peptide MccJ25. Using non-denaturing mass spectrometry, we show that McjD in complex with MccJ25 survives the gas phase. Partial delipidation of McjD resulted in reduced ATPase activity and thermostability as shown by circular dichroism, both of which could be restored upon addition of defined E. coli lipids. We have resolved a phosphatidylglycerol lipid associated with McjD at 3.4 Å resolution, whereas molecular dynamic simulations carried out in different lipid environments assessed the binding of specific lipids to McjD. Combined, our data show a synergistic effect of zwitterionic and negatively charged lipids on the activity of McjD; the zwitterionic lipids provide structural stability to McjD, whereas the negatively charged lipids are essential for its function.
Keywords: ABC transporter, circular dichroism (CD), lipid, membrane protein, membrane transport, molecular dynamics, structural biology, structure-function, delipidation, non-denaturing mass spectrometry
Introduction
Biological membranes contain hundreds of lipid types, which differ in acyl chain length and degree of unsaturation, and head group polarity, size, and charge (1). Such a complex lipid environment is essential to maintain the structural integrity and function of membrane proteins, which often have defined binding sites for lipids (2). Annular lipids are usually defined as the first lipid shell that surrounds the protein surface spanning the membrane (3). The interactions established with the protein, although weak and usually not essential for protein function, allow for decreased exchange rates with the bulk lipids, thus promoting the formation of the lipid annulus that masks the hydrophobic surface of the protein (4). Lower exchange rates are found for specific lipids, tightly bound between transmembrane (TM)6 helices or crevices (4). Specific lipids are associated with the activity of membrane proteins, and their loss during protein purification usually results in aggregation or reduced transport and enzymatic activity.
In recent years, there has been an increase in the numbers of membrane protein structures determined at medium to high resolution. Some of these structures show resolved lipids, which most probably are involved in specific interactions because they have survived the purification and crystallization processes. In many cases, no correlation between the structure and function of the specific lipids has been made. For the sarcoplasmic reticulum Na,K-ATPase (5), Ca-ATPase (SERCA) (6), and the bacterial secondary active betaine transporter BetP from Corynebacterium glutamicum (7), the influence of specific lipids on the activity of these proteins has clearly been demonstrated.
ABC transporters are one of the largest superfamilies found in eukaryotes, archaea, and prokaryotes. They are powered by the hydrolysis of ATP and can be classified as exporters and importers, the latter found only in bacteria. ABC exporters are associated with multidrug resistance in both bacterial and eukaryotic cells. To date, there are a few structures of ABC exporters, but none of these structures show well resolved specific lipids bound to them. Moreover, despite the importance of lipids, no detailed studies have linked their function to a specific lipid class. For example, the ABC exporter TmrAB, from Thermus thermophilus, is associated with phosphatidylglycerol (PG) and lipid A, as revealed by mass spectrometry (8). However, electron density in the low resolution electron cryo-microscopy single particle reconstruction envelope of TmrAB was assigned to the detergent micelle (9). In the low resolution crystal structure of the eukaryotic P-glycoprotein, no lipids were observed (10, 11), but non-denaturing mass spectrometry identified tightly associated lipids and a clear dependence of ligand and lipid binding (12).
To understand how specific lipids modulate the activity of ABC exporters, we have selected the antibacterial peptide ABC transporter McjD from E. coli (13). McjD is involved in the export of the antibacterial peptide microcin J25 (MccJ25), conferring immunity to the producing strain toward the peptide. MccJ25 is a plasmid-encoded, ribosomally synthesized peptide consisting of 21 amino acids with a unique lasso structure (14, 15). It is produced under conditions of nutrient exhaustion and exerts potent antibacterial activity against closely related species. Inside the cell, MccJ25 inhibits the bacterial RNA polymerase (16, 17). The uptake of MccJ25 into the target cell is facilitated by the outer membrane protein FhuA, dependent on the TonB pathway (18, 19), and the inner membrane protein SbmA in E. coli (18, 20). We have previously determined the high resolution structure of McjD (13), and we observed electron density that could be attributed to specific lipids. However, the electron density was not strong enough to model in structures of lipids. We have also shown that detergent-purified and proteoliposome-reconstituted McjD hydrolyzes ATP with a rate of 35 and 75 nmol/min/mg McjD, respectively (13), suggesting that lipids are important for the ATPase activity.
In light of these observations, we have combined functional and structural studies to characterize a specific class of lipids associated with the function of the ABC exporter McjD. Using non-denaturing and tandem mass spectrometry, we have identified specific lipids, phosphatidylethanolamine (PE) and PG, that are tightly bound to McjD and have survived the purification and remained associated to the protein in the gas phase. Because the E. coli inner membrane consists mainly of PE, PG, and cardiolipin (CARD), we performed basal and ligand-induced ATPase assays in detergent-destabilized small unilamellar vesicles with different lipid compositions, and we showed that both PE and PG rescued the ligand-induced ATPase activity of a delipidated McjD protein. These data were further verified by measuring the melting curves in the different lipids. Our structural data at 3.4 Å resolution revealed one resolved PG lipid. MD simulations performed on McjD embedded in different lipid environments provided molecular insights into the lipid organization in the proximity of the protein. Although zwitterionic (PE) and negatively charged lipids (PG) exchange to interact with the transporter, positively charged residues located on the protein surface provide preferable interactions sites for PG and CARD.
Results
Mass Spectrometry of McjD Reveals Co-purified Phospholipids and Lipopolysaccharides
Following expression and purification of McjD from E. coli, we used non-denaturing mass spectrometry to determine the oligomeric state of the protein and presence of any bound lipids. The mass spectrum of McjD solubilized in n-dodecyl β-d-maltopyranoside (DDM) resulted in a very broad peak even under relatively harsh activation conditions (supplemental Fig. S1). Mass spectral peaks of this width are usually assigned to the presence of endogenous lipids co-purified with the protein in the DDM micelle. These lipids resist delipidation with DDM implying that they have a relatively high affinity for McjD, and therefore, their interactions are preserved during the purification process. To reduce this heterogeneity of lipid binding, we carried out a detergent exchange from DDM into octyl glucose neopentyl glycol (OGNG). In this detergent, it was possible not to strip the micelle but also to resolve charge states of McjD (Fig. 1A). The deconvolution of these charge states showed a mass of 133 kDa, which is in agreement with the theoretical mass of dimeric McjD. A second series of peaks are assigned to adducts of mass ∼650–800 Da, likely phospholipids (discussed later). In addition to phospholipids, a second unexpected series of peaks with adduct masses of ∼3.3 kDa was observed. We attribute these adducts to lipopolysaccharides (LPS), in agreement with a recently published study where it was shown that MscL, an inner membrane protein, has the tendency to bind to outer membrane lipids or lipopolysaccharides (21). Given that LPS are larger than phospholipids or CARD, consisting of several hydrocarbon chains, their size implies hydrophobic binding to the surface of the protein. In agreement with this observation, our lasso-peptide binding experiment also shows that LPS does not obstruct the binding pocket but likely binds to the surface of the protein (Fig. 1B). Addition of ATP/Mg2+ did not show any displacement of the LPS (supplemental Fig. S1), and this is also consistent with LPS binding to the protein during purification rather than as a substrate, as shown recently for the ABC transporter TmrAB, which displaces lipid A on addition of ATP/Mg2+ (8). The presence of LPS is therefore likely irrelevant to the function of the protein because McjD is known as an antimicrobial peptide transporter.
FIGURE 1.
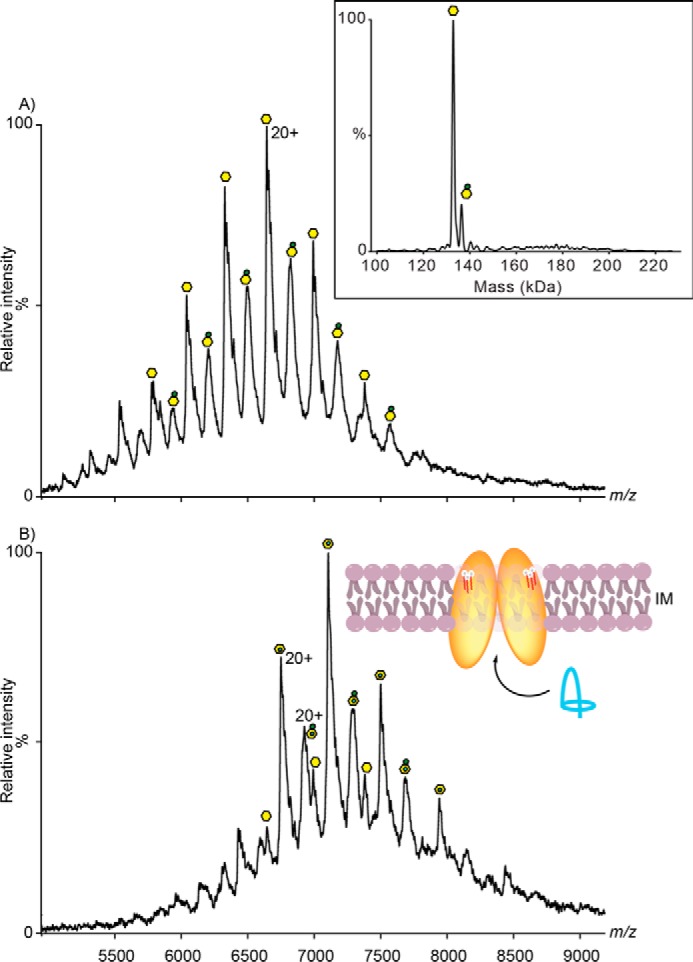
Mass spectrum of McjD. A, mass spectrum of McjD dimer complex exchanged in OGNG detergent acquired on a Q-ToF mass spectrometer. Charge states are assigned to the apo complex (yellow hexagon), phospholipids (yellow hexagon with red adduct), and possibly binding of 3.2–3.5-kDa species of LPS (yellow hexagon with green adduct). In the inset, the deconvoluted mass spectrum of McjD is shown (confirming the mass of the protein). B, McjD incubated with 10 μm lasso-peptide Mccj25 for 10 min. New peaks emerged as adduct to the apo- and LPS-bound McjD showing that binding of lasso peptide is independent of LPS presence. Insets are the position of McjD in the inner membrane and transport of lasso-peptide MccJ25 (shown in blue ribbon) to the periplasmic space. LPS are shown as white spheres and red sticks.
To answer the question whether binding of lipids to McjD has an effect on the function and stability of the protein, we investigated the presence of different lipids in DDM purified and OGNG-treated samples, and we identified them using tandem mass spectrometry (supplemental Fig. S1 and Table 1). The tandem mass spectrometry for the DDM purified McjD revealed a total of 11 lipid species of which two were negatively charged and nine were zwitterionic (Table 1). Treatment of McjD with OGNG resulted in the loss of several zwitterionic lipids (Table 1). We performed three replicate analyses of McjD preparations used for x-ray crystallization and found essentially similar results. Interestingly, we found several species of lipopolysaccharides, ranging from 3.3 to 3.4 kDa, consistent with the denaturing mass spectra of McjD. However, because of low signal intensity, we were unable to confirm the presence of LPS. Our lipid identification results suggest that McjD has selectivity toward zwitterionic and negatively charged lipids of varied chain lengths.
TABLE 1.
List of identified phospholipid species bound to lipidated and delipidated McjD
| DDM (lipidated) | OGNG (delipidated) |
|---|---|
| PG(18:1/18:1) | PG(18:1/18:1) |
| PG(16:0/18:1) | PG(16:0/18:1) |
| PE(14:0/14:0) | PE(14:0/14:0) |
| PE(16:0/12:0) | |
| PE(14:0/17:1) | |
| PE(16:0/16:1) | |
| PE(18:1/16:1) | |
| PE(16:1/16:1) | |
| PE(16:0/14:0) | PE(16:0/14:0) |
| PE(18:1/18:1) | PE(18:1/18:1) |
| PE(16:0/18:1) | PE(16:0/18:1) |
Influence of Different Lipids on the ATPase Activity of McjD
The mass spectrometry lipid identification and analysis showed the presence of PE and PG lipids (Table 1). Because these lipids have been retained, their roles could be to (i) stabilize the overall architecture of the protein or (ii) to have an effect on the activity of McjD. We investigated the effect of the different lipid types identified by mass spectrometry on the ATPase activity of McjD by partially delipidating it using OGNG (see “Materials and Methods”); McjD treated using this protocol will be referred to as delipidated McjD and the DDM-purified protein as DDM-lipidated. Extended delipidation of McjD resulted in aggregated/precipitated protein and total loss of the ATPase activity. Partial delipidation of McjD resulted in the loss of different chain length PE lipids (Table 1) and retention of PG. The basal ATPase activity of delipidated McjD was 20.77 ± 1.47 nmol/min/mg McjD, which is around 30% less active relative to the DDM-lipidated protein, 28.50 ± 0.39 nmol/min/mg McjD (Fig. 2A). When the delipidated protein was supplemented with detergent-destabilized small unilamellar vesicles composed of E. coli polar lipid extract (PL) (with a molar ratio of McjD to lipid of 1:870 mol/mol), the McjD basal ATPase activity was restored to similar levels as for the DDM-lipidated protein. Because the E. coli PL extract is a mixture of PE, PG, and CARD (6.7:3.2:0.1), the ATPase activity of delipidated McjD was measured in the presence of different detergent-destabilized small unilamellar vesicles composed of different lipids; we did not measure the activity in the presence of CARD because it was not detected in our analysis. PG was not able to fully restore the basal ATPase activity, whereas PE was able to restore it and also shows a slightly elevated ATPase activity, 33.05 ± 1.33 nmol/min/mg McjD, relative to the DDM-lipidated protein, 28.50 ± 0.39 nmol/min/mg McjD (Fig. 2A), which is in agreement with the loss of PE lipids upon delipidation. Because PE showed slightly elevated basal ATPase activity, we investigated which of its lipid components were responsible for the increased activity. PE is mostly composed of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (16:0) (33.5% of the total PE composition) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (18:1) (34% of the total PE composition) in lipid preparations from E. coli. The mass spectrometry analysis identified different fragments of PE (Table 1), with variants of 1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine (16:1) (9% of the total PE composition) and 18:1. Because not all mixed acyl variants are commercially available, we decided to assess only the effect of pure 16:1 PE and 18:1 PE lipids on the basal ATPase activity. In the presence of 16:1 PE or 18:1 PE, the basal ATPase activity was restored, but neither of them showed an elevated ATPase activity as did the PE mixture, thus suggesting a synergistic role of the different PE lipids present in the mixture in the McjD activity. We have previously shown that Hoechst33342 (Hoechst) can stimulate the ATPase activity of detergent-purified McjD, and it can also be transported by McjD in proteoliposomes (13). In this study, Hoechst stimulated the DDM-lipidated and delipidated McjD by 2.1- and 1.6-fold, respectively, relative to the basal ATPase activity of the delipidated McjD (Fig. 2A). When delipidated McjD was supplemented with lipids, the ATPase activity in the presence of Hoechst almost doubled (calculated relative to the basal ATPase activity of delipidated McjD). The ligand stimulated ATPase activity in the presence of pure PE 18:1, and pure PE 16:1 was not measured because PE did not show enhanced stimulation compared with the other lipids. In the presence of Hoechst, the slightly higher (2.3-fold) ATPase activity in PL suggests a synergistic role of PE and PG in the activity and probably stability of McjD. Overall, in the presence of all the lipids, the ligand-induced ATPase activity was restored compared with the delipidated protein.
FIGURE 2.

Effect of lipids on the activity and secondary structure of McjD. A, basal (gray bars) and ligand-induced (100 μm Hoechst) (red bars) ATPase activities were measured at the different lipid environments. Delipidated McjD (OGNG-treated) shows reduced basal ATPase activity relative to the lipidated protein (DDM-purified). The ATPase activity of the delipidated McjD is rescued in the presence of PL extract but not in pure PG. ATPase measurements were performed from three different protein preparations. Error bars are shown for all measurements (mean S.E.; n = 3). B, far-UV CD spectra of McjD in various lipid conditions at 20 °C. C, melting curves of far-UV CD spectra of McjD in various lipid conditions monitored at 224 nm. D, melting curves of α-helix content of McjD in various lipid conditions.
Influence of Different Lipids on the Structure and Stability of McjD
Because the basal and ligand-stimulated ATPase activity of the delipidated protein was restored in the presence of various lipids, we sought to establish whether the different lipid environments stabilize the overall protein architecture by measuring the secondary structure and melting temperature (Tm) of McjD. The secondary structure of delipidated and lipid-supplemented McjD was monitored by Synchrotron Radiation Circular Dichroism (SRCD). The normalized far UV-CD spectra of DDM-lipidated McjD showed a typical α-helical structure, with an α-helix content of ∼68% (Fig. 2B and Table 2). This is in agreement with our previously published crystal structure in DDM (PDB code 4PL0), based on the Dictionary of Secondary Structure of Proteins (DSSP) algorithm (22, 23) used to calculate its secondary structure. Delipidation of McjD (in OGNG) resulted in a less folded structure with a higher content of β-strand (26%) and a significant reduction in α-helical content (29%) compared with lipidated McjD (in DDM) (68%) (Table 2). McjD supplemented with PL, PE, and PG showed similar CD features to that in DDM, with a comparable α-helix content of 64–68% (Table 2), suggesting that the lipids are capable of reverting the loss of the secondary structure. Slight differences in α-helical content in PL may be due to the presence of a small ratio of CARD.
TABLE 2.
Secondary structure estimation from CD spectra of McjD in various lipid conditions using Contin algorithm
The secondary structure of the McjD crystal structure (PDB codes 4PL0) was calculated using DSSP.
| Secondary structure estimation (%) | DDM | OGNG | PL | PG | PE | 4PL0 (DSSP) |
|---|---|---|---|---|---|---|
| α-Helix | 68.3 | 29.1 | 64.2 | 66.8 | 68.5 | 62 |
| β-Strand | 2.9 | 26.4 | 5.4 | 2.9 | 3.9 | 10 |
| Turns | 10.0 | 19.9 | 12.6 | 11.2 | 11.1 | 16 (turns and bend) |
| Unordered | 18.8 | 24.6 | 17.8 | 19 | 16.4 | 12 |
| Standard deviation | 0.012 | 0.043 | 0.037 | 0.021 | 0.033 |
We also measured the thermal stability of McjD in the different lipid environments by SRCD as a function of secondary structure loss upon heating. DDM-lipidated McjD has a mean S.E. of 67.2 °C, and removal of lipids by OGNG destabilizes McjD dramatically, reducing its Tm to 48.6 °C (Fig. 2, C and D, and Table 3). When McjD was supplemented with PL, the thermal stability was recovered, and its Tm increased to 65.2 °C (Table 3). This trend of stability was also observed in the presence of PE, whose Tm (68.9 °C) is the closest to that of DDM-lipidated McjD and with PG, which has two transitions, the second one being the higher Tm at 74.3 °C. PG has another lower thermal transition at 13.1 °C (Table 3). This trend of thermal denaturation is mirrored in the α-helix contents of the McjD in the various lipid conditions. The α-helix component in McjD in various lipids (PL, PG, and PE) gave higher Tm (>70 °C) compared with McjD in DDM (67.2 °C), suggesting that other secondary structure components of McjD were likely perturbed before the α-helix content was affected.
TABLE 3.
Tm of McjD in various lipid conditions monitored from far-UV CD spectra, at 224 nm and as a function of % α-helix, and nano-DSF as a ratio of 350:/330 nm
ND indicates not determined value.
| Lipids used | Tm (ΔΔϵ224 nm) °C | Tm (% α-helix) °C | Tm (350/330 nm) °C |
|---|---|---|---|
| DDM | 67.2 | 66.1 | 53.7 |
| Polar lipids (PL) | 65.2 | 71.4 | 63.2 |
| OGNG | 48.6 | 46.9 | Tm1 ND; Tm2 71.4 |
| PG | Tm1 13.1; Tm2 74.3 | Tm1 9.4; Tm2 76.0 | 54.4 |
| PE | 68.9 | 72.8 | 68.5 |
| PE 16:1 | Not measured | Not measured | 65.7 |
| PE 18:1 | Not measured | Not measured | 63.2 |
Influence of Lipids on the Local Environment of Tryptophans
We also measured the stability of the delipidated and lipid-supplemented McjD using differential scanning fluorimetry (nano-DSF) by monitoring the intrinsic tryptophan fluorescence. McjD contains two tryptophans, Trp-167 and Trp-247, which are located in the head group region of the lipid bilayer. Changes in their local environment upon denaturation would affect their intrinsic fluorescence. The Tm of the DDM-lipidated McjD was measured to be 53.7 ± 0.3 °C (Fig. 3A and Table 3). The delipidated McjD displayed a biphasic profile (Fig. 3B and Table 3), suggesting that the protein was rather unstable, partially unfolded, at the beginning of the experiment. Because the assay started at 20 °C, we could not establish the first unfolding Tm event, but a secondary Tm was measured at 71.4 ± 0.5 °C (Fig. 3B). Supplementing the delipidated protein with PL showed an enhanced Tm stabilization, 63.2 ± 0.1 °C (Fig. 3C and Table 3) relative to the lipidated protein as well as absence of the biphasic profile (Fig. 3B). The two main components of the polar lipid extract PE and PG displayed Tm of 68.5 ± 0.3 and 54.4 ± 0.2 °C, respectively (Fig. 3, D and E, and Table 3). The PE shows a small degree of biphasic profile, which could correspond to some partially unstable protein. Finally, the Tm of McjD in the presence of 16:1 and 18:1, the two components of PE, were 65.7 ± 0.3 and 63.2 ± 0.3 °C (Fig. 3, F and G, and Table 3). All the lipids appear to have rescued/reduced the biphasic profile that the delipidated protein displayed, suggesting a possible role in stabilizing the local environment of tryptophans.
FIGURE 3.
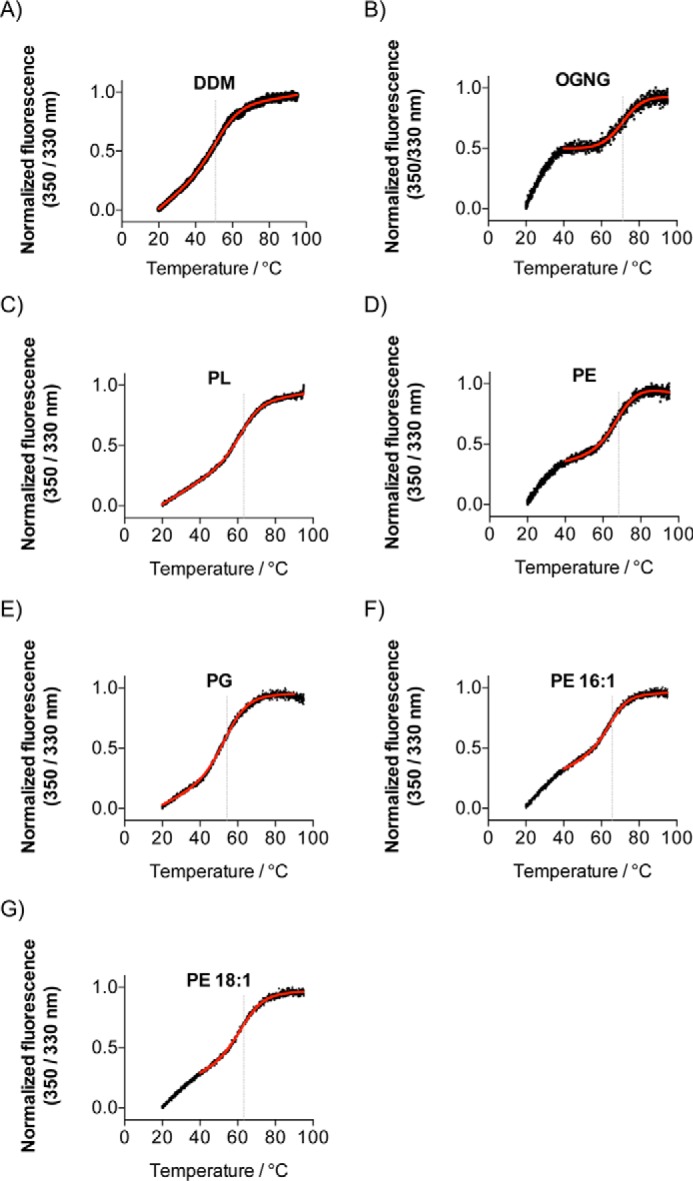
Effect of lipids on the local environment of tryptophans. A–G, melting temperature profile of the delipidated McjD (B) is biphasic, suggesting unstable protein, which recovers in the presence of the lipids. All lipids appear to enhance the stability of McjD relative to the lipidated protein (DDM purified, A). All measurements were performed in triplicate, and the modified Boltzmann sigmoidal Equation 1 is fitted in the mean value of the data. Gray line indicates the calculated Tm values.
Structurally Resolved Bound Lipids
Our current biochemical data show that the ATPase activity of McjD is strongly correlated and dependent toward the presence of lipids. We have previously determined the crystal structure of McjD, and we observed some electron density that could correspond to lipids. The electron density was not continuous, and it did not provide us with confidence to build any lipids. A structural study to identify bound lipids on SERCA employed the measurement and inclusion of low resolution diffraction spots in the refinement and map calculations (6). We followed a similar approach, and we used the same purified McjD batch for crystallization as for the mass spectrometry to compare the identified lipids with the crystal structure. We determined the crystal structure of McjD in the presence of AMPPNP and MgCl2 at 3.4 Å resolution, including low diffraction data to 60 Å resolution (Table 4). The overall structure is identical to our previously published one; McjD is in a nucleotide-bound outward occluded conformation. Inspection of electron density maps revealed the presence of elongated positive |Fo| − |Fc| electron density at 2.0 σ near the elbow helix and between TM3 and 4 (Fig. 4, A and B) of one subunit; the electron density was assigned as a PG molecule. We excluded the presence of DDM, as its alkyl chain is not long enough to occupy the electron density. Among all the lipids that were identified in the mass spectrometry, only 1,2-di-(9E-octadecenoyl)-sn-glycero-3-phospho-(1′-sn-glycerol) (PG (18:1/18:1) (9.7% of the total PG composition)) was long enough to fit in the electron density. In addition, the cleft of the resolved lipid electron density is positively charged (Fig. 4C), thus further suggesting that a negatively lipid-like PG is most likely to occupy this site. However, we cannot exclude that the lipid might be PE, because a PE head group can also be refined within the electron density. In the refined structure, the PG lipid (18:1/18:1) is weakly associated with residues from TM3 and 4 (Fig. 4B); Arg-186 is within hydrogen bond distance with the glycerol moiety of PG (18:/18:1), which is further stabilized by weak electrostatic interactions with Thr-185. The 9E-octadecenoyl acyl chain makes hydrophobic contacts with Ile-155, Ile-158, and Phe-174. We also observed some weak electron density between the elbow helix and TM5 that may correspond to either the phosphoethanolamine head group of PE or to tightly bound maltoside head groups of DDM. As described previously, the non-denaturing mass spectrometry and lipid analysis also identified an LPS molecule. Some positive |Fo| − |Fc| electron density at 2.5 σ is present between the elbow helix and TM4 of the opposite monomer. The observed electron density is in close proximity to charged residues from TM4 that could bind the sugar moieties of the lipid A molecule. However, because the acyl chains are not stabilized by hydrophobic contacts as they point away from the transmembrane domains (TMDs), this electron density is likely not associated with lipid A, although it probably corresponds to “well ordered” detergent/lipid mixture. If lipid A molecules were specifically bound to McjD, we would expect a tighter association with the TMDs as in the structure of the outer membrane β-barrel ferrichrome receptor FhuA (19).
TABLE 4.
Data collection and refinement statistics
Values in parentheses refer to data in the highest resolution shell.
| Data collection statistics | |
| Beamline | Diamond Light Source I24 |
| Space group | P212121 |
| Resolution (Å) | 67.61–3.42 (3.51–3.42) |
| Cell dimensions | a = 82.9, b = 109.1, c = 233.1 |
| No. of reflections | 103723 (7200) |
| No. of unique reflections | 29066 (2111) |
| Completeness (%) | 99.0 (98.9) |
| Redundancy | 3.6 (3.4) |
| Rmerge | 0.073 (0.76) |
| Mean (I/σ(I)) | 11.6 (2.1) |
| Mn(I) half-set correlation CC(1/2) (%) | 0.99 (0.91) |
| Refinement statistics | |
| Rwork (%) | 25.35 (29.98) |
| Rfree (%) | 27.18 (30.42) |
| Protein atoms | 9107 |
| AMPPNP | 62 |
| Mg2+ | 2 |
| Lipids | 51 |
| B-factors | |
| Protein | 125 |
| AMPPNP | 126 |
| Ion | 88 |
| Lipids | 184 |
| Root mean square deviations from ideal values | |
| Bonds (Å) | 0.01 |
| Angle (°) | 1.07 |
| Ramachandran plot outliers (%) | 0.44 |
FIGURE 4.

Structurally resolved PG lipid bound to McjD. A, positive |Fo| − |Fc| electron density (pink mesh, 2.0 σ) was observed for the PG head group after molecular replacement and refinement. The PG lipid was not included in the refinement but is shown for clarity. PG is shown as sticks with carbon in orange, oxygen in red, and nitrogen in blue. McjD is shown as green ribbons. B, final 2|Fo| − |Fc| electron density map contoured at 1 σ after inclusion of PG in the refinement. C, cartoon representation of McjD in association with PG (orange sticks). The electrostatic surface of one McjD molecule is also displayed. The negatively charged head group of PG is bound within a positively charged region of McjD, whereas the acyl chain is found within a hydrophobic groove. The surfaces are colored from blue (positively charged regions) to red (negatively charged regions). Hydrophobic regions are shown as white. AMPPNP is shown in black sticks. D, composite omit map contoured at 0.8 σ (blue mesh).
Molecular Simulations in Different Lipid Environment
To investigate the structural organization of the lipid bilayer around McjD, we performed CG MD simulations of the transporter in four lipid environments, i.e. pure palmitoyloleoyl-PE (POPE), pure palmitoyloleoyl-PG (POPG), a mixture of 70% POPE and 30% POPG, and a mixture of 67% POPE, 23% POPG, and 10% CARD. This choice of lipids allowed us to study the interactions between the protein and some of the hallmark components of the E. coli inner membrane.
Membrane Organization in the Proximity of the Protein
We first estimated the number of lipids in the proximity of the protein based on the number of phosphate head groups within a distance cutoff of 0.7 nm (24). The number of lipids in contact with the protein converges for all the simulation systems within the first ∼5 μs (supplemental Fig. S3). All the systems are characterized by a higher number of lipids around the protein in the lower leaflet of the membrane, correlating with the wider shape of transporter in the cytosolic side. For the systems with 100% POPE and 100% POPG lipids (supplemental Fig. S3), the number of lipids in the upper and lower leaflet differs by about four and two molecules, respectively. In the binary mixture (supplemental Fig. S3), the number of both POPE and POPG lipids decreases when compared with the pure bilayers. In the ternary mixture (supplemental Fig. S3), the CARD molecules also establish contacts with the protein, with a significant difference between lower and upper leaflets, thus altering the profile of POPE and POPG lipids within the same cutoff. To interpret these changes in the number of lipids in contact with the protein, we calculated the time-averaged lipid densities for the phosphate head groups of each lipid type. Fig. 5 shows such densities calculated for each system during the last 10 μs of the simulations, together with the average number of lipids molecules found within the contact cutoff distance during the last 10 μs. The 1st and 2nd panels of Fig. 5, A–D, represent the lipid densities (calculated on the phosphate head groups only) in the upper and lower leaflet for each system, respectively. In both the 100% PE and 100% PG systems, in the proximity of the protein, the lipid density shows the same organization, and in the binary and ternary mixtures, the density of different lipid types overlap. This is an indication that, although POPE lipids are the most abundant species in our simulation setup, lipid exchange occurs with regard to the interactions established by lipids of different types with the protein. In fact, changes in the number of POPE and POPG molecules are observed for the binary and ternary mixture when compared with the pure bilayers (4th panel of Fig. 5, A–D, and supplemental Fig. S3).
FIGURE 5.

Lipid organization in proximity of McjD. For each of the simulation systems: a, protein-POPE; b, protein-POPG; c, protein-POPE-POPG; d, protein-POPE-POPG-CARD, the 1st two panels represent the lipid density (POPE, cyan; POPG, magenta; CARD, yellow) calculated using the phosphate head groups for the upper (1st panel) and lower (2nd panel) leaflet. The McjD crystal structure was superimposed to each CG system, and it is shown as gray cartoons. The 3rd panel is a focused view on the PG lipid co-crystallized with McjD. The results of the density calculations performed on the full lipid molecules are shown. The last panel shows the average number of lipids and corresponding standard deviation calculated during the last 10 μs of the simulations within a contact cutoff distance of 0.7 nm from the protein.
A focused view on the lipid-binding site identified via protein crystallization, in combination with the densities calculated on the full lipid molecules, is shown (Fig. 5, A–D, 3rd panel). Time-averaged lipid densities for the three lipid types (POPE, POPG, and CARD) used in the simulations are found in the region corresponding to the crystallographic binding site and in particular in the region surrounding the elbow helix (residues 4–16) and the hydrophobic groove formed by TM3, TM4, and TM6, which accommodates one of the lipid tails of the crystallographic lipid. The lipid density identified around the elbow helix can be linked to the presence of a cluster of positively charged residues, such as Lys-4, Lys-6, Arg-186, and Arg-311 (supplemental Fig. S4), which could facilitate the binding of negatively charged lipid head groups (POPG and CARD) or the binding of zwitterionic lipids (POPE) via the phosphate moiety of their headgroup.
Protein-Lipid Interactions
For each residue of the TMDs, we calculated, for each frame of the trajectory, the total number of contacts established with the phosphate head group of the lipids, where a contact is defined when the phosphate head group is within 0.7 nm of any bead of a given protein residue (supplemental Fig. S5). This analysis showed preferable interaction sites with some of the TMD residues and highlighted how positively charged residues strongly interact with the lipid head groups (supplemental Fig. S5). The majority of such residues is located on the intracellular side of the membrane (supplemental Fig. S4). Similar interaction patterns were found for both POPE and POPG in all the simulation systems. Overall, for the two pure bilayer systems, POPG showed higher contacts than POPE, and this behavior was observed also in the binary system (supplemental Fig. S5). However, for the ternary bilayer, the presence of CARD molecules decreases the number of contacts of the POPG molecules to a higher extent than the number of contacts of the POPE molecules (supplemental Fig. S5 and Fig. 5D, panel 4). This might suggest a competition between POPG and CARD molecules stronger than between POPE and CARD. Overall, the positively charged residues in proximity of both the elbow helices are associated with higher number of contacts in the simulation systems tested in this study. The combination of (i) positive charges to coordinate the binding of the lipid phosphate group and (ii) grooves on the protein surface to accommodate the lipid tails might be correlated with the isolation and crystallization of McjD with PG lipids.
Discussion
In this study, we have evidence for the synergistic role of specific lipids on the modulation of the structure and activity of the ABC exporter McjD. The tandem mass spectrometry of McjD has identified various zwitterionic PE lipids, negatively charged PG lipids and LPS associated with McjD. These lipids are tightly associated with McjD, because they have survived the detergent extraction from the membrane, the purification process, and the gas phase, and they likely represent specific lipids. Using non-denaturing mass spectrometry, we have shown that McjD can bind the lasso antibacterial peptide MccJ25 without interference from the lipids or LPS. From the various lipids identified, we sought to explore the role of each class of lipids on the structure and function of McjD. Delipidation of McjD resulted in loss of PE lipids with retention of PG (Table 1), suggesting a functional role of negatively charged lipids. Partial delipidation of McjD reduced its basal ATPase activity by 30%, which could be restored with the addition of different classes of lipids, such as PL extract and PE (including PE 16:1 and PE 18:1), although addition of PG did not restore the basal ATPase activity of McjD. These data suggest that zwitterionic lipids are able to provide the correct hydrophobic environment where the transporter can bind and hydrolyze ATP effectively. However, the basal ATPase activity only measures the futile hydrolysis of ATP, and it is only a measure of ATP turnover. We have previously demonstrated that the ATPase activity of McjD can be stimulated in the presence of the antibacterial lasso peptide MccJ25 and Hoechst (13). The ligand Hoechst stimulated the ATPase activity of McjD in the presence of all the different lipids. Interestingly, McjD supplemented with PG lipids appears to have a higher relative ligand-induced ATPase activity compared with the other lipids. In this study, we did not observe significant stimulation or inhibition of the basal ATPase activity by lipids as shown for the Na,K-ATPase (5, 25). From a molecular point of view, this could suggest preferential interactions between McjD and negatively charged lipids.
Our MD simulations indicate that a belt of positively charged residues distributed on the surface of McjD at the protein-lipid head group interface favors interactions with the negatively charged lipid molecules, POPG and CARD, thus reducing the number of contacts with the POPE lipids (Fig. 5 and supplemental Figs. S3–S5). However, zwitterionic lipids, like POPE, also play structural and functional roles in the bacterial inner membrane; they can facilitate the insertion of proteins in the lipid bilayer (26); they can affect protein function, as shown for the E. coli lactose permease LacY (27), or they can be localized in the annular region of membrane proteins together with PG lipids (28). Indeed, for McjD, the lipid organization, described in terms of lipid density (Fig. 5), identifies both zwitterionic and negatively charged lipids in the proximity of the protein and highlights lipid exchange in the binary and tertiary mixtures.
Our thermostability measurements in the different lipids by nano-DSF and SRCD further suggest a synergy between zwitterionic and negatively charged lipids. The different melting temperatures measured by the two techniques suggest a more complex relationship between the stabilization of the overall structure of McjD by lipids and the local environment of the two tryptophans, Trp-167 and Trp-247, in the presence of detergent and/or lipids. Despite the differences, both techniques are in close agreement about the stabilizing role of lipids on the local or overall structure. The SRCD of the delipidated McjD confirmed the structural stabilization role of PE lipids and subsequently reduced ATPase activity due to loss of helical content (i.e. structural integrity and stability) that is reversible upon addition of detergent-destabilized small unilamellar vesicles composed of PL, PE, or PG lipids. The data are also in very close agreement with the lipid exchange (PE-PG-CARD) observed in MD simulations.
The structurally resolved lipid in our McjD structure was assigned as PG (18:1/18:1), and it probably acts as a specific lipid. We can speculate that the PG stabilizes the transporter in the nucleotide-bound outward occluded state in addition to the reported salt bridges (13); the lipid is found between TM3 and 4 that are part of the bundle of helices that undergo conformational changes during the transport cycle. Lipids are known to modulate the structure of membrane proteins and for their role in stabilizing a given conformational state (29, 30). Structures of SERCA in different conformations with resolved lipids have been reported (Ref. 31 lists all the SERCA states with different lipids), where lipids are found in state-dependent and state-independent sites (four distinct sites, namely A–D). In the low calcium affinity state E2, PE lipids occupy sites A and B, where lipids in site A have been proposed to stabilize this state by keeping TM2 and 4 apart. Transition from E2 to the high calcium affinity state E1 results in a structural conformation along TM2 and 4 and displacement of the PE lipid (31); in the E1 state, PC lipids are found in sites C and D.
All our data suggest a synergistic role of PE and PG lipids that are reflected during the transport cycle from an inward- to an outward-facing conformation, measured as a function of substrate-induced ATPase activity, similar to the stabilization of SERCA at the different states; specific lipids might interchange in specific surface grooves to facilitate this transition, as shown by the elevated ATPase activity upon addition of PL lipids. Crystal structures of McjD in different conformations with resolved lipids can provide further support for the role of lipids on its activity.
Materials and Methods
Protein Purification and Crystallization
McjD was purified and crystallized as before with no modifications (13). The same purified protein preparations were used for mass spectrometry, ATPase assays, thermostability measurements, and crystallization to have an identical sample to correlate all the data. Three different protein preparations were used for the functional studies. The protein concentration was determined by UV280 and BCA protein assay. In brief, 10 mg/ml purified McjD (in 20 mm Tris, pH 7.5, 150 mm NaCl, and 0.03% DDM) was incubated with 10 mm AMPPNP and 2.5 mm MgCl2 at room temperature for 30 min prior to crystallization. Crystals were grown at 293 K using the vapor diffusion method by mixing protein and reservoir solution at 1:1. Crystals were grown from a precipitant solution containing 100 mm sodium citrate, pH 5.5, 45 mm NaCl, 28% PEG 400, and 1% nonyl-glucopyranoside. The crystals were directly frozen into liquid nitrogen, and diffraction screening and data collection were performed at Diamond Light Source synchrotron.
MccJ25 Purification
MccJ25 was produced from cultures of E. coli K12 MC4100 harboring the plasmid pTUC202 and purified as described previously (32).
Tandem Mass Spectrometry of McjD
McjD protein was digested with trypsin (1:20 units) for 15 h at 37 °C in a thermomixer (Eppendorf). The digest was dried in a SpeedVac until complete dryness and re-dissolved in 35% acetonitrile aqueous solution. The tryptic digest mixture was loaded onto a pre-equilibrated C18 column (Acclaim PepMap 100, C18, 75 μm × 15 cm; Thermo Scientific) and separated with a linear gradient of 35–100% acetonitrile at a flow rate of 300 nl/min. The nano-flow reversed-phase liquid chromatography (Dionex UltiMate 3000 RSLC nano System, Thermo Scientific) was directly coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Scientific) via a dynamic nanospray source. Typical MS conditions were spray voltage of 1.6 kV and capillary temperature of 160 °C.
The LTQ-Orbitrap XL was set up in negative ion mode and in data-dependent acquisition mode to perform five MS/MS scans per MS scan. Survey full-scan MS spectra were acquired in the Orbitrap (m/z 350–2,000) with a resolution of 60,000.
OGNG-treated McjD
The McjD protein samples solubilized in DDM were buffer-exchanged to 200 mm ammonium acetate supplemented with 0.02% DDM using Bio-Spin columns (Bio-Rad). For OGNG detergent, exchange samples were diluted 200-fold in 200 mm ammonium acetate supplemented with 0.16% OGNG and incubated for 30 min at 4 °C. Prolonged incubation initiated protein aggregation/precipitation. The OGNG-diluted samples were centrifuged at 12,000 × g for 15 min to remove any possible aggregation/precipitation and then concentrated using ultrafiltration device (Amicon) with 100-kDa cutoff to 2.0 mg/ml final protein concentration.
MccJ25 in Complex with McjD
MccJ25 lasso peptide was added to OGNG-solubilized McjD sample at room temperature and incubated for 10 min. Peptide stock solution was prepared in DMSO at 20 mm concentration. The stock solution was diluted to 100 μm concentration in 200 mm ammonium acetate buffer supplemented with 0.16% OGNG. The final concentration of peptide and protein was 8 μm in reaction mixture.
Non-denaturing Mass Spectrometry
Non-denaturing mass spectra were acquired on a previously modified Q-TOF2 mass spectrometer for the transmission of high mass and also modified for high collision energy in collision-induced dissociation cells (33, 34). Samples were introduced into the ion source via in-house prepared gold-coated capillaries under static nanospray. Q-ToF was operated under following instrumental parameters as follows: capillary voltage 1.4–1.7 kV; cone voltage 150 V; extractor 4 V; collision-induced dissociation energy 220–350 V; and source backing pressure 4.5–6 mbar.
ATPase Assays in Detergent/Lipid Mixtures
ATPase activities were measured in 0.1% DDM solution using a coupled assay as before (EnzCheck, Molecular Probes) (13). The inactive E506Q mutant, which retains 2% ATPase activity relative to the wild type McjD, was used to subtract background degradation of ATP (13). All ATPase activity data were measured at 25 °C. The McjD concentration was 1.5 μm for all experiments. ATPase measurements were performed at 1 mm ATP. For ligand-induced ATPase activity assays, Hoechst33342 was dissolved in water and was added to a final concentration of 100 μm. All the lipids were purchased from Avanti Polar Lipids. Powder lipids were rehydrated in a buffer consisting of 20 mm Tris, pH 7.5, 150 mm NaCl at a concentration of 1 mg/ml. The opaque lipid stocks were bath sonicated for 10–15 min until they were clear. The small unilamellar vesicles were destabilized by the addition of 0.2% DDM. Delipidated McjD was added to a final concentration of 0.1 mg/ml in each lipid mixture to give a molar ratio of McjD to lipid of 1:870 (mol/mol). The protein/lipid/detergent mixtures were incubated at room temperature for 30 min and immediately used in the ATPase assays. All lipids and protein/lipid/detergent mixtures were freshly made. All measurements were performed in triplicate (mean ± S.E., n = 3).
SRCD Measurements
SRCD experiments were performed using a nitrogen-flushed Module B end-station spectrophotometer at B23 Synchrotron Radiation CD Beamline at the Diamond Light Source (35, 36). McjD samples at 0.1 mg/ml were prepared in 20 mm Tris, pH 7.5, 8 mm NaCl containing 0.03% DDM or lipids PE, PG, or PL. Using sample volumes of 30 μl, scans were acquired using an integration time of 1 s, path length of 0.02 cm, and 1.2 nm bandwidth. For thermal stability studies, McjD samples were incubated at 5 °C, and spectra were obtained over a range of temperatures starting at 5 °C and increasing incrementally to 90 °C, with 2 min equilibration time followed by a return to 20 °C in one step of 15 min equilibration time (supplemental Fig. S2). Results obtained were processed using CDApps (37) and OriginLab with Tm calculated using Boltzmann equation. Secondary structure estimation from CD spectra was carried out using CDApps using Continll algorithm (38).
Melting Temperature Assays
The Tm of McjD in detergent and lipid/detergent mixtures was measured by nano-DSF using a Prometheus NT.48 instrument (NanoTemper Technologies GmbH). 10 μl of protein mixture at 0.1 mg/ml in different lipids, prepared as above, was loaded into nano-DSF grade capillaries. Intrinsic tryptophan fluorescence at emission wavelengths of 330 and 350 nm was measured continuously with a temperature gradient of 20–95 °C increasing at a rate of 1 °C/min. All measurements were performed in triplicate (mean ± S.E., n = 3). The 350/330 nm ratio of tryptophan fluorescence was calculated, and data were analyzed and plotted in GraphPad PRISM. The Tm was calculated by fitting the data to a modified Boltzmann sigmoidal Equation 1 to take into account the sloping baselines.
 |
where Y is fluorescence ratio 350 nm/330 nm; X is temperature degrees Celsius; V50 is the Tm; top and bottom denote the fluorescence before and after transition; slope describes the steepness of the curve; and m1 and m2 are the gradient of the sloping top and bottom baselines, respectively.
Data Collection
Diffraction data to 3.4 Å resolution were collected on I24 at Diamond Light Source at a wavelength of 0.968 Å using a Pilatus3 6M detector and a modified beam stop to be able to measure the low resolution data (6); the usual beam stop measuring 800 μm diameter was replaced with a smaller one measuring 700 × 600 μm, to maximize the chance of recording low angle (low resolution) reflections. The diffraction data were processed using xia2 (39). Further processing was performed using the CCP4 suite (40). The resolution of the data and anisotropy analysis were evaluated by half-dataset correlation coefficient in Aimless (41). The space group was determined to be P212121 with two copies of McjD in the asymmetric unit. The data collection statistics are summarized in Table 4.
Structure Solution and Refinement
The structure was determined by molecular replacement in Phaser (42) using our previously published McjD structure (PDB code 4PL0) (13) as search model. All model building was performed in Coot (43). Initial refinement was carried out in REFMAC5 (44) and at later stages in Buster (45). The structure was refined with restraints against the high resolution structure 4PL0.
After rigid body and restrained refinement, extra electron density corresponding to two AMPPNP molecules and two MgCl2 ions were identified. Additional unaccounted positive |Fo| − |Fc| and 2|Fo| − |Fc| electron densities were observed at the surface of the protein (Fig. 4A) that correspond to lipid molecules. Strong |Fo| − |Fc| electron density was observed between TM3 and 4, and the elbow helix that corresponds to PG. Inclusion of the head group in the refinement allowed us to observe extra electron density for the acyl chain. A PE molecule could also be modeled and refined within this electron density, but based on our biochemical data, we chose to build it as PG. Composite omit maps were generated in CCP4 to remove any model bias (Fig. 4D). Inclusion of the low resolution data has also allowed us to visualize electron density that probably corresponds to the mixed lipid/detergent micelle that surrounds the protein.
The final model has an Rwork of 25.35% and an Rfree of 27.18%. The refinement statistics are summarized in Table 4. The McjD structure has 92.93% of the residues in the favored Ramachandran region and has five outliers as calculated by MolProbity (46).
The coordinates and structure factors of McjD with bound PG have been deposited in the Protein Data Bank with PDB code 5EG1.
Molecular Dynamics Simulations
Simulation Setup
A CG model of the McjD crystal structure (PDB code 4PL0 (13)) was generated according to the MARTINI force field (47). To restrain the protein tertiary structure, an elastic network was applied on the backbone beads using a cutoff distance of 9 Å, with a force constant of 500 kJ mol−1 nm−2 (48). The CG protein model was then inserted into four different bilayers, with a ratio of ∼1–500 for protein and lipids. The first and the second bilayer consist of 507 POPE and POPG lipids, respectively. The third bilayer is composed of 70% POPE and 30% POPG lipids, for a total of 507 lipid molecules. Finally, the fourth system consists of 67% POPE, 23% POPG, and 10% CARD, for a total of 506 lipids. The POPE and POPG parameters correspond to the Martini 2.0 lipid definition (49), whereas the CARD parameters (total charge of −2) are from Dahlberg and Maliniak (50), with updated bead type definition according to the latest version of the MARTINI force field (47).
To determine the degree of insertion of the protein in the lipid bilayer, we used the 4PL0 entry of the OPM database (51). The four membrane-protein systems were then generated using Insane (52) and additional in-house software. After protein insertion, the system was solvated using the non-polarizable MARTINI water model (53), and sodium and chloride ions were added to neutralize the system and to reach a final concentration of 150 mm. Each system was initially energy-minimized for 10,000 steps with position restraints applied to all the protein beads (force constant of 1000 kJ mol−1 nm−2). A 3-ns-long equilibration was performed in multiple steps, with position restraints on all the protein beads first, using a time step of 10 fs, v-rescale thermostat (time constant of 0.2 ps), and a semi-isotropic pressure of 1 bar maintained with the Berendsen barostat (time constant for coupling of 0.2 ps) (54). The position restraints were then applied only to the backbone beads, and the time step was set to 20 fs, with a time constant for pressure and temperature coupling of 5.0 and 1.0 ps, respectively. An additional 2-ns-long equilibration was performed after removing the position restraints from the backbone, and by setting the time constant for temperature coupling to 2.0 ps.
Following this initial setup, each system was simulated for 40 μs, at 310 K, with a time step of 20 fs, using the v-rescale thermostat (time constant of 2.0 ps) (55), and the Parrinello-Rahman barostat (time constant of 12 ps) (56). The nonbonded interactions were treated with a shift function using a Coulomb and Lennard-Jones cutoff of 1.2 nm. The neighbor list was updated every 10 steps. Frames were saved every 400 ps. The simulations were carried out using GROMACS, version 4.6.7 (57).
Analyses
Analyses were performed using GROMACS (57), in-house software (to perform the 3D density analysis and to calculate the lipid-protein contacts), and PyMOL Molecular Graphics System, version 1.7.2.
3D Density Analysis
To calculate the lipid density around the protein, we first processed the simulation trajectory to center the protein in the box, remove rotations and translations in x and y, and fit progressively on the backbone beads of the protein. The system was then placed into a 3D grid with voxels of 1 Å3. For each voxel, we calculated the average density from the last 10 μs of the simulation, using the phosphate group heads of each lipid type or the full lipid molecules, as described in the text. All the voxels with a density value that is above 99% of the maximum observed density value were displayed as volume maps with PyMOL.
Protein-Lipid Contacts
To estimate the number of contacts between the TMD residues (chain A, residues 9–331; chain B, residues 4–331) and lipids, we defined a protein-lipid contact when the phosphate head group of a given lipid is found within 0.7 nm of any bead of a given protein residue. For the given residue, the total number of contacts is then calculated as the sum of all the contacts established with the phosphate group heads satisfying the cutoff, for each frame of the trajectory. These calculations were performed using the MDTraj library (58) and were conducted separately for each lipid type.
Author Contributions
C. V. R. and K. B. designed and managed the overall project. H. G. C., D. A., and K. B. grew crystals, collected data, built and refined the structure. H. G. C. and P. B. purified protein for mass spectrometry analysis, ATPase assays, and SRCD. S. Z. and S. R. produced and purified the MccJ25. R. H. performed SRCD measurements and analysis. V. C. and D. P. T. performed and analyzed molecular simulations. S. M. and C. V. R. performed mass spectrometry measurements and analysis. C. V. R. and K. B. wrote the manuscript with help from the other authors.
Supplementary Material
Acknowledgments
We are grateful to NanoTemper Technologies GmbH and Dr. James Wilkinson for kindly providing the Prometheus NT.48 instrument for the nano-DSF measurements; we also thank Dr. Wilkinson for help with data analysis. We thank C. Bechara (University of Oxford) for help with the LC-MS experiments. We thank the Diamond Light Source for beam time allocation and access. We thank Dr. Gurpreet Singh, Department of Biological Sciences, University of Calgary, for suggestions and help with the programming component of the MD simulations analyses. MD simulations were carried out on WestGrid/Compute Canada facilities. Membrane Protein Lab is funded by Wellcome Trust Grant WT/099165/Z/12/Z (to Professor So Iwata). The Oxford University Mass Spectrometry facility is funded by ERC IMPRESS Grant 26851 and Medical Research Council Grant 98101.
This work was supported in part by Biotechnology and Biological Sciences Research Council Grant BB/H01778X/1 (to K. B.) and by Canadian Institutes for Health Research Grant MOP-62690 (to the D. P. T. laboratory). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S5.
The atomic coordinates and structure factors (code 5EG1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TM
- transmembrane
- PG
- phosphatidylglycerol
- DDM
- n-dodecyl β-d-maltopyranoside
- OGNG
- octyl glucose neopentyl glycol
- PL
- polar lipid
- AMPPNP
- adenosine 5′-(β,γ-imido)triphosphate
- PDB
- Protein Data Bank
- POPE
- palmitoyloleoyl-PE
- POPG
- palmitoyloleoyl-PG
- CARD
- cardiolipin
- SRCD
- Synchrotron radiation circular dichroism
- nano-DSF
- nano-differential scanning fluorimetry
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- TMD
- transmembrane domain
- DSSP
- Dictionary of Secondary Structure of Proteins.
References
- 1. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee A. G. (2004) How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 1666, 62–87 [DOI] [PubMed] [Google Scholar]
- 3. Lee A. G. (2011) Lipid-protein interactions. Biochem. Soc. Trans. 39, 761–766 [DOI] [PubMed] [Google Scholar]
- 4. Contreras F. X., Ernst A. M., Wieland F., and Brügger B. (2011) Specificity of intramembrane protein-lipid interactions. Cold Spring Harb. Perspect. Biol. 3, a004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haviv H., Habeck M., Kanai R., Toyoshima C., and Karlish S. J. (2013) Neutral phospholipids stimulate Na,K-ATPase activity: a specific lipid-protein interaction. J. Biol. Chem. 288, 10073–10081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sonntag Y., Musgaard M., Olesen C., Schiøtt B., Møller J. V., Nissen P., and Thøgersen L. (2011) Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nat. Commun 2, 304. [DOI] [PubMed] [Google Scholar]
- 7. Koshy C., Schweikhard E. S., Gärtner R. M., Perez C., Yildiz O., and Ziegler C. (2013) Structural evidence for functional lipid interactions in the betaine transporter BetP. EMBO J. 32, 3096–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bechara C., Nöll A., Morgner N., Degiacomi M. T., Tampé R., and Robinson C. V. (2015) A subset of annular lipids is linked to the flippase activity of an ABC transporter. Nat. Chem. 7, 255–262 [DOI] [PubMed] [Google Scholar]
- 9. Kim J., Wu S., Tomasiak T. M., Mergel C., Winter M. B., Stiller S. B., Robles-Colmanares Y., Stroud R. M., Tampé R., Craik C. S., and Cheng Y. (2015) Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517, 396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., and Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szewczyk P., Tao H., McGrath A. P., Villaluz M., Rees S. D., Lee S. C., Doshi R., Urbatsch I. L., Zhang Q., and Chang G. (2015) Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 71, 732–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marcoux J., Wang S. C., Politis A., Reading E., Ma J., Biggin P. C., Zhou M., Tao H., Zhang Q., Chang G., Morgner N., and Robinson C. V. (2013) Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. U.S.A. 110, 9704–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choudhury H. G., Tong Z., Mathavan I., Li Y., Iwata S., Zirah S., Rebuffat S., van Veen H. W., and Beis K. (2014) Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl. Acad. Sci. U.S.A. 111, 9145–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosengren K. J., Clark R. J., Daly N. L., Göransson U., Jones A., and Craik D. J. (2003) Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J. Am. Chem. Soc. 125, 12464–12474 [DOI] [PubMed] [Google Scholar]
- 15. Wilson K. A., Kalkum M., Ottesen J., Yuzenkova J., Chait B. T., Landick R., Muir T., Severinov K., and Darst S. A. (2003) Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J. Am. Chem. Soc. 125, 12475–12483 [DOI] [PubMed] [Google Scholar]
- 16. Semenova E., Yuzenkova Y., Peduzzi J., Rebuffat S., and Severinov K. (2005) Structure-activity analysis of microcinJ25: distinct parts of the threaded lasso molecule are responsible for interaction with bacterial RNA polymerase. J. Bacteriol. 187, 3859–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vincent P. A., Bellomio A., de Arcuri B. F., Farías R. N., and Morero R. D. (2005) MccJ25 C-terminal is involved in RNA-polymerase inhibition but not in respiration inhibition. Biochem. Biophys. Res. Commun. 331, 549–551 [DOI] [PubMed] [Google Scholar]
- 18. Mathavan I., and Beis K. (2012) The role of bacterial membrane proteins in the internalization of microcin MccJ25 and MccB17. Biochem. Soc. Trans. 40, 1539–1543 [DOI] [PubMed] [Google Scholar]
- 19. Mathavan I., Zirah S., Mehmood S., Choudhury H. G., Goulard C., Li Y., Robinson C. V., Rebuffat S., and Beis K. (2014) Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat. Chem. Biol. 10, 340–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corbalan N., Runti G., Adler C., Covaceuszach S., Ford R. C., Lamba D., Beis K., Scocchi M., and Vincent P. A. (2013) Functional and structural study of the dimeric inner membrane protein SbmA. J. Bacteriol 195, 5352–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reading E., Walton T. A., Liko I., Marty M. T., Laganowsky A., Rees D. C., and Robinson C. V. (2015) The effect of detergent, temperature, and lipid on the oligomeric state of MscL constructs: insights from mass spectrometry. Chem. Biol. 22, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 23. Joosten R. P., te Beek T. A., Krieger E., Hekkelman M. L., Hooft R. W., Schneider R., Sander C., and Vriend G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res. 39, D411–D419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalli A. C., Sansom M. S., and Reithmeier R. A. (2015) Molecular dynamics simulations of the bacterial UraA H+-uracil symporter in lipid bilayers reveal a closed state and a selective interaction with cardiolipin. PLoS Comput. Biol. 11, e1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Habeck M., Haviv H., Katz A., Kapri-Pardes E., Ayciriex S., Shevchenko A., Ogawa H., Toyoshima C., and Karlish S. J. (2015) Stimulation, inhibition, or stabilization of Na,K-ATPase caused by specific lipid interactions at distinct sites. J. Biol. Chem. 290, 4829–4842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dowhan W., and Bogdanov M. (2009) Lipid-dependent membrane protein topogenesis. Annu. Rev. Biochem. 78, 515–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vitrac H., Bogdanov M., and Dowhan W. (2013) Proper fatty acid composition rather than an ionizable lipid amine is required for full transport function of lactose permease from Escherichia coli. J. Biol. Chem. 288, 5873–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Picas L., Montero M. T., Morros A., Vázquez-Ibar J. L., and Hernández-Borrell J. (2010) Evidence of phosphatidylethanolamine and phosphatidylglycerol presence at the annular region of lactose permease of Escherichia coli. Biochim. Biophys. Acta 1798, 291–296 [DOI] [PubMed] [Google Scholar]
- 29. Laganowsky A., Reading E., Allison T. M., Ulmschneider M. B., Degiacomi M. T., Baldwin A. J., and Robinson C. V. (2014) Membrane proteins bind lipids selectively to modulate their structure and function. Nature 510, 172–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martens C., Stein R. A., Masureel M., Roth A., Mishra S., Dawaliby R., Konijnenberg A., Sobott F., Govaerts C., and Mchaourab H. S. (2016) Lipids modulate the conformational dynamics of a secondary multidrug transporter. Nat. Struct. Mol. Biol. 23, 744–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drachmann N. D., Olesen C., Møller J. V., Guo Z., Nissen P., and Bublitz M. (2014) Comparing crystal structures of Ca2+-ATPase in the presence of different lipids. FEBS J. 281, 4249–4262 [DOI] [PubMed] [Google Scholar]
- 32. Zirah S., Afonso C., Linne U., Knappe T. A., Marahiel M. A., Rebuffat S., and Tabet J. C. (2011) Topoisomer differentiation of molecular knots by FTICR MS: lessons from class II lasso peptides. J. Am. Soc. Mass Spectrom. 22, 467–479 [DOI] [PubMed] [Google Scholar]
- 33. Benesch J. L., Ruotolo B. T., Sobott F., Wildgoose J., Gilbert A., Bateman R., and Robinson C. V. (2009) Quadrupole-time-of-flight mass spectrometer modified for higher-energy dissociation reduces protein assemblies to peptide fragments. Anal. Chem. 81, 1270–1274 [DOI] [PubMed] [Google Scholar]
- 34. Sobott F., Hernández H., McCammon M. G., Tito M. A., and Robinson C. V. (2002) A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 74, 1402–1407 [DOI] [PubMed] [Google Scholar]
- 35. Jávorfi T., Hussain R., Myatt D., and Siligardi G. (2010) Measuring circular dichroism in a capillary cell using the b23 synchrotron radiation CD beamline at diamond light source. Chirality 22, E149–E153 [DOI] [PubMed] [Google Scholar]
- 36. Hussain R., Jávorfi T., and Siligardi G. (2012) Circular dichroism beamline B23 at the Diamond Light Source. J. Synchrotron Radiat. 19, 132–135 [DOI] [PubMed] [Google Scholar]
- 37. Hussain R., Benning K., Myatt D., Javorfi T., Longo E., Rudd T. R., Pulford B., and Siligardi G. (2015) CDApps: integrated software for experimental planning and data processing at beamline B23, Diamond Light Source. Corrigendum. J. Synchrotron Radiat. 22, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sreerama N., and Woody R. W. (2004) On the analysis of membrane protein circular dichroism spectra. Protein Sci. 13, 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 43, 186–190 [Google Scholar]
- 40. Collaborative and Computational Project No. 4. (1994) The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. D. Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 41. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 44. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 45. Blanc E., Roversi P., Vonrhein C., Flensburg C., Lea S. M., and Bricogne G. (2004) Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D Biol. Crystallogr. 60, 2210–2221 [DOI] [PubMed] [Google Scholar]
- 46. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Jong D. H., Singh G., Bennett W. F., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., and Marrink S. J. (2013) Improved parameters for the Martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 [DOI] [PubMed] [Google Scholar]
- 48. Periole X., Cavalli M., Marrink S. J., and Ceruso M. A. (2009) Combining an elastic network with a coarse-grained molecular force field: structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 5, 2531–2543 [DOI] [PubMed] [Google Scholar]
- 49. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., and de Vries A. H. (2007) The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111, 7812–7824 [DOI] [PubMed] [Google Scholar]
- 50. Dahlberg M., and Maliniak A. (2010) Mechanical properties of coarse-grained bilayers formed by cardiolipin and zwitterionic lipids. J. Chem. Theory Comput. 6, 1638–1649 [DOI] [PubMed] [Google Scholar]
- 51. Lomize M. A., Lomize A. L., Pogozheva I. D., and Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 [DOI] [PubMed] [Google Scholar]
- 52. Wassenaar T. A., Ingólfsson H. I., Böckmann R. A., Tieleman D. P., and Marrink S. J. (2015) Computational lipidomics with insane: a versatile tool for generating custom membranes for molecular simulations. J. Chem. Theory Comput. 11, 2144–2155 [DOI] [PubMed] [Google Scholar]
- 53. Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., and Marrink S. J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 [DOI] [PubMed] [Google Scholar]
- 54. Berendsen H. J., Postma J. P., Vangunsteren W. F., Dinola A., and Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 55. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 56. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single-crystals–a new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]
- 57. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 58. McGibbon R. T., Beauchamp K. A., Harrigan M. P., Klein C., Swails J. M., Hernández C. X., Schwantes C. R., Wang L. P., Lane T. J., and Pande V. S. (2015) MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.