

Abstract
TNF is a master pro-inflammatory cytokine whose pathogenic role in inflammatory disorders has long been attributed to induction of pro-inflammatory mediators. TNF also activates cell survival and death pathways, and recent studies demonstrated that TNF also causes inflammation by inducing cell death. The default response of most cells to TNF is survival and NF-κB-mediated up-regulation of pro-survival molecules is a well-documented protective mechanism downstream of TNFR1. Recent studies revealed existence of an NF-κB-independent cell death checkpoint that restricts cell demise by inactivating RIPK1. Disruption of this checkpoint leads to RIPK1 kinase-dependent death and causes inflammation in vivo. These revelations bring complexity to the control of TNF-induced cell death, and suggest clinical benefit of RIPK1 inhibitors in TNF-driven human inflammatory disorders.
The uncertain biological role of TNF-mediated cell death
Tumor necrosis factor (TNF) is well established as a major driver of inflammation and is a pharmacological target in several inflammatory disorders. However, despite its initial discovery over 40 years ago [1, 2], TNF-mediated cell death has long remained an in vitro phenomenon and its physiological and patho-physiological roles uncertain. The clinical success of anti-TNF biologics such as adalimunab, etanercept and infliximab in treating inflammatory disorders has been attributed to their effectiveness in blocking TNF from binding to its receptors TNFR1 and TNFR2. It is thought that this blockade reduces inflammation because TNFR1 can no longer activate the MAPKs and canonical NF-κB pathways, which normally would have collectively led to the transcriptional up-regulation of pro-inflammatory genes that underlie the inflammatory pathology [3]. But it is now becoming increasingly clear from recent studies that TNF can also promote and exacerbate inflammation by inducing cell death, in the form of apoptosis or necroptosis [4]. The inflammatory response can result from the sensing by pattern recognition receptors (PRRs) of damage associated molecular patterns (DAMPs) exposed or released by the dying cells. In addition, or alternatively, the inflammatory response can also originate from the loss of barrier function caused by the death of epithelial cells and the subsequent sensing of pathogen associated molecular patterns (PAMPs) present on microbes that have breached the barrier. This connection between cell death and inflammation provides the impetus to understand how TNF-mediated cell death is regulated, in the hope of identifying new therapeutic targets for the treatment of a growing list of inflammatory pathologies. Engagement of TNFR1 by TNF results in the sequential assembly of a membrane bound primary signaling complex (complex I, see Glossary) that drives gene activation and of a secondary cytoplasmic complex (Complex II) that mediates cell death [5, 6]. Nevertheless, in most cell types, activation of TNFR1 does not induce death but instead triggers a robust pro-survival response. Thus, while TNF has the capability to induce cell death, this response is suppressed unless some cell death checkpoints are disrupted. The difficulty in studying the function of TNF-mediated cell death is due to an incomplete molecular understanding of how TNF is able to dictate which of two divergent cellular responses, cell survival versus cell demise, is triggered. Early studies demonstrated that NF-κB plays a determinant role in this cell fate decision [7]. However, subsequent tissue culture studies revealed the existence of an early NF-κB-independent cell death checkpoint in the TNFR1 signaling pathway, whose physiological relevance was demonstrated in two recently published studies [8–13]. In this review, we will discuss the two TNFR1 cell death checkpoints (Box 1 & 2) and describe how the new findings on the early NF-κB-independent cell death checkpoint may open doors for new therapeutic opportunities for the treatment of some inflammatory disorders and cancers. These studies indicate that our ability to understand the physiological and patho-physiological roles of TNF-dependent cytotoxicity may finally be at hand.
Trends Box.
TNF can cause inflammation by activating NF-κB transcriptional responses, as well as by inducing cell death, in the form of apoptosis and necroptosis.
The NF-κB-mediated up-regulation of pro-survival molecules serve as a late cell death checkpoint protecting cells from TNF-mediated RIPK1-independent apoptosis.
A receptor proximal NF-κB-independent cell death checkpoint protects the cells from TNF-mediated RIPK1 kinase-dependent apoptosis and necroptosis.
This early checkpoint inactivates RIPK1 via a two-step mechanism that promotes ubiquitylation of RIPK1 first and phosphorylation of RIPK1 second.
Inactivating the kinase activity of RIPK1 prevents inflammation in TNF-driven murine models of inflammation; RIPK1 kinase inhibitors may thus have great clinical benefit in TNF-mediated human pathologies.
The late NF-κB-dependent cell death checkpoint
The membrane-bound TNFR1 complex I (also known as known as TNFR1-SC) forms within seconds following engagement of TNFR1 by TNF, and drives expression of pro-survival molecules via activation of the canonical NF-κB pathway (Figure 1). A network of poly-ubiquitin chains tightly regulates the dynamic assembly of complex I and the subsequent activation of the NF-κB pathway. These ubiquitin chains are required for the activation of the TAK1-IKK kinase cascade that ultimately results in the translocation of the NF-κB heterodimer p50/p65 to the nucleus for pro-survival gene expression. In brief, the activated receptor independently recruits TRADD and RIPK1 via homotypic death domain (DD) interactions. TRADD then serves as a platform for the recruitment of TRAF2 and/or TRAF5, which subsequently attract cIAP1 and cIAP2 to the receptor complex [14, 15]. RIPK1, and possibly other components of the complex, are conjugated with K63-, K11- and K48-linked poly-ubiquitin chains by cIAP1/2 [16–21]. The ubiquitin chains generated by cIAP1/2 allow further recruitment of LUBAC (composed of HOIP, HOIL-1 and SHARPIN), which adds complexity to the ubiquitin network by conjugating RIPK1, NEMO, TRADD and TNFR1 with M1-linked poly-ubiquitin chains [15, 17, 22, 23]. The ubiquitin chains added to the different complex I components by cIAP1/2 and LUBAC serve as scaffolds for the recruitment, activation and retention of the TAB2/3-TAK1 and NEMO-IKKα-IKKβ kinase complexes. These kinases are brought to the ubiquitin chains via the ubiquitin-binding domain (UBD) present in TAB2/3, which specifically binds K63-linked chains, and NEMO, which preferentially binds M1-linked chains [16, 24–28]. It is not completely understood why signaling molecules in complex I are modified with ubiquitin chains of different linkages. One recent study reported that K63-linked chains are good substrate for M1-linked poly-ubiquitylation [29], suggesting a model in which hybrid K63/M1-linked poly-ubiquitin chains on RIPK1 allow close proximity between the TAB2/3-TAK1 and NEMO-IKKα-IKKβ complexes, and thereby facilitate TAK1-catalyzed activation of IKKα and IKKβ (Figure 1). Phosphorylation of IKKβ by TAK1 then results in the IKKβ-mediated phosphorylation of IκBα, a signal for its ubiquitylation-dependent proteasomal degradation. IκBα degradation permits translocation of the NF-κB heterodimer p50/p65 to the nucleus where it drives transcription of multiple pro-inflammatory and pro-survival genes [30]. In order to ensure a transient and controlled TNF response, the ubiquitylation-dependent activation of NF-κB requires active repression. This repression is in part realized by deubiquitylases (DUBs) that dismantle the ubiquitin network associated with complex I. A20 was initially proposed to repress the NF-κB pathway by removing the K63-linked poly-ubiquitin chains attached to RIPK1 and by promoting RIPK1 proteasomal degradation by conjugating it with K48-linked chains [31]. These molecular functions of A20 have however been challenged by more recent studies [23, 32–34]. CYLD, another DUB, is reported to limit NF-κB activation by removing K63- and M1-linked poly-ubiquitin chains on several complex I components (including TRAF2, NEMO and RIPK1), thereby disrupting the ubiquitin scaffold required for the recruitment and activation of the TAB2/3-TAK1 and NEMO-IKKα-IKKβ complexes [23, 35–38].
Figure 1. Two sequential cell death checkpoints in the TNFR1 pathway.
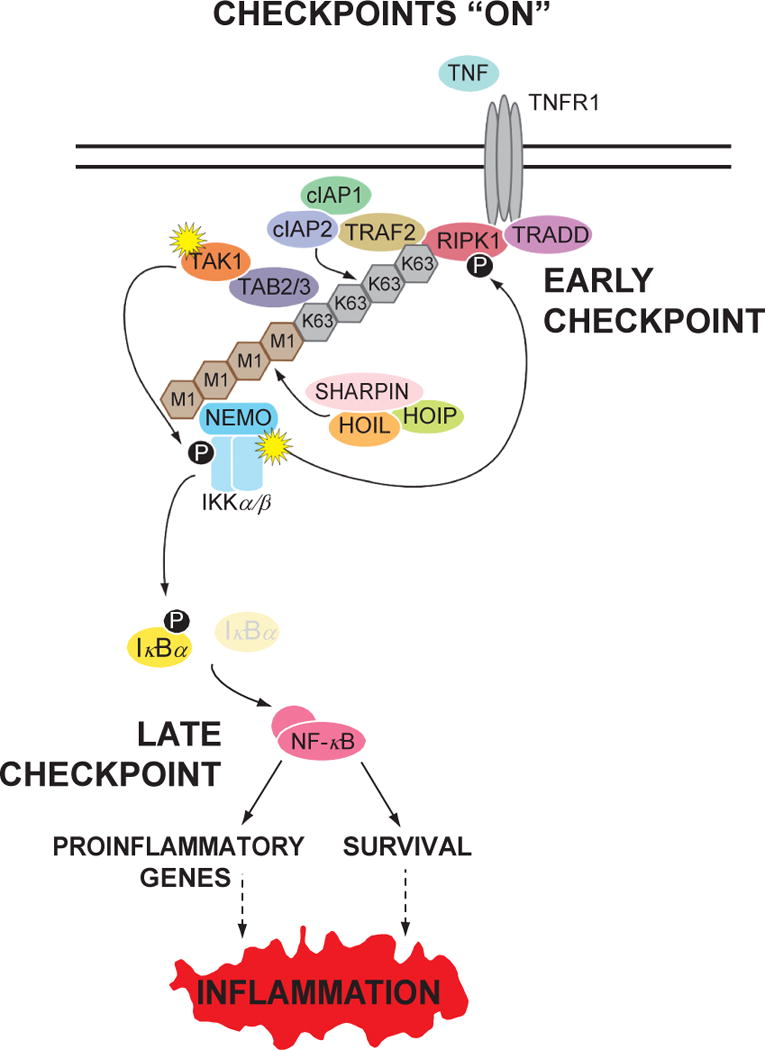
Most cells are protected from TNF-mediated cell death due to the presence two sequential cell death checkpoints that are turned on upon initiation of signaling from TNFR1. The early checkpoint is initiated by the ubiquitylation of RIPK1 by the TRAF2-cIAP1/2 and LUBAC E3 ligases. This process does not require gene transcription and occurs within seconds after ligand binding. Ubiquitylation of RIPK1 functions as a scaffold to recruit the TAB2/3-TAK1 and NEMO-IKKα/β kinase complexes to activate the latter. Within a few minutes after stimulation, NEMO-IKKα/β then phosphorylates RIPK1, which prevents RIPK1 from associating with and activating downstream death-signaling molecules. Under these conditions, RIPK1 functions in a survival-signaling mode. Active NEMO-IKKα/β also leads to the late checkpoint by phosphorylating IκBα, resulting in its degradation and induction of NF-κB-dependent gene transcription. The late checkpoint is dependent on the NF-κB-mediated induction of pro-survival genes, which occurs within an hour after stimulation and serves to reinforce the early checkpoint and to inhibit the formation of death-signaling complexes. Since NF-κB also induces a number of pro-inflammatory genes, this leads to an inflammatory response.
The pro-survival molecules induced by NF-κB counteract the death signal emerging from TNFR1 in multiple ways. The cytosolic complex II assembles slowly and transiently following TNF stimulation. It requires dissociation of TRADD from complex I and its subsequent association with FADD, which serves as a platform for Caspase-8 recruitment, Caspase-8 activation and apoptosis induction [6, 39]. cFLIP, a NF-κB-inducible gene product, is a Caspase-8 homolog that lacks catalytic activity and competes with FADD binding. Accordingly, under NF-κB transcriptionally active conditions, cFLIP levels are high and the transient formation of complex II is inhibited. In contrast, when NF-κB activity is repressed and cFLIP levels low, Caspase-8 is activated and the cells succumb by apoptosis [40]. In line with a role for RIPK1 in NF-κB activation, RIPK1 deficient cells succumb by apoptosis upon single TNF stimulation [25, 41–43]. Nevertheless, RIPK1 deficiency does not affect TNF-mediated NF-κB activation in every cell type, thereby suggesting that RIPK1 can generate a pro-survival signal via an NF-κB-independent mechanism [44–46]. Interestingly, although reported as part of complex II, RIPK1 was demonstrated to be dispensable for TNF-mediated apoptosis in conditions where the induced expression of pro-survival molecules by NF-κB is inhibited. This was demonstrated following genetic deletion of the NF-κB component p65, the expression of a dominant negative degradation-resistant form of IκBα (IκBαSR) that prevent nuclear translocation of p50/p65 or in presence of cycloheximide (CHX), which inhibits translation of the mRNAs encoding pro-survival molecules [6, 11, 13, 47]. The TNF-mediated TRADD-, FADD- and Caspase-8-dependent apoptotic complex that forms in NF-κB inhibited conditions has been defined as complex IIa [48] (Figure 2). Other NF-κB-dependent gene products can inhibit death elsewhere in the pathway. For instance, BCL-2 is a well established NF-κB-dependent gene that can block the mitochondria-dependent death pathway (for review see [49]). Thus, the late NF-κB-dependent checkpoint is a transcription-dependent genetic reprogramming of the cell to achieve a more permanent death-resistant state.
Figure 2. Disruption of the checkpoints leads to cell death via different mechanisms.
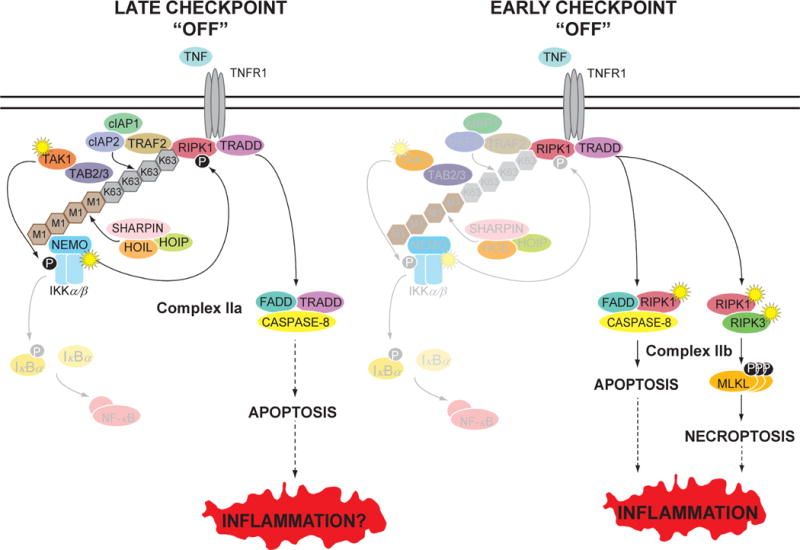
(Left panel) If the late NF-κB-dependent checkpoint is disrupted, this leads to FADD/Caspase-8-dependent apoptosis. This is caused by the failure to replenish cFLIP, an NF-κB-inducible gene that antagonizes Caspase-8, allowing Caspase-8 to undergo auto-catalytic processing and activation. In this case, the activation of Caspase-8 and apoptosis does not require RIPK1. This death-signaling complex has been termed complex IIa. TNF-mediated apoptosis resulting from disruption of the late checkpoint, via the combined deletion of REL-A, REL-B and c-REL in the gut, resulted in only minimal inflammation in vivo. (Right panel) If the early NF-κB-independent checkpoint is disrupted, RIPK1 converts to a death-signaling mode where it associates with Caspase-8 to initiate apoptosis. This death-signaling complex has been termed complex IIb. If Caspase-8 is not available, RIPK1 can associate with RIPK3 to initiate necroptosis. In both forms of cell death, the enzymatic activity of RIPK1 is required. In vivo studies have shown that disruption of the early checkpoint and subsequent RIPK1 kinase-dependent cell death to be highly inflammatory. RIPK1 kinase-dependent necroptosis may cause inflammation via the release of DAMPs. RIPK1 kinase-dependent apoptosis may cause inflammation via the loss of barrier function thereby facilitating microbial intrusion or via incomplete removal of excessive apoptotic debris, which then undergoes secondary necrosis to generate DAMPs. In both panels, the loss of the checkpoint is indicated by the faded molecules, signifying their absence or lack of activity.
The early NF-κB-independent cell death checkpoint
In contrast to the late cell death checkpoint, the early checkpoint in the TNFR1 pathway is not dependent on transcription. The first evidence of the existence of an additional NF-κB-independent cell death checkpoint came from studies using RIPK1-deficient cells reconstituted with WT RIPK1 or with a form of RIPK1 that is mutated in its ubiquitin acceptor site (K377R) [8, 25]. As mentioned above, RIPK1 ubiquitylation is required for full NF-κB activation, and it was therefore not surprising to see the RIPK1-K377R reconstituted cells undergo apoptosis following TNF stimulation. The interesting part came from the observation that the RIPK1-deficient cells reconstituted with the RIPK1-K377R mutant were still more sensitive to TNF-induced death than those reconstituted with wild type RIPK1, even in the setting of NF-κB blockade [8]. These results demonstrate that RIPK1 ubiquitylation additionally regulates another cell death checkpoint than NF-κB in the TNFR1 pathway. This notion was further supported by studies showing that perturbation in RIPK1 ubiquitylation, obtained by depletion of cIAP1/2 or LUBAC, also resulted in increased sensitivity to TNF-mediated death [47, 50–55]. The sensitization was demonstrated to result from aberrant complex II formation and apoptosis induction. However, contrary to apoptosis originating from complex IIa assembly in NF-κB inhibited conditions, cell death in the absence of cIAP1/2 or LUBAC was greatly dependent on RIPK1 and its kinase activity. The cytosolic RIPK1-FADD-Caspase-8 complex was therefore defined as complex IIb (Figure 2) to distinguish it from complex IIa [48]. These observations led to the model that ubiquitylation of RIPK1 signals survival by functioning as a brake on its pro-death tendency, which is revealed when this post-translational modification is disrupted. In line with a role of ubiquitylation in preserving RIPK1 in a survival mode, loss of CYLD and therefore sustained ubiquitylation was shown to block complex IIb formation and apoptosis induction [47]. A20 was also recently reported to protect cells from TNF-mediated apoptosis by binding to M1-linked poly-ubiquitin chains and protecting them from CYLD-mediated degradation [23]. The importance of LUBAC-dependent ubiquitylation in limiting RIPK1-dependent cell death was also demonstrated in vivo. Indeed, a null mutation in the mouse gene encoding the LUBAC component SHARPIN results in a severe multi-organ inflammation phenotype, called chronic proliferative dermatitis, which is driven by TNF-mediated apoptosis and that can be completely prevented by crossing the mice with RIPK1 kinase-inactive knockin mice [53, 55].
Additional studies however demonstrated that while the conjugation of ubiquitin molecules to RIPK1 may serve to initiate this early cell death checkpoint, there are additional regulatory mechanisms subsequent to this modification. It was shown that TAK1, NEMO and IKKα/β are also key regulators of this checkpoint. Loss/inhibition of TAK1, NEMO or IKKα/β leads to RIPK1 kinase-dependent apoptosis but without affecting RIPK1 ubiquitylation in complex I [10, 11, 13]. Indeed, the addition of K63- and M1-linked poly-ubiquitin chains to RIPK1 precedes recruitment of the TAB2/3-TAK1 and NEMO-IKKα-IKKβ complexes. The pro-survival functions of TAK1, NEMO and IKKα/β in RIPK1 inactivation were also demonstrated independent of their roles in NF-κB. In the case of NEMO, this pro-survival function was even demonstrated to be dependent on its ability to associate with ubiquitylated RIPK1 [10]. In line with this, CYLD repression protects cells from TNF-mediated RIPK1 kinase-dependent apoptosis in absence of cIAP1/2 but not in TAK1 inhibited conditions, confirming regulation of RIPK1 killing potential downstream of its ubiquitylation [11]. It was demonstrated that ubiquitylation and IKKα/IKKβ-mediated phosphorylation of RIPK1 occur sequentially, and that any conditions affecting proper IKKα/IKKβ activation – such as absence of cIAP1/2, SHARPIN, TAK1 or NEMO – results in defective phosphorylation of RIPK1 in complex I leading to complex IIb assembly and RIPK1 kinase-dependent apoptosis [11]. Thus, IKKα/IKKβ-mediated phosphorylation of RIPK1 in complex I inhibits RIPK1 from initiating cell death, pointing to another layer of complexity in the regulation of this early NF-κB-independent cell death checkpoint. Importantly, this checkpoint was recently demonstrated to be critical for regulating inflammation in vivo. It was elegantly shown, using complex genetic mouse models, that NEMO has an NF-κB-independent role in protecting epithelial cells in the colon from TNF-mediated RIPK1 kinase-dependent cell death [12]. NEMO deficiency in intestinal epithelial cells (IEC) led to RIPK1 kinase-dependent death and subsequent microbiota-driven colitis [12]. The same group also reported an NF-κB-independent role of NEMO in protecting liver parenchymal cells from RIPK1 kinase-mediated apoptosis independently of death receptor activation [56]. These in vivo results are consistent with earlier observations obtained from a NEMO-deficient cell line as discussed above [10]. In line with the role of NEMO is recruiting IKKα/IKKβ to complex I and subsequent IKKα/IKKβ-mediated phosphorylation and inactivation of RIPK1 [13], these NEMO-deficient cells/tissues would be expected to have defective RIPK1 phosphorylation. In contrast to the severe mucosal inflammation observed following deletion of NEMO in the IEC, the combined deletion of the three transcriptional activators in the NF-κB family (REL-A, REL-B and c-REL) in the same tissue resulted in only minimal IEC cell death and inflammation. These results therefore demonstrate the presence of this NF-κB-independent cell death checkpoint in vivo, which protects cells from RIPK1 kinase-dependent apoptosis. Further, disruption of this early checkpoint has a more profound pathological consequence than disruption of the late NF-κB-dependent checkpoint.
Apart from apoptosis induction, the early cell death checkpoint was also shown to protect cells from necroptosis [9, 11, 13], a regulated form of necrosis that prevails when Caspase-8 activation fails or is inhibited, and whose physiological relevance has been demonstrated in various pathological conditions (viral infection, myocardial infarction and stroke, atherosclerosis, ischemia-reperfusion injury, pancreatitis, inflammatory bowel disease, systemic inflammatory response syndrome) [4, 57]. TNF-mediated necroptosis relies on RIPK1 kinase–dependent assembly of the necrosome, a cytosolic death complex with RIPK3 and MLKL as core components [4] (Figure 2). Necroptosis was initially suggested to occur upon recruitment of RIPK3 to a RIPK1-FADD-Caspase-8 complex, but the necrosome can also form independently of FADD and Caspase-8. It is presently unclear whether the necrosome originates from an inactive complex IIb or whether it forms in parallel. Nevertheless, the fact that the early TNFR1 cell death checkpoint protects cells from both RIPK1 kinase-dependent apoptosis and necroptosis suggests that this checkpoint serves to maintain RIPK1 in a kinase “repressed” state, thereby preventing unnecessary death.
The molecular complexity in regulating the early checkpoint
While the basic framework of this early checkpoint has now emerged, the molecular details of its regulation remain unclear and are likely to be complex. Since ubiquitylation of RIPK1 is crucial in the early checkpoint, the regulation of its ubiquitin editing machinery provides a first layer of complexity. The E3 ligases TRAF2/5, cIAP1/2 and LUBAC, and the DUBs A20 and CYLD, are likely to be regulated by post-transcriptional means. In this regard, Caspase-8 in its pro-survival role can proteolyze and remove CYLD, which serves to maintain ubiquitylation of RIPK1 [58]; illustrating how a ubiquitin-modifying enzyme can be regulated by another upstream enzyme. Furthermore, it was also recently reported that the DUB activities of A20 and CYLD could be activated by phosphorylation [59, 60]. It is also known that the TRAF2-cIAP1/2 E3 ligase complex can undergo K48-linked auto-ubiquitylation and subsequent proteasomal degradation, for instance when it is recruited to TNFR2 [61, 62]. However, this auto-degradation process does not seem to happen upon TRAF2-cIAP1/2 recruitment to TNFR1 where instead, this complex catalyzes K63-linked ubiquitylation [18, 50]. Why this E3 ligase complex functions in a non-degradative manner in TNFR1 signaling is unclear.
It is also unclear how phosphorylation of RIPK1 by IKKα/IKKβ leads to the inhibition of RIPK1 kinase-dependent death. Phosphorylation could directly alter its enzymatic activity or, alternatively, affect binding of RIPK1 to the death-inducing complex components or even prevent RIPK1 from dissociating from complex I. NIK, another NF-κB-related kinase, was also shown to phosphorylate RIPK1 in vitro [63]. In contrast to IKKα/β-mediated phosphorylation, which is inhibitory for RIPK1’s death signaling role, NIK-dependent phosphorylation has been suggested to have the opposite effect and to be required for TNF-mediated complex IIb assembly in cIAP1/2-depletion conditions. It is currently unclear how NIK-mediated phosphorylation activates RIPK1. Thus, the death-signaling function of RIPK1 could be both positively and negatively regulated by phosphorylation, as previously suggested [64], and characterization of phospho-acceptor site mutants will likely provide greater insights. Once the early checkpoint is disrupted, the substrate(s) phosphorylated by RIPK1 to propagate apoptosis or necroptosis has not been definitively identified. An interesting possibility is that RIPK1 autophosphorylates itself and the resultant conformational changes enhance binding of RIPK1 to the death complex components. Thus, multiple post-translational mechanisms, including ubiquitination, phosphorylation and proteolysis, regulate the early checkpoint and they remain to be fully characterized.
It is also important to mention that the conditions identified so far that inhibit the early NF-κB-independent checkpoint and promote RIPK1 kinase-dependent death (depletion/inhibition of cIAP1/2, LUBAC, TAK1, NEMO and IKKα/IKKβ) also affect the late NF-κB-mediated checkpoint to varying degrees. Indeed, all these proteins are also required for full NF-κB activation; it is therefore currently unknown whether a gain of function in RIPK1’s death signaling capability is sufficient to execute cell death or whether it also requires a down-modulation of NF-κB in concert. The identification of the IKK-mediated phosphorylation sites on RIPK1 should help to clarify this point. While the early checkpoint clearly operates in a NF-κB-independent manner, the late NF-κB checkpoint does function to sustain the early checkpoint, via the genes it up-regulates such as TRAF2 and cIAP1/2. Finally, it is also intriguing to note that caspase inhibition is sufficient to switch complex IIa-mediated RIPK1-independent apoptosis to necrosome-mediated RIPK1 kinase-dependent necroptosis. Activation of RIPK1 under these circumstances presumably originates from stabilization of a de-ubiquitylated form of RIPK1. Indeed, both RIPK1 and CYLD are substrates of Caspase-8 [58]. The fact that depletion/inhibition of cIAP1/2, TAK1, NEMO or IKKα/IKKβ still sensitizes cells to RIPK1 kinase-dependent necroptosis suggest that these proteins regulate RIPK1 activation at a different layer than Caspase-8 [9, 11, 13, 65, 66].
Physiological function of the early cell death checkpoint
The in vitro and in vivo inactivation of components of the TNFR1 pathway by means of pharmacological inhibitors or genetic approaches has allowed us to identify two main cell death checkpoints in the pathway. The fact that most cell types are resistant to TNF demise indicates that the “on” position of these checkpoints represent the default mode. It is now important to identify the physiological conditions that turn off each of these checkpoints. Contrary to the late NF-κB-mediated checkpoint, the early checkpoint is post-translationally regulated, indicating that it may provide a more facile and rapid way to regulate the fate of a cell stimulated by TNF. In the best-documented example, ligation of TNFR2 leads to the degradation of a pool of TRAF2-cIAP1/2 that switches the TNFR1 response [61, 62]. Ligation of TNFR1 leads to survival whereas simultaneous ligation of TNFR1 and TNFR2 leads to RIPK1 kinase-dependent death. Other members of the TNFRSF have been reported to have an even stronger effect than TNFR2, probably due to the bigger pool of TRAF2-cIAP1/2 that they degrade [63, 67, 68]. These observations suggest that TNF most probably triggers RIPK1 kinase-dependent death in an in vivo inflammatory context, where many ligands of the TNFRSF are generated at the same time.
The existence of the early NF-κB-independent cell death checkpoint and the fact that its disruption leads to RIPK1-dependent cell death raises the question as to why this cellular response has evolved. The most likely reason, postulated several decades ago, was that TNF-mediated cytotoxity evolved to provide a survival advantage against infectious diseases [69]. Therefore, we speculate that the early checkpoint may also serve to ‘sense’ the presence of pathogens if they encode or generate signals that disrupt the checkpoint. For instance, pathogens may encode inhibitors to block IKKs in an attempt to prevent pro-inflammatory gene expression. This would however render infected cells sensitive to RIPK1 kinase-dependent cell death and thereby facilitate elimination of the infected cells. Furthermore, RIPK1 kinase-dependent cell death has been shown to be inflammatory serving to further alert the immune response [4, 12, 53, 56, 70]. To date, the few concrete examples of a beneficial role for TNF-mediated cytotoxicity in anti-microbial responses are limited to viruses that encode caspase inhibitors and activate necroptosis [71]. The evidence that TNF-mediated apoptosis has a role in anti-microbial responses remains lacking. It is widely believed that apoptotic death is non-inflammatory, as opposed to necroptotic death. However, the recent studies of NEMO and the SHARPIN mutant mice now indicate that RIPK1-dependent apoptotic death can be inflammatory. A substantial proportion of the IEC-specific knockout of NEMO that were crossed to the Ripk3 knockouts, which can undergo only TNF-mediated apoptosis, still developed robust mucosal inflammation [12, 53]. Similarly, the skin inflammation in the SHARPIN-deficient mice was not fully reversed by crossing to the Ripk3−/− strain but was reversed with the additional deletion of Fadd in keratinocytes or one copy of Caspase-8 in the germline [55, 72]. The continued inflammation of the NEMO and SHARPIN-deficient models on the Ripk3−/− background, which cannot undergo necroptosis, strongly suggests that apoptosis can be inflammatory. This could come about from the loss of barrier function subsequent to epithelial cell death, the release of inflammatory mediators from dying cells or the failure to ‘mop’ up cellular debris due to excessive apoptosis leading to secondary necrosis. The cellular/tissue context where apoptosis is occurring is likely to be a key determinant of inflammation and the possibility exists that RIPK1-dependent apoptosis may be qualitatively different from other forms of apoptosis. The evolutionary conservation of the RIPK1-dependent apoptosis pathway provides the strongest argument that this TNF-mediated death response is likely to be important in anti-microbial defense. With the availability of mouse strains with genetic modifications that activate or inhibit RIPK1 kinase-dependent cell death, the role of this checkpoint in anti-microbial responses can now be addressed.
Clinical relevance of the early cell death checkpoint
In human inflammatory pathologies caused by TNF (such as rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease, psoriasis, hidradenitis suppurativa and asthma), it is an accepted paradigm that the inflammation is due to TNF-dependent induction of pro-inflammatory signaling pathways including the NF-κB and other MAPK pathways. The demonstration that the inflammation in the SHARPIN-deficient and IEC-specific NEMO-deficient mouse strains can be reversed by a kinase-inactive allele of RIPK1 provides formal proof that TNF-driven cell death can cause inflammation when the early checkpoint is disrupted [12, 53]. In light of this, it is highly probable that the TNF-dependent inflammation in some human patients may not be due to excessive NF-κB activation but rather be caused by dysregulated cell death due to genetic and/or environment perturbations in the early checkpoint. The role of TNF-driven cell death in the pathogenesis of IBD, rheumatoid arthritis and psoriasis should now be examined more closely and if shown to be involved, RIPK1 kinase inhibitors may be of therapeutic value. These RIPK1 inhibitors could be delivered topically or orally, avoiding the complications associated with injection or intravenous administration of anti-TNF biologics. RIPK1 inhibitors could potentially be of benefit to two groups of patients with monogenic defects. A proportion of EDA-ID (anhidrotic ectodermal dysplasia with immunodeficiency) patients with a genetic defect in NEMO develop colitis that is amenable to anti-TNF therapy [73–75]. Considering that these patients are already immunodeficient, anti-TNF therapy may not be the most appropriate treatment and in light of the data from the IEC-specific knockout of NEMO, RIPK1 kinase blockade may be preferable. A potential advantage of RIPK1 inhibitors to anti-TNF biologics is that they may not have as much of a global effect on suppressing immunity. Indeed, they block RIPK1-dependent death without affecting the NF-κB arm of the TNF response. While patients with genetic defects in SHARPIN have not been described to date, patients with defects in the HOIL-1 and HOIP components of LUBAC have been described [76, 77] and they share some of the phenotype present in the SHARPIN mutant mouse. Based on the SHARPIN studies, there is a possibility that the clinical defects in the HOIL-1 and HOIP patients could be due to inappropriate RIPK1-dependent cell death and if so, blockade of RIPK1 may be a therapeutic strategy.
Cancer is another disease where the early cell death checkpoint is likely to have a critical role. Despite its given name, TNF has largely failed as a single agent in anti-tumor therapy and this failure reflects the fact that TNF also has a potent pro-inflammatory effect and only a low dose of systemically administered TNF is tolerated. This failure could also be explained by the fact that with a few exceptions, most transformed cell lines are not susceptible to killing by TNF itself. The presence of the early checkpoint instead provides a survival signal to tumor cells. Indeed, SMAC mimetics, which disrupt the early checkpoint by causing the degradation of cIAP1/2, sensitizes tumor cells to RIPK1-dependent death [50, 51], and they are now in clinical trials for anti-cancer therapy [78]. With increasing understanding of the regulation of this early cell fate toggle in the TNFR1 pathway and the development of pharmacological disruptors of this checkpoint (e.g., SMAC mimetics), TNF may yet live up to its given name as a tumor necrosis factor.
Concluding Remarks
The emerging data point toward a critical role of the early NF-κB-independent cell death checkpoint in regulating TNF-driven inflammation. However, a lot remains unknown regarding the mechanistic regulation and the physiological regulators of this checkpoint (see Outstanding Questions). Presumably, the ability of this checkpoint to rapidly regulate cellular survival or demise provides an evolutionary advantage against microbial infections but this has remained poorly documented. Furthermore, the role this checkpoint may play in human inflammatory disorders remains to be clarified. This checkpoint is also functional in tumor cells but its role in tumorigenesis has not been examined thoroughly. Thus the study of this early checkpoint is in its early stages and much work remains to be done.
Outstanding Questions.
What are the post-translational mechanisms that regulate the catalytic activities of ubiquitin-modifying enzymes in the early NF-κB-independent cell death checkpoint? What mechanisms regulate the ubiquitylation of RIPK1 that prevents it from becoming a death-signaling molecule? How is control by E3 ligases such as TRAF2/5, cIAP1/2 and LUBAC and by deubiquitylases such as A20 and CYLD regulated?
While it is known that NEMO-IKK α/β is recruited to ubiquitylated RIPK1 and phosphorylates RIPK1 to inhibit its death-signaling function, how does phosphorylation turn off RIPK1? Is it due to an allosteric regulation of kinase activity? Does it involve the compartmentalization of RIPK1 at specific sub-cellular sites or its access to partner proteins?
What substrates phosphorylated by RIPK1 lead to kinase-mediated induction of cell death?
When and under what circumstances do other receptors undergo the TNFR1 switch from survival to death to initiate RIPK1-dependent cell death?
What microbial-derived products and infectious agents could encode products to modulate this early checkpoint to counter downstream inflammatory gene transcription and trigger RIPK1-dependent cell death? How might this be advantageous to the host?
Could the evidence from mouse studies that indicates that disruption of the early checkpoint and the ensuing RIPK1-dependent cell-death could cause inflammation also be a cause of human inflammatory disorders?
Could modulating this checkpoint be a therapeutic strategy in circumstances where oncogenes and tumors co-op this checkpoint to enhance survival during transformation?
Acknowledgments
We thank Diana Legarda for generating the figure. A.T.T. is supported by National Institutes of Health (NIH) grants AI052417, AI104521 and DK072201 and is a recipient of a Senior Research Award from the Crohn’s and Colitis Foundation. M. J. M. B. is supported by Belgian Science Policy (BELSPO) grant IAP7/32 and Research Foundation Flanders (FWO) grants G017212N, G013715N.
Glossary
- Biologics
Are any pharmaceutical product manufactured in, or extracted from, a living system, such as microorganisms, plant cells or animal cells. They are large and complex molecules, or even mixtures of molecules. Biologics are also known as biopharmaceuticals or biological
- Pattern recognition receptors (PRRs)
Represent an array of innate immune receptors that initiate the inflammatory response. They are expressed by cells of the immune system and/or barrier cells that line the outside world. PRRs detect a variety of conserved and invariant microbial components, called pathogen-associated molecular patterns (PAMPs), and endogenous intracellular molecules that are modified, released or exposed by dying cells, called damage-associated molecular patterns (DAMPs). PRRs have been classified into different families according to their subcellular localization, molecular structure and ligand repertoire
- Complex I
This macro-molecular complex is formed rapidly at the receptor upon binding of TNF. It contains TNFR1, TRADD, TRAF2, cIAP1/2, LUBAC, RIPK1, TAB2/3-TAK1 and NEMO-IKKα/β. This complex forms when TRAF2-cIAP1/2 and LUBAC ubiquitylate RIPK1, which then serves as a platform for the recruitment of the TAB2/3-TAK1 and NEMO-IKKα/β kinase complexes. Complex I regulates both cell death checkpoints, first by phosphorylating RIPK1 to inhibit its death-signaling function in the early checkpoint, and subsequently in the late checkpoint by phosphorylating I-κBα to activate NF-κB
- Complex IIa
At later points after TNFR1 ligation, a cytosolic complex containing TRADD, FADD, Caspase-8 and cFLIP is formed. If NF-κB (i.e., the late checkpoint) is blocked, cFLIP expression level diminishes and Caspase-8 undergoes auto-catalysis to initiate apoptosis. This death-signaling complex is termed Complex IIa and while RIPK1 may physically be present in this complex in low amounts, it is not required for the activation of Caspase-8 and apoptosis. If in addition to NF-κB, Caspase-8 is also blocked, Complex IIa can induce necroptosis and in this case, RIPK1 is required to activate RIPK3
- Complex IIb
This cytosolic death-signaling complex is formed when the early checkpoint is disrupted. The composition of this complex is likely similar to Complex IIa but it differs in the amount and requirement for RIPK1 and its kinase activity, which activates Caspase-8 via a mechanism that is poorly understood. Contrary to Complex IIa, TRADD is not required for the activation of Caspase-8. Blockade of Caspase-8 can cause RIPK1 to associate with RIPK3 to initiate necroptosis. It should be noted that all disruptions of the early checkpoint known to date also affected the late NF-κB-dependent checkpoint to varying degrees. Therefore, the loss of pro-survival gene transcription may contribute to the death-signaling capability of Complex IIb
- Poly-ubiquitin chains
Two non-degradative forms of poly-ubiquitin chains are necessary for the early checkpoint. RIPK1 is modified with both K63-linked poly-ubiquitin chains catalyzed by the TRAF2-cIAP1/3 E3 ligase complex, and with M1-linked poly-ubiquitin chains catalyzed by the LUBAC E3 ligase complex consisting of HOIL-1, HOIP and SHARPIN. Mix K63-linked and M1-linked poly-ubiquitin chains may be present. Un-conjugated K63-linked poly-ubiquitin chains may also be involved
- SMAC mimetics
SMAC/Diablo is a polypeptide released from mitochondria of cells undergoing apoptosis. SMAC mimetics are synthetic compounds that bind to cIAP1/2 causing them to undergo auto-ubiquitylation with K48-linked chains resulting in their degradation. These are the first described pharmacological disruptors of the early checkpoint
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kolb WP, Granger GA. Lymphocyte in vitro cytotoxicity: characterization of human lymphotoxin. Proc Natl Acad Sci U S A. 1968;61:1250–1255. doi: 10.1073/pnas.61.4.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruddle NH, Waksman BH. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism. J Exp Med. 1968;128:1267–1279. doi: 10.1084/jem.128.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nature reviews. Rheumatology. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 5.Hsu H, et al. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 6.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 7.Van Antwerp DJ, et al. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell MA, et al. Ubiquitination of RIP1 regulates an NF-kappaB-independent cell-death switch in TNF signaling. Curr Biol. 2007;17:418–424. doi: 10.1016/j.cub.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Donnell MA, et al. NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS One. 2012;7:e41238. doi: 10.1371/journal.pone.0041238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Legarda-Addison D, et al. NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ. 2009;16:1279–1288. doi: 10.1038/cdd.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dondelinger Y, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20:1381–1392. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlantis K, et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-kappaB-Dependent and -Independent Functions. Immunity. 2016;44:553–567. doi: 10.1016/j.immuni.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dondelinger Y, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60:63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 14.Vince JE, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284:35906–35915. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Dynek JN, et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010;29:4198–4209. doi: 10.1038/emboj.2010.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 18.Varfolomeev E, et al. c-IAP1 and c-IAP2 Are Critical Mediators of Tumor Necrosis Factor {alpha} (TNF{alpha})-induced NF-{kappa}B Activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahoney DJ, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci U S A. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertrand MJ, et al. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 21.Park SM, et al. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004;566:151–156. doi: 10.1016/j.febslet.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 22.Tokunaga F, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 23.Draber P, et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell reports. 2015;13:2258–2272. doi: 10.1016/j.celrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanayama A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–548. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Ea CK, et al. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 26.Wu CJ, et al. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 27.Komander D, et al. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009;10:466–473. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahighi S, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Emmerich CH, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A. 2013;110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayden MS, Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Seminars in immunology. 2014;26:253–266. doi: 10.1016/j.smim.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 32.Mevissen TE, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De A, et al. The deubiquitinase activity of A20 is dispensable for NF-kappaB signaling. EMBO Rep. 2014;15:775–783. doi: 10.15252/embr.201338305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu TT, et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity. 2013;38:896–905. doi: 10.1016/j.immuni.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright A, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–716. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Kovalenko A, et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 37.Trompouki E, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 38.Brummelkamp TR, et al. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 39.Salvesen GS, Walsh CM. Functions of caspase 8: the identified and the mysterious. Semin Immunol. 2014;26:246–252. doi: 10.1016/j.smim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuchiya Y, et al. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. International journal of molecular sciences. 2015;16:30321–30341. doi: 10.3390/ijms161226232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513:95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 43.Kelliher MA, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 44.Blackwell K, et al. Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IkappaB kinase activation. Mol Cell Biol. 2013;33:1901–1915. doi: 10.1128/MCB.01416-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gentle IE, et al. In TNF-stimulated cells, RIPK1 promotes cell survival by stabilizing TRAF2 and cIAP1, which limits induction of non-canonical NF-kappaB and activation of caspase-8. J Biol Chem. 2011;286:13282–13291. doi: 10.1074/jbc.M110.216226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong WW, et al. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17:482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, et al. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 48.Wilson NS, et al. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 49.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 50.Bertrand MJ, et al. cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 51.Petersen SL, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12:445–456. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moulin M, et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012;31:1679–1691. doi: 10.1038/emboj.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berger SB, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peltzer N, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell reports. 2014;9:153–165. doi: 10.1016/j.celrep.2014.08.066. [DOI] [PubMed] [Google Scholar]
- 55.Rickard JA, et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. 2014;3 doi: 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondylis V, et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell. 2015;28:582–598. doi: 10.1016/j.ccell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Linkermann A, Green DR. Necroptosis. The New England journal of medicine. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Donnell MA, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wertz IE, et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature. 2015;528:370–375. doi: 10.1038/nature16165. [DOI] [PubMed] [Google Scholar]
- 60.Thein S, et al. IKK regulates the deubiquitinase CYLD at the postsynaptic density. Biochem Biophys Res Commun. 2014;450:550–554. doi: 10.1016/j.bbrc.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, et al. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature. 2002;416:345–347. doi: 10.1038/416345a. [DOI] [PubMed] [Google Scholar]
- 62.Fotin-Mleczek M, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- 63.Boutaffala L, et al. NIK promotes tissue destruction independently of the alternative NF-kappaB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ. 2015;22:2020–2033. doi: 10.1038/cdd.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McQuade T, et al. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem J. 2013;456:409–415. doi: 10.1042/BJ20130860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 67.Vince JE, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol. 2008;182:171–184. doi: 10.1083/jcb.200801010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varfolomeev E, et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- 69.Old LJ. Tumor necrosis factor (TNF) Science. 1985;230:630–632. doi: 10.1126/science.2413547. [DOI] [PubMed] [Google Scholar]
- 70.Yatim N, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350:328–334. doi: 10.1126/science.aad0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mocarski ES, et al. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology. 2015;479–480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumari S, et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife. 2014;3 doi: 10.7554/eLife.03422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pai SY, et al. Allogeneic transplantation successfully corrects immune defects, but not susceptibility to colitis, in a patient with nuclear factor-kappaB essential modulator deficiency. J Allergy Clin Immunol. 2008;122:1113–1118 e1111. doi: 10.1016/j.jaci.2008.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanson EP, et al. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allergy Clin Immunol. 2008;122:1169–1177 e1116. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mizukami T, et al. Successful treatment with infliximab for inflammatory colitis in a patient with X-linked anhidrotic ectodermal dysplasia with immunodeficiency. J Clin Immunol. 2012;32:39–49. doi: 10.1007/s10875-011-9600-0. [DOI] [PubMed] [Google Scholar]
- 76.Boisson B, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boisson B, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. 2015;212:939–951. doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]