

Abstract
The cardiac troponin I (cTnI) R145W mutation is associated with restrictive cardiomyopathy (RCM). Recent evidence suggests that this mutation induces perturbed myofilament length-dependent activation (LDA) under conditions of maximal protein kinase A (PKA) stimulation. Some cardiac disease-causing mutations, however, have been associated with a blunted response to PKA-mediated phosphorylation; whether this includes LDA is unknown. Endogenous troponin was exchanged in isolated skinned human myocardium for recombinant troponin containing either cTnI R145W, PKA/PKC phosphomimetic charge mutations (S23D/S24D and T143E), or various combinations thereof. Myofilament Ca2+ sensitivity of force, tension cost, LDA, and single myofibril activation/relaxation parameters were measured. Our results show that both R145W and T143E uncouple the impact of S23D/S24D phosphomimetic on myofilament function, including LDA. Molecular dynamics simulations revealed a marked reduction in interactions between helix C of cTnC (residues 56, 59, and 63), and cTnI (residue 145) in the presence of either cTnI RCM mutation or cTnI PKC phosphomimetic. These results suggest that the RCM-associated cTnI R145W mutation induces a permanent structural state that is similar to, but more extensive than, that induced by PKC-mediated phosphorylation of cTnI Thr-143. We suggest that this structural conformational change induces an increase in myofilament Ca2+ sensitivity and, moreover, uncoupling from the impact of phosphorylation of cTnI mediated by PKA at the Ser-23/Ser-24 target sites. The R145W RCM mutation by itself, however, does not impact LDA. These perturbed biophysical and biochemical myofilament properties are likely to significantly contribute to the diastolic cardiac pump dysfunction that is seen in patients suffering from a restrictive cardiomyopathy that is associated with the cTnI R145W mutation.
Keywords: contractile protein, heart, molecular dynamics, phosphorylation, troponin, contractility, length-dependent activation, myocardium, myofilament
Introduction
Cardiomyopathies are diseases of the heart muscle that are either acquired or inherited (1). Several forms of the disease are formally recognized, including dilated cardiomyopathy, hypertrophic cardiomyopathy (HCM),2 and restrictive cardiomyopathy (RCM) cardiomyopathy. Numerous mutations in sarcomeric proteins have been identified that associate with, and may indeed be causal to, inherited cardiomyopathy, notably the giant elastic protein titin (TTN) in dilated cardiomyopathy (2) and myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) in HCM (3). RCM is a rare form of cardiomyopathy characterized by severe diastolic dysfunction secondary to significant increased ventricular diastolic stiffness but without the prominent increase in wall thickness that is commonly seen in HCM (4). RCM is associated with a poor prognosis (1), and the inherited form is believed to be caused by mutations in various sarcomeric proteins, including cardiac troponin I (TNNI3) (5).
Troponin-I (TnI) is the inhibitory component of troponin, the trimeric thin filament-associated protein complex that mediates calcium-initiated contraction in striated muscle (see Fig. 1). The molecular mechanism underlying this action is believed to be detachment of the inhibitory peptide (IP) domain of TnI from its binding site on actin upon binding of activating Ca2+ ions to troponin C (TnC) (6). The latter event induces an increased affinity between the N-terminal regulatory domain of TnC and the switch peptide (SP) domain of TnI. The cardiac TnI (cTnI) SP domain is located in a region just N-terminal from the IP domain (6). Threonine 143 (Thr-143), a residue that lies in the center of the IP domain in cTnI, is a substrate for members of the protein kinase C family. This residue has been shown to play a critical role in sarcomere length modulation of cardiac myofilament function, the cellular basis of the Frank-Starling law of the heart (7). Phosphorylation at this target is reported to be altered in human heart failure (8). Mutations of arginine 145 (Arg-145), just two residues N-terminal from this PKC target (see Fig. 1), have been identified as causal to both HCM (R145G and R145Q) and RCM (R145W) (9–12). These HCM and RCM mutations have been characterized both for their in vitro and in vivo impacts, albeit more extensively for the R145G HCM mutation; the general consensus is that the RCM R145W mutation induces the most severe phenotype, at least in vitro, that manifests as an increase in myofilament Ca2+ sensitivity that is greater than that seen for either the R145G or R145Q HCM mutation (9–12). The drastic increase in myofilament Ca2+ sensitivity associated with the R145W mutation is speculated to cause an inability of actomyosin cross-bridges to fully disengage throughout diastole. That phenomenon may explain the increased ventricular diastolic stiffness and consequent severe diastolic dysfunction that is commonly seen in patients that carry this specific RCM-associated mutation despite the absence of ventricular hypertrophy or altered systolic function (1).
FIGURE 1.
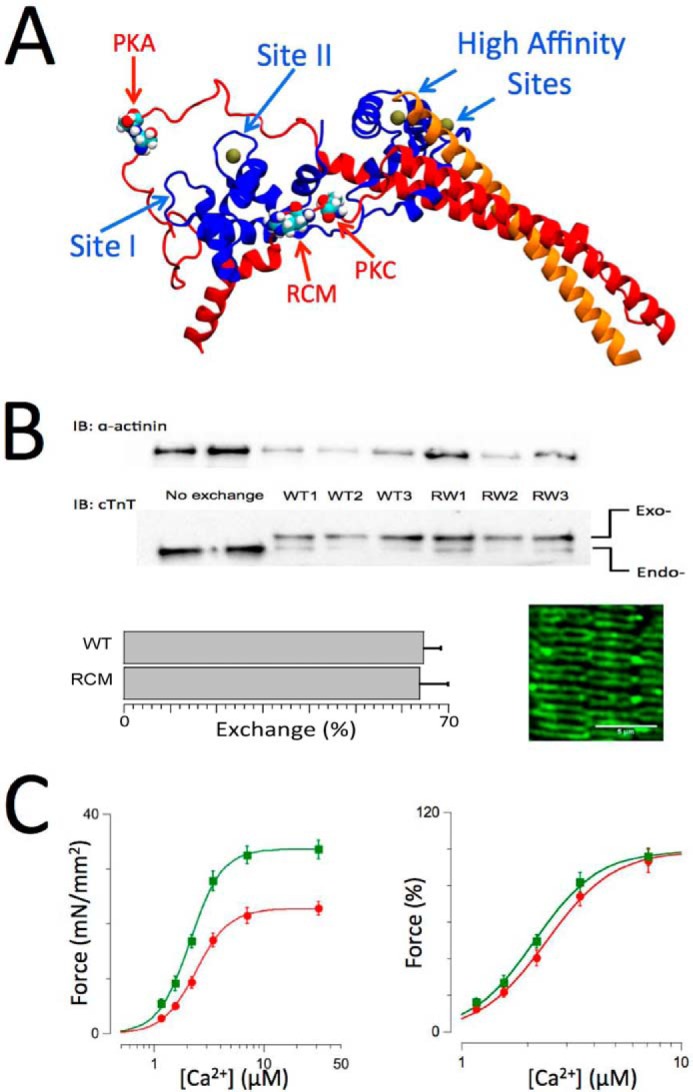
Cardiac troponin structure, exchange, and functional measurements. A, structure of troponin based on the crystal structure by Takeda et al. (46). cTnI is shown in red, cTnC is shown in blue, and cTnT is shown in orange. Ca2+ ions (solid balls) are shown bound to the high affinity sites and the single cardiac low affinity at Site II. The locations in cTnI of the PKA (Ser-23/Ser-24) and protein kinase C (Thr-143) phosphorylation motifs and the RCM mutation (R145W) are as indicated. B, endogenous troponin (Endo-) was exchanged for recombinant human troponin (Exo-) containing either WT, RCM mutation, or PKA/PKC phosphomimetic cardiac troponin I. Inclusion of an N-terminal myc tag in cTnT allowed for separation of the endogenous and exogenous cTnT on SDS-PAGE/Western blotting using a cTnT-specific antibody (bottom gels), whereas anti-α-actinin Western blotting was used as a loading control (upper gels). Exchange for recombinant troponin (in this case WT or RCM) resulted in ∼68% exchange (bar plots; mean ± S.E.; WT, n = 8; RCM, n = 12). The image shows an example of a confocal microscopy scan of a WT recombinant troponin-exchanged muscle probed with a myc-specific primary antibody followed by a mouse Alexa Fluor-488 secondary antibody; the double banded pattern indicates incorporation of the recombinant troponin into the thin filaments on either side of the Z-disk. C, force-[Ca2+] relationships (n = 12) recorded on WT recombinant troponin-exchanged isolated skinned multicellular human cardiac muscles at short (SL = 2.0 μm; red) and long (SL = 2.3 μm; green) sarcomere lengths. Force was either normalized to muscle cross-sectional area and expressed as millinewtons (mN)/mm2 (left panel) or normalized to maximum force in each individual muscle (right panel). An increase in SL induced both an increase in maximum force and an increase in myofilament Ca2+ sensitivity, indicative of myofilament LDA. Data are presented as mean ± S.E. Error bars reflect S.E. IB, immunoblotting.
A recent study reported that some sarcomeric contractile protein mutations perturb myofilament length-dependent activation (LDA) (13). In that comprehensive study, cardiac muscle samples from a large cohort of patients harboring a wide range of confirmed HCM/RCM-associated sarcomeric protein mutations were compared with mutation-negative or donor control samples (13). The HCM/RCM samples displayed reduced contractile protein phosphorylation profiles, a finding that is consistent with previous reports on human cardiomyopathies (14). In many cases, but notably not all, depressed LDA was reversed by treatment of the skinned myocytes with protein kinase A (PKA). Cardiac LDA has been shown to be modulated by contractile protein phosphorylation by most (7, 15–18) although not all (19, 20) reports. Moreover, selective introduction of a PKA phosphomimetic isoform of cTnI into the cardiac sarcomere increases LDA (15, 17, 21–24), a finding we confirm in the present study on isolated multicellular skinned human myocardium. Sequeira et al. (13) concluded that perturbed LDA in human HCM/RCM is due to (i) reduced PKA activity and the associated contractile protein hypophosphorylation or (ii) in cases where PKA treatment was unable to restore LDA toward control levels by other as of yet undetermined molecular mechanisms. The latter group prominently included the cTnI R145W RCM-associated mutation studied here.
Some cardiac disease-causing mutations have been shown to cause a blunting of the myofilament Ca2+ desensitization that is normally associated with PKA-mediated phosphorylation (22, 25, 26). Accordingly, it is plausible that the perturbed length sensitivity reported for the R145W mutation (13) resulted from such a “PKA uncoupling” phenomenon as opposed to a primary and direct impact of the mutation on sarcomeric LDA. The aim of the present study was to test this hypothesis. Endogenous troponin was exchanged in isolated skinned human myocardium for recombinant troponin containing the cTnI R145W RCM mutation, PKA/PKC phosphomimetic charge mutations (S23D/S24D and T143E), or various combinations of these residue substitutions. Phosphomimetics were chosen in the present study because kinase-mediated genuine residue phosphorylation is generally only possible in limited situations, such as those involving reconstituted protein or peptide systems and unique kinase-specific target sites. Myofilament Ca2+ sensitivity of force, tension cost, LDA, and single myofibril activation/relaxation kinetic parameters were measured in the troponin-exchanged muscle preparations. Our results show that both R145W and T143E uncouple the impact of S23D/S24D phosphomimetic on myofilament function, including myofilament Ca2+ sensitivity, single myofibril relaxation dynamics, and LDA. Moreover, the presence of the cTnI R145W mutation eliminated the ability of either PKCα or PKCϵ to phosphorylate the cTnI Thr-143 target residue but notable not PKA-mediated phosphorylation at the cTnI Ser-23/Ser-24 targets. Molecular dynamics (MD) simulations revealed a marked reduction in the interactions between cardiac troponin C (cTnC) helix C (residues 56, 59, and 63) and cTnI (residue 145) in the presence of either the RCM mutation or PKC phosphomimetic in cTnI.
Our results suggest that the RCM-associated cTnI R145W mutation induces a structural state that is similar to, albeit more extensive than, that induced by PKC-mediated phosphorylation of cTnI Thr-143. This structural conformational change induces increased myofilament Ca2+ sensitivity and uncoupling from the impact of cTnI PKA-mediated phosphorylation. However, the structural impact does not include (directly) myofilament length-dependent activation. These altered biophysical and biochemical cardiac myofilament properties are likely to significantly contribute to the diastolic cardiac pump dysfunction that is seen in cTnI R145W RCM patients.
Results
Impact of Isolated PKA/PKC/RCM on Myofilament Ca2+ Sensitivity
Fig. 2A summarizes force-[Ca2+] relationships recorded on troponin-exchanged multicellular skinned human muscles containing wild-type (WT) (black circles), RCM (green squares), PKC (red up triangles), or PKA (blue down triangles) cTnI. These data confirm, in the human sarcomere, that the R145W RCM mutation induces a drastic increase in myofilament Ca2+ sensitivity. In contrast, the PKC phosphomimetic at residue Thr-143 induced a relatively modest increase in myofilament Ca2+ sensitivity, consistent with previously reported data in skinned human single myocytes (23). Conversely, the PKA phosphomimetic at residues Ser-23/Ser-24 induced a significant desensitization of the multicellular skinned human muscles, consistent with previous reports in both rodent (15–17, 22, 24) and human (21, 23) isolated myocardium.
FIGURE 2.
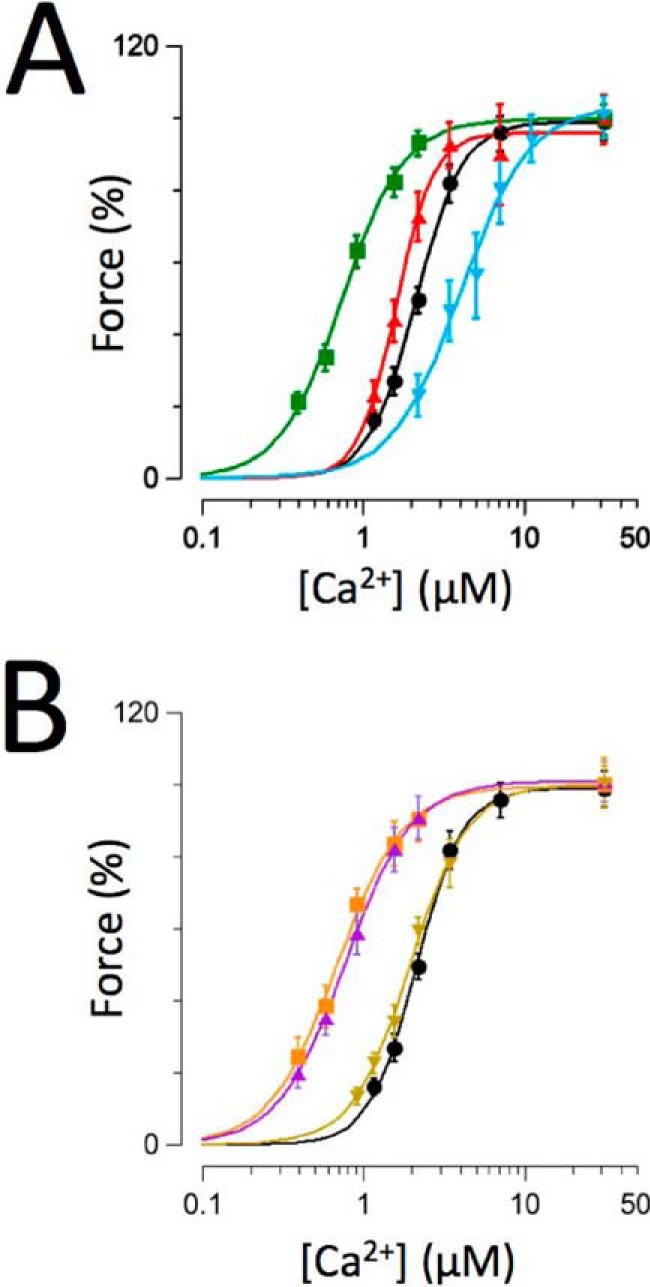
Impact of recombinant troponin on force-[Ca2+] relationships. Normalized force-[Ca2+] relationships were recorded on human recombinant troponin-exchanged muscles containing WT (black circles; n = 12), RCM (green squares; n = 7), PKC (red up triangles; n = 9), or PKA (blue down triangles; n = 8) (A). B, WT (transcribed from A), RCM + PKA (purple up triangles; n = 6), RCM + PKC (orange squares; n = 5), and PKA + PKC (brown down triangles; n = 6). Experiments were performed at 25 °C. Data are presented as mean ± S.E. Error bars reflect S.E.
PKA/PKC/RCM Interactions
Fig. 2B summarizes force-[Ca2+] relationships recorded on muscles that were exchanged with troponin that contained within cTnI combinations of either the RCM disease mutation or the PKA/PKC charge mutations or PKA + PKC as compared with the WT troponin exchange (the WT data are transcribed from Fig. 2A, black circles). Presence of the R145W mutation rendered the human cardiac sarcomere relatively unresponsive to the simultaneous presence of either the PKC (orange squares) or PKA (purple up triangles) phosphomimetic charge mutation. Likewise, introduction of the PKC phosphomimetic virtually eliminated the desensitization that otherwise occurred upon introduction of the PKA phosphomimetic (brown down triangles). The relatively large (+1.3 μm) desensitization that was seen upon introduction of the PKA phosphomimetic alone was no longer observed upon the simultaneous additional introduction of either the RCM mutation (+0.1 μm) or the PKC phosphomimetic (+0.3 μm). Likewise, the modest increase in calcium sensitivity (−0.4 μm) induced by introduction of the PKC phosphomimetic was markedly reduced (−0.04 μm) in the presence of the RCM mutation. These data support the notion that both the RCM-associated disease mutation and, to a slightly lesser extent, the PKC phosphomimetic “uncouple” the impact of PKA-mediated cTnI Ser-23/Ser-24 phosphorylation on (human) myofilament function.
Myofilament Length-dependent Activation Interactions
The impact of sarcomere length (SL) on myofilament function is illustrated in Fig. 1C (in this case, WT troponin-exchanged muscles), confirming myofilament LDA in the multicellular (i.e. “trabecula-like”) isolated human skinned muscles used in the present study. The average fit parameters obtained in all experimental groups are summarized in Fig. 3. The PKA phosphomimetic introduced at residues Ser-23/Ser-24 induced a significant increase in myofilament LDA (as indexed by the ΔEC50 parameter (Fig. 3A, right panel)), consistent with previous reports in both rodent (15, 17, 22, 24) and human (21, 23) isolated myocardium. However, in contrast to overall Ca2+ sensitivity (Fig. 3A, left panel), myofilament LDA was not affected by the introduction of the R145W mutation (RCM). Simultaneous introduction of the PKA phosphomimetic (RCM + PKA) induced a slight, but significant, reduction in LDA compared with WT exchange. Moreover, introduction of the PKC phosphomimetic alone was without impact on LDA, consistent with a previous report on skinned human single myocytes (23), as was the combined introduction of either PKC + PKA or RCM + PKC. Note that the latter combination is unlikely to occur physiologically due to the destruction of the PKC phosphorylation motif in R145W (see below). Thus, presence of the T143E phosphomimetic rendered myofilament LDA insensitive to the presence of the Ser-23/Ser-24 phosphomimetic. Moreover, in the presence of the RCM mutation, introduction of the PKA phosphomimetic induced an opposite response, that is a reduction in LDA, albeit at greatly reduced magnitude (+0.6 μm WT versus WT + PKA; −0.04 μm RCM versus RCM + PKA). That is, introduction within the cTnI IP domain of either RCM mutation or PKC phosphomimetic effectively “uncoupled” the physiological response that is otherwise observed upon introduction of the PKA phosphomimetic.
FIGURE 3.

Impact of human recombinant troponin exchange on myofilament function. Average fit parameters were obtained from force-[Ca2+] relationships indexing myofilament Ca2+ sensitivity (EC50; A, left bars) and length-dependent activation of Ca2+ sensitivity (A; ΔEC50; right bars), maximum force development (Fmax; B, left bars); and length dependence of force development (ΔFmax; B, right bars), the Hill coefficient, an index of cooperativity (nHill; C, right bars). C, left bars, summarizes tension cost, the slope of the ATPase-force relationship, an index that is proportional to cross-bridge detachment rate. Experiments were performed at 25 °C. Data are presented as mean ± S.E. *, p < 0.05 versus WT (number of samples as in Fig. 2). Error bars reflect S.E.
Maximum Force, Tension Cost, and Cooperativity of Force
Fig. 3B summarizes average maximum force development at SL = 2.3 μm (Fmax; left panel) and the average impact of SL on Fmax (ΔFmax; right panel). Presence of the RCM mutation induced a modest increase in (Fmax) that was not affected by the simultaneous presence of the PKA phosphomimetic. Unlike myofilament Ca2+ sensitivity and LDA (Figs. 2 and 3A), there were no differences in ΔFmax between the experimental groups (Fig. 3B, right panel) or in tension cost, an index of actomyosin cross-bridge detachment rate (Fig. 3C, left panel). These data indicate that the PKA/PKC phosphomimetics and the RCM mutation affect myofilament function primarily by their impacts on thin filament Ca2+ regulation, not by alterations in the kinetics of the actomyosin cross-bridge cycle. This conclusion is further supported by the rate of rapid release-restretch force redevelopment (ktr) data obtained from single myofibril experiments (see below). Finally, presence of the RCM mutation induced a modest reduction in the nH parameter, the steepness of the force-[Ca2+] relationship that indexes cooperative myofilament activation (Fig. 3C, right panel). This small, albeit significant, decrease in nH was observed irrespective whether the PKA or PKC phosphomimetic was co-introduced with the RCM mutation.
Single Myofibril Activation/Relaxation Kinetics
The single myofibril technique allows for measurement of dynamic Ca2+ activation/relaxation kinetics of force (27–29). In addition, following Ca2+ activation when steady-state activation was reached, a rapid release-restretch maneuver was imposed to measure ktr, an index that is proportional to the sum of the apparent cross-bridge attachment and detachment rates (30, 31). The rates of Ca2+ activation (kCa) and ktr were similar in the present study in all experimental groups, consistent with previous reports (29). This finding indicates that the dynamics of Ca2+ binding to cTnI and the subsequent thin filament activation are sufficiently fast to not be rate-limiting for force development. There were no differences between the experimental groups for either the kCa or the ktr parameter (Fig. 4A), indicating that neither the RCM mutation nor the PKA/PKC phosphomimetics directly affect actomyosin cross-bridge cycling kinetics. Upon the rapid removal of activating Ca2+, force relaxation has been shown to proceed in two distinct phases: (i) an initial slow and close to linear phase, which is likely related to the initial detachment rate of mechanically loaded cross-bridges, followed by (ii) a rapid pseudo-exponential phase, which likely reflects rapid detachment of cross-bridges induced by sarcomere length inhomogeneity (29). Here we confirm this phenomenon in the troponin-exchanged human single myofibrils as is illustrated in Fig. 4B. There were no differences in the rapid phase of force relaxation between the experimental groups (data now shown). The rate of the linear phase of force relaxation (klin) was slower in the presence of the RCM mutation but only upon the additional introduction of the PKA phosphomimetic (Fig. 4C, left panel). Introduction of the PKA phosphomimetic alone significantly reduced the duration of the slow linear relaxation phase (Tlin), whereas introduction of the RCM mutation together with the PKA phosphomimetic significantly prolonged Tlin. Likewise, the PKA uncoupling phenomenon seen for EC50 and ΔEC50 (Fig. 3A) was also apparent for the Tlin slow force relaxation parameter (Fig. 4C). The duration of the slow force relaxation phase is determined by the time at which an abrupt transition takes place where marked sarcomere length inhomogeneity, caused by alternating stretches and releases of individual serially connected sarcomeres, induces rapid relaxation. The Tlin parameter is presumed to reflect the tendency of an ensemble of attached cross-bridges to resist detachment (29) and may be related to myofilament length-dependent activation.
FIGURE 4.
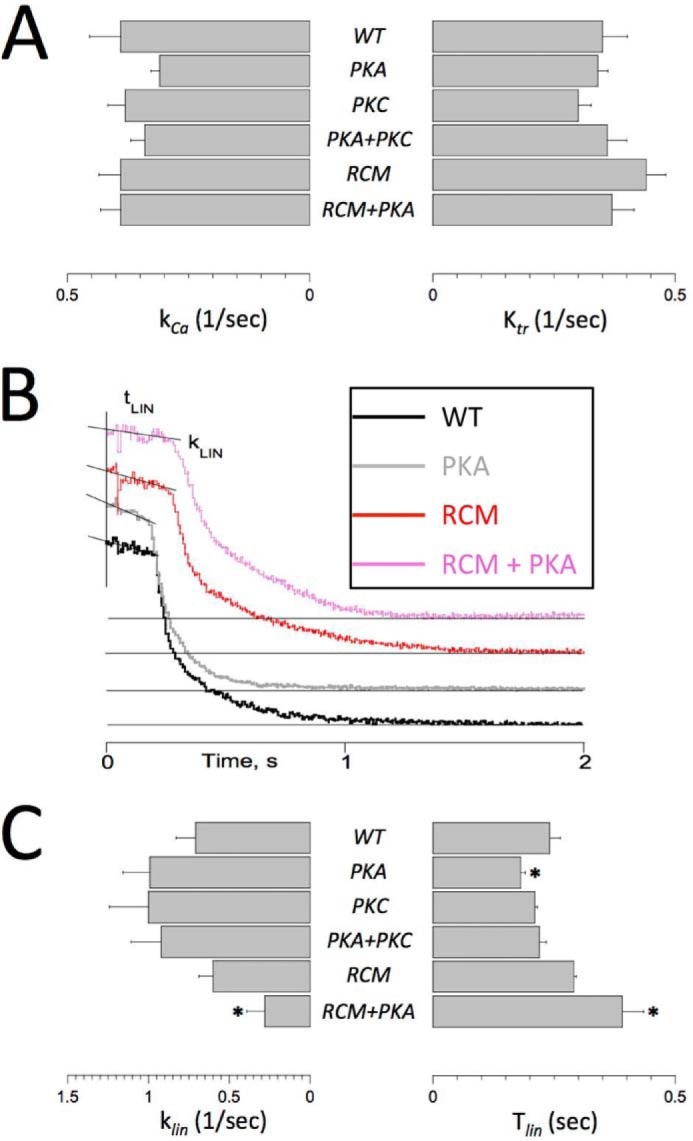
Single myofibril activation/relaxation kinetics. Single myofibril activation/relaxation and release-restretch kinetics were measured in human recombinant troponin-exchanged isolated single myofibrils. A summarizes the average rate of force development upon a rapid (<5-ms) switch of bathing [Ca2+] from pCa = 10 to pCa = 4.5 (kCa; left panel) and the rate of force redevelopment following a rapid release-restretch maneuver during steady-state force development (ktr). There were no significant differences between the experimental groups, indicating that neither RCM nor PKA/PKC directly affect cross-bridge cycle kinetics. B, original recordings of force relaxation upon a rapid switch of bathing [Ca2+] from pCa = 4.5 to pCa = 10. Force relaxation took place in two distinct phases, a slow almost linear decline followed by a rapid pseudo-exponential decline back toward baseline (indicated by the thin horizontal lines); note that the force traces have been offset vertically to aid visualization of the individual traces. Two parameters of the slow force relaxation, klin and the Tlin, were measured as indicated; the rate of fast relaxation was estimated by exponential fit of the force data (not shown). C, average slow force relaxation fit parameters (klin and Tlin). Experiments were performed at 15 °C. Data are presented as mean ± S.E. *, p < 0.05 versus WT (RCM, n = 7; all other groups, n = 6). Error bars reflect S.E.
In Vitro PKCα-, PKCϵ-, and PKA-mediated Phosphorylation
The R145W mutation involves replacement of a positively charged arginine by a relatively bulky neutral tryptophan positioned just two residues N-terminal to the Thr-143 PKC phosphorylation target site. Hence, it is possible that this mutation may disrupt the PKC phosphorylation motif. The data shown in Fig. 5 indicate that this is indeed the case both for PKCϵ (Fig. 5A) and for PKCα (Fig. 5B) but notably not the phosphorylation of Ser-23/Ser-24 mediated by PKA (Fig. 5C). That is, presence of the RCM mutation effectively renders the Thr-143 PKC target site phospho-null, and therefore the recombinant RCM + PKC cTnI construct that we tested in the present study, although of interest in itself, is nevertheless physiologically unlikely to occur in vivo.
FIGURE 5.

Impact of the R145W RCM mutation on PKC- and PKA-mediated phosphorylation. The ability of PKC or PKA to phosphorylate recombinant human troponin was tested in vitro. Human recombinant troponin (250 μg/ml) was treated for 120 min at 37 °C with either PKCϵ (A), PKCα (B), or PKA (C) using 0–10 μg/ml kinase protein as indicated (b represents boiled in A). The reaction was quenched by addition of SDS sample buffer. Western blots were probed for TnT (top gels; loading controls) or site-specific phosphoantibodies (bottom gels) against Thr(P)-143 (PKC; A and B) or cTnI Ser(P)-23/Ser(P)-24 (PKA; C). Presence of the RCM mutation rendered the Thr-143 PKC phosphorylation motif inactive but notably not the Ser-23/Ser-24 PKA motif. These experiments were performed in triplicate.
Molecular Dynamics Simulation
To shed light on the possible molecular mechanisms underlying the impact of the RCM mutation, we conducted MD simulations of troponin in the presence of either cTnI T143E or cTnI R145W. Data were averaged over the course of the last 100 ns in production runs for three independent MD simulation runs. Fig. 6 shows differential contact map profiles between the cTnI IP/SP domains (ordinate) and cTnC (abscissa) in the presence of the PKC phosphomimetic (Fig. 6A) or the RCM mutation (Fig. 6B); in both panels, after the WT contact map is subtracted, the differential map is displayed in a color map where blue represents a decrease and red represents an increase in contact frequency. We defined contact based on the distance between residues; 5 Å was used as the cutoff. Fig. 6C summarizes the average contact frequencies recorded among the three cTnC helix C residues. Contacts significantly decreased upon introduction of the T143E PKC phosphomimetic and were virtually eliminated in the presence of the RCM mutation. The reduced interaction frequency between cTnI residue 145 and the cTnC helix C residues may also explain a trend toward increased mobility that was noted in the low affinity Ca2+ binding domain of cTnC (data not shown).
FIGURE 6.
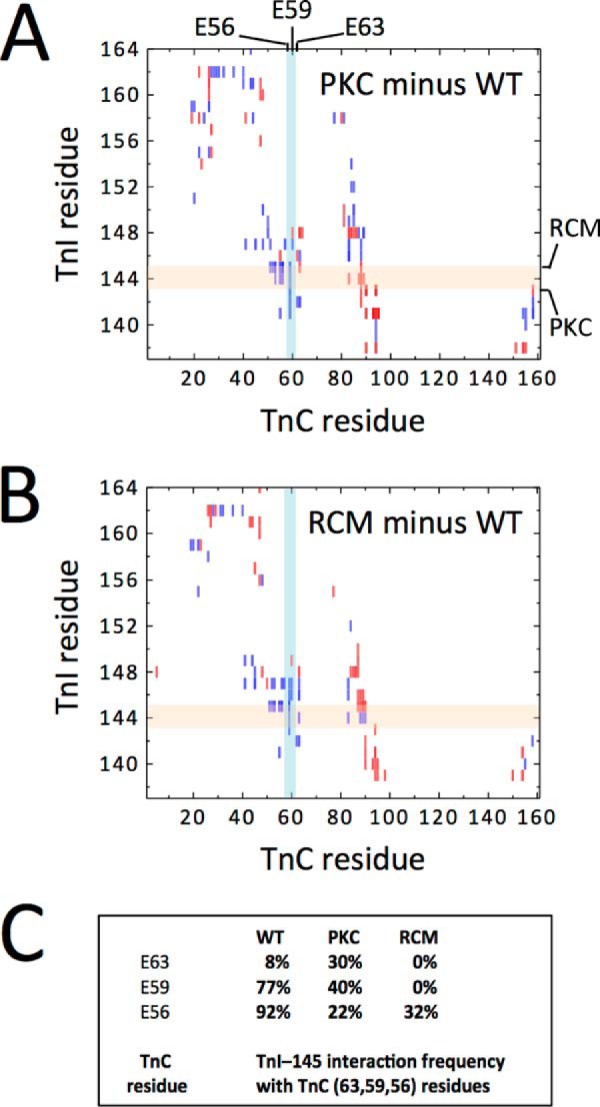
Interaction between cTnC and cTnI. Contact maps between cTnC (abscissa) and cTnI (ordinate) simulated for the PKC phosphomimetic (A) or the RCM mutant (B) were calculated from three independent production runs. In each panel, a differential map is constructed by subtracting the WT contact map and subsequently color-coded such that blue represents a decrease and red represents an increase in contact frequency. C summarizes contact frequencies between cTnI residue 145 and cTnC helix C Glu-56, Glu-59, and Glu-63 as indicated by the lightly shaded colored bars. PKC phosphomimetic significantly reduced contact between these residues, whereas the RCM R145W mutation virtually eliminated all contacts.
Discussion
In the present study, we investigated the RCM-associated mutation cTnI R145W in terms of in vitro human myofilament function and the interaction with PKA/PKC-mediated cTnI phosphorylation. The main new findings of our study are that human cTnI R145W 1) by itself induces a modest increase in maximum Ca2+-saturated force development, a drastic increase in myofilament Ca2+ sensitivity, and prolonged/slowed kinetics of force relaxation; 2) disrupts the cTnI Thr-143 PKC phosphorylation motif; 3) uncouples the functional impact of cTnI PKA-mediated phosphorylation, 4) by itself does not affect myofilament LDA, and 5) disrupts structural protein/protein interactions between the cTnI IP domain and the C-Helix domain of cTnC as determined by MD simulation.
Sarcomeric mutations associated with familial HCM or RCM are frequently associated with increased Ca2+ sensitivity in reconstituted in vitro protein assays (12, 32–34), at the isolated myofilament level (9–11, 26, 35), or in human samples (13). Three separate mutations have been identified in cTnI at residue 145; two (R145G and R145Q) are associated with HCM, and one (R145W) is associated with RCM. As noted above, although all three mutations were reported to increase myofilament Ca2+ sensitivity, this was most severe for the R145W RCM-associated mutation (9–12). To our knowledge, this is the first report in which human recombinant R145W-containing troponin was exchanged for endogenous troponin in the human sarcomere. The results of this study are generally consistent with those from in vitro reconstituted protein and isolated experimental animal skinned myocardium studies, including the drastic increase in Ca2+ sensitivity (9). Either a decrease (9) or, as in the present study, an increase (36) in maximum force development has been reported for the RCM-associated cTnI R145W mutation; most reports on the HCM-associated cTnI R145G mutation show a decrease in this parameter (10, 11, 35, 37). Whether this reflects a genuine difference between the HCM-associated R145G mutation and the R145W RCM-associated mutation that we examined in the present study remains to be determined. The slight increase in maximum force persisted in the simultaneous presence of the PKA but notably not the PKC phosphomimetic (Fig. 3B). Likewise, the cooperativity of force development was reduced in the presence of the RCM mutation regardless of simultaneous presence of either PKA or PKC phosphomimetic (Fig. 3C). These data suggest an impact of the RCM mutation on actomyosin interaction kinetics and/or interactions between attached and force-generating cross-bridges. However, the absence of alterations in either the tension cost (Fig. 3C) or the kCa and ktr parameters (Fig. 4A) suggests that actomyosin cycling kinetics were not affected by this mutation. Hence, the molecular mechanisms underlying the impact of the RCM mutation on maximum force development and the cooperativity of force development are not clear at this time, and further mechanistic insights are difficult to derive from the present study. Regardless, our results demonstrate that the presence of R145W within the IP domain of cTnI in the human sarcomere, by itself, is sufficient to induce a drastic increase in myofilament Ca2+ sensitivity, a modest increase in maximum force development, and a modest decrease in the cooperativity of Ca2+ activation.
None of the residue substitutions within cTnI affected (i) the kinetics of force development or force redevelopment in single myofibrils or (ii) tension cost in multicellular skinned preparations. Similar single rodent myofibril kinetic data were previously reported for the R145G HCM mutation (26, 35). These results indicate that neither the RCM mutation nor the phosphomimetic substitutions affect actomyosin cross-bridge cycling kinetics. Rather, and combined with the force relaxation data discussed below, it appears that these cTnI modifications predominantly operate via modulation of thin filament regulatory processes. Moreover, the rate of Ca2+-induced force development was similar to the rate of force redevelopment at steady-state maximum Ca2+ activation, consistent with most (29, 38) but not all (26) previous reports. The implication of this observation is that the kinetics of actomyosin cross-bridge cycling are rate-limiting for force development and that the kinetics of Ca2+ thin filament regulation must be much faster.
Introduction of R145W + PKA into cTnI induced reduced kinetics of force relaxation in the single myofibrils, a phenomenon that has also previously been reported for the R145G HCM-associated mutation in rodent single myofibrils (26). Introduction of the PKA S23D/S24D phosphomimetic alone accelerated relaxation kinetics, consistent with results reported for rodent single myofibrils (26). We previously reported unaltered relaxation kinetics in human single myofibrils upon treatment with PKA (27), a finding seemingly inconsistent with the present findings on human myofibrils. However, PKA treatment results in phosphorylation of not only cTnI but also myosin-binding protein C and titin. The impact of PKA-mediated phosphorylation of those target residues on relaxation kinetics is not known, but those alternate phosphorylation events may explain the different results obtained in the present study.
Neither Ca2+ activation kinetics, force redevelopment kinetics, nor tension cost was impacted by cTnI R145W. Therefore, it is likely that the impact on force relaxation kinetics seen here in the single myofibril was the result of altered thin filament regulation. Regardless of the underlying molecular mechanism, slowed and delayed force relaxation coupled with the drastic increase in myofilament Ca2+ sensitivity is likely to contribute to relaxation abnormalities at the level of the whole heart and may additionally lead to persistent cross-bridge activity throughout the diastolic phase of the cardiac cycle. Both myofilament-based phenomena are expected to induce the diastolic cardiac pump dysfunction that is seen in RCM patients in the notable absence of ventricular hypertrophy.
Presence of the R145W RCM mutation rendered the human sarcomere unresponsive to charge phosphomimetic mutations introduced at Ser-23/Ser-24. Similar “uncoupling” has previously been reported for a range of sarcomeric mutations both in isolated skinned muscle (22, 25, 26) and for in vitro reconstituted protein biochemical studies (34). Here, we demonstrate that a similar uncoupling phenomenon exists in the human sarcomere and with regard to myofilament LDA. In WT troponin-exchanged myocardium, treatment with PKA or introduction of the PKA S23D/S24D phosphomimetic induced a large increase in LDA, consistent with previous reports by others (24) and us (15). We recently reported that increased LDA upon PKA phosphorylation results from independent contributions by myosin-binding protein C (∼65%) and cTnI (35%), whereas PKA-mediated phosphorylation of titin appears to decrease LDA (15). The latter is most likely due to increased titin compliance upon PKA-mediated phosphorylation. Titin strain directly impacts myofilament Ca2+ sensitivity via strain-dependent modulation of thick and thin filament structure via an unknown molecular mechanism (39). Isolated human myocardium presents with variable levels of phosphorylation at the PKA target sites and, moreover, in cardiac diseases is often associated with significant reduction of PKA activity. Consequently, PKA-mediated contractile protein phosphorylation is expected to be substantially reduced in cardiac disease. Therefore, to allow comparisons to be made between different samples, PKA treatment is often used to maximally phosphorylate all contractile protein PKA target residues, some of which may actually induce opposite myofilament functional effects. Here we circumvented that confounding factor by using recombinant troponin exchange on skinned muscle derived from a single human donor cardiac left ventricular septum sample. Thus, all experiments were performed on an identical sarcomere structure and phosphorylation status; moreover, we selected a sample with relatively low contractile phosphorylation as judged by ProQ Diamond SDS-PAGE. Using this approach, we found no direct impact of RCM cTnI R145W on myofilament LDA. Rather, the mutation induced a virtually complete PKA uncoupling such that the normal PKA-induced increase in LDA was no longer observed. Of interest, introduction of the PKC phosphomimetic at residue 143 likewise induced such uncoupling. Of note, both R145W and T143E introduce a relative negative charge alteration within the cTnI IP domain and may, therefore, share the same molecular pathway (see below). We demonstrate here that the cardiac TnI RCM-associated R145W mutation renders the cTnI Thr-143 target inaccessible to PKC-mediated phosphorylation. Hence, patients carrying this mutation are unable to modulate sarcomere function via PKC phosphorylation at this target site. Of note, both PKC phosphorylation and the RCM R145W mutation increased myofilament calcium sensitivity, but the shift was much larger for the RCM mutation. Thus, our data indicate that either introduction of a negative charge at the Thr-143 residue or the neutral charge introduced for the positive residue by the RCM mutation at residue 145 significantly blunts (or sometimes even reverses) the impact of PKA-mediated phosphorylation on both calcium sensitivity and myofilament LDA. Thus, these data suggest that a physiological function of PKC phosphorylation at Thr-143 may be to disengage the physiological impact of PKA-mediated phosphorylation on myofilament function. However, unlike the phosphorylation event on the Thr-143 residue that can be reversed by protein phosphatase action, the R145W mutation is permanent, and its impact, therefore, is physiologically irreversible. Hence, presence of the R145W mutation nullifies the ability to “activate” PKA modulation by dephosphorylation of Thr-143.
To provide structural insight into potential molecular mechanisms, we performed MD simulations of troponin containing WT, R145W, or T143E residue substitutions. In general, upon introduction of either R145W or T143E, we observed increased mobility of cTnC within the low affinity Site II Ca2+ binding domain. Of note, previous MD simulations have reported altered intramolecular interactions within cTnI between the N terminus and the IP domain upon introduction of the PKA S23D/S24D phosphomimetic, and this was disrupted by the presence of the HCM-associated cTnI R145G mutation (26, 40, 41). However, we did not see such intramolecular interactions in preliminary MD simulation runs. This preliminary result is consistent with a recent MD study by Gould and co-workers (42) who reported that this intramolecular interaction is both rare and not affected by PKA-mediated phosphorylation. Accordingly, we focused instead on the impact of R145W and T43E on dynamic interactions between cTnI residue 145 and helix C of cTnC (residues Glu-56, Glu-59, and Glu-63). We found a marked reduction in the interaction frequency between these residues upon replacement of (i) the positively charged arginine residue (Arg-145) with a neutral tryptophan or (ii) the neutral threonine residue (Thr-143) with a negatively charged aspartic acid. Both substitutions caused substantial reductions in interaction frequency between cTnI and cTnC at these residues (Fig. 6). These results may explain the observed increases in myofilament Ca2+ sensitivity (modest for T143E and drastic for R145W). Fig. 7 shows snapshots from the MD simulation for WT (Fig. 7A), T143E phosphomimetic (Fig. 7B), and R145W mutation (Fig. 7C). Both T143E and to an even greater extent R145W induce a movement of residue 145 (and the surrounding IP domain) away from helix C in cTnC and a straightening out of this section of the cTnI IP domain. It is possible that this is the result of a disruption of the interactions between the negative glutamic acids (Glu-56, Glu-59, and Glu-63) in cTnC helix C and the positive arginine (Arg-145) on cTnI in WT. Introduction of a negatively charged glutamic acid at residue 143, close to Arg-145, may provide a repellent force sufficiently large to also blunt the interactions between cTnC helix C and Arg-145. This would explain the similar, albeit of different magnitude, functional impact of either T143E or R145W at the myofilament level. How such a destabilization of the cTnI IP domain leads to increased myofilament Ca2+ sensitivity and, possibly, increased cTnC Ca2+ binding (26) cannot be determined from these MD simulations. Likewise, how introduction of either T143E or R145W within cTnI disrupts PKA-induced modulation of LDA cannot be determined from our MD simulations.
FIGURE 7.
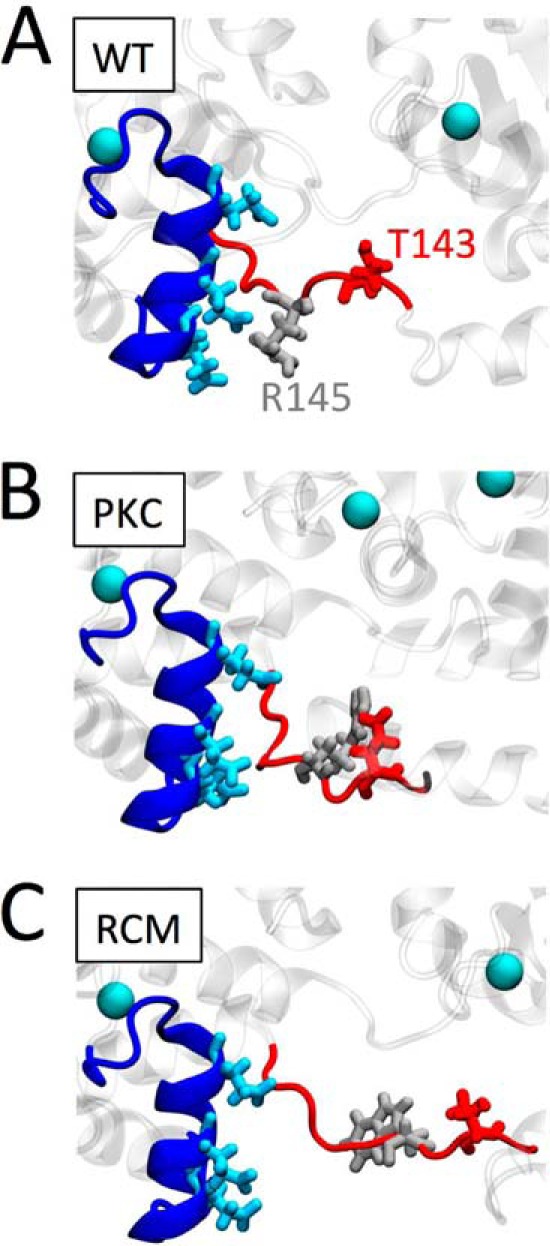
Proposed structural impact of the PKC phosphomimetic or the RCM disease mutation. Enlarged structural detail of troponin shows cTnC (dark blue), highlighting helix C Glu-56, Glu-59, and Glu-63 (light blue). cTnI is shown in red; WT Thr-143 (A and C) or the PKC phosphomimetic Glu-143 (B) is shown in dark red. WT arginine Arg-145 (A and B) or its mutation to Trp-145 is shown in light gray. Ca2+ ions are illustrated as light blue balls. Introduction of the negatively charged PKC phosphomimetic at cTnI position 143 causes this region of cTnI to sample structural states away from negatively charged Glu-56, Glu-59, and Glu-63 within the cTnC helix C domain. Replacing the positively charged arginine residue at cTnI position 145 by a neutral, relatively large tryptophan in the RCM-associated mutation further exacerbates the structural conformational changes in this region. We propose that these structural changes lead to increased myofilament Ca2+ sensitivity and uncoupling of the functional impacts mediated by cTnI PKA phosphorylation.
Several limitations need to be considered in interpreting our results. First, we relied on charge phosphomimetics to simulate the negative charge that normally accompanies protein phosphorylation. This approach has been used previously both by others and us (43). In that study, similar results were obtained using either charge phosphomimetics or genuine phosphorylation (43). The use of phosphomimetics for troponin is supported by MD simulations (40). As mentioned, kinase-mediated genuine residue phosphorylation is generally only possible in limited situations, such as those involving reconstituted protein or peptide systems. However, some reports have indicated differential functional impacts depending on whether glutamic acid or aspartic acid charge phosphomimetics were used (44). Likewise, non-neutral functional impacts of the alanine substitution that is commonly used as a phospho-null phosphomimetic have been reported (44). A major advantage of using a phosphomimetic substitution, however, is that it allows for site/residue-specific insights regarding the functional impacts of phosphorylation post-translational modifications that could not otherwise be obtained. Because multiple PKA and PKC target residues reside within the cardiac sarcomere, experiments that rely on kinase treatment may be difficult to interpret. Second, although we accomplished >68% troponin exchange, we cannot be certain that residual endogenous troponin influenced our myofilament biophysical functional results. Likewise, the potential influence of post-translational modifications of other non-exchanged endogenous contractile proteins cannot be determined from the current study. However, use of a single human sample throughout the entire study ensured that comparisons between groups were derived from experiments that always started from an identical sarcomeric background prior to recombinant troponin exchange.
In conclusion, the restrictive cardiomyopathy- associated cTnI mutation R145W induced a drastic increase in myofilament Ca2+ sensitivity. The mutation caused uncoupling from the impact of PKA phosphorylation but did not by itself affect myofilament length-dependent activation. The molecular mechanisms underlying this phenomenon appear to include altered interactions between the IP domain of cTnI and helix C of cTnC. The observed perturbed myofilament properties are likely to contribute significantly to the diastolic dysfunction that is seen in cTnI R145W RCM patients.
Experimental Procedures
Human Samples
Non-age/sex-matched, de-identified hearts were procured via the National Disease Research Interchange. The University of Michigan Institutional Review Board approved tissue procurement and processing, and the Institutional Review Board at Loyola University Chicago approved the protocol for the use of de-identified human donor hearts. Prior to explant, hearts were flushed with ice-cold cardioplegia solution and brought to the laboratory on ice <12 h postexplant. Sections of left ventricular tissue were then immediately frozen in liquid N2 and stored at −80 °C until use. Myofilament fractions of septal tissue samples were assayed by SDS-PAGE/ProQ Diamond staining. A single left ventricular septum sample (∼10 g), displaying relatively low levels of overall contractile protein phosphorylation, was selected and divided in ∼50-mg aliquots while still frozen under liquid N2 and again stored −80 °C until further use. Adoption of this protocol assured that comparisons among the various groups in the present study were made on the basis of an invariable baseline sarcomere contractile protein status prior to troponin exchange.
Expression, Purification, and Reconstitution of Recombinant Troponin
The methods used to clone, express, and purify the troponins and reconstitution of the recombinant troponin complex have been previously described (45). Briefly, human DNA clones of cTnC, myc-tagged cTnT, and various cTnI clones were transformed into Rosetta BL21(DE3) Escherichia coli bacteria (Novagen) and grown at 37 °C overnight. The cTnI clones used were: WT, S23D/S24D (PKA), T143E (PKC), S23D/S24D/T143E (PKA + PKC), R145W (RCM), S23D/S24D/R145W (RCM + PKA), and T143E/R145W (RCM + PKC). The locations of the phosphomimetic charge mutations and the RCM mutation within the cardiac troponin complex are illustrated in Fig. 1A (46). High protein yield was achieved by isopropyl 1-thio-β-d-galactopyranoside induction. Proteins were isolated by lysis and centrifugation followed by column chromatography purification (45). Troponins were stored separately in 6 m urea buffer at −80 °C. The troponin complexes were formed by mixing equimolar amounts of (WT or mutated) cTnI, cTnC, and myc-cTnT in 6 m urea, FPLC desalting, and finally FPLC purification (RecourceQ). Fractions containing the purified troponin complex were dialyzed to a final ionic strength of 180 mm in Ca2+-containing activating solution to which 50 mm BDM was added.
Skinned Multicellular Isolated Human Myocardium
Permeabilized human cardiac tissue was obtained as described previously (47). Briefly, frozen tissue was cut into 2–4-mm pieces in ice-cold relaxing solution followed by homogenizing at low speed (1,000 rpm, 3 s) three times (Power Gen 700D, Fisher Scientific) in relaxing solution. The preparation was allowed to settle, and the supernatant was discarded after which the tissue was resuspended in relaxing solution. The tissues were permeabilized overnight in 2% Triton X-100 at 4 °C. The tissues were then extensively washed in fresh ice-cold relaxing solution, stored on ice, and used within 8 h.
Exchange for Recombinant Troponin
Multicellular strips of skinned myocardium were selected for appearance under a dissecting microscope. Muscles were attached to aluminum T-clips on each end, slightly stretched, and next immobilized using insect needles pinned through the T-clips to the bottom of a Sylgard-coated well in a 96-well plate. The muscles were incubated for 3–4 h at room temperature in exchange buffer (100 μl) that was composed of activating solution, 50 mm BDM (to prevent contraction), and 2.5 mg/ml troponin while gently agitating on an orbital shaker (∼1 rpm). Exchange at high free [Ca2+] (∼30 μm) and room temperature assured >65% exchange efficiency (see Fig. 1B). Following exchange, muscles were washed three times with relaxing solution.
Western Blotting Analysis of Recombinant Troponin Exchange
The Western blotting method to measure the extent of recombinant troponin exchange has been reported previously (48). Briefly, isolated skinned myocardium following exchange was washed one additional time with relaxing solution to which 2 mg/ml BSA was added to reduce unspecific binding of recombinant troponin. Next, muscles were solubilized in 8 m Tris, urea denaturing buffer, and 2× Laemmli buffer was added. Total protein homogenates were separated by 10% SDS-PAGE (20 min, 80 V; 1 h, 140 V; room temperature) and blotted onto nitrocellulose membrane (2 h, 300 mA, 4 °C). The presence of the myc tag on the N terminus of cTnT allows for separation from endogenous cTnT (see Fig. 1B). Use of a pan-reactive cTnT antibody (CT3, Santa Cruz Biotechnology) allows for quantitative measurement of both endogenous cTnT and myc-tagged cTnT (which co-migrates with actin under these SDS-PAGE conditions) using the chemiluminescence approach (ECL, Bio-Rad). The exchange efficiency was calculated as the percentage ratio of exogenous myc-cTnT (top band) relative to total cTnT (top + bottom bands), which was quantified using the ImageJ gel densitometry analysis toolkit. Presence of the N-terminal myc tag on cTnT has been shown to not affect the cardiac force-[Ca2+] relationship (48). The average exchange efficiency was ∼68% for all groups (WT and RCM groups are shown in Fig. 1B). The inset of Fig. 1B shows confocal analysis of a WT exchange muscle probed with a mouse anti-myc antibody followed by Alexa Fluor-488 mouse secondary antibody (Sigma); the double banded appearance in this image is consistent with selective troponin exchange into the thin filament of the cardiac sarcomere.
Simultaneous Measurements of Isometric Tension and ATPase Activity
Following troponin exchange, skinned muscles were attached to a force transducer (KG4A, World Precision Instruments, Sarasota, FL) and high speed length controller (model 315C, Aurora Scientific Inc., Ontario, Canada) via the aluminum T-Clips. Muscle dimensions were determined using an ocular micrometer mounted in the dissection microscope (resolution, ∼10 μm). SL was measured by laser diffraction as described previously (47) and adjusted to either 2.0 or 2.3 μm. Isometric force and ATPase activity were measured at 25 °C at various levels of Ca2+ activation as described previously (47). Briefly, the isolated muscle was exposed to a range of activating calcium via solutions obtained by appropriate mixing of activating and relaxing solutions, and the force generated and ATP consumed were measured simultaneously. The activating solution contained 20 mm Ca2+-EGTA, 1.55 mm potassium propionate, 6.59 mm MgCl2, 100 mm BES, 5 mm sodium azide, 1 mm DTT, 10 mm phosphoenolpyruvate, 0.01 mm oligomycin, 0.1 mm PMSF, and 0.02 mm A2P5 as well as protease inhibitor mixture (Sigma). The relaxing solution was identical except it contained 20 mm EGTA, 21.2 mm potassium propionate, and 7.11 mmMgCl2. The preactivating solution was identical except it contained 0.5 mm EGTA, 19.5 mm HDTA, and 21.8 mm potassium propionate. All solutions contained 1.0 mg/ml pyruvate kinase and 0.05 mg/ml lactate dehydrogenase (Sigma) and had an ionic strength of 180 mm, 5 mm MgATP, and 1 mm free magnesium. ATPase activity was measured by a UV-coupled optical absorbance enzyme assay as described previously (47). Force was normalized to muscle cross-sectional area and expressed as milliNewtons/mm2. ATPase activity was normalized to muscle volume (in mm3) and expressed as pmol/s/muscle volume. Tension cost was derived from the slope of the ATPase-force relationship, which yields a rate parameter (in s−1) that is proportional to the cross-bridge detachment rate (31).
Single Myofibril Activation/Relaxation Kinetics
Single myofibrils were prepared as described previously (27, 28). Briefly, overnight skinned muscle (prepared as above) was homogenized in ice-cold relaxing solution at 18,000 rpm for 9 s. After centrifugation (200 × g), the myofibril pellet was resuspended in 100 μl of exchange buffer containing 2.5 mg of recombinant troponin, 50 mm BDM, and ∼50 μm free Ca2+ and placed on an orbital shaker (∼1 rpm) for 3–4 h at room temperature. After three washes with relaxing solution, myofibrils were kept at 4 °C for up to 2 days until use. A droplet of myofibril suspension was placed into the experimental chamber for subsequent mounting to fire-polished glass microtools in relaxing solution containing 10 mm EGTA. The attached myofibril was then superfused with alternating solution streams of relaxing and activating solutions (1 mm EGTA) emanating from a double-barreled pipette (∼180 μl/min) that was mounted on a translation stage capable of rapid (<5-ms) solution switches. This approach allows for the measurement of steady-state [Ca2+]-saturated force as well as the rates of Ca2+-induced force activation and relaxation. Experiments were performed at SL = 2.3 μm. kCa, ktr, and biphasic relaxation (klin and the rapid pseudo-exponential phase) together with the Tlin were analyzed by linear and exponential curve fitting using offline custom in house-written software (Labview). The compositions of bath, relaxing, and activating solutions were as described previously (28). Single myofibril experiments were performed at 15 °C.
PKA- and PKC-mediated Phosphorylation of Recombinant Troponin in Vitro
Recombinant troponin containing WT, PKC, PKA, or RCM cTnI was suspended in relaxing solution (250 μg/ml) and treated with PKCα, PKCϵ, or PKA (0, 1, or 10 μg/ml) at 37 °C for 120 min at which time the reaction was quenched by the addition of SDS sample buffer. Following SDS-PAGE and transfer to nitrocellulose membranes, Western blotting analysis (as above) was performed using the cTnT-specific antibody (CT3) as loading control, and phosphospecific antibodies against rabbit cTnI Ser-22/Ser-23 or cTnI Thr-144 (Abcam, Cambridge, MA).
Molecular Dynamics Simulations
The cardiac troponin model was based on the core crystal structure reported by Takeda et al. (46) and modified as described previously (26, 40, 41). The troponin model contained cTnC residues 1–161, cTnI residues 1–171, cTnT residues 236–285, and three Ca2+ ions. The following cTnI mutations were introduced: T143E to mimic PKC-mediated phosphorylation and R145W, the RCM-associated mutation. MD simulations were performed with GROMACS (49, 50) using the CHARMM36 all-atom parameter set (51) and TIP3P water. After minimization, each model was solvated in a rhombic dodecahedron box of water with the size of the box chosen such that the minimal distance to the wall was 1.0 nm, resulting in a minimal distance between periodic images of at least 2.0 nm. Within cTnC, two Ca2+ ions were placed at the high affinity sites, and a single Ca2+ ion was placed at the low affinity binding site (Site II). K+ and Cl− ions were added to the solution to neutralize the charge of the system and to produce 150 mm ionic strength. 1,000 steps of minimization of both water and ions were performed with the protein backbone atoms restrained using a force constant of 1,000 kJ/mol/nm2. The particle mesh Ewald method (52, 53) was used to calculate long range electrostatic interactions, whereas a cutoff of 12 Å was used for short range interactions. van der Waals interactions were reduced to zero by switch truncation that was applied between 10 and 12 Å. MD simulations were carried out with an integration time step of 2 fs. To heat the system from 0 K to the target temperature (300 K) and reach the target pressure (1 bar), the Berendesen method was used with 0.1-ps relaxation time (54). After 1-ns equilibration, the production run was performed in the constant number, pressure and temperature (NPT) ensemble using the Nose-Hoover thermostat (55, 56) and the Parrinello-Rahman barostat (57, 58) with relaxation times of 1.0 ps. Each independent production run was started with a different set of assigned velocities corresponding to 300 K. The atom coordinates of the trajectories were saved every 1 ps; three independent production runs for each system (WT, PKC, and RCM) were carried out for 150 ns each.
Data Analysis
All data are presented as mean ± S.E. Each individual force-calcium and ATPase-calcium relationship was fit to a modified Hill equation.
where Y is either the force or the ATPase parameter, Ymax is the Ca2+-saturated maximum that Y can attain, nH is the Hill coefficient (an index of cooperativity), and EC50 is the [Ca2+] at which Y is half-maximal (an index of myofilament Ca2+ sensitivity). ΔEC50 was calculated as the difference in EC50 at SL = 2.0 and 2.3 μm (an index of myofilament LDA). The impact of SL on the force-[Ca2+] relationship in WT troponin-exchanged muscles, both in terms of absolute force (left panel) and relative normalized force (right panel), is illustrated in Fig. 1C.
Statistical Analysis
Statistical analysis was performed using Systat 13 (Systat Software, Inc.). The statistical analyses of the force/ATPase-[Ca2+] fit parameters were performed in all groups by one-way analysis of variance as appropriate. Statistical significance was defined as p < 0.05.
Author Contributions
A. V. D., N. S., S. L. R., and P. P. d. T. designed the experiments. A. V. D., N. S., M. Z., and J. L. M. performed research. A. V. D., S. L. R., N. S., and P. P. d. T. analyzed results. A. V. D., S. L. R., and P. P. d. T. wrote the paper.
Acknowledgments
We are indebted to Drs. Yuanhua Cheng and Stefan Lindert for sharing the troponin Protein Data Bank structure (40) used in the MD simulations. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant ACI-1053575. This research was supported by equipment and facilities provided by National Institute of Health Grant “Loyola Research Computing Core” 1G20RR030939.
This work was supported in part by National Institutes of Health Grants HL62426 and HL75494 (to P. P. d. T.) and HL092321 (to S. L. R.) and American Heart Association Grant 13POST14510047 (to A. V. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HCM
- hypertrophic cardiomyopathy
- RCM
- restrictive cardiomyopathy
- TnI
- troponin I
- IP
- inhibitory peptide
- TnC
- troponin C
- SP
- switch peptide
- cTnI
- cardiac TnI
- LDA
- myofilament length-dependent activation
- SL
- sarcomere length
- cTnC
- cardiac troponin C; c
- cTnT
- cardiac troponin T
- Fmax
- maximum Ca2+-saturated developed force
- EC50
- [Ca2+] at which force development is 50% of Fmax
- MD
- molecular dynamics
- BDM
- butanedione monoxime
- kCa
- rate of Ca2+ activation
- ktr
- rate of rapid release-restretch force redevelopment
- klin
- rate of the linear phase of force relaxation
- Tlin
- duration of the slow linear relaxation phase
- BES
- 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid
- A2P5
- diadenosine pentaphosphate
- HDTA
- hexamethylenediaminetetraacetic acid.
References
- 1. Towbin J. A. (2014) Inherited cardiomyopathies. Circ. J. 78, 2347–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., Seidman J. G., Seidman C., Granzier H., Labeit S., Frenneaux M., and Thierfelder L. (2002) Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 30, 201–204 [DOI] [PubMed] [Google Scholar]
- 3. Seidman C. E., and Seidman J. G. (2011) Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res. 108, 743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sen-Chowdhry S., Syrris P., and McKenna W. J. (2010) Genetics of restrictive cardiomyopathy. Heart Fail. Clin. 6, 179–186 [DOI] [PubMed] [Google Scholar]
- 5. Mogensen J., Kubo T., Duque M., Uribe W., Shaw A., Murphy R., Gimeno J. R., Elliott P., and McKenna W. J. (2003) Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 111, 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kobayashi T., and Solaro R. J. (2005) Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 67, 39–67 [DOI] [PubMed] [Google Scholar]
- 7. de Tombe P. P., Mateja R. D., Tachampa K., Ait Mou Y., Farman G. P., and Irving T. C. (2010) Myofilament length dependent activation. J. Mol. Cell. Cardiol. 48, 851–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang P., Kirk J. A., Ji W., dos Remedios C. G., Kass D. A., Van Eyk J. E., and Murphy A. M. (2012) Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 126, 1828–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yumoto F., Lu Q.-W., Morimoto S., Tanaka H., Kono N., Nagata K., Ojima T., Takahashi-Yanaga F., Miwa Y., Sasaguri T., Nishita K., Tanokura M., and Ohtsuki I. (2005) Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem. Biophys. Res. Commun. 338, 1519–1526 [DOI] [PubMed] [Google Scholar]
- 10. Gomes A. V., and Potter J. D. (2004) Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol. Cell. Biochem. 263, 99–114 [DOI] [PubMed] [Google Scholar]
- 11. Davis J., Wen H., Edwards T., and Metzger J. M. (2008) Allele and species dependent contractile defects by restrictive and hypertrophic cardiomyopathy-linked troponin I mutants. J. Mol. Cell. Cardiol. 44, 891–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kobayashi T., and Solaro R. J. (2006) Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J. Biol. Chem. 281, 13471–13477 [DOI] [PubMed] [Google Scholar]
- 13. Sequeira V., Wijnker P. J., Nijenkamp L. L., Kuster D. W., Najafi A., Witjas-Paalberends E. R., Regan J. A., Boontje N., Ten Cate F. J., Germans T., Carrier L., Sadayappan S., van Slegtenhorst M. A., Zaremba R., Foster D. B., et al. (2013) Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ. Res. 112, 1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van der Velden J., and de Tombe P. P. (2014) Heart failure: a special issue. Pflugers Arch. 466, 1023–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar M., Govindan S., Zhang M., Khairallah R. J., Martin J. L., Sadayappan S., and de Tombe P. P. (2015) Cardiac myosin binding protein C and troponin-I phosphorylation independently modulate myofilament length dependent activation. J. Biol. Chem. 290, 29241–29249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Konhilas J. P., Irving T. C., Wolska B. M., Jweied E. E., Martin A. F., Solaro R. J., and de Tombe P. P. (2003) Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J. Physiol. 547, 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hanft L. M., Biesiadecki B. J., and McDonald K. S. (2013) Length dependence of striated muscle force generation is controlled by phosphorylation of cTnI at serines 23/24. J. Physiol. 591, 4535–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hanft L. M., and McDonald K. S. (2009) Sarcomere length dependence of power output is increased after PKA treatment in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 296, H1524–H1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Velden J., de Jong J. W., Owen V. J., Burton P. B., and Stienen G. J. (2000) Effect of protein kinase A on calcium sensitivity of force and its sarcomere length dependence in human cardiomyocytes. Cardiovasc. Res. 46, 487–495 [DOI] [PubMed] [Google Scholar]
- 20. Rao V. S., Korte F. S., Razumova M. V., Feest E. R., Hsu H., Irving T. C., Regnier M., and Martyn D. A. (2013) N-terminal phosphorylation of cardiac troponin-I reduces length-dependent calcium sensitivity of contraction in cardiac muscle. J. Physiol. 591, 475–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wijnker P. J., Foster D. B., Tsao A. L., Frazier A. H., dos Remedios C. G., Murphy A. M., Stienen G. J., and van der Velden J. (2013) Impact of site-specific phosphorylation of protein kinase A sites Ser23 and Ser24 of cardiac troponin I in human cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 304, H260–H268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biesiadecki B. J., Kobayashi T., Walker J. S., Solaro R. J., and de Tombe P. P. (2007) The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 100, 1486–1493 [DOI] [PubMed] [Google Scholar]
- 23. Wijnker P. J., Sequeira V., Foster D. B., Li Y., Dos Remedios C. G., Murphy A. M., Stienen G. J., and van der Velden J. (2014) Length-dependent activation is modulated by cardiac troponin I bisphosphorylation at Ser23 and Ser24 but not by Thr143 phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 306, H1171–H1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nixon B. R., Thawornkaiwong A., Jin J., Brundage E. A., Little S. C., Davis J. P., Solaro R. J., and Biesiadecki B. J. (2012) AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J. Biol. Chem. 287, 19136–19147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papadaki M., Vikhorev P. G., Marston S. B., and Messer A. E. (2015) Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 108, 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng Y., Rao V., Tu A.-Y., Lindert S., Wang D., Oxenford L., McCulloch A. D., McCammon J. A., and Regnier M. (2015) Troponin I mutations R146G and R21C alter cardiac troponin function, contractile properties, and modulation by PKA-mediated phosphorylation. J. Biol. Chem. 290, 27749–27766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker J. S., Walker L. A., Margulies K., Buttrick P., and de Tombe P. (2011) Protein kinase A changes calcium sensitivity but not crossbridge kinetics in human cardiac myofibrils. Am. J. Physiol. Heart Circ. Physiol. 301, H138–H146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dvornikov A. V., Dewan S., Alekhina O. V., Pickett F. B., and de Tombe P. P. (2014) Novel approaches to determine contractile function of the isolated adult zebrafish ventricular cardiac myocyte. J. Physiol. 592, 1949–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stehle R., Solzin J., Iorga B., and Poggesi C. (2009) Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 458, 337–357 [DOI] [PubMed] [Google Scholar]
- 30. Brenner B., and Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Tombe P. P., and Stienen G. J. (2007) Impact of temperature on cross-bridge cycling kinetics in rat myocardium. J. Physiol. 584, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindhout D. A., Li M. X., Schieve D., and Sykes B. D. (2002) Effects of T142 phosphorylation and mutation R145G on the interaction of the inhibitory region of human cardiac troponin I with the C-domain of human cardiac troponin C. Biochemistry. 41, 7267–7274 [DOI] [PubMed] [Google Scholar]
- 33. Takahashi-Yanaga F., Morimoto S., Harada K., Minakami R., Shiraishi F., Ohta M., Lu Q. W., Sasaguri T., and Ohtsuki I. (2001) Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 33, 2095–2107 [DOI] [PubMed] [Google Scholar]
- 34. Deng Y., Schmidtmann A., Redlich A., Westerdorf B., Jaquet K., and Thieleczek R. (2001) Effects of phosphorylation and mutation R145G on human cardiac troponin I function. Biochemistry 40, 14593–14602 [DOI] [PubMed] [Google Scholar]
- 35. Kruger M., Zittrich S., Redwood C., Blaudeck N., James J., Robbins J., Pfitzer G., and Stehle R. (2005) Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. J. Physiol. 564, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wen Y., Xu Y., Wang Y., Pinto J. R., Potter J. D., and Kerrick W. G. (2009) Functional effects of a restrictive-cardiomyopathy-linked cardiac troponin I mutation (R145W) in transgenic mice. J. Mol. Biol. 392, 1158–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lang R., Gomes A. V., Zhao J., Housmans P. R., Miller T., and Potter J. D. (2002) Functional analysis of a troponin I (R145G) mutation associated with familial hypertrophic cardiomyopathy. J. Biol. Chem. 277, 11670–11678 [DOI] [PubMed] [Google Scholar]
- 38. Poggesi C., Tesi C., and Stehle R. (2005) Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 449, 505–517 [DOI] [PubMed] [Google Scholar]
- 39. Ait-Mou Y., Hsu K., Farman G. P., Kumar M., Greaser M. L., Irving T. C., and de Tombe P. P. (2016) Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc. Natl. Acad. Sci. U.S.A. 113, 2306–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng Y., Lindert S., Kekenes-Huskey P., Rao V. S., Solaro R. J., Rosevear P. R., Amaro R., McCulloch A. D., McCammon J. A., and Regnier M. (2014) Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys. J. 107, 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lindert S., Cheng Y., Kekenes-Huskey P., Regnier M., and McCammon J. A. (2015) Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys. J. 108, 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zamora J. E., Papadaki M., Messer A. E., Marston S. B., and Gould I. R. (2016) Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Phys. Chem. Chem. Phys. 18, 20691–20707 [DOI] [PubMed] [Google Scholar]
- 43. Sumandea M. P., Pyle W. G., Kobayashi T., de Tombe P. P., and Solaro R. J. (2003) Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J. Biol. Chem. 278, 35135–35144 [DOI] [PubMed] [Google Scholar]
- 44. Liu B., Lopez J. J., Biesiadecki B. J., and Davis J. P. (2014) Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS One 9, e86279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang M., Martin J. L., Kumar M., Khairallah R. J., and de Tombe P. P. (2015) Rapid large-scale purification of myofilament proteins using a cleavable His6-tag. Am. J. Physiol. Heart Circ. Physiol. 309, H1509–H1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeda S., Yamashita A., Maeda K., and Maéda Y. (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41 [DOI] [PubMed] [Google Scholar]
- 47. Witayavanitkul N., Ait Mou Y., Kuster D. W., Khairallah R. J., Sarkey J., Govindan S., Chen X., Ge Y., Rajan S., Wieczorek D. F., Irving T., Westfall M. V., de Tombe P. P., and Sadayappan S. (2014) Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J. Biol. Chem. 289, 8818–8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chandra M., Rundell V. L., Tardiff J. C., Leinwand L. A., De Tombe P. P., and Solaro R. J. (2001) Ca2+ activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am. J. Physiol. Heart Circ. Physiol. 280, H705–H713 [DOI] [PubMed] [Google Scholar]
- 49. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 50. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. MacKerell A. D. Jr., Feig M., and Brooks C. L. 3rd (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 [DOI] [PubMed] [Google Scholar]
- 52. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 53. Ulrich E., Lalith P., Max L. B., Tom D., Hsing L., and Lee G. P. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 54. Berendsen H., and Postma J. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684 [Google Scholar]
- 55. Hoover W. G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 56. Nosé S. (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52, 255–268 [Google Scholar]
- 57. Nosé S., and Klein M. (1986) Constant-temperature-constant-pressure molecular-dynamics calculations for molecular solids: application to solid nitrogen at high pressure. Phys. Rev. B Condens. Matter 33, 339–342 [DOI] [PubMed] [Google Scholar]
- 58. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 [Google Scholar]