

Abstract
Endonuclease V (EndoV) is an enzyme with specificity for inosines in nucleic acids. Whereas the bacterial homologs are active on both DNA and RNA, the mammalian variants only cleave RNA, at least when assayed with recombinant proteins. Here we show that ectopically expressed, as well as endogenously expressed human (h)EndoV, share the same enzymatic properties as the recombinant protein and cleaves RNA with inosine but not DNA. In search for proteins interacting with hEndoV, polyadenylate-binding protein C1 (PABPC1) was identified. The association between PABPC1 and hEndoV is RNA dependent and furthermore, PABPC1 stimulates hEndoV activity and affinity for inosine-containing RNA. Upon cellular stress, PABPC1 relocates to cytoplasmic stress granules that are multimolecular aggregates of stalled translation initiation complexes formed to aid cell recovery. Arsenite, as well as other agents, triggered relocalization also of hEndoV to cytoplasmic stress granules. As inosines in RNA are highly abundant, hEndoV activity is likely regulated in cells to avoid aberrant cleavage of inosine-containing transcripts. Indeed, we find that hEndoV cleavage is inhibited by normal intracellular ATP concentrations. The ATP stores inside a cell do not overlay stress granules and we suggest that hEndoV is redistributed to stress granules as a strategy to create a local environment low in ATP to permit hEndoV activity.
Keywords: ATP, nitrosative stress, ribonuclease, RNA, RNA processing, stress granule, endonuclease V, PABPC1, deamination, inosine
Introduction
Deamination of adenosine (A) to inosine (I) is a reaction that occurs spontaneously in cells and is enhanced by exposure to nitrosative agents formed as a response to inflammation, infection, or from the environment (1, 2). Both DNA and RNA as well as unincorporated (deoxy)ribonucleotides are subjected to deamination (3). Inosine is read as guanosine (G) by cellular proteins, and in DNA this event is thus considered mutagenic (3, 4). Consequences of adenosine deamination in RNA depend on which type and part of a transcript is deaminated and may result in altered coding, splicing, stability, and structure of an RNA or in intracellular relocalization (5).
In addition to arbitrary deamination events, inosine is also introduced in RNA by specific enzymes in a highly regulated manner to increase transcriptomic diversity (6). Responsible enzymes are the adenosine deaminases acting on RNA (ADARs)2 that catalyze A to I editing on mRNA and non-coding (nc)RNA including long ncRNA, micro (mi)RNA and small interfering (si)RNA. A to I editing is abundant in higher eukaryotes and edited sites amount to more than 100 million and are spread over the majority of human genes (7). Defective editing is linked to various human diseases including neurological disorders, infections, and cancer (8–11). Also some tRNAs undergo A to I editing at the anticodon wobble adenosine A34, a reaction that is catalyzed by adenosine deaminases acting on tRNA (ADATs) (12). ADATs are homologs to the ADARs and their editing is essential for protein synthesis (12, 13).
Until recently, no enzymatic activity specific for inosine in RNA was known. However, endonuclease V (EndoV), a well characterized protein for repair of inosines in DNA in Escherichia coli and some other bacteria (12, 14, 15), turned out to also incise RNA with inosine (16, 17). This applies also for human (h)EndoV, which in contrast to the bacterial variants, appears not to cleave at inosine in DNA or only with low efficiency (17, 18). The reaction catalyzed by EndoV is cleavage of the second phosphodiester bond 3′ to inosine in an Mg2+-dependent manner (15). Cleavage of RNA was catalyzed with similar efficiencies for E. coli (Ec) EndoV and hEndoV, and kinetics were comparable with EcEndoV on DNA, suggesting that RNA is the preferred substrate for hEndoV (16, 17).
Consistent with a substrate preference for RNA, cellular distribution of hEndoV is mainly cytoplasmic (19). A robust in vitro activity for recombinant hEndoV has been demonstrated, however, the function of hEndoV at the cellular level is not yet known. To get insight into hEndoV biology, we have performed biochemical analyses of endogenous and ectopically expressed hEndoV and find the same inosine-RNA specificity. Moreover, we find that hEndoV interacts with polyadenylate-binding protein C1 (PABPC1) in vitro and that both proteins relocate to cytoplasmic stress granules in cultured human cells upon exposure to various toxic agents. We suggest that this is a mechanism for the cell to regulate hEndoV activity.
Results
Human EndoV Has Inosine-specific Ribonuclease Activity When Ectopically Expressed in Human Cells
Human EndoV has been identified as a ribonuclease with specificity for inosine-containing RNA (16, 17). As this result was obtained with recombinant enzyme purified from E. coli, we tested whether hEndoV showed the same catalytic activity when expressed in human cells. Stably transfected T-REx 293 cell lines with ectopic expression of FLAG-hEndoV and the catalytic inactive site-specific mutant D52A (17) were established. Cell-free cytoplasmic extracts (S100) were made and Western blot analyses revealed similar levels of expression for the two FLAG-hEndoV variants (data not shown). To test for activity, an RNA oligonucleotide (IIUI) corresponding to part of the α-tropomyosin mRNA known to be highly deaminated by ADARs (20) was used as substrate. In FLAG-hEndoVwt extracts, a robust cleavage was observed both with the single- and double-stranded IIUI substrates (Fig. 1A, upper and middle panels) demonstrating that hEndoV is active at inosines in RNA also when expressed in human cells. RNA without inosine was not cleaved by these extracts (Fig. 1A, lower panel). As expected, the FLAG-hEndoV(D52A) mutant was inactive toward the IIUI RNA substrates (Fig. 1A). Inosine cleavage was not seen in extract from non-transfected T-REx 293 cells, indicating that endogenous hEndoV activity is absent or below the detection limit (Fig. 1A). Use of higher amounts of extracts gave only unspecific degradation of the substrates (data not shown).
FIGURE 1.

Activity and affinity for inosine-containing RNA for ectopically expressed FLAG-hEndoV. A, FLAG-hEndoVwt (hEVwt) and mutant D52A (hEVD52A) were overexpressed in T-REx 293 cells, protein extracts were prepared and assayed for activity toward ss- and dsRNA substrates containing multiple inosines (IIUI). Protein extract was also made from cells without any overexpression (T-REx). Increasing amounts of extracts were tested (10–30-100 and 30–100 ng for the T-REx extract) and recombinant (r)hEndoV ((r)hEV) (30 nm) was included as a positive control. An RNA substrate without inosine (dsCtr) was also tested. Cleavage products were analyzed on a 20% denaturing polyacrylamide gel. B, single- and double-stranded IIUI substrates (upper panels) as well as control RNA oligonucleotides (ss/dsCtr; lower panels) were incubated with FLAG-hEndoVwt and FLAG-hEndoV(D52A) protein extracts (1–2-4 μg; 2–4 μg for the T-REx extract) on ice. Recombinant (r)hEndoV (150 nm) was included as a positive control. Free and bound fractions were separated on native 10% polyacrylamide gels. −, no enzyme added. Middle panels: a fraction of the EMSA reactions with ss/dsIIUI were analyzed for cleavage by 20% denaturing PAGE as in A. (r)hEndoV assayed under condition for activity are positive controls for cleavage (most to the right).
To assess hEndoV binding to substrate, electrophoretic mobility shift assays were performed. The EMSAs revealed weak shifts with the FLAG-hEndoVwt extracts with the single-stranded IIUI substrate (Fig. 1B, upper left panel). For the FLAG-hEndoV(D52A) extract, strong bands were detected demonstrating that the inactive D52A mutant makes more stable contacts with the inosine-RNA substrate than hEndoVwt. No binding of endogenous hEndoV (T-REx) was detected in EMSAs using the ssIIUI substrate (Fig. 1B, upper left panel). With the double-stranded IIUI substrate, shifts were only seen with the FLAG-hEndoV(D52A) extract (Fig. 1B, upper right panel). Fractions of the EMSA reaction mixtures were run on denaturing gels to test for cleavage. No cleavage was detected even for recombinant hEndoV (Fig. 1B, middle panels), demonstrating that the EMSA condition does not allow for hEndoV activity. Using control RNA substrates without inosine (ss/dsCtr), no specific bands corresponding to hEndoV were seen, however, some unspecific shifts of lower mobility were visible in all three extracts at comparable levels, demonstrating the similarity of the extracts (Fig. 1B, lower panels).
Endogenously Expressed hEndoV Cleaves Inosine-containing RNA
To identify potential activity of endogenous hEndoV (without overexpression), a larger amount of T-REx 293 cells were cultivated and a cytoplasmic (S100) protein extract prepared. The extract was run through a Superdex 75 column followed by ResQ and ResS chromatography (Fig. 2A). A corresponding extract from cells overexpressing FLAG-hEndoVwt was run in parallel and used for identifications of fractions rich in hEndoV as we assumed that FLAG-hEndoV and endogenous hEndoV have similar chromatographic properties. After ResS purification, the FLAG-hEndoV peak was identified and the corresponding fractions for the T-REx extract were tested. Cleavage products of similar size as those generated by FLAG-hEndoV and recombinant hEndoV were found in ResS fractions 10–14 when using the single-stranded IIUI substrate, demonstrating the endogenous hEndoV also has affinity for inosines in RNA (Fig. 2B).
FIGURE 2.

Activity of endogenously expressed hEndoV. A, purification scheme for endogenously expressed hEndoV using T-REx 293 cells as starting material. ResQ, Resource Q; ResS, Resource S chromatography; Fr, fraction; FT, flow-through. B, flow-through (QFT and SFT) and partly purified fractions (fraction 10–16) after ResS chromatography were probed for inosine-RNA (ssIIUI) cleavage activity. 1 μl from each fraction was used in the assays and recombinant (r)hEndoV (30 nm) as positive control. C, immunoprecipitation followed by Western blot analysis of endogenous hEndoV. Fractions 10 and 11 after ResS were diluted in IP buffer and supplemented with 2 μg of polyclonal (α1) or monoclonal (α2) hEndoV antibody. Precipitated material was run on SDS-PAGE, the gel was blotted onto a nylon membrane and proteins were detected with a third hEndoV antibody (polyclonal; α3). Recombinant hEndoV ((r)hEV, 20 ng) was included as a positive control in the Western blot (WB) analysis. Molecular mass marker (M) with sizes (in kDa) is shown to the left. D, cleavage products for endogenous hEndoV (Q6, ResQ Fr 6 T-REx) on single- and double-stranded IIUI substrates were run on 20% sequencing gels alongside with recombinant (r)hEV. Markers were RNA oligonucleotides of 10 and 13 residues. The sequence of the IIUI substrate and the preferred cleavage position for hEndoV is shown above the gel picture.
To verify that cleavage in Fig. 2B is executed by hEndoV, immunoprecipitations with a commercially available antibody as well as a monoclonal antibody raised against hEndoV were performed on peak fractions (Fig. 2, A and B). The precipitated materials were subjected to Western blot analysis and probing with a polyclonal hEndoV antibody identified endogenous hEndoV in the fractions (Fig. 2C). Recombinant hEndoV migrates slightly faster than the endogenous protein, which could be due to post-translational modifications not present in the recombinant version or alternatively that human cells express a different isoform. Human ENDOV mRNA is known to exist in numerous splice variants (19).
As the IIUI substrates contain multiple inosines, several cleavage sites for hEndoV could exist. To obtain higher resolution, a sequencing gel was run where the single- and double-stranded IIUI substrates were run adjacent to RNA markers of known sizes. The gel revealed that for both endogenous and recombinant hEndoV, the preferred cleavage position was 3′ to the middle insoine (I10) with some nicking also next to the furthermost 5′ inosine (I9) especially for the double-stranded substrate (Fig. 2D). It appears that expression both in human and E. coli cells give active and comparable hEndoV enzymes suggesting that there are no extensive post-translational modifications of hEndoV that are critical for enzymatic activity.
Initially EndoV homologs were characterized as DNA repair enzymes, however, recombinant hEndoV apparently does not share this property (16, 17). We tested whether this was true also for hEndoV expressed in human cells. Activity assays revealed no cleavage of DNA with inosine, neither after overexpression nor with endogenously expressed hEndoV (data not shown).
Analysis of hENDOV mRNA Levels
Our data demonstrate that endogenous hEndoV is an inosine-specific ribonuclease that is expressed at a low level at least in the cell line used originating from kidney epithelium. However, expression levels of hENDOV mRNA do vary among different tissues as previously demonstrated by transcript profiling (19). That work demonstrated expression of hENDOV in 12 different tissues where cervix, colon, and adrenal gland had low/no expression. Highest hENDOV transcript levels were found in pancreas and testis possibly suggesting a vital function for hEndoV in these organs.
We compared the amount of hENDOV mRNA in three different cell lines: T-REx 293 (kidney), HeLa (cervix), and HAP1 (myeloid) by qRT-PCR. The levels of hENDOV mRNA were normalized to GAPDH and tested for quality control by agarose gel electrophoresis (Fig. 3). We found that both HeLa and HAP1 cells had significantly less hENDOV mRNA than T-REx 293 cells (about 50 and 20% of the levels of T-Rex, respectively; Fig. 3). Exposure of the cells to arsenite or nitrite did not significantly change the amount of hENDOV transcripts in either of the cell lines (Fig. 3). It appears that hENDOV mRNA levels vary between various cell lines and tissues, but the steady-state levels of the hENDOV mRNA are not altered in response to stress.
FIGURE 3.
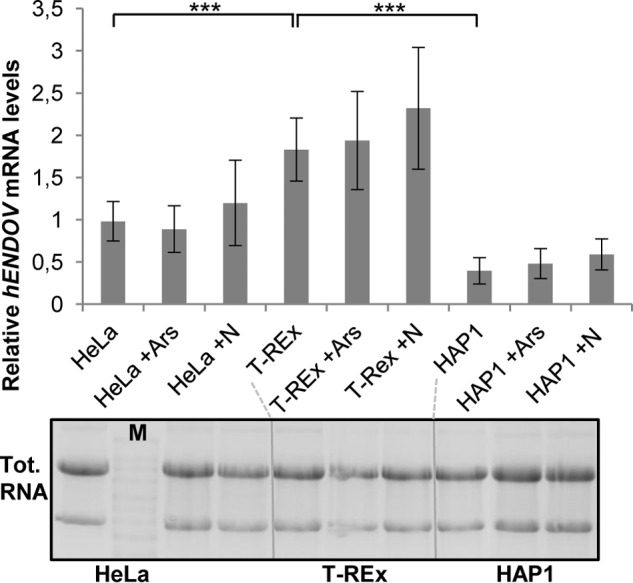
Expression of hENDOV mRNA. Human ENDOV transcript levels were measured by qRT-PCR in HeLa, T-REx 293, and HAP1 cells treated with or without arsenite (Ars) or nitrite (N). Values relative to GAPDH were calculated and the results presented are the average of 6 qRT-PCRs using cDNA from three independent experiments. Error bars represent standard deviations and statistical significance was determined by Student's t test; ***, p < 0.002. Total RNA used as starting material for cDNA synthesis was analyzed by denaturing agarose gel electrophoresis (samples loaded in the same order as shown for the qRT-PCR). Marker (M) is the Millenium RNA marker (Ambion).
RNA-dependent Interaction between hEndoV and PABPC1
To learn more about the cellular function of hEndoV, we searched for protein partners interacting with hEndoV by co-immunoprecipitation (co-IP) experiments coupled to MS analysis. We used cytoplasmic extracts made from cells overexpressing the FLAG-hEndoV(D52A) mutant taking advantage of its more stable interaction with inosine-containing RNA (Fig. 1B). MS analysis of the immunoprecipitated material revealed various ribosomal proteins as well as proteins known to be involved in different aspects of RNA metabolism (Table 1). Among the hits, a protein that caught our interest was PABPC1, which is a protein that binds poly(A) tails of mRNAs and is involved in various aspects of RNA metabolism including regulation of mRNA translation and stability in the cytoplasm (21). To verify the interaction, co-IP using anti-FLAG beads and the hEndoV(D52A) extract was repeated and the precipitated material subjected to Western blot analysis using an antibody against PABPC1. The co-IP confirmed the interaction between hEndoV(D52A) and PABPC1 (Fig. 4A, upper panel). Co-IP with an extract with overexpressed FLAG-hEndoVwt also precipitated PABPC1, showing that the protein-protein interaction is not a consequence of the amino acid change introduced in D52A (Fig. 4A). Given that both PABPC1 and hEndoV bind RNA, we next sought to determine whether the interaction was RNA dependent. Treatment of the extracts with RNase completely abolished co-IP of PABPC1 by both FLAG-hEndoVwt and D52A, demonstrating that the interaction relied on RNA (Fig. 4A, upper panel). For the T-REx 293 extracts, co-IP gave a weak band for PABPC1, indicating some unspecific binding of PABPC1 to the anti-FLAG beads. Also this signal was lost by RNase treatment (Fig. 4A, upper panel). Reprobing of the membrane with a hEndoV antibody confirmed that both FLAG-hEndoVwt and D52A were precipitated (Fig. 4A, lower panel).
TABLE 1.
25 top proteins identified by MS after co-IP with FLAG-hEndoV(D52A)
Ribosomal proteins are omitted from the list.
| T test p value | T test difference | Protein name | Gene name | Peptide counts (all) |
|---|---|---|---|---|
| 2.52E-05 | 3.70734 | Endonuclease V | ENDOV | 21;17;17;14;11;8;8;7;6;6;6;6;6;5;4;2;2;1 |
| 5.26E-06 | 3.00836 | X-box-binding protein 1 | XBP1 | 1;1;1;1 |
| 4.0E-06 | 2.98271 | Insulin-like growth factor 2 mRNA-binding protein 1 | IGF2BP1 | 15;11 |
| 2.30E-05 | 2.30214 | Polyadenylate-binding protein 1; polyadenylate-binding protein 3 | PABPC1;PABPC3 | 22;21;21;21;11;11;7;6;6;6;5;5;5;4;4;4;4; 3;3;3;2;1;1;1;1;1 |
| 0.000628637 | 2.10569 | Insulin-like growth factor 2 mRNA-binding protein 3 | IGF2BP3 | 6;2;1 |
| 0.000185407 | 1.90331 | Nuclease-sensitive element-binding protein 1 | YBX1 | 8;7;4;2 |
| 0.000237113 | 1.77645 | Insulin-like growth factor 2 mRNA-binding protein 2 | IGF2BP2 | 4;4;4;3;3;3;3 |
| 0.0430461 | 1.75115 | Mesencephalic astrocyte-derived neurotrophic factor | MANF | 1;1 |
| 0.00680958 | 1.74702 | Polyadenylate-binding protein 4 | PABPC4 | 16;16;16;16;12;7;4;3;3;2;2;2;2 |
| 0.0270908 | 1.65183 | La-related protein 1 | LARP1 | 9;8;6;3;3;2;1;1;1;1;1;1 |
| 0.016685 | 1.59752 | Histone H3; histone H3.3C; histone H3.2; histone H3.1t; histone H3.3; histone H3.1 | H3F3B;H3F3A;H3F3C;HIST2H3A; HIST3H3;HIST1H3A | 2;2;2;2;2;2;2;2;2;2 |
| 0.000384325 | 1.54604 | Guanine nucleotide-binding protein subunit β2-like 1 | GNB2L1 | 1;1;1;1;1;1;1;1;1;1;1;1;1 |
| 0.000118816 | 1.5447 | Y-box-binding protein 3 | YBX3 | 7;6;6;2 |
| 0.0150847 | 1.5411 | Double-stranded RNA-binding protein Staufen homolog 1 | STAU1 | 7;7;7;6;2;1 |
| 5.38E-06 | 1.49226 | La-related protein 7 | LARP7 | 5;5;4;3;2;2 |
| 0.00318245 | 1.48783 | Protein mago nashi homolog;Protein mago nashi homolog 2 | MAGOHB;MAGOH | 1;1;1;1;1 |
| 0.00348873 | 1.48094 | Eukaryotic translation initiation factor 6 | EIF6 | 2;1 |
| 0.000175999 | 1.36854 | Uncharacterized protein C7orf50 | C7orf50 | 1;1 |
| 0.000544839 | 1.34037 | Putative helicase MOV-10 | MOV10 | 4;4;2 |
| 0.000166308 | 1.23335 | tRNA (cytosine(34)-C(5))-methyltransferase | NSUN2 | 14;12;8;6 |
| 0.00509954 | 1.23131 | 7SK snRNA methylphosphate capping enzyme | MEPCE | 6;4 |
| 0.000423126 | 1.2146 | Nuclear cap-binding protein subunit 1 | NCBP1 | 2 |
| 0.00378886 | 1.18884 | Protein BUD31 homolog | BUD31 | 3;2;1;1 |
| 0.00114663 | 1.17908 | FACT complex subunit SPT16 | SUPT16H | 5 |
| 0.0128086 | 1.14924 | Hepatoma-derived growth factor-related protein 3 | HDGFRP3 | 3;2 |
FIGURE 4.

Interaction between hEndoV and PABPC1. A, protein extracts were made from T-REx 293 cells expressing FLAG-hEndoVwt and FLAG-hEndoV(D52A). FLAG-hEndoVwt and D52A were isolated using anti-FLAG beads and co-precipitated material was separated by SDS-PAGE. Extract without hEndoV overexpression (T-REx) was used as control for unspecific binding to the beads and input represents the starting material (1.5 mg). Addition of RNase to the samples is indicated (−/+). After blotting, the membrane was probed with a PABPC1 antibody (upper panel). The membrane was stripped and reprobed with a hEndoV antibody (polyclonal; in house)(lower panel). Gray lines are drawn to separate the three extracts. B, upper panel, (r)hEndoV (0.2 nm) was incubated with the single-stranded IIUI substrate and increasing amounts of PABPC1, RRM1–4, MLLE, GST (0.6–1.8–3.6 nm), or BSA (3.6 nm) and activity was measured by denaturing PAGE. Samples without (r)hEndoV (−) contained protein (3.6 nm) as indicated. Middle panel, (r)hEndoV (70 nm) was incubated with the single-stranded IIUI substrate and increasing amounts of the same proteins as above (70–200-700 nm) or a fixed amount of BSA (700 nm) and a band shift assay was performed. Samples without (r)hEndoV (−) contained protein (700 nm) as indicated. Lower panel: a fraction of the EMSA reactions was analyzed for cleavage by 20% denaturing PAGE. C, cleavage and band shift in B was quantified in two-three separate experiments with two parallels in each. Error bars indicate S.D. D, yet another fraction of the EMSA reactions was run on a separate native EMSA gel, blotted onto a nylon membrane, and subjected to Western analysis. The membrane was probed first with PABPC1 antibody (upper panel) prior to hEndoV antibody (polyclonal, in house).
To test if there is a functional interaction between hEndoV and PABPC1, recombinant proteins were purified and activity assays performed. The single-stranded IIUI substrate was incubated with a fixed amount of hEndoV (0.2 nm) and increasing amounts of PABPC1 (0.6 to 3.6 nm). Addition of PABPC1 stimulated hEndoV activity and a 3-fold molar excess of PABPC1 increased cleavage from 17 to 22%. With further addition of PABPC1, more cleavage was seen and with 20-fold excess of PABPC1 (3.6 nm), 32% of the substrate was incised (Fig. 4, B and C).
PABPC1 consists of four RNA-binding domains (RRM1–4) followed by a linker and a conserved C-terminal MLLE domain (21, 22). The RRM domains mediate circularization of mRNA required for protein translation, whereas the MLLE domain mediates binding to PABP-binding proteins. To identify specific regions involved in hEndoV interaction, the two domains RRM1–4 and MLLE were expressed separately in E. coli, purified, and tested in activity assays. The RRM1–4 protein stimulated hEndoV activity comparable with full-length PABPC1, whereas the MLLE domain had no effect (Fig. 4, B, upper panel, and C). As controls, pure GST and BSA were included where none had effect on hEndoV activity. The highest amount of protein partners alone (3.6 nm) did not result in any cleavage (Fig. 4B, upper panel).
To study potential effects on substrate binding, EMSAs were performed. Also here PABPC1 had a positive effect on hEndoV evident as stronger binding to the RNA substrate. Without PABPC1, 70 nm hEndoV shifted 15% of the substrate, which increased to 45% with a 10-fold excess of PABPC1 (700 nm) (Fig. 4B, middle panel, and C). A small increase in hEndoV binding was observed by addition of RRM1–4 and MLLE (33 and 31%), but the effect was not as strong as seen for full-length PABPC1. GST in 10-fold excess only had a marginal effect. 700 nm of the protein partners alone did not give any visible band shifts using the IIUI substrate (Fig. 4B, middle panel). The EMSA conditions allowed only for substrate binding and not cleavage (Fig. 4B, lower panel). No supershift indicative of a complex with both hEndoV and PABPC1 was observed, suggesting that the contact between the two proteins is weak or transient. In fact, Western blot analysis of the bound material only identified hEndoV in the shifts and not PABPC1 (Fig. 4D).
hEndoV Colocalizes with PABPC1 in Stress Granules
PABPC1 and hEndoV both have a cytoplasmic localization in cells (17, 19, 23). Previous research has shown that upon cellular stress, PABC1 accumulates in cytoplasmic granules referred to as stress granules (24). These granules contain a selection of RNAs and proteins and are multimolecular aggregates of stalled translational pre-initiation complexes. They are formed in response to certain stress factors to temporarily reduce translation of housekeeping genes and optimize translation of stress-response genes with the aim to increase cell survival (25). We tested for redistribution of hEndoV and possible colocalization with PABPC1 in stress granules in human cells by the use of a T-REx 293 cell line stably transfected with GFP-hEndoVwt. The cells were cultured in the presence of arsenite, which is the most commonly used inducer of stress granules (24), and probed with antibodies against the stress granule marker G3BP (26) as well as PABPC1. Fluorescence microscopy showed that after arsenite exposure, hEndoV was found in discrete cytoplasmic foci overlapping with G3BP (Fig. 5A). Whereas most of the G3BP proteins accumulated in stress granules, a substantial part of hEndoV remained evenly distributed in the cytoplasm. Merged images revealed that hEndoV staining overlapped with PABPC1 in stress granules indicating a connection between hEndoV and PABPC1 also in intact cells (Fig. 5A).
FIGURE 5.

hEndoV and PABPC1 colocalize in cytoplasmic stress granules. A, T-REx 293 cells stably transfected and B, HEK 293T cells transiently transfected with GFP-hEndoVwt were left untreated or exposed to arsenite (0.5 mm, 30 min), fixed, and processed for confocal microscopy. Cells were stained with PABPC1 (cyan) or G3BP (red) antibodies and DAPI (blue) to visualize the nuclei. GFP-hEndoV is shown in green. Localization of proteins was observed by confocal microscopy (Leica SP8) using a ×40 oil objective. Bar, 5 μm. Representative images are shown. C, the Manders' coefficients for colocalization (tM1 and tM2 ± S.D.) were determined for cells stably or transiently expressing GFP-hEndoVwt. tM1 ± S.D. expresses the degree of colocalization of the green channel (GFP-hEndoV) with the red/cyan channel (G3BP/PABPC1). tM2 ± S.D. expresses the degree of colocalization of the red/cyan channel with the green channel.
To determine the degree of colocalization between the different proteins, Manders' coefficients (tM1 and tM2 ± S.D.) were calculated. This analysis revealed that 55% of the hEndoV-positive pixels colocalized with the PABPC1-positive pixels (tM1 = 0.55) and opposite, 59% of the PABPC1-positive pixels overlapped with hEndoV-positive pixels (tM2 = 0.59) (Fig. 5C). The corresponding values for hEndoV and G3BP are tM1 = 0.52 and tM2 = 0.42.
To test another expression system, HEK 293T cells were transiently transfected with a vector expressing GFP-hEndoVwt. Also here GFP-hEndoV fusion proteins accumulated in arsenite-induced stress granules colocalizing with PABPC1 (tM1 = 0.25 and tM2 = 0.27) and G3BP (tM1 = 0.36 and tM2 = 0.36) (Fig. 5, B and C). Also in HeLa cells, GFP-hEndoVwt was found in arsenite-induced stress granules (data not shown). In summary, our data suggest an RNA-dependent physical and functional interaction between hEndoV and PABPC1 in human cells.
Analysis of hEndoV-containing Stress Granules
Redistribution of hEndoV to stress granules was further elucidated, first by analyzing FLAG-tagged instead of GFP-tagged proteins. T-REx 293 cells stably expressing FLAG-hEndoVwt were exposed to arsenite and examined by fluorescent microscopy. The images showed that FLAG-hEndoVwt also was found in stress granules overlapping with both G3BP and PABPC1 (Fig. 6, A and E). In the cytoplasm, closely associated with stress granules are processing bodies (P-bodies) involved in RNA degradation (27). Cells were stained with the P-body marker Dcp1. FLAG-hEndoVwt staining did not overlap with the Dcp1 positive P-bodies, but were often seen juxtaposed (Fig. 6B).
FIGURE 6.

Induction of hEndoV-containing stress granules by various stress. A, T-REx 293 cells ectopically expressing FLAG-hEndoVwt were cultured in the absence or presence of arsenite (0.5 mm, 30 min) before processing for visualization of hEndoV (green) and G3BP (red) (left panels) or hEndoV (green) and PABPC1 (red) (right panels) or (B) hEndoV (green) and Dcp1 (red). C, T-REx 293 FLAG-EndoVwt cells were cultured in the presence of nitrite (200 mm, 1 h) or D, transiently transfected with poly(I:C) (500 ng, 7 h) and processed for imaging of hEndoV (green) and G3BP/PABPC1 (red). Mock-treated cells were also included. Nuclei were counterstained using DAPI (blue) and colocalization (yellow) is shown in Merge. Localization of proteins was observed by confocal microscopy (Leica SP8) using a ×40 oil objective. Bar, 5 μm. E, Manders' coefficients for colocalization of hEndoV and G3BP (left columns) or hEndoV and PABPC1 (right columns).
Stress granules are assembled in response to certain toxic agents and the protein/RNA composition may vary (27–29). Arsenite generates oxidative stress and we expanded the analysis to include sodium nitrite (nitrosative stress). Nitrite exposure led to formation of stress granules in T-REx 293 cells and probing with a hEndoV-antibody confirmed sorting of hEndoV to the granules. Also here hEndoV colocalized with PABPC1 (Fig. 6, C and E). Viral infections may also induce stress granules, and transfection of cells with long double-stranded RNA (poly(I:C)) can be used as a mimic (30). Poly(I:C) transfection gave stress granules in a subset of cells not seen after mock transfection (Fig. 6D). These granules were fewer and bigger compared with those induced by arsenite treatment. Also the poly(I:C)-induced stress granules harbored hEndoV. For all treatments there was a substantial overlap between hEndoV and G3BP or PABPC1 (Fig. 6E).
To evaluate the contribution of hEndoV activity to stress granule formation, FLAG-hEndoVwt and D52A expressing cells were cultured and prepared for microscopy. The inactive hEndoV(D52A) mutant was sorted similarly as the wild type enzyme with an even cytoplasmic staining in untreated cells and accumulation in stress granules upon arsenite exposure (Fig. 7, A and B). Stress granules were quantified with respect to number and size and there were no differences in the assembly of stress granules between the two cell types (Fig. 7C). To exclude possible contribution from endogenous hEndoV, a knock-out human cell line was established (HAP1 ENDOV−). Also in these HAP1 cells lacking hEndoV, stress granules appeared similar as in the corresponding wild type cells (Fig. 7, D and E). Hence, hEndoV activity is not crucial for formation of stress granules. However, there was a tendency that the average size of the stress granules was smaller in the ENDOV− cells (Fig. 7E).
FIGURE 7.

Stress granule assembly is independent of hEndoV activity. A, T-REx 293 cells ectopically expressing FLAG-hEndoV(D52A) were cultured in the absence or presence of arsenite (0.5 mm, 30 min) before processing for visualization of hEndoV (green) and G3BP (red). Nuclei were counterstained using DAPI (blue) and colocalization (yellow) is shown in Merge. Localization of proteins was observed by confocal microscopy (Leica SP8) using a ×40 oil objective. Bar, 5 μm. B, Manders' coefficients for colocalization of FLAG-hEndoV(D52A) and G3BP and C, the average number and size of stress granules per cell in A. Error bars represent the S.D. calculated from 20 randomly selected cells in each cell type. Stress granules were defined as G3BP foci measuring >0.1 μm. Representative images are shown. D, stress granules were induced in HAP1wt and ENDOV− cells and analyzed as above (E).
Previous research has shown that two proteins linked to inosine-containing RNA, the ADAR1 and Tudor staphylococcal nuclease (Tudor-SN), also relocate to stress granules after arsenite treatment (31). To test possible colocalization of these proteins with hEndoV, T-REx 293 cells overexpressing FLAG-hEndoVwt were treated with arsenite and probed with antibodies against ADAR1 or Tudor-SN. ADAR1 is known to exist in two forms: a nuclear form (p110) that is constitutively expressed or an inducible cytoplasmic form (p150) (32). The antibody used here detects both forms. We find colocalization of hEndoV with both ADAR1 and Tudor-SN in stress granules (Fig. 8, A–C). ADAR1 is mainly nuclear, but upon arsenite treatment weak accumulation in cytoplasmic stress granules is evident (Fig. 8A). Tudor-SN is mainly cytoplasmic and after exposure to arsenite clear spots overlapping with stress granules are seen (Fig. 8B). In contrast to the colocalization data, the co-IP experiment coupled with MS analysis did not identify Tudor-SN or ADAR1 as physical interacting partners of hEndoV (data not shown and Table 1). Treatment of the T-REx 293 cells with arsenite prior to extract preparation to induce stress did not change this. Furthermore, none of them were detected in Western blotting of the FLAG-hEndoVwt or D52A precipitated material using ADAR or Tudor-SN antibodies (data not shown). It might be that the proteins appear close to each other in stress granules without a direct interaction or alternatively, the interaction is too weak or transient to be detected in co-IP.
FIGURE 8.
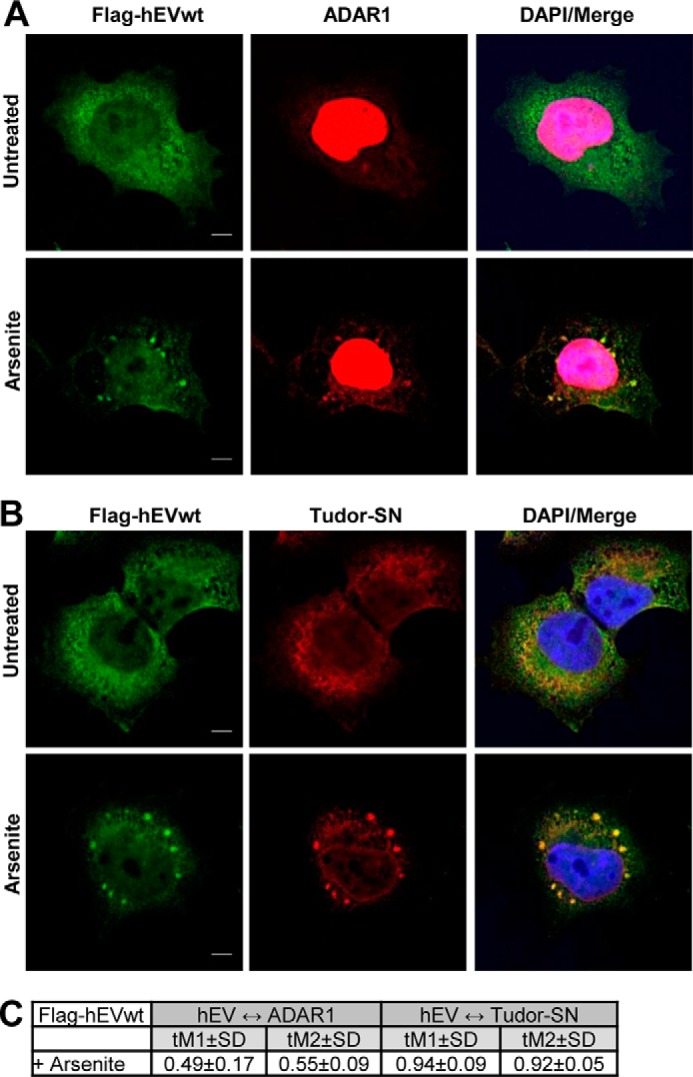
ADAR1 and Tudor-SN colocalize with hEndoV in stress granules. T-REx 293 cells ectopically expressing FLAG-hEndoV were cultured in the presence or absence of arsenite (0.5 mm, 30 min). Subsequently, cells were fixed and immunofluoroscence experiments were performed using hEndoV (green) and (A) ADAR1 (red) or (B) Tudor-SN (red) antibodies. DRAQ5 (blue) was used to counterstain nuclei, and colocalization (yellow) is shown in Merge. Localization of proteins was observed by confocal microscopy (Zeiss) using a ×63 oil objective.
hEndoV Activity Is Inhibited at Physiological Concentration of ATP
Formation of stress granules could be a strategy for the cell to create a local environment with a high concentration of related proteins, cofactors, or other supplements, and/or contrary, a way to exclude certain factors. Taken into account the abundance of inosines in RNA and the potent ribonuclease activity, hEndoV activity is most likely regulated to avoid uncontrolled degradation of inosine-containing RNA in the cell. For example, divalent ions (Mg2+/Mn2+) are known cofactors critical for hEndoV cleavage. Among the other metabolites in the cells, ATP is abundant reaching millimolar concentrations in most cell types (33). Furthermore, the intracellular ATP concentration is known to fluctuate and for example, under certain stress conditions, like oxidation and arsenite exposure, a large reduction is seen (34, 35). Therefore, ATP was included in activity assays for hEndoV and intriguingly, 1 mm ATP almost completely inhibited hEndoV activity (Fig. 9A). Titration experiments revealed a narrow window between 0.2 and 0.5 mm ATP where hEndoV activity was steeply reduced. Guanosine triphosphate (GTP), another purine nucleotide, as well as the pyrimidine nucleotide thymidine triphosphate (TTP) had a similar inhibiting effect on hEndoV activity (Fig. 9B). It appears that it is not specific binding of the adenine base into the active site of hEndoV that causes the inhibiting effect. It could be that the phosphates bring in negative charge in the active site that disturbs the coordination of molecules necessary for catalysis. As ATP is 6–10-fold more abundant in the cytosol than the other nucleotides (33) and known to fluctuate during stress, it could therefore have a master regulatory role.
FIGURE 9.

ATP inhibits hEndoV activity. A, recombinant hEndoV (3 nm) was incubated with increasing amounts of ATP (0.1–1 mm) and activity toward the double-stranded IIUI substrate was analyzed. Reaction products were separated by denaturing PAGE. B, recombinant hEndoV (2 nm) was incubated with nucleotides as indicated, activity was analyzed as in A and cleavage was quantified. C, partly purified endogenous hEndoV (QFr6T-REx) and overexpressed FLAG-hEndoV (QFr6EVwt) after Resource Q chromatography were diluted as indicated and assayed for activity toward single-stranded IIUI with or without ATP (1 mm). Dilution factors are indicated. −, no enzyme added. D, T-REx 293 cells were incubated with BODIPY FL ATP (20 μm, 3 h), treated with arsenite (0.5 mm, 30 min), and prepared for confocal microscopy. Cells were stained with G3BP (red) antibody for stress granules and DAPI (blue) for visualization of the nuclei. BODIPY FL ATP is green. The merge images are an overlay of all three stains. Confocal images are acquired by a ×40 oil objective (Leica SP8). Bar, 5 μm.
We also tested the activity of endogenously expressed hEndoV (QFr6T-REx from Fig. 2A) with ATP. The weak cleavage found in the T-REx 293 extract disappeared completely when ATP was added to the reaction mixture (Fig. 9C). Inhibition with ATP was also seen for partly purified FLAG-hEndoV (QFr6FLAGEVwt) (Fig. 9C). These results demonstrate that the cleavage at inosines seen in the T-REx 293 extract (Fig. 2) indeed is accomplished by hEndoV.
If inhibition of hEndoV by ATP also occurs in vivo, it means that under physiological ATP concentration, hEndoV is inactive given that ATP is evenly distributed throughout the cells. To get an impression of ATP storage in cells, T-REx 293 cells were cultured in media supplemented with fluorescently labeled ATP (BODIPY FL ATP). Cells were treated with arsenite to induce stress granules and prepared for microscopy. The images demonstrate that ATP is found throughout the cytoplasm with enrichment in specific vesicles not overlapping with the stress granules (Fig. 9D). Hence, relocalization of hEndoV to stress granules could represent a way for the cell to create a local environment with reduced ATP levels allowing hEndoV to be active.
Discussion
In this study we demonstrate that hEndoV expressed in human cells, either endogenously or ectopically, shares the same inosine-RNA cleavage properties as recombinant hEndoV purified from E. coli. Furthermore, we show that the hEndoV activity is modulated by the ATP level and that exposure of cells to certain toxic agents results in redistribution to cytoplasmic stress granules. In stress granules, hEndoV colocalizes with PABPC1, which also physically and functionally interacts with hEndoV.
Analysis of RNA at single nucleotide resolution has identified several million positions where adenosine is deaminated to inosine (7). These are spread throughout most of the transcriptome, hence most transcripts are potential substrates for hEndoV. To avoid uncontrolled degradation, hEndoV activity must be kept under strict regulation in the cell. Our data suggest that under normal intracellular ATP concentrations, hEndoV is inactive. Previous studies have demonstrated that in response to cellular insults such as methylnitronitrosoguanidine, arsenite, hydrogen peroxide, or menadione, intracellular ATP levels drop significantly, probably due to activation of poly(ADP-ribose) polymerase, which depletes energy stores in the cells (35–37). Dependent on dose and exposure time, ATP stores may be reduced with 70–80%, which gives an ATP concentration that alleviates hEndoV inhibition in vitro. An activation of hEndoV by such a mechanism makes sense, as these harmful agents, or at least some of them, introduce deamination damage to RNA, which indeed is a substrate for hEndoV.
Redirection of hEndoV to stress granules after cellular insult could also help create a local environment with low ATP concentration allowing hEndoV to be active. Furthermore, formation of molecular aggregates such as stress granules is an efficient way to gather proteins and enzymes with related biochemical properties. Stress granules comprise stalled translational initiation complexes, small ribosomal proteins, mRNAs, and a collection of other proteins. The purpose is to temporarily suppress synthesis of housekeeping proteins to enhance translation of stress-response specific mRNAs to aid cellular recovery (38). Also, stress granules serve as a site for storage and triage of selected mRNA and they are dynamically linked to processing bodies (P-bodies), which are granules involved in RNA decay (27). A function for hEndoV in stress granules could be to initiate degradation of inosine-containing mRNAs and funnel these transcripts to neighboring P-bodies. In this way reassembly of initiation complexes to polysomes to start translation of transcripts with damage is avoided. Accordingly, formation of stress granules after NaNO2 exposure that contain hEndoV, appears beneficial as NaNO2 induces deamination damage that is a substrate for hEndoV.
Yet another function of stress granules is to sequester certain proteins not to enhance their activity, but rather making them inaccessible. For example, signaling molecules (39) and apoptosis regulatory factors are redirected to stress granules with the proposed intention to inhibit apoptosis (40, 41). In agreement with this, agents that induce stress granules are generally not very genotoxic. On the other hand, agents like methyl methanesulfonate, methylnitronitrosoguanidine, and UV radiation that cause severe DNA damage do not stimulate stress granule formation, but rather apoptosis is induced with the aim to eradicate damaged cells (41). This could also be the situation for hEndoV; redistribution to stress granules removes hEndoV from the cytoplasm upon cellular stress to avoid cleavage of inosine-containing RNA when ATP levels drop.
Another protein, ADAR1, is also shown to be directed to stress granules after arsenite exposure (31). An attractive model for degradation of specific mRNAs could be a concerted action of ADAR1 and hEndoV, where ADAR1 deaminates adenosines, which subsequently are cleaved by hEndoV. Our immunocytochemistry experiments revealed colocalization in stress granules, substantiating possible concerted action between hEndoV and ADAR1. However, in the co-IP experiments, we were not able to identify ADAR1 as an interacting partner of hEndoV. Furthermore, we neither detected Tudor-SN as a partner for hEndoV. A previous report shows that Tudor-SN stimulates hEndoV activity (16) and also that Tudor-SN degrades inosine-containing RNA (42). Further experiments are needed to clarify possible cooperation between ADAR1, Tudor-SN, and hEndoV in selective degradation of inosine-containing RNA.
PABPC1 is an abundant protein in the cells involved in multiple aspects of mRNA processing and translation. We find an interaction between hEndoV and PABPC1 that is strictly RNA dependent. It could be that the RNA/PABPC1 (or RNA/hEndoV) complex rather than the pure protein is recognized. However, we cannot exclude the possibility that the RNA only functions as a carrier of both proteins. In an attempt to map any direct interaction, we performed pulldown experiments with recombinant proteins. Unfortunately, both hEndoV and PABPC1 were very sticky and we were not able to remove unspecific binding to the beads making the results unreliable (data not shown). The association between hEndoV and PABPC1 identified in co-IP did not depend on arsenite treatment, suggesting that the two proteins are close also under noninduced conditions. This is evident by the cytoplasmic staining of both proteins in untreated cells (Figs. 5 and 6). However, microscopic analysis did not reveal specific overlap until the cells were exposed to arsenite. PABPC1 is previously shown to aggregate in stress granules, but is not critical for stress granule assembly as shown for TIAR and G3BP (25, 43, 44). A role for PABPC1 could be to target hEndoV to stress granules, either for sequestering or to bring hEndoV to an environment allowing activity. As PABPC1 stimulates hEndoV activity, we favor the last hypothesis (21, 22). Further analyses of mutated or truncated versions of these proteins are needed to directly map domains involved in the interaction.
Experimental Procedures
Cell Lines, Culturing, and Transfection
Flp-In T-REx 293 cells were from Invitrogen (hereafter referred to as T-REx 293), whereas HeLa S3 (human cervix epithelial cells) and HEK 293T cells were purchased from American type Culture Collection (ATCC).
Stable Cell Lines with Wild Type (wt) FLAG-hEndoV
FLAG-hEndoV open reading frame (ORF: CCDS54172.1/NP_775898) was PCR amplified from pCMV-Tag2-hEndoVwt (synthesized by GenScript) and cloned into pcDNA 5/FRT/TO vector (Invitrogen) using KpnI and NotI restriction sites. The pcDNA 5/FRT/TO-FLAG-hEndoVwt construct was verified by DNA sequencing. For generation of stable expression clones T-Rex 293 cells were co-transfected with pcDNA 5/FRT/TO-FLAG-hEndoVwt and pOG44 (Invitrogen) using X-tremeGENE 9 DNA transfection reagent (06365787001, Roche Applied Science) and cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Sigma), 1× GlutaMAX (Gibco), 1× penicillin-streptomycin (Lonza), and 7.5 μg/ml of blasticidin (R210-01, Life Technologies) for 48 h. Cells were then fed with selection medium (DMEM with supplements as above and 200 μg/ml of hygromycin B (400052-20, Life Technologies) for 3–4 days (media refreshed after 24 h), until colonies were formed. After hygromycin B selection, the colonies were pooled and screened for tetracycline-regulated expression of FLAG-hEndoVwt.
Stable Cell Lines with EGFP-hEndoVwt
The hEndoV ORF was released from pGFP-hEndoVwt (19) by ApaI and EcoRI restriction digestion and cloned into pENTR2B vector (Gateway, Life Technologies) in the EcoRI restriction site. The hEndoV ORF was then transferred to pDest-FRT/TO-EGFP (referred to as GFP hereafter) vector (45) by Gateway LR clonase reaction. The pDestEGFP-Flp-In-FRT/TO-hEndoVwt construct was confirmed by DNA sequencing and used for transfection of T-REx 293 cells to generate stable cell lines.
Stable Cell Lines with FLAG/GFP-hEndoV(D52A)
pcDNA 5/FRT/TO-FLAG-hEndoV(D52A) or pDest-FRT/TO-EGFP-hEndoV(D52A) constructs were made by QuikChange II Site-directed Mutagenesis Kit (Aglient) according to the manufacturer's instructions using pcDNA 5/FRT/TO-FLAG-hEndoVwt or pDest-FRT/TO-EGFP-hEndoVwt as template and primers CAGAGGGTCGGGGGCGTTGCTGTGTCCTTCGTGAAAGGGG and CCCCTTTCACGAAGGACACAGCAACGCCCCCGACCCTCTG. Stable cell lines were generated as described for FLAG-hEndoVwt.
Establishment of hENDOV Knock-out Cell Line
A hENDOV knock-out cell line was purchased from Horizon Genomics, established using the CRISPR/Cas9 system in a near-haploid human cell line (HAP1) derived from a patient with chronic myelogenous leukemia. Two hENDOV knock-out clones were obtained bearing a frameshift mutation caused by a 2-bp deletion in exon 2. The mutations were verified through PCR and Sanger sequencing by the manufacturer.
Transient Transfection
HEK 293T cells were transiently transfected with EGFP-hEndoV (19) and EGFP N-vectors (Clontech) using X-tremeGene 9 DNA tranfection agent according to the manufacturer's protocol.
Human cells (except HAP1 cell lines) were cultured in DMEM supplemented as described above and kept at 37 °C, and 5% CO2. In addition, 7.5 μg/ml of blasticidin and 200 μg/ml of hygromycin B or 100 μg/ml of Zeocin (R250–05, Life Technologies) were added in culturing media for all T-REx 293 cell lines according to the Flp-InTM T-RExTM manual. To induce FLAG- or GFP-hEndoV expression, to the cultures we also added 1 μg/ml of tetracycline (87128, Sigma) or 1 μg/ml of doxycycline (D6750, Sigma), for 24 to 48 h. The HAP1 cell lines were cultured in IMDM (Gibco) with 10% FBS and 1× penicillin/streptomycin.
For experiments on localization of ATP stores, cells were incubated with 20 μm BODIPY FL ATP (12410, Life Technologies) for 3 h prior to fixation. BODIPY FL ATP excitation was 488 nm and an emission wavelength of 500–550 nm. For stress granule formation, cells were either exposed to 0.5 mm sodium arsenite (Sigma; hereafter referred to as arsenite) for 30 min or 200 mm sodium nitrite (NaNO2, Merck) in 0.1 m sodium acetate, pH 5.5, for 1 h. For polyinosinic-polycytidylic acid (poly(I:C); P9582, Sigma) transfection, cells were pre-treated with Lipofectamine® 2000 Transfection Reagent (11668-019, Life Technologies). Cells were fixed 7 h after adding poly(I:C) and Lipofectamine alone was used in mock transfections. To induce stress granules, 500 ng of poly(I:C) was used. Vectors expressing GFP-hEndoV (19) and GFP alone were transiently transfected in HeLa cells using X-tremeGENE.
RNA Analysis
Total RNA was isolated from T-REx 293, HeLa S3, and HAP1 cells, with and without exposure to arsenite or nitrite as above, using TRIzol reagent (15596, Life Technologies) according to the manufacturer's instructions. The quality of the RNA was checked by formaldehyde-agarose gel electrophoresis and Bioanalyzer 2100 (Agilent). All samples had RNA Integrity numbers between 9 and 10. cDNA was generated from 1 μg of total RNA using the Quantitect Reverse Transcription Kit (205313, Qiagen). Human ENDOV mRNA levels were determined by qRT-PCR using primers amplifying exons 2 to 3 (200 nm), Power SYBR Green PCR master mix (A25778, Life Technologies), and the Step One Plus Real-time PCR system (Applied Biosystem) according to the kit and system instructions. All samples were run in duplicates/triplicates and melting point analyses were performed to confirm the specificity of the PCR. The glyceraldehyde-3-phosphate dehydrogenase gene (hGAPDH) was used as the reference gene for normalization, and untreated HeLa S3 RNA was used to generate standard curves. The qRT-PCRs were repeated 6 times using cDNA from three independent experiments. Primers used are listed in Table 2.
TABLE 2.
qRT-PCR primers used in the study
| 5′→3′ sequence: | Description: |
|---|---|
| TTGACGTGTCCTTCGTGAAA | Forward exon 2 hENDOV |
| ATGCGGCTCTCCTCATACAC | Reverse exon 3 hENDOV |
| CCACATCGCTCAGACACCAT | Forward exon 2 hGAPDH |
| GCGCCCAATACGACCAAAT | Reverse exon 3 hGAPDH |
Immunofluorescence and Antibodies
Cells were grown on 12-mm fibronectin-coated (20 μg/ml, F1141; Sigma) coverslips until 70–80% confluence and exposed to the appropriate stress as indicated. Cells were washed in PBS, before fixation in 4% paraformaldehyde in PBS for 15 min. After washing in PBS, cells were quenched in PBS, 0.1 m glycine for 10 min, permeabilized in PBS, 0.1% Triton for 10 min, and incubated in blocking buffer (PBS, 0.5% bovine serum albumin (BSA)) for 30 min. All subsequent labeling steps were performed in blocking buffer. Cells were incubated for 1 h with primary antibodies against hEndoV (ab69400; Abcam), G3BP (611126; BD Biosciences), PABPC1 (ab21060; Abcam), Dcp1 (ab4781; Abcam), Tudor-SN (sc-166676; Santa Cruz), or ADAR1 (sc73408; Santa Cruz). In addition, two different hEndoV antibodies were raised using recombinant hEndoV (purified as described in Vik et al. (17)) as immunogenes: monoclonal (supernatant from hybridoma cells) and polyclonal hEndoV (affinity purified rabbit serum). Both antibodies were detected down to 10 ng of recombinant hEndoV and gave no background signals when up to 250 μg of T-REx cell extracts were analyzed by SDS-PAGE (data not shown). Specific signals for endogenous hEndoV was not seen using 25–250 μg of extract, indicating a low endogenous level of hEndoV (data not shown). However, ectopic expression of hEndoV gave a signal in Western blot analysis (data not shown). Cells were washed in PBS, and further incubated for 1 h with Alexa 488-, 594-, or 647-conjugated anti-mouse or anti-rabbit IgG antibodies (Life Technologies) for detection. After washing with PBS, nuclei were stained with DAPI (D1306; Life Technologies) or DRAQ5 (DR50050; Biostatus) and coverslips were mounted with Mowiol (475904; Sigma). Confocal images were acquired with Carl Zeiss LSM 510 CLSM laser scanning microscope with a ×63/1.1 NA oil immersion objective (Jena, Germany), or Leica TCS SP8 equipped with a ×40/1.3 NA oil immersion objective using ×6 zoom. Representative images are shown.
Quantification of Colocalization
To determine the degree of colocalization, Manders' coefficients (tM) were calculated using the Costes' thresholding method and Coloc2 plugin in ImageJ/Fiji on background-corrected images. In our analysis tM1 expresses the degree of colocalization of the green channel with the red/cyan channel. tM2 expresses the degree of colocalization of the red/cyan channel with the green channel. Each tM value represents the average tM calculated for 9–12 cells from separate experiments and is accompanied by the corresponding S.D.
Quantification of Stress Granules
To quantify the number and size of the stress granules in each cell, 20 cells from each cell line were randomly selected. The occurrence of stress granules was determined on the basis of G3BP staining. Image analyses and quantification of stress granules (size >0.1 μm) were performed using ImageJ/Fiji software. All images are taken in one Z-plane in a similar position within the sample for comparison.
Preparation of Cytoplasmic (S100) Extracts
S100 extracts were prepared according to Mayeda and Krainer (46). Cells were cultured to ∼80% confluence, harvested, and washed with ice-cold PBS twice. After centrifugation (1,800 × g, 5 min), cells were resuspended in 5 cell pellet volumes of buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT and protease inhibitor mixture (PIC, P8340, Sigma)). Cells were incubated for 10 min on ice before centrifugation as above. The supernatant was transferred to new tubes and 0.11 volumes of buffer B (300 mm HEPES, pH 7.9, 1.4 m KCl, and 30 mm MgCl2) was added. The lysate was centrifuged (21,500 × g, 1 h, 4 °C) and the cleared lysate dialyzed against buffer C (20 mm HEPES, pH 7.9, 5% glycerol, 100 mm KCl, 0.2 mm EDTA, 0.5 mm PMSF, and 0.5 mm DTT; MWCO 6,000–8,000) overnight at 4 °C. S100 extracts were used for purification of endogenous hEndoV.
Preparation of Cytoplasmic Extracts for Co-IP
Cells were cultured and centrifuged as above, and resuspended in 1 cell pellet volume of lysis buffer (100 mm KCl, 5 mm MgCl2, 10 mm HEPES, pH 7.0, 0.5% Nonidet P-40, 1 mm DTT, 100 units/ml of RNaseOUT (10777-019, Thermo Fisher Scientific), 400 μm vanadyl ribonucleoside complex (S1402S, New England BioLabs), and PIC). The cell suspension was incubated on ice (5 min) and frozen at −80 °C for a minimum of 24 h. After thawing, the cell suspension was cleared by centrifugation (15,000 × g, 15 min) and the lysate collected.
Co-immunoprecipitation
10 μl of Anti-FLAG® M2 magnetic beads (M8823, Sigma) were washed twice with NT2 buffer (1 ml, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm MgCl2, 0.5% Nonidet P-40) and beads were collected with a magnetic rack. Beads were resuspended in NT2 and 1.5 mg of lysate, EDTA (20 mm), DTT (1 mm) was added and the volume adjusted to 1 ml with NT2. Reactions were added 200 units of RNaseOUT and 400 μm vanadyl ribonucleoside complex, except those treated with RNase. The lysate beads suspensions were incubated overnight at 4 °C with gentle agitation before unbound material was removed. Beads were resuspended in 1 ml of NT2 with 300 mm NaCl and 100 ng of RNaseA (Sigma) if RNase treated. RNaseA-treated samples were incubated at 37 °C for 15 min, with gentle agitation. All samples were washed (4× NT2 with 300 mm NaCl) and either 150 μl of 1× PBS (for mass spectrometry (MS) analysis) or 20 μl of 2× Laemmli buffer (Bio-Rad) for Western blot analysis were added. After denaturation at 95 °C for 5 min, proteins were separated by PAGE (10%, Mini-PROTEAN, Bio-Rad), transferred to PVDF membranes (Bio-Rad), and the membranes blocked with PBS with 0.05% Tween 20 (PBS-T) and 5% skimmed milk (1 h, room temperature). After incubation with primary antibody (PABPC1, ab21060, Abcam, and rabbit hEndoV, polyclonal in house) in PBS-T with 5% skimmed milk (1 h, room temperature), the membranes were washed (3× 10 min) with PBS-T prior to incubation with secondary antibody (rabbit IgG, horseradish peroxidase (HRP)-linked whole antibody, GE Healthcare) for 1 h and washing as above. Substrate (SuperSignalTM West Femto, Thermo Fisher Scientific) was added and the signals were analyzed with Image LabTM version 5.1 software from Bio-Rad.
Immunoprecipitation of Endogenous hEndoV
Fractions 9 and 12–15 (0.5-ml fractions) from Resource S (ResS) chromatography (see below for purification procedure) were pooled and diluted to 3.6 ml with 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, and 0.5% Nonidet P-40, to reduce the total salt concentration. The suspension was divided in two and 2 μg of hEndoV antibody added (monoclonal in house or polyclonal ab69400, Abcam). The protein-antibody suspensions were incubated (4 °C, overnight) with gentle agitation. 20 μl of Protein A/G PLUS-agarose (sc-2003, Santa Cruz Biotechnology) was added and incubated for another hour. The immunoprecipitates were harvested by centrifugation at 1000 × g for 3 min and washed 3 times with 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, and 0.5% Nonidet P-40. The samples were separated by PAGE (10%) and Western blot analysis was performed as described above, with mouse IgG, HRP-linked whole antibody (GE Healthcare) as secondary antibody.
Partial Purification of Endogenous hEndoV
As the endogenous level of hEndoV is below the detection limit in activity and Western blot analysis, an extract with overexpression of FLAG-hEndoV was used as a guide during purification for selection of fractions with endogenous hEndoV. We assume that these two proteins have similar chromatographic properties. S100 cytoplasmic cell-free extracts were made from ∼2×109 T-REx 293 cells with and without overexpression of FLAG-hEndoV. The extracts were run on a Superdex 75 column in buffer C (see “Preparation of Cytoplasmic (S100) Extracts”). Activity and Western blot analysis identified the FLAG-hEndoV peak in fractions 5–7 (no activity was found for T-REx extract). Fractions 6 and 7 were kept separately and diluted to reduce the KCl concentration to 50 mm and loaded on a Resource Q (ResQ) column. Proteins were eluted with a salt gradient (buffer C + 1 m KCl). Analyses of the ResQ fractions showed that for fraction 6 from Superdex 75, hEndoV bound weakly to ResQ, whereas for Superdex fraction 7, hEndoV appeared in the flow-through. This difference is likely due to different ionic strengths in the two fractions. The ResQ flow-through (from fraction 7) was run on a ResS column and eluted with buffer C with 1 m KCl.
Purification of Recombinant Proteins
Human EndoV was purified as previously reported (17) after expression in a modified version of E. coli ER 2566 (ER2566 Δnfi ΔrnhB) where the endogenous nfi and rnhB genes had been inactivated by transformation with the kanamycin-deletion cassettes.3 The PABPC1 reading frame (NP_002559.2; codon optimized for expression in E. coli) was cloned into the NcoI and XhoI sites of pET28b in-frame with the C-terminal His tag (GenScript). The PABPC1 open reading frame is 1911 nucleotides and translates into a protein of 636 amino acids with a calculated molecular mass of 70.7 kDa. pET28b-PABPC1 was transformed in the ER2566 Δnfi ΔrnhB strain and cells were grown in Lysogeny broth (LB) medium with kanamycin (50 mg/liter) at 37 °C until A600 reached 0.4–0.5. The temperature was lowered to 16 °C, protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and cells were incubated at 16 °C overnight. Cells were harvested by centrifugation, and the cell pellet dissolved in sonication buffer (buffer D: 50 mm Tris, pH 8.0, 300 mm NaCl, 10 mm imidazole, and 10 mm β-mercaptoethanol (β-ME)). Cells were sonicated (3 × 30 s) followed by centrifugation (13,000 rpm, 30 min). The extract was applied to a nickel-nitrilotriacetic acid-agarose column and after washing with buffer D with 50 mm imidazole, His-PABPC1 fusion protein was eluted using 300 mm imidazole in buffer D. Two PABPC1 deletion constructs were made (GenScript): one expressing the N-terminal RNA binding motifs (RRM1–4 amino acids 1–370, Mr 41.6) and one for the C-terminal MLLE domain (amino acids 498–636, Mr 14.9). The RRM1–4 construct has a C-terminal His tag, whereas the MLLE construct has an N-terminal GST tag giving a fusion protein with a predicted mass of 41 kDa. Both proteins were expressed as described for full-length PABPC1. His-RRM1–4 was purified with nickel-nitrilotriacetic acid-agarose as above following Superdex 75 chromatography in buffer D (omitting imidazole). GST-MLLE was purified with glutathione-Sepharose 4B according to the manufacturer's recommendations (GE Healthcare). For all proteins, peak fractions were pooled, glycerol was added to 20%, and stored at −80 °C.
Activity and Electrophoretic Mobility Shift Assays
Oligonucleotide substrates were from Eurofins (DNA) and Midland Certified (RNA) (Table 3). The oligonucleotides were end-labeled using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (3000 Ci/mmol, Amersham Biosciences). Double-stranded (ds) substrates were generated by annealing the labeled single-stranded (ss) oligonucleotide to a complementary strand. 10 μl of reaction mixture contained 1 nm substrate, the amount of enzymes as indicated, and the reaction buffer (10 mm Tris-HCl, pH 7.5, 0.5 mm MnCl2, 50 mm KCl, 1 mm DTT, and 5% glycerol). Reactions proceeded at 37 °C for 20 min and were stopped by adding 10 μl of formamide loading dye (80% formamide, 10 mm EDTA, 0.1% xylene cyanol and bromphenol blue). The oligonucleotides were denaturated at 52 °C for 2 min, and the reaction products were separated on 20% polyacrylamide/urea gels at 200 V for 1 h in 1× taurine buffer. The radiolabeled fragments were visualized by phosphorimaging (Typhoon 9410 Variable Mode Imager) and ImageQuant TL was used for quantification. All assays were performed 2–3 times and representative experiments are shown.
TABLE 3.
Oligonucleotide substrates used in the study
| 5′→3′ sequence: | DNA/RNA | Description |
|---|---|---|
| ACUGGACAAAUACUCCGAGG | RNA | Control (Ctr) |
| CCUCGGAGUAUUUGUCCAGU | RNA | Complementary oligo for Ctr |
| ACUGGACA[rI][rI]U[rI]CUCCGAGG | RNA | IIUI |
| CCUCGGAGU[rI]UUUGUCCAGU | RNA | Complementary oligo for IIUI |
| CCGTAGAGCTAC[dI]GATCGGTCACCG | DNA | I-DNA |
| CGGTGACCGATCTGTAGCTCTACGG | DNA | Complementary oligo for I-DNA |
In electrophoretic mobility shift assays (EMSA), enzymes (0.25–1 μm) and substrates (1 nm) were mixed in band shift buffer (40 mm Tris-HCl, pH 8.5, 10 mm CaCl2, 10 mm DTT, 6% glycerol). tRNA (1 ng/μl) was included as a competitor. Samples were incubated on ice for 15 min, DNA loading buffer (Thermo Scientific) was added prior to separation on 10% native polyacrylamide gels in 1× taurine with 5 mm CaCl2 at 100 V for 40 min in room temperature. The enzyme-substrate complexes were visualized as above.
Author Contributions
I. A., B. D., and M. B. conceived the study, M. S. N., E. S. V., N. B., C. F., and I. A. performed the experiments, M. S. N. and I. A. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Rune J. Forstrøm and Jens Erik Eriksson for technical assistance. The pDestEGFP-Flp-In-FRT/TO vector was a gift from Endalkachew A. Alemu.
This work was supported by Norwegian Cancer Society Grant 5739724 (to E. S. V.), The Research Council of Norway Grant 250474 (to N. B.), South-Eastern Norway Regional Health Authorities Grants 2014034 and 2015095 (to B. D.), grants from the Erasmus+ program/EU (to N. B.) and the Molecular Life Science program at UiO (to M. S. N.). The authors declare that they have no conflicts of interest with the contents of this article.
M. S. Nawaz, E. S. Vik, A. Galashevskaya, P. H. Backe, I. Rosnes, C. Fladeby, B. Dalhus, H. E. Krokan, G. Slupphaug, P. Schär, M. Bjørås, and I. Alseth, unpublished data.
- ADAR
- adenosine deaminases acting on RNA
- ncRNA
- non-coding RNA
- EndoV
- endonuclease V
- PABPC1
- polyadenylate-binding protein C1
- qRT
- quantitative real-time
- co-IP
- co-immunoprecipitation.
References
- 1. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 [DOI] [PubMed] [Google Scholar]
- 2. Prestwich E. G., Mangerich A., Pang B., McFaline J. L., Lonkar P., Sullivan M. R., Trudel L. J., Taghizedeh K., and Dedon P. C. (2013) Increased levels of inosine in a mouse model of inflammation. Chem. Res. Toxicol. 26, 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pang B., McFaline J. L., Burgis N. E., Dong M., Taghizadeh K., Sullivan M. R., Elmquist C. E., Cunningham R. P., and Dedon P. C. (2012) Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proc. Natl. Acad. Sci. U.S.A. 109, 2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yasui M., Suenaga E., Koyama N., Masutani C., Hanaoka F., Gruz P.,Shibutani S., Nohmi T., Hayashi M., and Honma M. (2008) Miscoding properties of 2′-deoxyinosine, a nitric oxide-derived DNA adduct, during translesion synthesis catalyzed by human DNA polymerases. J. Mol. Biol. 377, 1015–1023 [DOI] [PubMed] [Google Scholar]
- 5. Alseth I., Dalhus B., and Bjørås M. (2014) Inosine in DNA and RNA. Curr. Opin. Genet. Dev. 26, 116–123 [DOI] [PubMed] [Google Scholar]
- 6. Mannion N., Arieti F., Gallo A., Keegan L. P., and O'Connell M. A. (2015) New insights into the biological role of mammalian ADARs: the RNA editing proteins. Biomolecules 5, 2338–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F. J., Rechavi G., Li J. B., Eisenberg E., and Levanon E. Y. (2014) A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 24, 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen L., Li Y., Lin C. H., Chan T. H., Chow R. K., Song Y., Liu M., Yuan Y. F., Fu L., Kong K. L., Qi L., Li Y., Zhang N., Tong A. H., Kwong D. L., et al. (2013) Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 19, 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawahara Y., Kwak S., Sun H., Ito K., Hashida H., Aizawa H., Jeong S. Y., and Kanazawa I. (2003) Human spinal motoneurons express low relative abundance of GluR2 mRNA: an implication for excitotoxicity in ALS. J. Neurochem. 85, 680–689 [DOI] [PubMed] [Google Scholar]
- 10. Rice G. I., Kasher P. R., Forte G. M., Mannion N. M., Greenwood S. M., Szynkiewicz M., Dickerson J. E., Bhaskar S. S., Zampini M., Briggs T. A., Jenkinson E. M., Bacino C. A., Battini R., Bertini E., et al. (2012) Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toth A. M., Li Z., Cattaneo R., and Samuel C. E. (2009) RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 284, 29350–29356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolf J., Gerber A. P., and Keller W. (2002) tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 21, 3841–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bass B. L. (2002) RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo G., Ding Y., and Weiss B. (1997) nfi, the gene for endonuclease V in Escherichia coli K-12. J. Bacteriol. 179, 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao M., Hatahet Z., Melamede R. J., and Kow Y. W. (1994) Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J. Biol. Chem. 269, 16260–16268 [PubMed] [Google Scholar]
- 16. Morita Y., Shibutani T., Nakanishi N., Nishikura K., Iwai S., and Kuraoka I. (2013) Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nat. Commun. 4, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vik E. S., Nawaz M. S., Strøm Andersen P., Fladeby C., Bjørås M., Dalhus B., and Alseth I. (2013) Endonuclease V cleaves at inosines in RNA. Nat. Commun. 4, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mi R., Alford-Zappala M., Kow Y. W., Cunningham R. P., and Cao W. (2012) Human endonuclease V as a repair enzyme for DNA deamination. Mutat. Res. 735, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fladeby C., Vik E. S., Laerdahl J. K., Gran Neurauter C., Heggelund J. E.,Thorgaard E., Strøm-Andersen P., Bjørås M., Dalhus B., and Alseth I. (2012) The human homolog of Escherichia coli endonuclease V is a nucleolar protein with affinity for branched DNA structures. PLoS One 7, e47466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scadden A. D., and Smith C. W. (2001) Specific cleavage of hyper-edited dsRNAs. EMBO J. 20, 4243–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mangus D. A., Evans M. C., and Jacobson A. (2003) Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 4, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xie J., Kozlov G., and Gehring K. (2014) The “tale” of poly(A) binding protein: the MLLE domain and PAM2-containing proteins. Biochim. Biophys. Acta 1839, 1062–1068 [DOI] [PubMed] [Google Scholar]
- 23. Görlach M., Burd C. G., and Dreyfuss G. (1994) The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp. Cell Res. 211, 400–407 [DOI] [PubMed] [Google Scholar]
- 24. Kedersha N. L., Gupta M., Li W., Miller I., and Anderson P. (1999) RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 147, 1431–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kedersha N., Ivanov P., and Anderson P. (2013) Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 38, 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tourrière H., Gallouzi I. E., Chebli K., Capony J. P., Mouaikel J., van der Geer P., and Tazi J. (2001) RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell Biol. 21, 7747–7760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke-Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., and Anderson P. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allemand E., Guil S., Myers M., Moscat J., Cáceres J. F., and Krainer A. R. (2005) Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc. Natl. Acad. Sci. U.S.A. 102, 3605–3610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buchan J. R., Yoon J. H., and Parker R. (2011) Stress-specific composition, assembly and kinetics of stress granules in Saccharomyces cerevisiae. J. Cell Sci. 124, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Field A. K., Tytell A. A., Piperno E., Lampson G. P., Nemes M. M., and Hilleman M. R. (1972) Poly I:C, an inducer of interferon and interference against virus infections. Medicine 51, 169–174 [DOI] [PubMed] [Google Scholar]
- 31. Weissbach R., and Scadden A. D. (2012) Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA. 18, 462–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. George C. X., and Samuel C. E. (1999) Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene 229, 203–213 [DOI] [PubMed] [Google Scholar]
- 33. Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 34. Gresser M. J. (1981) ADP-arsenate: formation by submitochondrial particles under phosphorylating conditions. J. Biol. Chem. 256, 5981–5983 [PubMed] [Google Scholar]
- 35. Osenbroch P. Ø., Auk-Emblem P., Halsne R., Strand J., Forstrøm R. J., van der Pluijm I., and Eide L. (2009) Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J. 276, 2811–2821 [DOI] [PubMed] [Google Scholar]
- 36. Ha H. C., and Snyder S. H. (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. U.S.A. 96, 13978–13982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yih L. H., Huang H. M., Jan K. Y., and Lee T. C. (1991) Sodium arsenite induces ATP depletion and mitochondrial damage in HeLa cells. Cell Biol. Int. Rep. 15, 253–264 [DOI] [PubMed] [Google Scholar]
- 38. Decker C. J., and Parker R. (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol. 4, a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Takahara T., and Maeda T. (2012) Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 47, 242–252 [DOI] [PubMed] [Google Scholar]
- 40. Arimoto-Matsuzaki K., Saito H., and Takekawa M. (2016) TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 7, 10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arimoto K., Fukuda H., Imajoh-Ohmi S., Saito H., and Takekawa M. (2008) Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332 [DOI] [PubMed] [Google Scholar]
- 42. Scadden A. D. (2005) The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat. Struct. Mol. Biol. 12, 489–496 [DOI] [PubMed] [Google Scholar]
- 43. Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G., Dember L. M., and Anderson P. (2004) Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tourrière H., Chebli K., Zekri L., Courselaud B., Blanchard J. M., Bertrand E., and Tazi J. (2003) The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160, 823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Alemu E. A., Lamark T., Torgersen K. M., Birgisdottir A. B., Larsen K. B., Jain A., Olsvik H., Øvervatn A., Kirkin V., and Johansen T. (2012) ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 287, 39275–39290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayeda A., and Krainer A. R. (1999) Preparation of HeLa cell nuclear and cytosolic S100 extracts for in vitro splicing. Methods Mol. Biol. 118, 309–314 [DOI] [PubMed] [Google Scholar]