

Abstract
Trans-membrane signaling involving a serine/threonine kinase (Stt7 in Chlamydomonas reinhardtii) directs light energy distribution between the two photosystems of oxygenic photosynthesis. Oxidation of plastoquinol mediated by the cytochrome b6f complex on the electrochemically positive side of the thylakoid membrane activates the kinase domain of Stt7 on the trans (negative) side, leading to phosphorylation and redistribution (“state transition”) of the light-harvesting chlorophyll proteins between the two photosystems. The molecular description of the Stt7 kinase and its interaction with the cytochrome b6f complex are unknown or unclear. In this study, Stt7 kinase has been cloned, expressed, and purified in a heterologous host. Stt7 kinase is shown to be active in vitro in the presence of reductant and purified as a tetramer, as determined by analytical ultracentrifugation, electron microscopy, and electrospray ionization mass spectrometry, with a molecular weight of 332 kDa, consisting of an 83.41-kDa monomer. Far-UV circular dichroism spectra show Stt7 to be mostly α-helical and document a physical interaction with the b6f complex through increased thermal stability of Stt7 secondary structure. The activity of wild-type Stt7 and its Cys-Ser mutant at positions 68 and 73 in the presence of a reductant suggest that the enzyme does not require a disulfide bridge for its activity as suggested elsewhere. Kinase activation in vivo could result from direct interaction between Stt7 and the b6f complex or long-range reduction of Stt7 by superoxide, known to be generated in the b6f complex by quinol oxidation.
Keywords: cytochrome, light-harvesting complex (antenna complex), redox signaling, serine/threonine protein kinase, signal transduction, state transitions, Stt7, cytochrome b6f
Introduction
“State transitions” in the context of photosynthesis describe the ability of photosynthetic organisms to adjust over the short term to different spectra and intensities of light illumination by reversibly phosphorylating the light-harvesting chlorophyll complex protein II, LHCII,3 thus changing the distribution of absorbed light energy between the two photosystems, PSII and PSI (1–11). The signal for activation of state transitions is initiated through the oxidation of plastoquinol by the cyt b6f complex (Fig. 1, A–C) on the lumen (electrochemically positive) side of the thylakoid membrane (11, 12). Through the trans-membrane activity of the kinase, plastoquinol oxidation is coupled to phosphorylation of the LHCII (light-harvesting chlorophyll complex II) protein on the stromal (electrochemically negative (n)) side of the thylakoid membrane, which facilitates lateral diffusion of the phosphorylated LHCII proteins in the thylakoid membrane (13), resulting in their redistribution from the PSII to the PSI reaction center complex. The Stt7 protein (Fig. 1D) that responds to the signal generated by plastoquinol oxidation and functions as the LHCII kinase was identified by screens for Chlamydomonas mutants deficient in state transitions (4, 6) and was found to be a 754 residue serine/threonine kinase located in the chloroplast thylakoid membrane as well as in the soluble fraction (6). An orthologous protein, STN7, present in Arabidopsis thaliana, also participates in LHCII phosphorylation and state transitions (6, 7). Description of the molecular events that underlie photosynthetic state transitions would contribute not only to the understanding of photosynthetic energy transduction but also, more broadly, to that of trans-membrane signaling mechanisms, most completely described at present for the G protein-coupled receptor system (14).
FIGURE 1.

Summary: structure aspects of the cytochrome b6f complex and Stt7 kinase. A, the cytochrome b6f complex from the filamentous cyanobacterium Nostoc PCC 7120 (PDB code 4OGQ), consisting of eight distinct subunits (shown as ribbons). Cyt b6 (white) and subunit IV (yellow) are polytopic subunits containing four and three trans-membrane helices with seven prosthetic groups per monomer. Cyt f (cyan) and the Rieske ISP (red) contain a single trans-membrane helix and large p side extrinsic domains. Four single-helix trans-membrane subunits, PetG (blue), PetL (green), PetM (orange), and PetN (light brown), form the peripheral boundary of each monomer. B, electron transfer within the trans-membrane domain involves heme bp, bn (blue/red sticks) and heme cn (black/red sticks) and, on the p side, a [2Fe-2S] cluster (brown/yellow sticks, spheres) linked to the ISP subunit and covalently linked heme f (black/red sticks). Two pigments, Chl-a (green/red/blue sticks) and a β-carotene (β-Car, yellow sticks), are present in single copies in the trans-membrane domain. C, the p side Qp site is marked by a quinol analog (PDB code 2E76), shown as thin black/red sticks. On the p side, the Qp site is covered by the [2Fe-2S] cluster in the extrinsic domain of the ISP subunit (red). F and G helices of subunit IV (pale yellow) are located at the periphery. The phytyl tail of the Chl-a (green/blue/red sticks) wrapped around the F helix gates quinone residence at the Qp site (24). A lipid molecule (brown/blue sticks) is bound within the niche formed by F and G helices adjacent to the chlorin ring of Chl-a. Other protein subunits are represented in surface mode. D, schematic of the domain architecture of Stt7 kinase from the unicellular green alga C. reinhardtii, for which a crystal structure is not available. The Stt7 polypeptide consists of an N-terminal 41-residue signal peptide sequence (red) that targets the polypeptide to the chloroplast and is followed by a p side regulatory domain (blue, residues 42–96), containing two conserved Cys residues at positions 68 and 73 (yellow circles) implicated in the activation/deactivation of Stt7 (15, 18, 19). The single hydrophobic domain (brown, 97–122) is proposed (9) to span the membrane, connecting the regulatory and C-terminal kinase (yellow, residues 123–754) domains, the latter located on the n (stroma) side of the membrane. In this study, kinase activity was found to be restricted to residues 124–549 (defined by the dashed line), with residues numbered according to UNIPROT entry Q84V18.
Binding of the cyt b6f complex to Stt7 has been demonstrated via co-immunoprecipitation (15). However, no evidence has yet been provided for an interaction between the cytochrome complex and Stt7 that perturbs a measurable physical or chemical property of either member of the putative complex. Purification of Stt7 from its native source is difficult because it is present in substoichiometric levels, ∼1:20 to cyt b6f in the unicellular green alga Chlamydomonas reinhardtii (15). A crystal structure has been obtained of its kinase domain from a homologous protein, MsStt7d, isolated from Micromonas sp. (16), with which the kinase domain of Stt7 shares 47% sequence similarity. However, the two kinases differ in N- and C-terminal amino acid sequences, implying a different mechanism of action. MsStt7d lacks the two cysteines, Cys68 and Cys73, near the N terminus, which have been proposed to be involved in redox-mediated regulation of Stt7 activity (16, 17). Consistent with this inference, targeting of the sulfhydryl groups of the Stt7 in thylakoid membranes with N-ethylmaleimide inhibited kinase activity (18). Regarding the redox state of the Cys68 and Cys73 residues, proposed to be responsible for p side regulation of activity, there has been a difference of opinion as to whether the reduced –SH (17) or oxidized disulfide, -S-S- of the two Cys are required for activity (15). Recently, it has been inferred that these two cysteine residues form a permanent disulfide dimer that does not change redox state during state transitions (19).
The Cytochrome b6f Complex
Crystal structures of the active cytochrome b6f complex have been obtained from the filamentous unicellular cyanobacteria Mastigocladus laminosus and Nostoc PCC 7120 (20–22) and the green alga C. reinhardtii (23). The cytochrome b6f complex is organized as a symmetric dimer of eight distinct integral protein subunits (Fig. 1, A and B): cytochrome b6 (cyt b6) with four trans-membrane helices (A–D), subunit IV with three trans-membrane helices (E and F), and the single trans-membrane helix of cytochrome f (cyt f), the Rieske iron-sulfur protein (ISP), and the small hydrophobic peptides PetG, PetL, PetM, and PetN. The cyt f and ISP subunits each contain a large extrinsic domain on the lumen (p) side of the membrane. The b6f complex contains seven prosthetic groups per monomer, including hemes bp, bn, and cn, associated with the cyt b6 polypeptide within the hydrophobic trans-membrane domain, and heme f and [2Fe-2S] cluster bound to the p side extrinsic domain of cytochrome f and the ISP subunit, respectively (Fig. 1B). Single chlorophyll a (Chl-a) and a β-carotene pigment molecules are located within the hydrophobic domain of each monomer of the dimeric complex (Fig. 1B). The quinol oxidation (Qp) site of the cytochrome b6f complex, which has been proposed to initiate state transitions (11, 12), lies at the end of a narrow portal whose entrance is defined by the C and F trans-membrane helices (Fig. 1C) and that extends to the intermonomer cavity. The Chl-a phytyl tail is inserted into the portal, suggesting that it can occlude plastoquinol/quinone passage in the channel (24), thus prolonging the residence time of p side quinol/semiquinone/quinone and providing a mechanism for increased production of superoxide (25), which is proposed in this study to provide the reductant required for state transitions.
Although the crystal structures of the cyt b6f complex (Fig. 1, A–C) and MsStt7 kinase domain have provided significant insights into the functions of the individual proteins, the molecular details of the mechanism of Stt7 kinase action, its interaction with the b6f complex, the mechanism of signal transduction from the Qp site of the b6f complex to the kinase, and the regulation of kinase activity remain incompletely defined.
Pulldown assays have previously demonstrated an interaction between inactive Stt7 and the Rieske or cytochrome f subunits of the b6f complex but not with the intact b6f complex (15). Recent yeast two-hybrid studies have inferred that Stt7 interacts with an exposed p side site of the Rieske [2Fe-2S] protein (19). This study documents a different approach to the problem. Stt7 was overexpressed and purified from Escherichia coli as a tetramer of 332 kDa, and a physical interaction between purified active Stt7 and the intact b6f dimer was demonstrated. The isolated Stt7 showed in vitro ATP hydrolysis activity under reducing conditions. This implies that Stt7 displays kinase activity when the sulfhydryl group of key cysteine residue(s) is reduced, in contrast to the inference reached previously that a disulfide S-S bond is essential for activity (15, 26).
Results
Domain and Amino Acid Sequence Analysis of Stt7
The Stt7 serine/threonine kinase is divided into four domains (9) (Fig. 1D): an N-terminal 41-residue signal peptide, a p side regulatory domain (residues 42–96), a putative single trans-membrane domain (residues 97–122), and a C-terminal kinase domain (residues 123–754). On the p side, two conserved cysteine residues at positions 68 and 73 have been suggested to be involved in sensing the redox state of the lumen (15). An analysis of the Stt7/STN7 sequences from a variety of eukaryotic organisms reveals that cysteine residues at positions 68 and 73 are conserved only in higher organisms (including C. reinhardtii).
The predicted trans-membrane domain, from residue 97–122 (6), 97VALLAPVLAYLFLPPGVLPGAIDYYI122, contains four proline residues (highlighted in the sequence in underlined bold italics), which is a large number for a trans-membrane domain. However, only proline residues 111 and 115 are conserved in the Stt7/STN7 sequence (Fig. 2A). The Stt7 sequence also shows the presence of a 112GXXP115 motif (where X is any amino acid) in the putative trans-membrane domain (Fig. 2A), which is characteristic of a hinge motif (27, 28). The motif for the hinge in Stt7/STN7 is conserved across photosynthetic organisms (Fig. 2A). Regarding other sequence motifs, the Stt7 trans-membrane sequence contains the 112GXXXG116 motif (where X is any amino acid) (Fig. 2A), which has been implicated in the dimerization of trans-membrane domains (29). Other than the Stt7 homolog from Micromonas sp., the dimerization motif is conserved in the putative trans-membrane domain in photosynthetic organisms (Fig. 2A).
FIGURE 2.

Conserved motifs in Stt7/STN7 kinase family members. A, within the trans-membrane domain, the GXXP motif, which encodes a hinge, is found to be conserved and is shown in a dashed box. The dimerization motif GXXXG is present in the Stt7/STN7 kinase enzyme of all species except Micromonas sp. B, the HRD motif, which is a key signature of kinases, is found to be highly conserved across Stt7/STN7 from all species. However, the DFG motif is replaced by the amino acids DLG, as shown in the figure.
The n side of Stt7 contains several characteristic features of kinases (Fig. 2B), including the 300HRD302 motif, which is found in eukaryotic kinases and is involved in proper spatial orientation of the substrate (30, 31). It is significant that the conserved DFG motif, which has been linked to kinase activation/inactivation (32, 33) is replaced by 321DLG323 in the C. reinhardtii Stt7 (Fig. 2B).
As noted, Stt7 is predicted to contain the kinase domain from residue 123–754 on the n side (Fig. 1D). An analysis of the Stt7 sequence (Uniprot accession no. Q84V18, C. reinhardtii) by PSIPRED (34) shows that two C-terminal segments, Ala510-Lys706 and Glu723-Leu749, are predicted to be disordered. The N-terminal 499 residues of Stt7 contain 6.8% glycine and 11.4% alanine. In contrast, residues 500–754 consist of 12.5% glycine and 19.6% alanine. Furthermore, the C-terminal portion of the enzyme from residue 722–754 consists of a relatively high concentration of charged residues, 11 negatively charged (aspartate and glutamate) and five positively charged (arginine and lysine). The abundance of charged residues and of glycine and alanine, two of the smallest amino acids, has been linked previously to intrinsic protein disorder (35). It is noted that the C-terminal disordered domain of Stt7 is found exclusively in C. reinhardtii (6).
Location of Cysteine Residues
In addition to the p side cysteine pair Cys68/Cys73 mentioned above, there are five other cysteine residues, of which Cys10 and Cys27 are located in the signal peptide, and Cys293, Cys308, and Cys387 are present in the kinase domain that is predicted to reside on the n side.
Purification of Full-length Stt7 and the Isolated Kinase Domain
The gene for the full-length 754-residue Stt7 with a C-terminal hexahistidine purification tag was overexpressed in the E. coli BL23 Rosetta strain, and the protein was purified to homogeneity using metal affinity cobalt-nitrilotriacetic acid followed by size exclusion chromatography. Stt7 is expressed in the soluble fraction as well as in association with thylakoid membranes in the native host C. reinhardtii (6). In our overexpression studies in E. coli, the protein was localized in the cytosolic fraction. Purified Stt7 from E. coli displays a molecular weight of ∼75 kDa, as determined by 12% SDS-PAGE (Fig. 3A). The identity of the purified protein was confirmed by proteomics mass spectrometry (Fig. 3, B and C). The average molecular mass was measured as 83,410 Da by electrospray ionization-MS, which is larger than calculated for the translated coding sequence (81,568.15 Da). The mass spectrum was quite heterogeneous, and the measured mass spread between 83,300 and 83,500 Da (Fig. 3C). The origin of the heterogeneity and extra mass of ∼1.7 kDa in the monomer is most like a consequence of the use in the protease inhibitor mixture of 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (184 Da), which binds to the hydroxyl group of serine and tyrosine, in the protease inhibitor mixture, required to prevent cleavage during the purification. In its native state, Stt7 was found to have a sedimentation coefficient of 11.75 svedberg unit by an analytical ultracentrifugation analysis, which corresponds to a tetramer of 332 kDa, having a Stokes radius of 6.74 nm (Fig. 3D). The molecular mass of the monomer (83 kDa) inferred from the analytical centrifugation analysis is consistent with the monomeric mass determined by electrospray ionization-MS.
FIGURE 3.

Characterization of purified Stt7 kinase. A, SDS-PAGE analysis of Stt7 shows a band near 75 kDa that corresponds to Stt7. The sample was further analyzed by mass spectrometry. B, isolated Stt7 was digested with trypsin, and peptides were analyzed by nano-liquid chromatography with automated data-dependent tandem mass spectrometry on an Orbitrap instrument (Thermo Scientific, Orbitrap XL, high-resolution Orbitrap MS1 and ion-trap MS2). Peptide MS data were matched to Swiss-Prot entries using Mascot software (Matrix Science). Peptides matched within 95% confidence limits are mapped onto the Stt7 sequence (red) within the trans-membrane domain. C, intact Stt7 protein was analyzed by reverse-phase liquid chromatography with ion-trap mass spectrometry (Thermo Scientific, LTQ with Ionmax source). The mass spectrum was deconvoluted to the zero-charge state using BioMultiView software (MDS Sciex). D, molecular weight of the purified Stt7 in its native state determined by analytical ultracentrifugation. A relatively large amount, 92.95% of purified Stt7, showed a peak at 11.75 S corresponding to a molecular weight of 332 kDa, which implies a tetrameric organization of the enzyme. E, under reducing conditions (blue trace), Stt7 eluted in a single peak centered at 10.7 ml. The fractions marked by the dotted lines were used for characterization of the enzyme by analytical ultracentrifugation. In contrast, under oxidizing conditions (red trace), Stt7 was mostly aggregated and eluted in the void volume of the chromatography column. Note that the absorbance values at 280 nm of the protein peaks under reducing and oxidizing conditions have been normalized to 1. F, the purified Stt7-KD band was analyzed by 12% SDS-PAGE, which corresponds to a molecular weight of 42 kDa. Different fractions of eluted Stt7-KD contain a similar quantity of protein. Therefore, a representative sample is shown on SDS-PAGE. MW, molecular mass (kilodalton); Std, molecular weight standards.
The presence of a reducing agent was found to be crucial for the stability of the isolated full-length Stt7 enzyme (Fig. 3E). In the absence of a reducing agent, because of protein aggregation, purified Stt7 eluted in the void volume of the S200 size-exclusion chromatography column (Fig. 3E) and was retained within the stacking gel of SDS-PAGE.
The isolated kinase domain (residues 124–549) was expressed as inclusion bodies in the E. coli BL23 Rosetta strain (Fig. 3F). The inclusion bodies were refolded by dilution in 6 m guanidinium hydrochloride. The refolded protein was found to be enzymatically active as a kinase. Deletion of the C-terminal segment (residues 550–754), which is predicted to be disordered and is found exclusively in the C. reinhardtii Stt7 (6), did not lead to a loss of kinase activity. This implies that the Stt7 C-terminal segment extending from residues 550–754 is not involved in catalysis.
Visualization of the Stt7 Tetramer by Negative Stain Electron Microscopy
A negative stain EM image of full-length Stt7 (Fig. 4A), shows an approximate 4-fold rotational symmetry. The projection image displayed a square-shaped tetramer (Fig. 4, B and C), of 10 × 10 nm and having a noticeable cavity of 1 nm in diameter at the center. The Stt7 particles are measured to be of 14-nm diameter, which matches the Stokes diameter of 13.5 nm determined from analytical ultracentrifugation.
FIGURE 4.

Electron microscopy analysis of the Stt7 tetramer. Diluted Stt7 (2 μg/ml) was absorbed on a carbon-coated 400-mesh copper grid and stained with uranyl acetate. Images were recorded on an FEI Tecnai transmission electron microscope operated at 200 kV at a magnification of ×71,000 and defocus of −1 μm. A, the 2D image shows a square-like organization of the Stt7 tetramer of ∼10 × 10 nm with an ∼14-nm diagonal length. B and C, magnified views of two tetramers of Stt7 highlighted in A with yellow boxes. The tetramers show a dark circular feature at the center that represents a hollow cavity of ∼1 nm diameter.
Enzymatic Activity of Stt7
The isolated full-length Stt7 enzyme was found to be competent as a kinase in vitro by catalyzing the hydrolysis of ATP to ADP, measured qualitatively using the ADP-glo kit supplied by Promega (Fig. 5A). The specific activity of purified Stt7 was 300–330 nmol of ATP converted to ADP/minute/mg of protein. Wells 1, 3, and 4 (Fig. 5A) were negative controls in which ATP/protein, ATP, and protein, respectively, were omitted. Lanes 2 and 5 (Fig. 5A) exhibited luminescence associated, respectively, with Stt7-dependent conversion of ATP to ADP, and the presence of ADP alone as a positive control. It was observed that the kinase activity of the full-length Stt7 was dependent on reducing conditions. The enzyme was found to be active in the presence of DTT or βME (Fig. 5A). Similarly, Stt7 kinase domain (Stt7-KD) and the cysteine double mutant (C68S/C73S) were found to be active under reducing conditions. Lanes 2 and 3 in Fig. 5, B and C, respectively, contained Stt7-KD and C68S/C73S. Lane 1 in Fig. 5, B and C, displayed negative controls, whereas lanes 3 and 2 in Fig. 5, B and C, respectively, contained ADP as a positive control. Comparable in vitro kinase activity obtained with the double mutant C68S/C73S implies that an S-S disulfide bond between cysteine residues at positions 68 and 73 is not essential for activity.
FIGURE 5.

Assay of kinase activity. A, qualitative luciferase assay of ATP hydrolysis activity of purified Stt7 kinase in 0.1 m phosphate buffer (pH 8). Reaction mixture (kinase buffer, ADP glo reagent, and ADP reaction mixture) was added to lanes containing the following: lane 1, H2O blank; lane 2, Stt7 (3 μg) in 2 mm βME and 400 μm ATP; lane 3, Stt7; lane 4, 400 μm ATP as a negative control; lane 5, 400 μm ADP as positive control. B, activity of the Stt7 kinase domain (Stt7-KD). Lanes 1 and 3 contained no protein and acted as negative and positive controls, respectively. Reaction mixture kinase buffer, ADP glo reagent, was added to all lanes. The concentration of ATP and ADP was 400 μm. C, activity of C68S/C73S double cysteine mutant of Stt7 kinase assayed with luciferase. Reaction mixture was added to wells containing C68S/C73S Stt7 (lane 3) and lanes 1 and 2 as negative and positive controls, respectively.
Determination of Secondary Structure
Isolated full-length Stt7 was analyzed by far-UV CD spectroscopy. The enzyme was found to be predominantly α-helical, with the CD spectra showing negative minima at 222 and 208 nm and a positive maximum at 195 nm (Fig. 6A). The estimated content of α helix, β sheet, and disordered region of 56% ± 7%, 7% ± 2%, and 37% ± 8% was determined by the CONTINLL, CDSSTR, and K2D algorithms in the DichroWeb package using an extended set of reference proteins (36–38). The relatively large content of disorder in the full-length Stt7 matches well with the sequence-based prediction of disorder, localized predominantly at the C terminus of the C. reinhardtii Stt7, from residues 500–754 (Fig. 1D).
FIGURE 6.
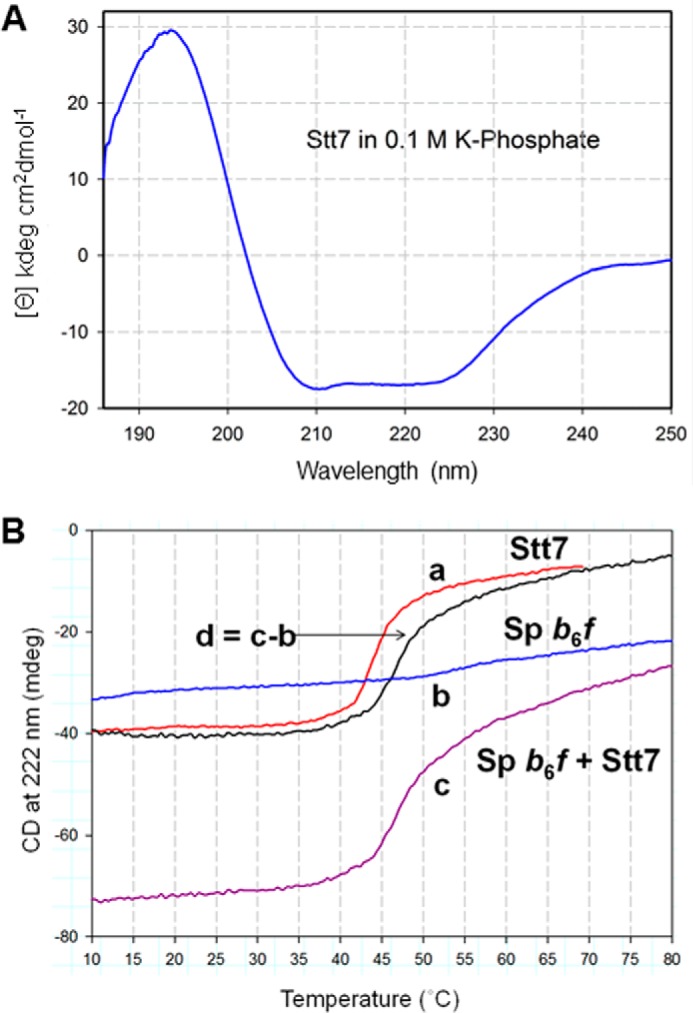
Far-UV circular dichroism spectra of Stt7. A, far-UV CD spectrum of purified Stt7 in 0.1 m phosphate buffer (pH 8), 12 mm βME, and 0.1% UDM. The spectrum with pronounced negative peaks at 208 and 222 nm shows that the protein is primarily α-helical. B, temperature dependence of ellipticity of Stt7 and b6f complex, measured at 222 nm in 0.1 m phosphate buffer (pH 8), 0.1% UDM of isolated Stt7 (a, red), spinach b6f complex (b, blue), and Stt7 together with the b6f complex (c, purple). d, thermally induced melting function of Stt7 in the presence of b6f (black), determined by the difference between functions c and b.
Evidence of Interaction between b6f and Stt7 from Far-UV CD Spectroscopy
Although it has been demonstrated previously that Stt7 in an inactive state interacts with components of the cyt b6f complex in the thylakoid membranes of C. reinhardtii (15), evidence of a physical interaction between the two isolated and purified protein complexes in their native and enzymatically active states has remained elusive. In this study, the interaction between the two isolated and enzymatically active proteins was investigated using far-UV CD spectroscopy (Fig. 6A) to probe intermolecular interactions that would affect the thermal stability, i.e. the temperature dependence, of the secondary structure of the Stt7. The yield of purified cyt b6f from C. reinhardtii was found to be limiting. PetD (subunit IV), which is proposed to be a junction of interaction with Stt7, shares 81% identity and 84% similarity between C. reinhardtii and spinach. Given the ease of purification of active dimeric cyt b6f from spinach, the interaction studies were conducted with purified dimeric spinach b6f. Isolated full-length Stt7 was mixed with purified intact cyt b6f, and the change in molar ellipticity at 222 nm was measured as a function of temperature (Fig. 6B). A net increase of 3.2 °C in melting temperature for Stt7 at 222 nm was observed in the Stt7-cyt b6f mixture (Fig. 6B), which implies stabilization of Stt7 caused by physical interaction with cyt b6f. The melting profile of Stt7 in solution containing 0.1% UDM detergent is derived from the difference between melting functions of (Stt7 + b6f) function (Fig. 6B, trace d) and that of the b6f complex alone (Fig. 6B, trace a).
Discussion
Summary of New Information Regarding the Properties of Stt7
This study provides a molecular characterization of purified Stt7, the enzyme that senses changes in trans-membrane redox poise across the thylakoid membrane and responds to quinol oxidation within the cyt b6f complex to activate photosynthetic state transitions by catalyzing the phosphorylation of the light-harvesting LHCII proteins. Purified full-length Stt7 was found to be enzymatically active in catalyzing the hydrolysis of ATP to ADP (Fig. 5A), indicating that the enzyme was properly folded and retained its native state. The enzymatic activity of the full-length Stt7 was found to be dependent on redox conditions and to require the presence of a reductant (Fig. 5A). Comparable activity (∼300–330 nmol of ATP converted to ADP/minute/mg of protein) activity was obtained for the Cys68 and Cys73-to-serine double mutant. (Fig. 5C). The enzymatically active, full-length Stt7 was purified as a tetramer with a molecular mass of 332 kDa (Fig. 3D). The thermal melting temperature of tetrameric Stt7, relative to Stt7 in the same concentration of UDM detergent, was increased by 3.2 °C in the presence of the intact cyt b6f complex, implying a significant energetic interaction between the two protein complexes (Fig. 6B). These are the first data that demonstrate the existence of a physical interaction that is involved in a model of redox control of Stt7 by the b6f complex.
Trans-membrane Domain Sequence of Stt7
Although the separation of redox control and enzymatic action on the two sides of the photosynthetic membrane imply a connecting trans-membrane structure of Stt7, the identity of this structure domain is not obvious. The amino acid sequence of the Stt7 kinase contains four proline residue at positions 102, 110, 111, and 115 within the putative trans-membrane domain (97VALLAPVLAYLFLPPGVLPGAIDYYI122). Prolines within trans-membrane domains have been associated with key functional roles in protein structure and dynamics, including the formation of hinges that provide conformational flexibility to the protein (27). As the functionally significant thiol groups are located predominantly within the p side domain, whereas the ATP-binding site is localized across the thylakoid membrane in the n side kinase domain, transfer of information from the n side ATP binding site to the p side thiol groups would involve a trans-membrane domain of Stt7. In this regard, it is important to note that proline 115 within the Stt7 trans-membrane domain is organized in a 112GXXP115 motif (Fig. 2A), which is characteristic of a flexible hinge (27, 28), and may be involved in transducing information across the membrane. The 112GXXP115 motif is conserved in Stt7/STN7 of higher organisms (Fig. 2A). An analysis of the Stt7 sequence reveals the presence of a dimerization motif, 112GXXXG116 (39), within the putative trans-membrane domain (Fig. 2A) that may be involved in the formation of a tetrameric Stt7.
Analysis of the n Side Kinase Domain Sequence of Stt7
The Stt7 kinase domain is localized on the n side of the thylakoid membrane (6, 15). The domain shares limited sequence conservation between photosynthetic organisms, especially at the C terminus (6, 16). A crystal structure of a homolog of the Stt7 kinase domain has been solved from Micromonas sp. alga (16). In this study, the kinase activity was localized within the amino acid segment 124–549 of Stt7. Moreover, the Stt7 C terminus (residues 500–754) is predicted to be disordered. As the C-terminal disordered domain is not a universal feature of the Stt7 kinase and is found exclusively in C. reinhardtii, it is proposed that the domain in its entirety is not involved in state transitions. Deletion of residues 550–754 (and residues 1–123) from Stt7 did not abrogate the kinase activity (Fig. 5B), indicating that the C-terminal domain is not directly involved in the structure-function of Stt7, as it pertains to state transitions. At present, the function of the Stt7 C-terminal disordered domain remains unknown.
Proposed Model I for a Physical Interaction Between the b6f Complex and Stt7
A recent proposal (19) suggested that the activation of the Stt7 kinase does not involve any net redox change of the p side cysteine residues Cys68 and Cys73. An experimental problem with assay of the oxidation state of the Cys68-Cys73 pair is that the disulfide redox potential is very negative, as in Refs. 26, 40, 41, and very strict anaerobic conditions would have to be maintained and checked to ensure reduction of the Cys68-Cys73 disulfide bond. The model also proposed that the Stt7 kinase is activated by a conformational change induced via an interaction with a p side domain of the Rieske ISP (19). There is a question as to whether the proposed Stt7 interactive residues, 51–104, in the ISP are accessible. In the cyt b6f structure from C. reinhardtii (PDB code 1Q90), residues 51–68 are buried within the hydrophobic domain of the membrane, implying a structure impediment to interaction with the Stt7 N-terminal domain, which is outside of the membrane in the p side aqueous phase. Moreover, the polyglycine hinge of the Rieske iron-sulfur protein subunit, proposed as a possible site of interaction between Stt7 and the cyt b6f complex, has a surface area of 340 Å2 (PDB code 4OGQ), which is small compared with the most frequent range of interfacial surface areas for interprotein interactions of 500–1000 Å2 (42). Alternatively, significant in vitro kinase activity is demonstrated in this study with purified Stt7 kinase under reducing conditions, which implies a role for the p side cysteine residues Cys68-Cys73.
Proposed Model II for Stt7 Kinase Activation via Reduction of the Cysteine 68 and Cysteine 73 Residues by Superoxide Generated from the b6f Qp Site
The LHCII phosphorylation activity associated with state transitions was reported to be sensitive to redox conditions prior to the identification of Stt7 (18). Subsequently, mutation analysis suggested an involvement of the p side residues, Cys68 and Cys73, in state transitions (15). In this study, in vitro assays of kinase activity of isolated and purified Stt7 and the Cys68 and Cys73-to-serine double mutant demonstrated the redox dependence, i.e. dependence on the reductant, dithiothreitol, of kinase activity (Fig. 5, B and C). Hence, it is inferred that an active kinase requires cysteine to be in the sulfhydryl state, i.e. no disulfide bridge between them. It has been suggested previously that the Stt7 kinase, with its single-pass trans-membrane domain, may be located proximal to the F and G helices of subunit IV of the cyt b6f complex (Fig. 1C) (39, 43–45). Such a complex between cyt b6f and Stt7 may place the p side extrinsic domain of Stt7 in proximity to the Qp site of cyt b6f (Fig. 7), which is the site of superoxide production from plastosemiquinone (25). Hence, in vivo, the superoxide (midpoint redox potential, −0.14 V (46)) may be a sufficiently strong reductant to reduce the disulfide bond between Cys68 and Cys73. Superoxide has been reported previously to reduce disulfide bonds in ion channels and pumps (26). The midpoint redox potential, Em, of the disulfide bond, which is pH-dependent, has been determined to be in the negative potential region, in the range of −0.12 to −0.47 V (40, 41, 47–49), where its chemical reduction requires strict anaerobic conditions. The potential of plastoquinone/plastosemiquinone is −0.10 to −0.15 V (4, 25, 49).
FIGURE 7.
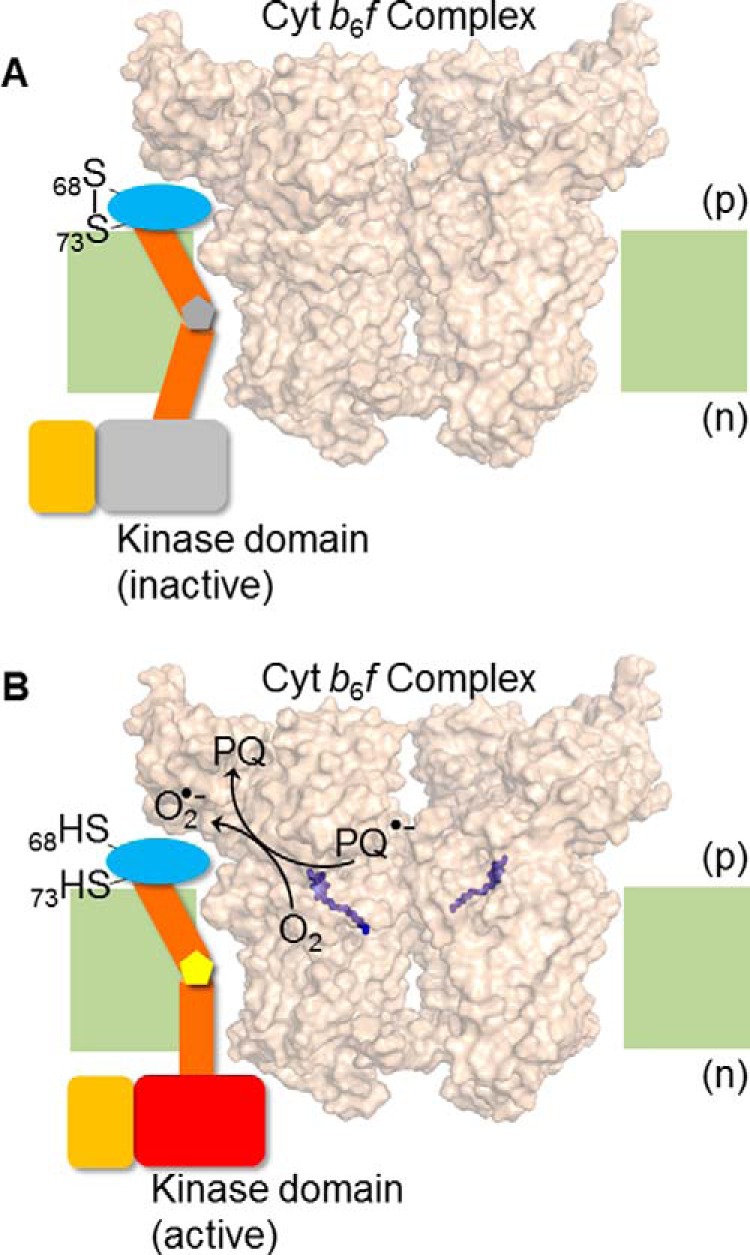
Hypothesis/model for trans-membrane activation of Stt7 kinase. Superoxide, O2˙̄, generated in the b6f complex via plastosemiquinone, reduces the disulfide bond between Cys68 and Cys73. Disulfide-sulfhydryl transition causes a conformation change in the Stt7 trans-membrane domain that is transmitted to the n side of the membrane through the proline hinge in the trans-membrane domain, thus activating the kinase on the n side. The p side domain containing Cys68 and Cys73, is shown as a blue oval, the trans-membrane domain as brown cylinders, and the proline hinge as a gray (A) and yellow (B) pentagon. The n side Stt7 kinase domain is shown in gray in the inactive state (A) and, when activated, as a red rectangle (B). The C-terminal disordered domain is shown as an orange rectangle. The cyt b6f complex (PDB code 4H13) is shown in surface representation, with the p side plastoquinol (Qp) binding site marked by the quinol analog tridecyl stigmatellin (purple sticks). Lipid bilayer, light green. (A) The Qp site of cyt b6f complex is unoccupied, Cys68 and Cys73 of Stt7 form a disulfide bond, and kinase remains inactive. (B) Upon binding and oxidation of plastoquinol at the Qp site, plastosemiquinone (PQ2˙̄) is generated which proposed to, serve as a reductant for molecular oxygen (O2), generating O2˙̄, which is oxidized to plastoquinone (PQ). As noted in the main text, the Stt7 trans-membrane domain has been proposed to occupy the niche between the peripheral F and G trans-membrane helices of subunit IV.
Hypothesis
A model is proposed, based on analysis of the sequence of the trans-membrane domain of Stt7 (Fig. 7), that also suggests a structural basis for signal transduction from the p to the n side. The trans-membrane domain of Stt7 contains four proline residues at positions 102, 110, 111, and 115 (97VALLAPVLAYLFLPPGVLPGAIDYYI122). Proline can exist in either cis or trans isomeric configuration with a large difference in the prolyl dihedral angle (50–52). Upon isomerization between cis and trans configurations, relatively large conformational changes are introduced in the polypeptide. It is suggested that the p side redox changes sensed by the Stt7 enzyme via the formation/breakage of the Cys68 and Cys73 disulfide bond, are communicated via the trans-membrane domain through isomerization of the trans-membrane proline residues, leading to the transduction of information across the membrane from the p to the n side. It is noted that isomerization of proline has been documented previously to be crucial to neurodegenerative disorders, cellular signaling, gating of ion channels, and control of access to enzyme active sites (53–58). It has been suggested previously that the Stt7 trans-membrane domain may be bound between the F and G trans-membrane helices of cyt b6f (43–45).
Tetrameric Organization of Stt7
It is significant to note that, previously, the Stt7 kinase pool was reported to be partitioned between the soluble and thylakoid membrane fractions (6). The membrane-bound fraction of Stt7 has been shown to be dimeric (19). In this study, the Stt7 kinase was isolated as a tetramer from the soluble fraction. It may be inferred that the tetrameric organization of Stt7 limits the exposure of the putative trans-membrane domain to the aqueous phase to prevent aggregation of the enzyme. In the membrane-bound fraction, it is expected that the hydrophobic trans-membrane domain of Stt7 will be exposed to the lipid acyl chains. The hydrophilic surface of Stt7, which protects the trans-membrane domain from aggregation in the soluble tetrameric state, will be excluded from the hydrophobic core of the lipid bilayer. The transition of Stt7 from a soluble to a membrane-bound form would require conformational changes in the enzyme that may be mediated by the conversion of a soluble Stt7 tetramer to a membrane-bound dimer.
Experimental Procedures
Cloning and Expression of the Stt7 Gene
The 2.2-kb nucleotide sequence that codes the full-length stt7 gene (C. reinhardtii, accession no. Q84V18) was cloned into expression vector pET28a at restriction sites NdeI and XhoI under the LacZ-inducible operon in E. coli DH5α cells. The plasmid was later transformed into BL21DE3 Rosetta cells for overexpression. Cysteines at the 68th and 73rd positions were mutated to serine using the forward and reverse primers 5′-GCAGCACCGTCCCAAGTTATGAAAAGTGGCGATATT-3′ and 5′-AATATCGCCACTTTTCATAACTTGGGACGGTGCTGC-3′, respectively.
Expression and Purification of Stt7 His-tagged Protein
Cultures of E. coli (BL21DE3 Rosetta) expressing Stt7 were grown at 37 °C, induced at an optical density of 0.6 with 100 μm isopropyl 1-thio-β-d-galactopyranoside, and grown overnight at 18 °C. The cells were harvested by centrifugation at 16,000 × g (10 min, 4 °C) and resuspended in lysis buffer (0.1 m potassium phosphate buffer, 400 mm NaCl, 12 mm citrate, and 2 mm βME (pH 8.0). The cells were lysed at 16,000 p.s.i. in a French pressure cell and then subjected to centrifugation (27,000 × g, 4 °C, 40 min). The clear supernatant thus obtained was loaded on a cobalt-nitrilotriacetic acid column pre-equilibrated with lysis buffer and washed (0.1 m potassium phosphate buffer, 400 mm NaCl, 12 mm citrate, 2 mm βME, and 2 mm imidazole (pH 8.0)). The protein was eluted with 0.1 m potassium phosphate buffer, 400 mm NaCl, 12 mm citrate, 2 mm βME, and 4 mm imidazole (pH 8.0), concentrated, and loaded on a S200 Superdex gel filtration column. All procedures were carried out at 4 °C. The eluted protein was analyzed by 12% SDS-PAGE.
Purification of Cytochrome b6f Complex
Spinach leaves were ground in 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, and 1 mm EDTA and sedimented at 11,000 × g (4 °C for 30 min). The sediment was washed with 10 mm Tris-HCl followed by 2 m NaBr, and the thylakoid membranes were suspended in TNES buffer (50 mm Tris-HCl, 100 mm NaCl, 1 mm EDTA, and 10% sucrose) supplemented with 30 mm n-octyl-β-d-glucopyranoside and 0.1% cholate. The soluble extract was precipitated sequentially with 38% and 65% ammonium sulfate to isolate b6f complex containing a native lipid complement. Further purification was achieved using a 10–32% sucrose density gradient in TNES buffer with a final detergent concentration of 0.05% UDM.
Assay of Kinase Activity
Catalysis by kinases involves the transfer of a phosphate group from an ATP molecule by ATP hydrolysis. ATPase activity was assayed using the ADP-glo kit (Promega). ATP hydrolysis activity of Stt7 was measured with 0.4 mm ATP in the presence of buffer (50 mm Tris-HCl, 5 mm Mg2+, and BSA) at room temperature for 15 min. The reaction mixture was incubated with ADP-glo reagent at room temperature for 40 min to consume unused ATP. Detection reagent was then added to convert ADP to ATP, detected in the presence of luciferase/luciferin, and luminescence was recorded with a Spectramax luminometer (Molecular Devices). This activity was determined from the amplitude of luminescence measured as a function of ADP concentration.
Far-UV CD Analysis
Far-UV CD spectra were measured in stirred cuvettes with an optical path length of 0.1 and 0.02 mm, respectively, in a Chirascan Applied Photophysics spectropolarimeter. Samples in phosphate buffer (pH 8), 20 °C, containing 0.1% UDM were used for analysis. Thermal denaturation profiles were obtained by measuring the amplitude of the CD signal at 222 nm over a temperature range of 10–95 °C.
Sequence Analysis
All Stt7/STN7 protein sequences were derived from the UNIPROT database: Q84V18, C. reinhardtii; C1EBN1, Micromonas sp., Q9S713; A. thaliana; A0A0K9QAH6,Spinacia oleracea; and A0A0B2PEE7, Glycine soja. Multiple sequence alignment was performed in Clustal Omega (59). The figures for sequence alignment were prepared in ESPript 3.0 (60). For prediction of disorder, the C. reinhardtii Stt7 sequence was analyzed in PSIPRED using the DISOPRED server (61). The DISOPRED server initially assigns an equal order-disorder probability to each amino acid position. The DISOPRED server performs a position-specific iterative basic local alignment search tool to generate a profile sequence. Then, using the BLAST results, the server performs an iterative search to calculate the disorder probability for each residue. The calculation is based on high-resolution crystal structures of related sequences deposited in the PDB, where the absence of coordinates from a chain is inferred to indicate disorder.
Mass Spectrometry
Intact protein mass spectrometry was performed as described previously (62) using a linear ion-trap mass spectrometer (LTQ, Thermo Electron) operated in positive ion mode. Mass accuracy for protein standards typically achieves a measurement accuracy better than 0.02%. For proteomics mass spectrometry, protein samples were alkylated, digested with trypsin, and analyzed by nanochromatography with tandem mass spectrometry on an hybrid linear ion trap/Orbitrap mass spectrometer as described previously (63). Mascot software (Matrix Science) was used to identify peptides using 10 ppm mass tolerance on high-resolution MS scan 1 and 0.5 Da mass tolerance on low resolution MS scan 2 for an overall 5% false discovery level.
Analytical Ultracentrifugation
Sedimentation velocity measurements were made with a ProteomeLab XL-A (Beckman Coulter) analytical ultracentrifuge. Purified Stt7 (400 μl, A280 = 0.6) in PBS buffer (pH 7.4), and the same volume of buffer as a blank reference was loaded in a double-sector analytical ultra-centrifugation sample cell assembled with a 12-mm epon-charcoal centerpiece and sapphire windows. The sample was loaded in a Ti-60 rotor, equilibrated at 20 °C in the rotor chamber for 2 h before centrifugation, and then centrifuged at 30,000 rpm at 20 °C. The progress of centrifugation was monitored by recording the absorbance at 280 nm. The sedimentation velocity boundaries were analyzed by the c(s) model in SEDFIT version 15.01b (64), which was also the source for the values of buffer density, viscosity, and protein-specific volume. The weight-average s value of each peak was created by c(s) analysis and converted to s(w,20) corresponding to standard conditions. The standard deviation of the molecular weight was calculated by c(M) analysis (65).
Electron Microscopy and Negative Stain EM
Homogeneous populations of Stt7 (2 μg/ml) were obtained by passing the purified protein through an S200 gel filtration column and collecting the selected peak fractions. The sample was applied to a freshly glow-discharged 400-mesh copper grid and washed with distilled water to remove excessive buffer without allowing the grid to dry. The grid was blotted and instantly stained with uranyl acetate on the side containing the sample. Excessive stain on the grid was removed by blotting, and the data were acquired on an FEI Tecnai transmission electron microscope operated at 200 keV.
Author Contributions
S. K. S. designed and conducted the experiments, analyzed the results, and wrote the manuscript. S. S. H. designed the experiments, conducted the sequence analysis, analyzed the results, and wrote the manuscript. S. D. Z. performed CD spectroscopy and analyzed the results. S. N. purified the Stt7 kinase domain from inclusion bodies and assayed its activity. J. P. W. performed mass spectroscopy, analyzed the results, and wrote the manuscript. W. C. performed mass spectroscopy. J. M. performed analytical ultracentrifugation, analyzed the results, and contributed the corresponding section under “Experimental Procedures.” W. A. C. proposed the project, analyzed the results, and wrote the manuscript.
Acknowledgments
We thank Prof. N. Noinaj for plasmid pRSF-1b, Prof. C. Gilpin for advice regarding EM images, and D. Cozzetto for discussions related to PSIPRED.
This work was supported by National Institutes of Health Grant GMS-038323 (to W. A. C.), Purdue University Center for Cancer Research Grant P30 CA23168 (to W. A. C.), and University of California, San Diego/Los Angeles NIDDK, National Institutes of Health Diabetes Research Center Grant P30 DK063491 (to J. P. W.). A preliminary account was presented at the meeting of the Biophysical Society, February 27-March 2, 2016, Los Angeles, CA ((2016) Biophys. J. 110, 91a). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LHCII
- light-harvesting chlorophyll complex protein II
- PSI
- photosystem I
- PSII
- photosystem II
- cyt
- cytochrome
- n side
- electrochemically negative side of the thylakoid membrane
- p side
- electrochemically positive side of the thylakoid membrane; iron-sulfur protein
- Chl-a
- chlorophyll a
- βME
- β-mercaptoethanol
- KD
- kinase domain
- UDM
- n-undecyl-β-d-maltopyranoside.
References
- 1. Horton P., Allen J. F., Black M. T., and Bennett J. (1981) Regulation of phosphorylation of chloroplast membrane polypeptides by the redox state of plastoquinone. FEBS Lett. 125, 193–196 [Google Scholar]
- 2. Allen J. F., Bennett J., Steinback K. E., and Arntzen C. J. (1981) Chloroplast protein-phosphorylation couples plastoquinone redox state to distribution of excitation-energy between photosystems. Nature 291, 25–29 [Google Scholar]
- 3. Barber J. (1982) Influence of surface-charges on thylakoid structure and function. Annu. Rev. Plant. Phys. 33, 261–295 [Google Scholar]
- 4. Kruse O., Nixon P. J., Schmid G. H., and Mullineaux C. W. (1999) Isolation of state transition mutants of Chlamydomonas reinhardtii by fluorescence video imaging. Photosynth. Res. 61, 43–51 [Google Scholar]
- 5. Wollman F. A. (2001) State transitions reveal the dynamics and flexibility of the photosynthetic apparatus. EMBO J. 20, 3623–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Depège N., Bellafiore S., and Rochaix J. D. (2003) Role of chloroplast protein kinase Stt7 in LHCII phosphorylation and state transition in Chlamydomonas. Science 299, 1572–1575 [DOI] [PubMed] [Google Scholar]
- 7. Bellafiore S., Barneche F., Peltier G., and Rochaix J. D. (2005) State transitions and light adaptation require chloroplast thylakoid protein kinase STN7. Nature 433, 892–895 [DOI] [PubMed] [Google Scholar]
- 8. Rochaix J. D. (2007) Role of thylakoid protein kinases in photosynthetic acclimation. FEBS Lett. 581, 2768–2775 [DOI] [PubMed] [Google Scholar]
- 9. Eberhard S., Finazzi G., and Wollman F. A. (2008) The dynamics of photosynthesis. Annu. Rev. Genet. 42, 463–515 [DOI] [PubMed] [Google Scholar]
- 10. Minagawa J., and Tokutsu R. (2015) Dynamic regulation of photosynthesis in Chlamydomonas reinhardtii. Plant J. 82, 413–428 [DOI] [PubMed] [Google Scholar]
- 11. Vener A. V., van Kan P. J., Rich P. R., Ohad I., and Andersson B. (1997) Plastoquinol at the quinol oxidation site of reduced cytochrome bf mediates signal transduction between light and protein phosphorylation: thylakoid protein kinase deactivation by a single-turnover flash. Proc. Natl. Acad. Sci. U.S.A. 94, 1585–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zito F., Finazzi G., Delosme R., Nitschke W., Picot D., and Wollman F. A. (1999) The Qo site of cytochrome b6f complexes controls the activation of the LHCII kinase. EMBO J. 18, 2961–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barber J. (1982) The control of membrane organization by electrostatic forces. Biosci. Rep. 2, 1–13 [DOI] [PubMed] [Google Scholar]
- 14. Manglik A., and Kobilka B. (2014) The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr. Opin. Cell Biol. 27, 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lemeille S., Willig A., Depege-Fargeix N., Delessert C., Bassi R., and Rochaix J. D. (2009) Analysis of the chloroplast protein kinase Stt7 during state transitions. PLoS Biol. 7, 664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo J., Wei X., Li M., Pan X., Chang W., and Liu Z. (2013) Structure of the catalytic domain of a state transition kinase homolog from Micromonas algae. Protein Cell 4, 607–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puthiyaveetil S. (2011) A mechanism for regulation of chloroplast LHC II kinase by plastoquinol and thioredoxin. FEBS Lett. 585, 1717–1721 [DOI] [PubMed] [Google Scholar]
- 18. Millner P. A., Widger W. R., Abbott M. S., Cramer W. A., and Dilley R. A. (1982) The effect of adenine nucleotides on inhibition of the thylakoid protein kinase by sulfhydryl-directed reagents. J. Biol. Chem. 257, 1736–1742 [PubMed] [Google Scholar]
- 19. Shapiguzov A., Chai X., Fucile G., Longoni P., Zhang L., and Rochaix J. D. (2016) Activation of the Stt7/STN7 kinase through dynamic interactions with the cytochrome b6f complex. Plant. Physiol. 171, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baniulis D., Yamashita E., Whitelegge J. P., Zatsman A. I., Hendrich M. P., Hasan S. S., Ryan C. M., and Cramer W. A. (2009) Structure-function, stability, and chemical modification of the cyanobacterial cytochrome b6f complex from Nostoc sp. PCC 7120. J. Biol. Chem. 284, 9861–9869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hasan S. S., and Cramer W. A. (2014) Internal lipid architecture of the hetero-oligomeric cytochrome b6f complex. Structure 22, 1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasan S. S., Yamashita E., Baniulis D., and Cramer W. A. (2013) Quinone-dependent proton transfer pathways in the photosynthetic cytochrome b6f complex. Proc. Natl. Acad. Sci. U.S.A. 110, 4297–4302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stroebel D., Choquet Y., Popot J. L., and Picot D. (2003) An atypical haem in the cytochrome b6f complex. Nature 426, 413–418 [DOI] [PubMed] [Google Scholar]
- 24. Hasan S. S., Proctor E. A., Yamashita E., Dokholyan N. V., and Cramer W. A. (2014) Traffic within the cytochrome b6f lipoprotein complex: gating of the quinone portal. Biophys. J 107, 1620–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baniulis D., Hasan S. S., Stofleth J. T., and Cramer W. A. (2013) Mechanism of enhanced superoxide production in the cytochrome b6f complex of oxygenic photosynthesis. Biochemistry 52, 8975–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peterson D. A., Archer S. L., and Weir E. K. (1994) Superoxide reduction of a disulfide: a model of intracellular redox modulation? Biochem. Biophys. Res. Commun. 200, 1586–1591 [DOI] [PubMed] [Google Scholar]
- 27. Sansom M. S., and Weinstein H. (2000) Hinges, swivels and switches: the role of prolines in signalling via transmembrane α-helices. Trends Pharmacol. Sci. 21, 445–451 [DOI] [PubMed] [Google Scholar]
- 28. Tieleman D. P., Shrivastava I. H., Ulmschneider M. R., and Sansom M. S. (2001) Proline-induced hinges in transmembrane helices: possible roles in ion channel gating. Proteins 44, 63–72 [DOI] [PubMed] [Google Scholar]
- 29. Senes A., Gerstein M., and Engelman D. M. (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol. 296, 921–936 [DOI] [PubMed] [Google Scholar]
- 30. Kannan N., and Neuwald A. F. (2005) Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J. Mol. Biol. 351, 956–972 [DOI] [PubMed] [Google Scholar]
- 31. Zhang L., Wang J. C., Hou L., Cao P. R., Wu L., Zhang Q. S., Yang H. Y., Zang Y., Ding J. P., and Li J. (2015) Functional role of histidine in the conserved his-x-asp motif in the catalytic core of protein kinases. Sci. Rep. 5, 10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hari S. B., Merritt E. A., and Maly D. J. (2013) Sequence determinants of a specific inactive protein kinase conformation. Chem. Biol. 20, 806–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schindler T., Bornmann W., Pellicena P., Miller W. T., Clarkson B., and Kuriyan J. (2000) Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942 [DOI] [PubMed] [Google Scholar]
- 34. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 41, W349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dyson H. J., and Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 [DOI] [PubMed] [Google Scholar]
- 36. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 37. Johnson W. C. (1999) Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35, 307–312 [PubMed] [Google Scholar]
- 38. Andrade M. A., Chacón P., Merelo J. J., and Morán F. (1993) Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 6, 383–390 [DOI] [PubMed] [Google Scholar]
- 39. Saif Hasan S., Yamashita E., and Cramer W. A. (2013) Transmembrane signaling and assembly of the cytochrome b6f-lipidic charge transfer complex. Biochim. Biophys. Acta 1827, 1295–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin T. Y., and Kim P. S. (1989) Urea dependence of thiol-disulfide equilibria in thioredoxin: confirmation of the linkage relationship and a sensitive assay for structure. Biochemistry 28, 5282–5287 [DOI] [PubMed] [Google Scholar]
- 41. Gilbert H. F. (1990) Molecular and cellular aspects of thiol-disulfide exchange. Adv. Enzymol. 63, 69–172 [DOI] [PubMed] [Google Scholar]
- 42. Chen J., Sawyer N., and Regan L. (2013) Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 22, 510–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zito F., Vinh J., Popot J. L., and Finazzi G. (2002) Chimeric fusions of subunit IV and PetL in the b6f complex of Chlamydomonas reinhardtii: structural implications and consequences on state transitions. J. Biol. Chem. 277, 12446–12455 [DOI] [PubMed] [Google Scholar]
- 44. Hasan S. S., and Cramer W. A. (2012) Lipid functions in cytochrome bc complexes: an odd evolutionary transition in a membrane protein structure. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 3406–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hasan S. S., Yamashita E., Ryan C. M., Whitelegge J. P., and Cramer W. A. (2011) Conservation of lipid functions in cytochrome bc complexes. J. Mol. Biol. 414, 145–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petlicki J., and van de Ven T. G. M. (1998) The equilibrium between the oxidation of hydrogen peroxide by oxygen and the dismutation of peroxyl or superoxide radicals in aqueous solutions in contact with oxygen. J. Chem. Soc. Faraday Trans. 94, 2763–2767 [Google Scholar]
- 47. Wunderlich M., and Glockshuber R. (1993) Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Soc. 2, 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huber-Wunderlich M., and Glockshuber R. (1998) A single dipeptide sequence modulates the redox properties of a whole enzyme family. Fold. Des. 3, 161–171 [DOI] [PubMed] [Google Scholar]
- 49. Krause G., Lundström J., Barea J. L., Pueyo de la Cuesta C., and Holmgren A. (1991) Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J. Biol. Chem. 266, 9494–9500 [PubMed] [Google Scholar]
- 50. Alric J., Pierre Y., Picot D., Lavergne J., and Rappaport F. (2005) Spectral and redox characterization of the heme ci of the cytochrome b6f complex. Proc. Natl. Acad. Sci. U.S.A. 102, 15860–15865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wedemeyer W. J., Welker E., and Scheraga H. A. (2002) Proline cis-trans isomerization and protein folding. Biochemistry 41, 14637–14644 [DOI] [PubMed] [Google Scholar]
- 52. Lu K. P., Finn G., Lee T. H., and Nicholson L. K. (2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629 [DOI] [PubMed] [Google Scholar]
- 53. Brazin K. N., Mallis R. J., Fulton D. B., and Andreotti A. H. (2002) Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 99, 1899–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lummis S. C., Beene D. L., Lee L. W., Lester H. A., Broadhurst R. W., and Dougherty D. A. (2005) Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 438, 248–252 [DOI] [PubMed] [Google Scholar]
- 55. Pastorino L., Sun A., Lu P. J., Zhou X. Z., Balastik M., Finn G., Wulf G., Lim J., Li S. H., Li X., Xia W., Nicholson L. K., and Lu K. P. (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature 440, 528–534 [DOI] [PubMed] [Google Scholar]
- 56. Sarkar P., Reichman C., Saleh T., Birge R. B., and Kalodimos C. G. (2007) Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell 25, 413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wulf G., Finn G., Suizu F., and Lu K. P. (2005) Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat. Cell Biol. 7, 435–441 [DOI] [PubMed] [Google Scholar]
- 58. Zhou X. Z., Kops O., Werner A., Lu P. J., Shen M., Stoller G., Küllertz G., Stark M., Fischer G., and Lu K. P. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol. Cell. 6, 873–883 [DOI] [PubMed] [Google Scholar]
- 59. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robert X., and Gouet P. (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jones D. T., and Cozzetto D. (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Whitelegge J. P., Zhang H., Aguilera R., Taylor R. M., and Cramer W. A. (2002) Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein: cytochrome b6f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol. Cell. Proteomics 1, 816–827 [DOI] [PubMed] [Google Scholar]
- 63. Patananan A. N., Capri J., Whitelegge J. P., and Clarke S. G. (2014) Non-repair pathways for minimizing protein isoaspartyl damage in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 289, 16936–16953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brown P. H., and Schuck P. (2006) Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90, 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]