

Abstract
Melanoma differentiation-associated gene 7 (MDA-7/IL-24) exhibits cytotoxic effects on tumor cells while sparing untransformed cells, and Bcl-x(L) is reported to efficiently block the induction of cell death by MDA-7/IL-24. The expression of Bcl-x(L) is regulated at the level of RNA splicing via alternative 5′ splice site selection within exon 2 to produce either the pro-apoptotic Bcl-x(s) or the anti-apoptotic Bcl-x(L). Our laboratory previously reported that Bcl-x RNA splicing is dysregulated in a large percentage of human non-small cell lung cancer (NSCLC) tumors. Therefore, we investigated whether the alternative RNA splicing of Bcl-x pre-mRNA was modulated by MDA-7/IL-24, which would suggest that specific NSCLC tumors are valid targets for this cytokine therapy. Adenovirus-delivered MDA-7/IL-24 (Ad.mda-7) reduced the viability of NSCLC cells of varying oncogenotypes, which was preceded by a decrease in the ratio of Bcl-x(L)/Bcl-x(s) mRNA and Bcl-x(L) protein expression. Importantly, both the expression of Bcl-x(L) and the loss of cell viability were “rescued” in Ad.mda-7-treated cells incubated with Bcl-x(s) siRNA. In addition, NSCLC cells ectopically expressing Bcl-x(s) exhibited significantly reduced Bcl-x(L) expression, which was again restored by Bcl-x(s) siRNA, suggesting the existence of a novel mechanism by which Bcl-x(s) mRNA restrains the expression of Bcl-x(L). In additional mechanistic studies, inhibition of SRC and PKCδ completely ablated the ability of MDA-7/IL-24 to reduce the Bcl-x(L)/(s) mRNA ratio and cell viability. These findings show that Bcl-x(s) expression is an important mediator of MDA-7/IL-24-induced cytotoxicity requiring the SRC/PKCδ signaling axis in NSCLC cells.
Keywords: apoptosis; cell death; cell signaling; RNA splicing; signal transduction; MDA-7/IL-24, Bcl-x; alternative splicing; non-small cell lung cancer
Introduction
A number of studies have demonstrated that the overexpression of Bcl-x(L) in cells confers apoptosis resistance as well as cooperates with oncogenic factors (e.g. c-Myc) in tumorigenesis (1–10). The regulation of Bcl-x(L) expression can be complex at times, consisting of both transcriptional and post-transcriptional processes. In regard to post-transcriptional processing/alternative splicing, the BCL-x gene, via alternative 5′ splice site (5′SS)5 selection within exon 2, produces either the Bcl-x(s) isoform through activation of an upstream/proximal 5′SS or the Bcl-x(L) isoform through activation of a downstream/distal 5′SS. A number of studies have demonstrated that Bcl-x(s), in contrast to Bcl-x(L), promotes apoptosis (9, 11–14). Hence, the alternative 5′SS selection of Bcl-x pre-mRNA emerged as a potential target for anti-cancer therapeutics. For example, Taylor et al. (15) demonstrated that Bcl-x 5′SS selection can be specifically modulated using antisense oligonucleotides specific against the Bcl-x(L) 5′ splice site. Treatment of cells with these oligonucleotides induced an increase in the expression of Bcl-x(s) and a decrease in the expression of Bcl-x(L), resulting in sensitization of NSCLC cells to chemotherapeutic agents (15). These findings were also demonstrated by Kole and co-workers (16) in additional cancer types as well as in vivo models. Thus, regulation of the 5′SS selection within the Bcl-x exon 2 is a critical factor in determining whether a cancer cell is susceptible or resistant to apoptosis in response to chemotherapy (15–19).
In cells, Bcl-x 5′SS selection is regulated by the generation of de novo ceramide in response to apoptotic stimuli such as the chemotherapeutic agent, gemcitabine (20, 21). More recent studies by Zhou and co-workers (22) and Chang et al. (23) verified these early findings and extended the list of chemotherapeutic agents to emetine, a potent protein synthesis inhibitor, and amiloride, a potassium-conserving diuretic. Later studies from our laboratory identified the RNA splicing factor, SAP155, as a regulator of the 5′SS selection of Bcl-x pre-mRNA (24, 25), and this RNA trans-factor was required for the effect of ceramide on the alternative 5′SS selection of Bcl-x pre-mRNA in NSCLC cells (24, 25).
In the present study, the role of melanoma differentiation-associated gene-7/interleukin-24 (MDA-7/IL-24) was examined in the context of Bcl-x 5′SS selection. MDA-7/IL-24 is a cytokine classified as a member of the IL-10 gene family that was initially identified through a subtraction hybridization approach using a differentiation therapy model of human melanoma (26). MDA-7/IL-24 potently inhibits cell growth and induces apoptosis in various epithelial cancers both in vitro and in vivo, including lung cancers (27). In contrast, MDA-7/IL-24 has shown no lethality toward normal cells (28).
The ability of MDA-7/IL-24 to inhibit cell growth of tumor cells and to induce apoptosis in tumor cells has been attributed, in part, to modulation of the expression of Bcl-x(L) (27, 29, 30). Specifically, a potential functional role for changes in Bcl-x(L) expression in adenovirus-delivered MDA-7/IL-24 (Ad.mda-7)-induced apoptosis was suggested by the finding that forced overexpression of Bcl-x(L) diminished the apoptotic effect of Ad.mda-7 in lung carcinoma cells (27, 29). The possible link to Bcl-x 5′SS selection was suggested in this mechanism as the induction of ceramide production plays a decisive role in MDA-7/IL-24-mediated apoptosis (31, 32).
In this study, we explored the hypothesis that MDA-7/IL-24 reduces the levels of Bcl-x(L) by modulating the 5′SS selection of Bcl-x pre-mRNA in a de novo ceramide-dependent manner. Indeed, we demonstrate that MDA-7/IL-24 induces the activation of the Bcl-x(s) 5′ splice site, thereby lowering the Bcl-x(L)/(s) ratio in NSCLC cells, and thus, instigating the down-regulation of Bcl-x(L). Surprisingly, this mechanism was ceramide-independent, but the loss of SAP155 expression was still observed. Furthermore, the expression of Bcl-x(s) mRNA was shown to be a major component in the ability of MDA-7/IL-24 to induce the loss of cell viability as well as induce the loss of Bcl-x(L) expression. Exploration of the signal transduction pathway mediating this distal mechanism in response to MDA-7/IL-24 identified the SRC/PKCδ signaling axis as critical. These findings, therefore, suggest that induction of Bcl-x(s) mRNA may prove an effective therapeutic avenue to enhance the cancer-specific killing of MDA-7/IL-24 treatment, which may be an effective treatment for NSCLC lung tumors presenting with a low Bcl-x(L)/(s) ratio.
Results
Ad.mda-7 Induces a Loss of Cell Viability in NSCLC Cells
Previously, MDA-7/IL-24 was reported to induce cytotoxic effects on NSCLC cell lines without affecting non-transformed counterparts (27, 28). Our initial studies confirmed this cytotoxic effect in regard to adenovirus-delivered MDA-7/IL-24 (Ad.mda-7) for several established NSCLC cell lines with differing oncogenotypes (A549, H838, and H1299) after 48 and 72 h (Table 1 and Fig. 1, A–C). Of note, no loss of viability was observed in these cell lines within ≤24 h of Ad.mda-7 treatment (data not shown). Importantly, Ad.mda-7 treatment had no significant effect on the viability of non-transformed, immortalized lung epithelial cells (HBEC-3KT cells; Fig. 1D). Hence, Ad.mda-7 elicits cytotoxicity in tumorigenic lung cells regardless of oncogenotype, while sparing non-cancerous lung cells as reported previously (27, 28).
TABLE 1.
Characterization of NSCLC cell lines
Characterization of the NSCLC cell lines utilized in this study is shown. For each cell line, their histology as well as Ras and p53 mutational status are represented.
| Cell line | NSCLC histology | Mutational status |
|---|---|---|
| A549 | Adenocarcinoma | WT p53, K-RasG12V mutation |
| H838 | Adenocarcinoma | p53 mutation, WT Ras |
| H1299 | Adenocarcinoma | p53 null, N-RasQ61K mutation |
FIGURE 1.

Ad.mda-7 induces the loss of cell viability in NSCLC cells, but in not non-transformed lung epithelial cells. Cells (1 × 104) were transduced with the indicated MOI (PFU/cell) of either ad.mda-7 or Ad.CMV control virus. After the indicated incubation time, the cells were assayed for cell viability using a WST-1 assay as described under “Experimental Procedures.” A, A549 cells. B, H838 cells. C, H1299 cells. D, HBEC-3KT cells. Data are expressed as mean of the percentage of viability ± S.D. Data are representative of six separate determinations on two separate occasions.
Ad.mda-7 Induces Alterations in the 5′ Splice Site Selection of Bcl-x Pre-mRNA
The loss of Bcl-x(L) expression is a required mechanism for MDA-7/IL-24-induced loss of cell viability in several cancer cell types (mesothelioma I-45xL, GBM glioblastoma, and prostate carcinoma cells) (29, 30–32). The alternative splicing of Bcl-x pre-mRNA is one method of regulating Bcl-x(L) expression. Indeed, alterations in Bcl-x splicing are sensitive to ceramide production, and MDA-7/IL-24 is reported to increase ceramide synthesis (20, 21, 24, 25, 33). Furthermore, the ceramide-sensitive RNA trans-factor, SAP155, promotes the formation of Bcl-x(L) mRNA, and siRNA targeting SAP155 results in decreases in the Bcl-x(L)/Bcl-x(s) mRNA ratio in NSCLC cells (25). Thus, we hypothesized that MDA-7/IL-24 treatment induces the down-regulation of SAP155 and the subsequent lowering of the Bcl-x(L)/Bcl-x(s) mRNA ratio prior to the observed loss of viability (less than 48 h). Consistent with this hypothesis, A549 cells treated with Ad.mda-7 for 24 h exhibited a reduction in SAP155 protein levels (Fig. 2A). Short-term treatment of NSCLC cells with Ad.mda-7 also induced a significant decrease in Bcl-x (L)/(s) mRNA ratios when compared with control Ad.CMV adenovirus (Fig. 2A). This effect of Ad.mda-7 was both concentration-dependent and stable for >36 h (Fig. 2, B and C). Ad.mda-7 also altered Bcl-x alternative splicing in H838 cells (Fig. 2D), demonstrating translatability to other NSCLC cell lines of differing oncogenotypes. To determine whether the effect was specific to NSCLC cells, the ovarian cancer cell lines (SKOV and DOV) were treated with either Ad.CMV or Ad.mda-7 (Fig. 2D). These cell lines also demonstrated significant changes in Bcl-x splicing. Importantly, Bcl-x alternative splicing was not affected by Ad.mda-7 in non-transformed HBEC-3KT cells, correlating with a lack of cytotoxicity induced by MDA-7/IL-24 (Fig. 2E). Lastly, we examined whether the effect of MDA-7/IL-24 on RNA splicing was specific for Bcl-x pre-mRNA. In this regard, we tested the capacity of MDA-7/IL-24 to affect the splicing ratios of Mcl-1(L)/(s) and CD44, but no effect on the alternative splicing of these pre-mRNAs was observed (Fig. 2F). Hence, MDA-7/IL-24 induces a reduction in the Bcl-x(L)/Bcl-x(s) mRNA ratio, which was not due to a generalized effect on constitutive RNA splicing.
FIGURE 2.

Ad.mda-7 induces the activation of the Bcl-x(s)/proximal 5′ splice site of Bcl-x pre-mRNA. A, cells (1.2 × 105) were transduced with 150 MOI of either ad.mda-7 or Ad.CMV control virus. After 24 h, total RNA and protein were extracted. Total protein was subjected to SDS-PAGE analysis and Western immunoblotting for MDA-7, SAP155, and β-actin (left panel). Total RNA was subjected to quantitative/competitive RT-PCR analysis (competitive qRT-PCR) of Bcl-x splice variants and the corresponding Bcl-x(L)/(s) mRNA ratios (Ratio (L)/(s)). The ratio of Bcl-x(L) to Bcl-x(s) mRNA was determined by densitometric analysis of RT-PCR fragments (p < 0.01, n = 6). Data are expressed as mean ± S.D. B and C, cells (1.2 × 105) were transduced with either the indicated MOI of either Ad.mda-7 or Ad.CMV control virus for 24 h (B) or 150 MOI of either Ad.mda-7 or Ad.CMV control virus for the indicated time (C). Total RNA was extracted and subjected to competitive qRT-PCR analysis of Bcl-x splice variants and the corresponding Bcl-x(L)/(s) mRNA ratios. Data are expressed as mean ± S.D. D and E, The indicated cells (1.2 × 105) were transduced with 150 MOI of either Ad.mda-7 or Ad.CMV control virus. After 24 h, total RNA was extracted and subjected to quantitative/competitive RT-PCR analysis as in panel A. Data are expressed as mean ± S.D. F, the same samples utilized in panel B were subjected to competitive qRT-PCR analysis of Mcl-1 and CD44 splice variants. For all panels, data are representative of six separate determinations on two separate occasions.
Ablation of Bcl-x(s) “Rescues” Ad.mda-7-elicited Cytotoxicity
As shown in this study, Ad.mda-7 reduces the cell viability on NSCLC cells and decreases the ratio of Bcl-x(L)/(s) mRNA. Although these data intriguingly correlate, they do not determine whether or not the Bcl-x splicing is a required mechanism for Ad.mda-7-induced killing or simply secondary to its pro-apoptotic effect. Furthermore, we posited the question: is it the loss of Bcl-x(L) or the production of Bcl-x(s) that plays a role in MDA-7/IL-24-induced loss of cell viability? To answer this question, we again treated NSCLC cells with Ad.mda-7, and first examined Bcl-x(L) expression. Indeed, it is currently known that MDA-7/IL-24 treatment reduces Bcl-x(L) expression, and that cells ectopically expressing Bcl-x(L) are resistant to MDA-7/IL-24-induced cytotoxicity (29). In line with these published observations, we observed that MDA-7/IL-24 reduced Bcl-x(L) protein levels in A549 cells after a 48-h incubation (Fig. 3A). The Ad.mda-7-elicited reduction in Bcl-x(L) protein was also evident in H838 cells, but not HBEC-3KT cells (Fig. 3, B and C). Surprisingly, Ad.mda-7 was not able to stimulate the detectable expression of Bcl-x(s) protein (Fig. 3, A–C). Other laboratories have also reported difficulty detecting endogenous Bcl-x(s), and typically, either 10-fold more protein extract or high ectopic expression of Bcl-x(s) is required to detect the protein (9, 15, 21, 34). Thus, it was unclear whether Bcl-x(s) mRNA or protein was sufficient to mediate the cytotoxic effects of MDA-7/IL-24.
FIGURE 3.
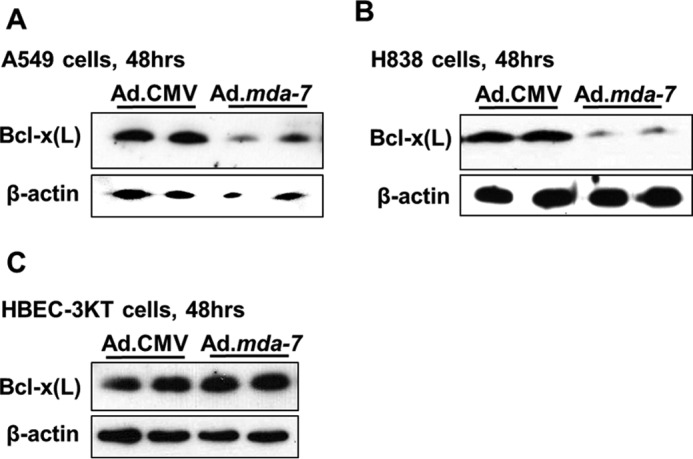
Ad.mda-7 induces the down-regulation of Bcl-x(L) in NSCLC cells, but not in non-transformed lung epithelial cells. The indicated cells (1.2 × 105) were transduced with 150 MOI of either Ad.mda-7 or Ad.CMV control virus. After 48 h, total protein was extracted and subjected to SDS-PAGE analysis/Western immunoblotting for Bcl-x(L) and β-actin. A, A549 cells. B, H838 cells. C, HBEC-3KT cells. Data are representative of four separate determinations on two separate occasions.
To address whether Ad.mda-7 exerts its cytotoxic effect via the production of Bcl-x(s), A549 cells were treated with the Bcl-x(s)-targeted siRNA followed by treatment with either Ad.mda-7 or Ad.CMV. As anticipated, MDA-7/IL-24-stimulated Bcl-x(s) mRNA levels were reduced by Bcl-x(s) siRNA (Fig. 4A). To examine whether the expression of Bcl-x(s) mRNA was a mediator of MDA-7-induced cell death, NSCLC cells were again treated with Bcl-x(s) siRNA, followed by either Ad.CMV or Ad.mda-7 for 48 and 72 h. Central to this study, NSCLC cells treated with Bcl-x(s) siRNA exhibited a significant increase in viability following Ad.mda-7 treatment when compared with NSCLC cells treated with control siRNA after 48 and 72 h (Fig. 4B). These data demonstrate that Ad.mda-7 induces cell death, at least in part, by the expression of Bcl-x(s) mRNA.
FIGURE 4.

Specific down-regulation of Bcl-x(s) mRNA significantly inhibits the loss of cell viability induced by Ad.mda-7. A, A549 cells (1.2 × 105) were transfected with Bcl-x(s) (siBcl-x(s), 100 nm) or control siRNA (siCON, 100 nm) and transduced with 150 MOI of either Ad.mda-7 or Ad.CMV control virus. After 48 h, total RNA was extracted and subjected to competitive (left panel) and quantitative (right panel) RT-PCR analysis of Bcl-x splice variants. The ratio of Bcl-x(L) to Bcl-x(s) mRNA was determined by densitometric analysis of RT-PCR fragments (p < 0.01, n = 6). The relative levels of Bcl-x(s) mRNA were determined as detailed under “Experimental Procedures” and normalized to 18s RNA. Data are expressed as mean ± S.D. and are representative of six separate determinations on two separate occasions. Scr siRNA, scrambled siRNA. B, A549 cells (1 × 104) were concomitantly treated as in panel A with the indicated MOI of either Ad.mda-7 or Ad.CMV control virus. After 48 or 72 h, the cells were assayed for cell viability using a WST-1 assay as described under “Experimental Procedures.” Data are expressed as mean of the percentage of viability of Ad.mda-7/Ad.CMV ± S.D. Data are representative of six separate determinations on two separate occasions. For the 48 h panel (left), * = p < 0.01 comparing lanes 3 and 4. For the 72 h panel (right), * = p < 0.01 comparing lanes 1 and 2 while ** = p < 0.01 comparing lanes 3 and 4.
The Expression of Bcl-x(s) mRNA Restrains the Expression of Bcl-x(L)
Our above data suggest that the expression of Bcl-x(s) is an important mechanism for MDA-7/IL-24-induced cell death. As mentioned previously, the Bcl-x(s) protein was not detected after Ad.mda-7 treatment. However, we did find that the levels of Bcl-x(L) protein were dramatically preserved by the down-regulation of Bcl-x(s) mRNA (Fig. 5A). Specifically, A549 cells were again treated with Ad.mda-7 in the presence or absence of Bcl-x(s) siRNA. Remarkably, NSCLC cells treated with Bcl-x(s) siRNA exhibited significantly enhanced levels of Bcl-x(L) protein when compared with control siRNA. These data demonstrate that Bcl-x(s) siRNA (i.e. specific removal of Bcl-x(s) mRNA) rescued the loss of Bcl-x(L) protein expression induced by Ad.mda-7 (Fig. 5A). This effect was not mediated at the mRNA level for Bcl-x(L), as the expression of Bcl-x(L) mRNA did not mimic the observed effects on the protein (data not shown). Thus, Bcl-x(L) protein expression is negatively regulated by Bcl-x(s) expression.
FIGURE 5.
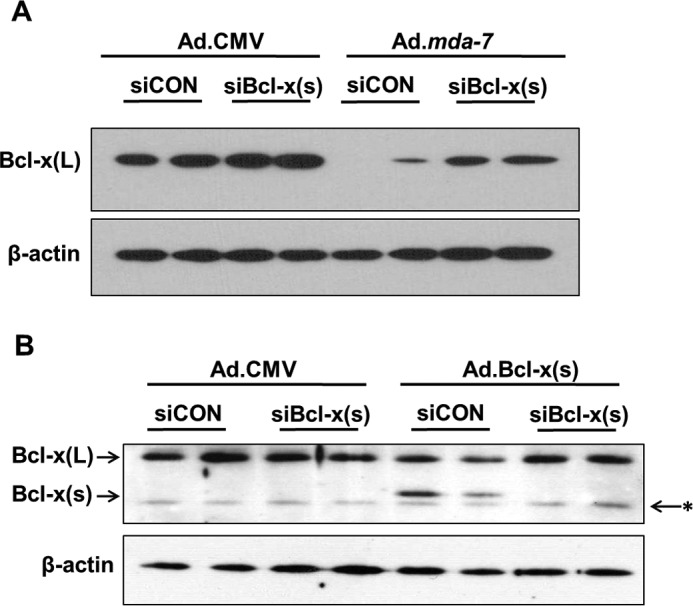
Specific down-regulation of Bcl-x(s) mRNA significantly inhibits the loss of Bcl-x(L) protein induced by Ad.mda-7 and Ad.Bcl-x(s). A, A549 cells (1.2 × 105) were transfected with Bcl-x(s) (siBcl-x(s), 100 nm) or control siRNA (siCON, 100 nm) and transduced with 150 MOI of either Ad.mda-7 or Ad.CMV control virus. After 48 h, total protein was extracted. Total protein was subjected to SDS-PAGE analysis and Western immunoblotting for Bcl-x(L) and β-actin. Data are representative of six separate determinations on two separate occasions. B, A549 cells (1.2 × 105) were transfected with Bcl-x(s) (100 nm) or control siRNA (100 nm) and transduced with 500 MOI of either Ad.Bcl-x(s) or Ad.CMV control virus. After 24 h, total protein was extracted. Total protein was subjected to SDS-PAGE analysis and Western immunoblotting for Bcl-x(L)/(s) and β-actin. * = nonspecific band or a different Bcl-x splice variant (Bcl-xγ). Data are representative of six separate determinations on two separate occasions.
To determine whether the expression of the coding sequence of Bcl-x(s) mRNA was mediating this effect on Bcl-x(L) expression, Ad.Bcl-x(s), which contains only the coding mRNA, was used to express this cDNA in A549 cells. The expression of Bcl-x(s) coding mRNA induced a significant (58%) reduction in the levels of Bcl-x(L) protein (Fig. 5B). More importantly, co-treatment with Bcl-x(s) siRNA completely blocked this effect, with no effect observed for Bcl-x(s) siRNA or coding cDNA on the levels of Bcl-x(L) mRNA (data not shown). These data demonstrate, for the first time, a novel mechanisms by which MDA-7/IL-24 induces cytotoxicity in tumor cells, at least in part, by generating Bcl-x(s) mRNA, which reduces Bcl-x(L) protein levels, and thereby, limits the cytoprotective effects of Bcl-x(L).
MDA-7/IL-24-induced Alterations in Bcl-x Pre-mRNA Are Independent of Ceramide-generating Pathways
As mentioned previously, MDA-7/IL-24 is known to induce ceramide synthesis via the de novo pathway (24, 25, 33), and Bcl-x 5′SS selection has been previously reported by our laboratory to be responsive to de novo ceramide production elicited by gemcitabine (20, 21, 24, 25). Therefore, we hypothesized that MDA-7/IL-24-induced reductions in the Bcl-x(L)/Bcl-x(s) splicing ratio required de novo ceramide production. Surprisingly, incubation of MDA-7/IL-24-treated A549 cells with fumonisin B1 or myriocin, two inhibitors of de novo ceramide synthesis, were unable to inhibit the effect of MDA-7/IL-24 on the Bcl-x(L)/Bcl-x(s) splicing ratio (Fig. 6, A and B). Additional inhibitors of sphingolipid synthesis such as acid and neutral sphingomyelinase did not affect the ability of MDA-7/IL-24 to reduce the Bcl-x(L)/Bcl-x(s) splicing ratio (data not shown). These data demonstrate that MDA-7/IL-24 affects the alternative splicing of Bcl-x pre-mRNA via a ceramide-independent pathway.
FIGURE 6.
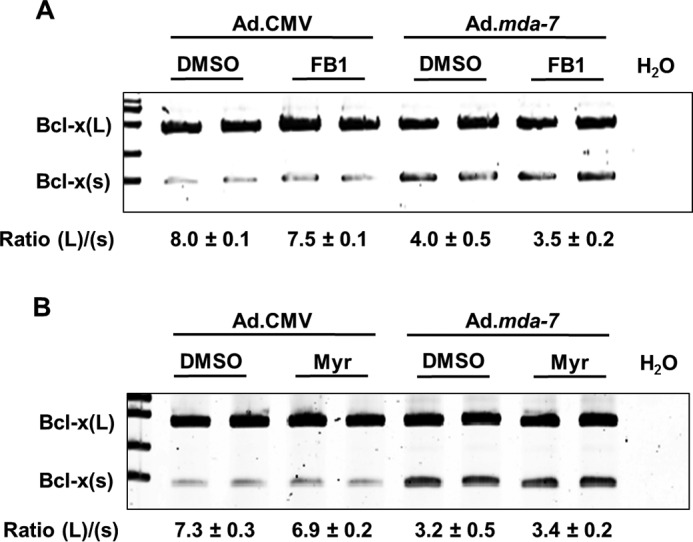
Ad.mda-7 induces the activation of the Bcl-x(s)/proximal 5′ splice site of Bcl-x pre-mRNA independent of de novo ceramide generation. A and B, A549 cells (1.2 × 105) were pre-treated with 100 μm fumonisin B1 (FB1) (A) or 100 nm myriocin (Myr) (B) for 4 h, followed by transduction with 150 MOI of either Ad.mda-7 or Ad.CMV control virus. After 24 h, total RNA was extracted and subjected to quantitative/competitive RT-PCR analysis of Bcl-x splice variants. The ratio of Bcl-x(L) to Bcl-x(s) mRNA (Ratio (L)/(s)) was determined by densitometric analysis of RT-PCR fragments (p < 0.01, n = 6). Data are expressed as mean ± S.D. and are representative of at least three separate determinations on two separate occasions
MDA-7/IL-24-induced Alterations in Bcl-x pre-mRNA Require the SRC/PKCδ Signaling Axis
Because the mechanism by which MDA-7/IL-24 induced the activation of the Bcl-x(s) 5′ splice site was independent of ceramide signaling, we undertook a broad-based approach to identify the signaling pathway required for MDA-7/IL-24 to affect Bcl-x RNA splicing. To identify a target pathway, we subjected A549 cells to an array of small molecule inhibitors (Table 2) targeting critical factors in signaling pathways related to MDA-7/IL-24-induced cell death (e.g. ER stress, SRC kinase, and protein kinase C (PKC) signaling pathways). Of these, the SRC inhibitor, Src-1, and the broad spectrum PKC inhibitor, Gö 6983, completely inhibited the ability of MDA-7/IL-24 to reduce the Bcl-x(L)/(s) mRNA ratio (Fig. 7, A and B). Interestingly, Inoue et al. (35) demonstrated that SRC signaling pathways were involved in MDA-7/IL-24 signaling, and other laboratory groups have demonstrated that the novel PKC isoform, PKCδ, is a downstream mediator of SRC signaling (70). For this reason, we treated NSCLC cells with rottlerin, a reported inhibitor of PKCδ (36), and specific PKCδ and SRC siRNA. Both the small molecule inhibitor of PKCδ and down-regulation of the enzyme by siRNA blocked the ability of MDA-7/IL-24 to reduce the Bcl-x(L)/(s) mRNA ratio and to rescue MDA-7-mediated cytotoxicity (Fig. 7, B and C). Taken together, these data indicate that MDA-7/IL-24-induced alterations in Bcl-x pre-mRNA splicing are dependent on the SRC/PKC-δ signaling axis (Fig. 7D), but independent of de novo ceramide generation.
TABLE 2.
List of Inhibitors
Signaling pathways and factors examined for involvement in the alternative splicing of Bcl-x pre-mRNA modulated by MDA-7/IL-24 as analyzed by small molecule inhibitors are shown. The table depicts the inhibitors and their respective concentrations utilized in this study. Signaling pathways were examined for effects on the ratio of Bcl-x(L)/(s) mRNA utilizing small-molecule inhibitors at doses well characterized in the scientific literature and previously utilized in studies on A549 cells (43–47). mTOR, mammalian target of rapamycin. N/C, no change; iPLA2, intracellular phospholipase A2.
| Inhibitor | Method of action | Concentration | Outcome |
|---|---|---|---|
| Salubrinal | ER stress inhibitor | 15 μm | N/C |
| Bromoenol lactone (BEL) | iPLA2 inhibitor | 20 μm | N/C |
| PKC Y translocation peptide | PKCϵ inhibitor | 100 μm | N/C |
| Rapamycin | mTOR inhibitor | 10 μm | N/C |
| Gö 6983 | Pan-PKC inhibitor | 100 nm | Inhibition |
| Rottlerin | PKCδ inhibitor | 50 μm | Inhibition |
| Src inhibitor-1 | Src inhibitor | 50 nm | Inhibition |
| PKCδ siRNA | PKCδ inhibitor | 100 nm | Inhibition |
| SRC siRNA | SRC inhibitor | 100 nm | Inhibition |
FIGURE 7.

Ad.mda-7 induces the activation of the Bcl-x(s)/proximal 5′ splice site of Bcl-x pre-mRNA via the SRC/PKCδ signaling axis. A, A549 cells were treated with Src inhibitor (SRC-1), pan-PKC-inhibitor (Gö 6983), or rottlerin (Rott) for the times indicated under “Experimental Procedures.” Cells were then exposed to Ad.mda-7 or Ad.CMV virus for 24 h. Cells were then harvested, and the ratio of Bcl-x(L)/(s) was determined. Veh, vehicle. B, A549 cells were transfected with either scrambled (si0), PKCδ (siPCKδ) or SRC siRNA (siSRC), and 48 h later, protein and RNA were harvested and the levels of SRC, PKC-δ, MDA-7, and actin, as well as the ratio of Bcl-x(L)/(s) mRNA, were determined. The ratio of Bcl-x(L) to Bcl-x(s) mRNA was determined by densitometric analysis of RT-PCR fragments. IB, immunoblot. Data are expressed as mean ± S.D. and are representative of three separate determinations on two separate occasions. C, A549 cells were exposed to either siRNA directed toward SRC or PKCδ for 48 h, and then exposed to MDA-7 for 24 h. Cells were then assayed using the WST-1 reagent as described under “Experimental Procedures.” Data are expressed as mean ± S.D. and are representative of six separate determinations on two separate occasions (* = p < 0.01, n = 6). D, schematic of how MDA7 suppresses cell survival by alteration of Bcl-x splicing via a SRC/PKCδ signaling pathway. Specifically, intracellular MDA7 expression promotes the activation of the Bcl-x(s) 5′ splice site via either an intracellular receptor event or ER stress to induce SRC and PKCδ activation, which may involve a direct or indirect effect of PKCδ on SAP155 (down-regulation) or other RNA trans-factors to up-regulate Bcl-x(s) level and down-regulate Bcl-x(L) level. As SAP155 is down-regulated by MDA-7 and cannot conclusively be determined as the regulatory RNA trans-factor, PKCδ is likely affecting Bcl-x 5′ splice site selection in an indirect fashion. The overall main theme of the study is that intracellular MDA-7 reduces cell viability through directly manipulating the level of anti-apoptotic Bcl-x(L) via affecting Bcl-x 5′ splice site selection, which requires the SRC/PKCδ signaling pathway.
Discussion
A number of studies have been published demonstrating that alternative splicing is often dysregulated in cancer (24, 25, 37–45). For example, modifications in the alternative splicing of apoptotic factors such as Bcl-x, caspase 9, and Mcl-1 have been linked to the resistance to chemotherapy of neoplastic cells (20, 21, 24, 25, 37, 38, 44, 45). In this study and for the first time, we show that MDA-7/IL-24 treatment of NSCLC cells alters the 5′SS selection of Bcl-x pre-mRNA, thereby increasing the pro-apoptotic Bcl-x(s) mRNA relative to anti-apoptotic Bcl-x(L) in A549 cells. The effect of MDA-7/IL-24 treatment on alternative splicing was specific to Bcl-x pre-mRNA, as Ad.mda-7 treatment had no effect on the ratio of Mcl-1(L)/Mcl-1(s) as well as any of the numerous CD44 splice variants. The effect of MDA-7/IL-24 on activating the Bcl-x(s) 5′SS also appeared to be independent of the oncogenotype (Table 1), as Ad.mda-7 also decreased the Bcl-x(L)/(s) ratio in H838 cells. The effect of MDA-7/IL-24 on Bcl-x 5′SS selection was also evident in other epithelial tumor cells such as the SKOV3 ovarian cancer cell line (p53 deletion, normal Ras), although to a lesser degree. Hence, the specific effect of MDA-7/IL-24 on the activation of the Bcl-x(s) 5′SS translates to multiple oncogenotypes as well as various solid tumor types, suggesting a broad-based mechanism that can be exploited for cancer therapeutics.
In regard to the therapeutic efficacy of MDA-7/IL-24, our findings in this study led to the hypothesis that the alternative 5′SS selection of Bcl-x pre-mRNA was an important mechanism for MDA-7/IL-24 to simply lower the expression of Bcl-x(L) and enhance the cytotoxic effects of this cytokine on cancer cells. In contrast to this hypothesis, we observed that siRNAs specifically targeting Bcl-x(s) significantly blocked the MDA-7/IL-24-induced cytotoxicity to NSCLC cells. These data initially suggested that the expression of Bcl-x(s), and not just the down-regulation of Bcl-x(L), induced by activation of the proximal 5′SS in exon 2, was important in the cytotoxic effects elicited by MDA-7/IL-24. What was most surprising in regard to the above findings is the lack of observed expression for Bcl-x(s) protein, although an increase in the mRNA is observed. Hence, the expression of Bcl-x(s) protein is unlikely to be mediating the cytotoxic effects of MDA-7/IL-24, and this study begins to clarify a decade-old question regarding the function of Bcl-x(s). In particular, previous studies suggested that Bcl-x(s) functions by dimerizing with Bcl-x(L) to sequester Bcl-x(L) from Bax association (34). Although the studies examining this interaction in vitro are compelling, albeit on occasion in opposition of earlier studies on Bcl-x(s) function (9, 34), the Bcl-x(s) protein itself is rarely observed in most cell types unless ectopically expressed (9, 15, 21, 34). In addition, 10-fold more protein extract has been required to detect Bcl-x(s) at the protein level in previously reported studies (15, 21). The findings from our laboratory over the years may even suggest that the Bcl-x(s) protein may have been misidentified as a separate Bcl-x splice variant (Fig. 5B) (see for example Refs. 24, 25). Regardless, the current study indicates that the expression of the Bcl-x(s) mRNA, not the protein, regulates the biological effects of MDA-7/IL-24-induced loss of cell viability by reducing the protein levels of Bcl-x(L). For example, NSCLC cells treated with Bcl-x(s) siRNA significantly rescued the reduction in Bcl-x(L) expression induced by Ad.mda-7 as well as treatment with Ad.Bcl-x(s). These data suggest that Bcl-x(s) mRNA functions to inhibit the expression of Bcl-x(L) rather than the Bcl-x(s) protein, directly antagonizing the function of the Bcl-x(L) protein.
The mechanism by which Bcl-x(s) coding mRNA elicits this effect remains unclear, although the data suggest that the effect occurs at the level of Bcl-x(L) protein synthesis or turnover/stability. One can surmise that the removal of the portion of exon 2 sequence encoding for the Bcl-x(L) mRNA induces the formation of a new RNA cis-element or hairpin structure that competes for the association of an RNA trans-factor or RNA-binding protein important in the synthesis of the Bcl-x(L) protein. Indeed, cytosolic polyadenylation binding proteins such as the CPEB family members play roles in regulating cytoplasmic polyadenylation, and thus, regulating the translation of proteins in response to cellular stress. These proteins bind specific RNA cis-elements, and Bcl-x(s) may simply act as a scavenger for an activating CPEB2, such as CPEB2B, which has roles in driving anoikis resistance and metastasis in triple negative breast cancer (43). Although this is a plausible mechanism, the effect of Bcl-x(s) mRNA on Bcl-x(L) protein expression may also occur at the post-translational level as there is a dramatic and rapid loss of Bcl-x(L) protein observed in response to Ad.mda-7. Indeed, Bcl-x(s) mRNA may also bind/sequester factors that stabilize the Bcl-x(L) protein, leading to the degradation of the protein. In support of this possibility, Fisher and co-workers (29) have shown that MDA-7/IL-24 can induce the loss of Bcl-x(L) at the post-translational level. Lastly, another possibility, albeit remote, exists in that Bcl-x(s) mRNA acts as a ribozyme for removal of the Bcl-x(L) protein. Although unlikely and not previously recorded for mammalian cells, several types of RNA mechanisms first thought to be limited to lower organisms have now been reported in mammalian cells such RNA trans-splicing and cytosolic RNA splicing (e.g. XBP-1 pre-RNA associated with the unfolded protein response) (44, 45). Overall, the ability of Bcl-x(s) mRNA to regulate the expression of Bcl-x(L) is novel and warrants future studies to identify this intriguing new mechanism.
In this study, we also delved into the signaling mechanism regulating the 5′SS selection of Bcl-x pre-mRNA in response to MDA-7/IL-24. Specifically, we show that SAP155, an RNA trans-factor reported by us as a major regulator of the 5′SS selection of Bcl-x pre-mRNA, was down-regulated by this cytokine (24, 25). Chalfant and co-workers (24, 25) and Fisher and co-workers (33) identified ceramide generation and subsequent activation of ceramide-induced protein phosphatases as a major mechanism by which certain chemotherapies reduced the survival of NSCLC cells. Furthermore, MDA-7/IL-24 induced cell death via a ceramide-dependent pathway in some cancer cell types (32, 33). Based on these studies, we examined the role ceramide in MDA-7/IL-24-induced reductions in Bcl-x(L)/Bcl-x(s) pre-mRNA ratios, but surprisingly, the ceramide synthase inhibitor, fumonisin B1, was unable to block MDA-7/IL-24-elicited alterations in Bcl-x alternative splicing. Furthermore, myriocin, which inhibits ceramide synthesis at the level of sphingolipid biosynthesis, had no effect on MDA-7/IL-24-induced changes in Bcl-x(L)/Bcl-x(s) mRNA ratios. Additional inhibitors of ceramide generation also had no effect. Therefore, MDA-7-induced changes in Bcl-X splicing must occur in a ceramide-independent manner.
The ceramide-independent nature of the signaling mechanism regulating Bcl-x RNA splicing in response to Ad.mda-7 led to a more broad-based approach and identified the SRC/PKCδ signaling pathway responsible for the modulation in Bcl-x RNA splicing in response to MDA-7/IL-24 (35). This was a novel finding as this is in contrast to previous signaling pathways reported to regulate Bcl-x RNA splicing in cancer such as the PI3K/PKCι pathway or PKCα signaling in non-cancerous cells (see for example Refs. 24, 25, 71, 72). One may surmise that MDA-7/IL-24 logically acts to drive terminal differentiation pathways, explaining the SRC activation paradigm as well as PKCδ, which are implicated in a number of differentiation processes. Indeed, our laboratories have previously shown that anoikis-resistant cancer cells induced SRC activation in response to MDA-7/IL-24, which correlated with significantly higher cell death (46). Thus, although SRC signaling is usually linked to pro-survival/oncogenic pathways, MDA-7/IL-24 may reinstate the normal cell cycle checkpoints, allowing for additional oncogenic stress (↑SRC activation) to induce cellular senescence and subsequent loss of viability for the cancer cell. Validation of this hypothesis may aid in explaining why SRC inhibitors did not show strong clinical effectiveness for breast and lung cancer as a single agent (47, 48). Overall, this study allows the postulation that multiple signaling pathways exist to modulate specific RNA trans-factors to repress or activate the Bcl-x(s) 5′SS, suggesting that this splicing event is a key distal point for numerous cell signaling pathways, and thus, may have broad roles in a number of biological functions as well as be exploited to enhance the killing of NSCLC cells.
In regard to the RNA trans-factor modulated by the SRC/PKCδ pathway, this study does not conclusively demonstrate that SAP155 is the mediator of the activation of the Bcl-x(s) 5′ splice site in response to MDA-7, but several key pieces of data implicate this RNA trans-factor in this paradigm. First, the levels of SAP155 are decreased in response to MDA-7 prior to an effect on Bcl-x RNA splicing, and our laboratory and others showed that this level of SAP155 reduction in cells would induce the activation of the Bcl-x 5′ splice site (25, 49). Second, MDA-7 did not significantly affect the levels of SAM68, hnRNP H, hnRNP K, and SRSF1, RNA trans-factors reported to affect Bcl-x RNA splicing in other cell systems (data not shown) (50, 51). Thus, SAP155 is a strong candidate as a regulating RNA trans-factor in this signaling cascade. Whether PKCδ directly affects the expression of SAP155 or another regulatory RNA trans-factor via phosphorylation or indirectly via additional downstream members of the signaling cascade is unknown. A study by White et al. (52) suggests that PKCδ and the RNA trans-factor, hnRNP K, are linked in another type of apoptosis mechanism. Although hnRNP K levels were not affected by MDA-7, one can also hypothesize that PKCδ may directly phosphorylate hnRNP K in response to MDA-7 to affect Bcl-x 5′ splice site selection. It is also plausible that PKCδ phosphorylates SAP155 as regulation of this RNA trans-factor by protein kinases and phosphatases has been reported (53–56). Furthermore, PKCδ has also been shown to translocate to the nucleus in response to apoptotic agonists (57), which localizes the enzyme in proximity to RNA splicing factors. Indeed, Cocco and co-workers (58) demonstrated that phosphoinositide-specific phospholipase Cβ 1 (PI-PLCβ1) will translocate to the nucleus with stimuli and is associated with RNA trans-factors (e.g. SRSF3). Thus, the enzyme that generates the activating second messenger lipid of PKCδ, diacylglycerol, is also localized to appropriate intracellular areas to mediate the direct regulation of RNA splicing. Future studies will focus on delineating the direct nature of this mechanism.
One open question from this study is whether MDA-7/IL-24 is driving cell death via receptor-mediated events using the IL-20R1/IL-20R2 and IL-22R1/IL-20R2 receptors or via intracellular pathways. For example, NSCLC cell lines have been reported to lack canonical cell surface IL-20/IL-22 receptor pairs for MDA-7/IL-24 such as the H1299 and A549 cell lines used in this study (59, 60). Accordingly, the MDA-7/IL-24 paracrine loop is not active in these NSCLC cells, and alternative intracellular signaling pathways involving ER stress have been implicated for Ad.mda-7 effects in other cancer cell types (e.g. prostate cancer cell lines) lacking the surface receptors (46, 59). Indeed, PKCδ activation has been implicated in response to ER stress (57), suggesting that Ad.mda-7 in NSCLC cells is inducing the activation of the Bcl-x(s) 5′ splice site via this pathway. On the other hand, our data demonstrating that inhibitors of ER stress pathways had no effect on Bcl-x RNA splicing (Table 2) argue against these pathways mediating the effects of Ad.mda-7, and thus, suggest a receptor-mediated pathway. Indeed, there is a lack of knowledge as to whether the MDA-7/IL-24 expressed intracellularly can act in an autocrine fashion outside of a paracrine mechanism due to differences in affinity for IL-20R homo-dimers. Furthermore, the MDA-7/IL-24 receptors may be sequestered internally in NSCLC, much like the FAS receptor in prostate cancer cells as shown by Norris and co-workers (61). Thus, a comprehensive study is necessary to delineate the current conundrum of whether the MDA-7/IL-24 receptors are sequestered intracellularly and active, and thus, can bind MDA-7/IL-24 expressed internally to mediate cell signaling events.
In conclusion, an important mechanism by which MDA-7/IL-24 elicits lethality to NSCLC cells has been identified (i.e. via altering the alternative splicing of Bcl-x pre-mRNA to increase Bcl-x(s) mRNA at the expense of Bcl-x(L) mRNA). Our data further suggest that the expression of Bcl-x(s) mRNA reduces the Bcl-x(L) available to bind Bax, or stabilize mitochondrial membranes, enhancing the cytotoxic effects of MDA-7/IL-24 on NSCLC cells. As several clinical trials for MDA-7/IL-24 are ongoing for the treatment of various cancers, our findings suggest that the modulation of Bcl-x 5′SS selection may promote the therapeutic efficacy of this cytokine. Furthermore, assaying the Bcl-x(L)/(s) mRNA ratio in patient tumors undergoing MDA-7/IL-24 therapy may serve as a biomarker for determining the efficacy of this therapy during treatment.
Experimental Procedures
Materials
DMEM, RPMI, FBS, and penicillin/streptomycin (100 units/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate) were obtained from Invitrogen Life Technologies. NSCLC cell lines of various oncogenotypes (Table 1), A549, H838, and H1299 cells, were obtained from ATCC (Manassas, VA). HBEC-3KT cells were a gracious gift of Drs. John Minna and Jerry Shay (University of Texas-Southwestern, Dallas, TX) (62). The Bcl-x(s) adenovirus, which was propagated as reported previously (34), was a gracious gift of Dr. Gabriel Nunez (University of Michigan, Ann Arbor, MI).
Cell Culture
A549, H838, and H1299 cells were grown in 50% RPMI 1640 and 50% Dulbecco's modified Eagle's medium supplemented with l-glutamine, 10% (v/v) fetal bovine serum, 100 units/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate. All NSCLC cell lines were used within 6 months and verified by the company via characteristic morphology consistent with epithelial origin, a positive result for epithelial cell marker cytokeratin 18, and where applicable, by mutational analysis and genotyping. HBEC-3KT cells were used within 6 months of receipt from the Minna and Shay laboratory, and were validated as described previously by them (62). These cells were cultured with keratinocyte serum-free medium containing bovine pituitary extract and recombinant epidermal growth factor (Life Technologies). All cells were maintained at less than 80% confluency under standard incubator conditions (humidified atmosphere, 95% air, 5% CO2, 37 °C).
Quantitative/Competitive RT-PCR Assays
Competitive qRT-PCR
Total RNA from cell lines was isolated using the RNeasy® Mini Kit (Qiagen Inc., Valencia, CA) according to the manufacturer's protocol. Total RNA (1 μg) was reverse-transcribed using Superscript III reverse transcriptase (SuperScriptTM First-Strand Synthesis System for RT-PCR, InvitrogenTM), and PCR was performed for Bcl-x splice variants as described previously (20, 21, 24, 25). The final PCR products were resolved by 5% Tris borate-EDTA acrylamide gel electrophoresis, stained with SYBR® Gold (InvitrogenTM), and visualized using a Molecular Imager® FX (Bio-Rad) with a 488-nm EX (530-nm BYPASS) laser. This assay is quantitative in regard to comparing the ratio of Bcl-x(L) to Bcl-x(s) mRNA between different samples.
Standard Real-time qRT-PCR
Total RNA and reverse transcription were undertaken as described above for quantitative/competitive RT-PCR. For real-time quantitative PCR, the levels of Bcl-x(L) and Bcl-x(s) mRNA were individually determined using quantitative PCR in accord with the TaqMan technology (Applied Biosystems) specific to Bcl-x(L), Bcl-x(s), and 18S RNA as described previously by us (24, 25). The cDNA was amplified using ABI 7900HT.
Western Immunoblotting
Cells were lysed using CelLyticTM lysis buffer (Sigma-Aldrich) supplemented with protease Inhibitor cocktail (Sigma-Aldrich). Protein samples (10 μg) were subjected to 10% SDS-PAGE, transferred to a PVDF membrane (Bio-Rad), and blocked in 5% milk/1× PBS-0.1% Tween (M-PBS-T) for 2 h. Primary antibodies were anti-MDA-7/IL-24 (1:1,000; GenHunter), anti-Bcl-x(L)/(s) (1:1,000; BD Biosciences), anti-SAP155 (1:2,000; Abcam), anti-Src (1:1,000; Sigma-Aldrich), anti-PKCδ (Cell Signaling, 1:1000), and anti-β-actin (1:1,000; Sigma-Aldrich). Secondary antibodies were HRP-conjugated goat anti-mouse or anti-rabbit (1:5,000; Sigma-Aldrich). Immunoblots were developed using Pierce enhanced chemiluminescence (ECL) reagents and Bio-Max film.
Small Interfering RNA Transfection
For inhibition of Bcl-x(s) expression, cell lines were transfected with Bcl-x(s) siRNA (5′-GCU UUG AAC AGG AUA CUU U-3′), or as a control, scrambled siRNA of the same length (Dharmacon, Lafayette, CO) or commercially available PKCδ or Src siRNA (Dharmacon) using DharmaFECT 1 transfection reagent (Dharmacon) as described previously by us (24, 25, 63–66). Briefly, cell lines were plated in 6-well tissue culture dishes and allowed to rest overnight. At 50% confluence, cells were transfected with siRNA (100 nm) using DharmaFECT 1 in Opti-MEM I reduced-serum medium. After 48 h, RNA and/or protein were isolated.
Small Molecule Inhibitor Studies
For these studies, A549 cells (1.2 × 105) were plated into 6-well tissue culture plates. The following day, medium was removed and replaced with the appropriate complete growth medium. Cells were subsequently treated with sham control (1:1000) or the appropriate concentration of active inhibitor (myriocin (100 nm, 4 h), fumonisin B1 (100 μm, 4 h) (Calbiochem), or Salubrinal (15 μm, 30 mins) (Tocris), bromoenol lactone (20 μm, 30 mins) (Sigma-Aldrich), rottlerin (50 μm, 30 mins) (Sigma-Aldrich), rapamycin (10 μm, 3 h) (Sigma-Aldrich), or Gö 6983 (100 nm, 30 mins) (Sigma-Aldrich), prior to the addition of Ad.control or Ad.mda-7 virus. After the designated incubation times, total RNA and/or protein was isolated (67–69).
WST-1 Assay
Cells (1.0 × 104) were plated into each well of a 96-well plate in 0.1 ml of medium. After 24 h at standard incubator conditions, the cells were transfected with siRNA and/or transduced with adenovirus as described previously (24, 25, 37, 38). After the designated incubation times, WST-1 reagent (Roche Applied Science) was added to the cells (final 1:10 dilution), and the cells were incubated along with the reagent for 30 min. The plates were then read against a blank using a microplate (ELISA) reader at 420–480 nm with the reference wavelength >600 nm.
Generation of Ad.5.mda-7 and Ad.5/3-mda-7
Recombinant serotype 5 and serotype 5/serotype 3 adenoviruses to express MDA-7/IL-24 (Ad.mda-7) or control (Ad.CMV empty vector) were generated as described elsewhere (29). Cells were infected with the designated adenoviruses at an appropriate multiplicity of infection (MOI) as indicated in the figure legends.
Statistical Analysis
Statistical differences between treatment groups were determined by either analysis of variance or a Newman-Keuls multiple comparison test. p values less than or equal to 0.05 were considered significant.
Author Contributions
N. T. V. and B. A. S. conceived and designed experimental studies, developed novel methodologies, acquired data, analyzed and interpreted data, and performed the initial writing of the manuscript. M. D. S., J. A. M., J. C. S., and M. M. G. acquired and analyzed RNA-seq (RNA sequencing) data, performed biostatistical analyses, verified all presented data, and provided technical support. A. Y. provided technical support and generated data for the initial MDA-7/IL-24 treatments. P. D., P. B. F., M. A. P., and C. E. C. acquired data, provided funding support, provided technical support, and performed data analysis/interpretation as related to the use and cell signaling of MDA-7/IL-24. C. E. C., M. A. P., and P. B. F. provided overall study supervision, provided funding support, generated hypotheses, conceived and designed the study, and edited and reviewed the manuscript.
Acknowledgments
Services and products in support of this project were generated by the VCU Massey Cancer Center Shared supported, in part, with funding from National Institutes of Health, NCI Cancer Center Support Grant P30 CA016059. National Institutes of Health Grant NH1C06-RR17393 was provided to Virginia Commonwealth University for renovation.
This work was supported by research grants from the Veteran's Administration (VA Merit Review, I BX001792 (to C. E. C.) and a Research Career Scientist Award, 13F-RCS-002 (to C. E. C.)); from the Vietnam Education Foundation (fellowship to N. T. V.); from the National Institutes of Health via Grants HL125353 (to C. E. C.), HD087198 (to C. E. C.), CA117950 (to C. E. C.), CA154314 (to C. E. C.), RR031535 (to C. E. C.), CA192613 (to P. D.), CA097318 (to P. B. F.), CA127641 (to P. B. F.), P01 CA104177 (to P. B. F.), and a T32 Post-Doctoral Fellowship (Postdoctoral Training Program in Cancer Biology, CA085159) (to B. A. S.); from the United States-Israel Binational Science Foundation via Grant BSF#2011360 (to C. E. C.); and from the National Foundation for Cancer Research (to P. B. F.). The authors declare that they have no conflicts of interest with the contents of this article. The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the National Institutes of Health.
- 5′SS
- 5′ splice site
- NSCLC
- non-small cell lung cancer
- ER
- endoplasmic reticulum
- MOI
- multiplicity of infection
- CPEB
- cytoplasmic polyadenylation element-binding protein
- qRT-PCR
- quantitative RT-PCR
- hnRNP
- heterogeneous nuclear ribonucleoprotein
- IL-20R
- interleukin 20 receptor.
References
- 1. Boise L. H., González-García M., Postema C. E., Ding L., Lindsten T., Turka L. A., Mao X., Nuñez G., and Thompson C. B. (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608 [DOI] [PubMed] [Google Scholar]
- 2. Boise L. H., and Thompson C. B. (1997) Bcl-x(L) can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc. Natl. Acad. Sci. U.S.A. 94, 3759–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coluccia A. M., Perego S., Cleris L., Gunby R. H., Passoni L., Marchesi E., Formelli F., and Gambacorti-Passerini C. (2004) Bcl-XL down-regulation suppresses the tumorigenic potential of NPM/ALK in vitro and in vivo. Blood 103, 2787–2794 [DOI] [PubMed] [Google Scholar]
- 4. Datta R., Manome Y., Taneja N., Boise L. H., Weichselbaum R., Thompson C. B., Slapak C. A., and Kufe D. (1995) Overexpression of Bcl-XL by cytotoxic drug exposure confers resistance to ionizing radiation-induced internucleosomal DNA fragmentation. Cell Growth Differ. 6, 363–370 [PubMed] [Google Scholar]
- 5. Dole M. G., Jasty R., Cooper M. J., Thompson C. B., Nuñez G., and Castle V. P. (1995) Bcl-xL is expressed in neuroblastoma cells and modulates chemotherapy-induced apoptosis. Cancer Res. 55, 2576–2582 [PubMed] [Google Scholar]
- 6. Finch A., Prescott J., Shchors K., Hunt A., Soucek L., Dansen T. B., Swigart L. B., and Evan G. I. (2006) Bcl-xL gain of function and p19ARF loss of function cooperate oncogenically with Myc in vivo by distinct mechanisms. Cancer Cell 10, 113–120 [DOI] [PubMed] [Google Scholar]
- 7. Johnson B. W., Cepero E., and Boise L. H. (2000) Bcl-xL inhibits cytochrome c release but not mitochondrial depolarization during the activation of multiple death pathways by tumor necrosis factor-α. J. Biol. Chem. 275, 31546–31553 [DOI] [PubMed] [Google Scholar]
- 8. Meyn R. E., Stephens L. C., Hunter N. R., and Milas L. (1995) Apoptosis in murine tumors treated with chemotherapy agents. Anti-cancer Drugs 6, 443–450 [DOI] [PubMed] [Google Scholar]
- 9. Minn A. J., Boise L. H., and Thompson C. B. (1996) Bcl-x(S) antagonizes the protective effects of Bcl-x(L). J. Biol. Chem. 271, 6306–6312 [DOI] [PubMed] [Google Scholar]
- 10. Minn A. J., Rudin C. M., Boise L. H., and Thompson C. B. (1995) Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 86, 1903–1910 [PubMed] [Google Scholar]
- 11. Ban J., Eckhart L., Weninger W., Mildner M., and Tschachler E. (1998) Identification of a human cDNA encoding a novel Bcl-x isoform. Biochem. Biophys. Res. Commun. 248, 147–152 [DOI] [PubMed] [Google Scholar]
- 12. Fang W., Rivard J. J., Mueller D. L., and Behrens T. W. (1994) Cloning and molecular characterization of mouse bcl-x in B and T lymphocytes. J. Immunol. 153, 4388–4398 [PubMed] [Google Scholar]
- 13. Shiraiwa N., Inohara N., Okada S., Yuzaki M., Shoji S., and Ohta S. (1996) An additional form of rat Bcl-x, Bcl-xβ, generated by an unspliced RNA, promotes apoptosis in promyeloid cells. J. Biol. Chem. 271, 13258–13265 [DOI] [PubMed] [Google Scholar]
- 14. Yang X. F., Weber G. F., and Cantor H. (1997) A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity 7, 629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor J. K., Zhang Q. Q., Wyatt J. R., and Dean N. M. (1999) Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat. Biotechnol. 17, 1097–1100 [DOI] [PubMed] [Google Scholar]
- 16. Mercatante D. R., Bortner C. D., Cidlowski J. A., and Kole R. (2001) Modification of alternative splicing of Bcl-x pre-mRNA in prostate and breast cancer cells: analysis of apoptosis and cell death. J. Biol. Chem. 276, 16411–16417 [DOI] [PubMed] [Google Scholar]
- 17. Deng G., Lane C., Kornblau S., Goodacre A., Snell V., Andreeff M., and Deisseroth A. B. (1998) Ratio of bcl-xshort to bcl-xlong is different in good- and poor-prognosis subsets of acute myeloid leukemia. Mol. Med. 4, 158–164 [PMC free article] [PubMed] [Google Scholar]
- 18. Mercatante D. R., Mohler J. L., and Kole R. (2002) Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 277, 49374–49382 [DOI] [PubMed] [Google Scholar]
- 19. Sumantran V. N., Ealovega M. W., Nuñez G., Clarke M. F., and Wicha M. S. (1995) Overexpression of Bcl-XS sensitizes MCF-7 cells to chemotherapy-induced apoptosis. Cancer Res. 55, 2507–2510 [PubMed] [Google Scholar]
- 20. Chalfant C. E., Ogretmen B., Galadari S., Kroesen B. J., Pettus B. J., and Hannun Y. A. (2001) FAS activation induces dephosphorylation of SR proteins: dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J. Biol. Chem. 276, 44848–44855 [DOI] [PubMed] [Google Scholar]
- 21. Chalfant C. E., Rathman K., Pinkerman R. L., Wood R. E., Obeid L. M., Ogretmen B., and Hannun Y. A. (2002) De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells: dependence on protein phosphatase-1. J. Biol. Chem. 277, 12587–12595 [DOI] [PubMed] [Google Scholar]
- 22. Boon-Unge K., Yu Q., Zou T., Zhou A., Govitrapong P., and Zhou J. (2007) Emetine regulates the alternative splicing of Bcl-x through a protein phosphatase 1-dependent mechanism. Chem. Biol. 14, 1386–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang W. H., Liu T. C., Yang W. K., Lee C. C., Lin Y. H., Chen T. Y., and Chang J. G. (2011) Amiloride modulates alternative splicing in leukemic cells and resensitizes Bcr-AblT315I mutant cells to imatinib. Cancer Res. 71, 383–392 [DOI] [PubMed] [Google Scholar]
- 24. Massiello A., Salas A., Pinkerman R. L., Roddy P., Roesser J. R., and Chalfant C. E. (2004) Identification of two RNA cis-elements that function to regulate the 5′ splice site selection of Bcl-x pre-mRNA in response to ceramide. J. Biol. Chem. 279, 15799–15804 [DOI] [PubMed] [Google Scholar]
- 25. Massiello A., Roesser J. R., and Chalfant C. E. (2006) SAP155 binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′ splice site selection of Bcl-x pre-mRNA. FASEB J. 20, 1680–1682 [DOI] [PubMed] [Google Scholar]
- 26. Jiang H., Lin J. J., Su Z. Z., Goldstein N. I., and Fisher P. B. (1995) Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 11, 2477–2486 [PubMed] [Google Scholar]
- 27. Saeki T., Mhashilkar A., Swanson X., Zou-Yang X. H., Sieger K., Kawabe S., Branch C. D., Zumstein L., Meyn R. E., Roth J. A., Chada S., and Ramesh R. (2002) Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene 21, 4558–4566 [DOI] [PubMed] [Google Scholar]
- 28. Saeki T., Mhashilkar A., Chada S., Branch C., Roth J. A., and Ramesh R. (2000) Tumor-suppressive effects by adenovirus-mediated mda-7 gene transfer in non-small cell lung cancer cell in vitro. Gene Ther. 7, 2051–2057 [DOI] [PubMed] [Google Scholar]
- 29. Lebedeva I. V., Sarkar D., Su Z. Z., Kitada S., Dent P., Stein C. A., Reed J. C., and Fisher P. B. (2003) Bcl-2 and Bcl-x(L) differentially protect human prostate cancer cells from induction of apoptosis by melanoma differentiation associated gene-7, mda-7/IL-24. Oncogene 22, 8758–8773 [DOI] [PubMed] [Google Scholar]
- 30. Hamed H. A., Yacoub A., Park M. A., Eulitt P., Sarkar D., Dimitrie I. P., Chen C. S., Grant S., Curiel D. T., Fisher P. B., and Dent P. (2010) OSU-03012 enhances Ad.mda-7-induced GBM cell killing via ER stress and autophagy and by decreasing expression of mitochondrial protective proteins. Cancer Biol. Ther. 9, 526–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhutia S. K., Dash R., Das S. K., Azab B., Su Z. Z., Lee S. G., Grant S., Yacoub A., Dent P., Curiel D. T., Sarkar D., and Fisher P. B. (2010) Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer Res. 70, 3667–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Su Z. Z., Lebedeva I. V., Sarkar D., Emdad L., Gupta P., Kitada S., Dent P., Reed J. C., and Fisher P. B. (2006) Ionizing radiation enhances therapeutic activity of mda-7/IL-24: overcoming radiation- and mda-7/IL-24-resistance in prostate cancer cells overexpressing the antiapoptotic proteins bcl-xL or bcl-2. Oncogene 25, 2339–2348 [DOI] [PubMed] [Google Scholar]
- 33. Sauane M., Su Z. Z., Dash R., Liu X., Norris J. S., Sarkar D., Lee S. G., Allegood J. C., Dent P., Spiegel S., and Fisher P. B. (2010) Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J. Cell. Physiol. 222, 546–555 [DOI] [PubMed] [Google Scholar]
- 34. Mitra R. S., Benedict M. A., Qian D., Foreman K. E., Ekhterae D., Nickoloff B. J., and Nuñez G. (2001) Killing of sarcoma cells by proapoptotic Bcl-X(S): role of the BH3 domain and regulation by Bcl-X(L). Neoplasia 3, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Inoue S., Branch C. D., Gallick G. E., Chada S., and Ramesh R. (2005) Inhibition of Src kinase activity by Ad-mda7 suppresses vascular endothelial growth factor expression in prostate carcinoma cells. Mol. Ther. 12, 707–715 [DOI] [PubMed] [Google Scholar]
- 36. Kontny E., Kurowska M., Szczepańska K., and Maśliński W. (2000) Rottlerin, a PKC isozyme-selective inhibitor, affects signaling events and cytokine production in human monocytes. J. Leukoc. Biol. 67, 249–258 [DOI] [PubMed] [Google Scholar]
- 37. Shultz J. C., Goehe R. W., Murudkar C. S., Wijesinghe D. S., Mayton E. K., Massiello A., Hawkins A. J., Mukerjee P., Pinkerman R. L., Park M. A., and Chalfant C. E. (2011) SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 9, 889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shultz J. C., Goehe R. W., Wijesinghe D. S., Murudkar C., Hawkins A. J., Shay J. W., Minna J. D., and Chalfant C. E. (2010) Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 70, 9185–9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng C., and Sharp P. A. (2006) Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol. Cell. Biol. 26, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karni R., de Stanchina E., Lowe S. W., Sinha R., Mu D., and Krainer A. R. (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 14, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valacca C., Bonomi S., Buratti E., Pedrotti S., Baralle F. E., Sette C., Ghigna C., and Biamonti G. (2010) Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 191, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warzecha C. C., Sato T. K., Nabet B., Hogenesch J. B., and Carstens R. P. (2009) ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnson R. M., Vu N. T., Griffin B. P., Gentry A. E., Archer K. J., Chalfant C. E., and Park M. A. (2015) The alternative splicing of cytoplasmic polyadenylation element binding protein 2 drives anoikis resistance and the metastasis of triple negative breast cancer. J. Biol. Chem. 290, 25717–25727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Wang J., Mor G., and Sklar J. (2008) A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 321, 1357–1361 [DOI] [PubMed] [Google Scholar]
- 45. Nekrutenko A., and He J. (2006) Functionality of unspliced XBP1 is required to explain evolution of overlapping reading frames. Trends Genet. 22, 645–648 [DOI] [PubMed] [Google Scholar]
- 46. Hamed H. A., Das S. K., Sokhi U. K., Park M. A., Cruickshanks N., Archer K., Ogretmen B., Grant S., Sarkar D., Fisher P. B., and Dent P. (2013) Combining histone deacetylase inhibitors with MDA-7/IL-24 enhances killing of renal carcinoma cells. Cancer Biol. Ther 14, 1039–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rothschild S. I., Gautschi O., Haura E. B., and Johnson F. M. (2010) Src inhibitors in lung cancer: current status and future directions. Clin. Lung Cancer 11, 238–242 [DOI] [PubMed] [Google Scholar]
- 48. Herold C. I., Chadaram V., Peterson B. L., Marcom P. K., Hopkins J., Kimmick G. G., Favaro J., Hamilton E., Welch R. A., Bacus S., and Blackwell K. L. (2011) Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin. Cancer Res. 17, 6061–6070 [DOI] [PubMed] [Google Scholar]
- 49. Moore M. J., Wang Q., Kennedy C. J., and Silver P. A. (2010) An alternative splicing network links cell-cycle control to apoptosis. Cell 142, 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paronetto M. P., Achsel T., Massiello A., Chalfant C. E., and Sette C. (2007) The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J. Cell Biol. 176, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Revil T., Pelletier J., Toutant J., Cloutier A., and Chabot B. (2009) Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J. Biol. Chem. 284, 21458–21467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. White M. C., Gao R., Xu W., Mandal S. M., Lim J. G., Hazra T. K., Wakamiya M., Edwards S. F., Raskin S., Teive H. A., Zoghbi H. Y., Sarkar P. S., and Ashizawa T. (2010) Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCδ to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 6, e1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eto K., Sonoda Y., and Abe S. (2011) The kinase DYRKIA regulates pre-mRNA splicing in spermatogonia and proliferation of spermatogonia and Sertoli cells by phosphorylating a spliceosomal component, SAP155, in postnatal murine testes. Mol. Cell. Biochem. 355, 217–222 [DOI] [PubMed] [Google Scholar]
- 54. Tanuma N., Kim S. E., Beullens M., Tsubaki Y., Mitsuhashi S., Nomura M., Kawamura T., Isono K., Koseki H., Sato M., Bollen M., Kikuchi K., and Shima H. (2008) Nuclear inhibitor of protein phosphatase-1 (NIPP1) directs protein phosphatase-1 (PP1) to dephosphorylate the U2 small nuclear ribonucleoprotein particle (snRNP) component, spliceosome-associated protein 155 (Sap155). J. Biol. Chem. 283, 35805–35814 [DOI] [PubMed] [Google Scholar]
- 55. Shi Y., Reddy B., and Manley J. L. (2006) PP1/PP2A phosphatases are required for the second step of Pre-mRNA splicing and target specific snRNP proteins. Mol. Cell 23, 819–829 [DOI] [PubMed] [Google Scholar]
- 56. de Graaf K., Czajkowska H., Rottmann S., Packman L. C., Lilischkis R., Lüscher B., and Becker W. (2006) The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem. 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Larroque-Cardoso P., Swiader A., Ingueneau C., Nègre-Salvayre A., Elbaz M., Reyland M. E., Salvayre R., and Vindis C. (2013) Role of protein kinase C δ in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 4, e520, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Piazzi M., Blalock W. L., Bavelloni A., Faenza I., D'Angelo A., Maraldi N. M., and Cocco L. (2013) Phosphoinositide-specific phospholipase C β 1β (PI-PLCβ1β) interactome: affinity purification-mass spectrometry analysis of PI-PLCβ1β with nuclear protein. Mol. Cell. Proteomics 12, 2220–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sieger K. A., Mhashilkar A. M., Stewart A., Sutton R. B., Strube R. W., Chen S. Y., Pataer A., Swisher S. G., Grimm E. A., Ramesh R., and Chada S. (2004) The tumor suppressor activity of MDA-7/IL-24 is mediated by intracellular protein expression in NSCLC cells. Mol. Ther. 9, 355–367 [DOI] [PubMed] [Google Scholar]
- 60. Su Z., Emdad L., Sauane M., Lebedeva I. V., Sarkar D., Gupta P., James C. D., Randolph A., Valerie K., Walter M. R., Dent P., and Fisher P. B. (2005) Unique aspects of mda-7/IL-24 antitumor bystander activity: establishing a role for secretion of MDA-7/IL-24 protein by normal cells. Oncogene 24, 7552–7566 [DOI] [PubMed] [Google Scholar]
- 61. Hyer M. L., Voelkel-Johnson C., Rubinchik S., Dong J., and Norris J. S. (2000) Intracellular Fas ligand expression causes Fas-mediated apoptosis in human prostate cancer cells resistant to monoclonal antibody-induced apoptosis. Mol Ther. 2, 348–358 [DOI] [PubMed] [Google Scholar]
- 62. Sato M., Vaughan M. B., Girard L., Peyton M., Lee W., Shames D. S., Ramirez R. D., Sunaga N., Gazdar A. F., Shay J. W., and Minna J. D. (2006) Multiple oncogenic changes (K-RASV12, p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 66, 2116–2128 [DOI] [PubMed] [Google Scholar]
- 63. Lamour N. F., Stahelin R. V., Wijesinghe D. S., Maceyka M., Wang E., Allegood J. C., Merrill A. H. Jr, Cho W., and Chalfant C. E. (2007) Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J. Lipid Res. 48, 1293–1304 [DOI] [PubMed] [Google Scholar]
- 64. Lamour N. F., Subramanian P., Wijesinghe D. S., Stahelin R. V., Bonventre J. V., and Chalfant C. E. (2009) Ceramide 1-phosphate is required for the translocation of group IVA cytosolic phospholipase A2 and prostaglandin synthesis. J. Biol. Chem. 284, 26897–26907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pettus B. J., Bielawska A., Spiegel S., Roddy P., Hannun Y. A., and Chalfant C. E. (2003) Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J. Biol. Chem. 278, 38206–38213 [DOI] [PubMed] [Google Scholar]
- 66. Pettus B. J., Bielawska A., Subramanian P., Wijesinghe D. S., Maceyka M., Leslie C. C., Evans J. H., Freiberg J., Roddy P., Hannun Y. A., and Chalfant C. E. (2004) Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 279, 11320–11326 [DOI] [PubMed] [Google Scholar]
- 67. Wiederrecht G. J., Sabers C. J., Brunn G. J., Martin M. M., Dumont F. J., and Abraham R. T. (1995) Mechanism of action of rapamycin: new insights into the regulation of G1-phase progression in eukaryotic cells. Prog. Cell Cycle Res. 1, 53–71 [DOI] [PubMed] [Google Scholar]
- 68. Boschelli D. H., Wang Y. D., Johnson S., Wu B., Ye F., Barrios Sosa A. C., Golas J. M., and Boschelli F. (2004) 7-Alkoxy-4-phenylamino-3-quinolinecar-bonitriles as dual inhibitors of Src and Abl kinases. J. Med. Chem. 47, 1599–1601 [DOI] [PubMed] [Google Scholar]
- 69. Peterman E. E., Taormina P. 2nd, Harvey M., and Young L. H. (2004) Gö 6983 exerts cardioprotective effects in myocardial ischemic/reperfusion. J. Cardiovasc. Pharmacol. 43, 645–656 [DOI] [PubMed] [Google Scholar]
- 70. Joseloff E., Cataisson C., Aamodt H., Ocheni H., Blumberg P., Kraker A. J., Yuspa S. H. (2002) Src family kinases phosphorylate protein kinase C delta on tyrosine residues and modify the neoplastic phenotype of skin keratinocytes. J Biol. Chem. 277, 12318–12323 [DOI] [PubMed] [Google Scholar]
- 71. Shultz J. C., Vu N., Shultz M. D., Mba M. U., Shapiro B. A., Chalfant C. E. (2012) The Proto-oncogene PKCι regulates the alternative splicing of Bcl-x pre-mRNA. Mol. Cancer Res. 10, 660–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Revil T., Toutant J., Shkreta L., Garneau D., Cloutier P., Chabot B. (2007) Protein kinase C-dependent control of Bcl-x alternative splicing. Mol. Cell Biol. 27, 8431–8441 [DOI] [PMC free article] [PubMed] [Google Scholar]