

Abstract
Large neutral amino acid transporter 1 (LAT1) is a solute carrier protein located primarily in the blood-brain barrier (BBB) that offers the potential to deliver drugs to the brain. It is also up-regulated in cancer cells, as part of a tumor’s increased metabolic demands. Previously, amino acid prodrugs have been shown to be transported by LAT1. Carboxylic acid bioisosteres may afford prodrugs with an altered physicochemical and pharmacokinetic profile than those derived from natural amino acids, allowing for higher brain or tumor levels of drug and/or lower toxicity. The effect of replacing phenylalanine’s carboxylic acid with a tetrazole, acylsulfonamide and hydroxamic acid (HA) bioisostere was examined. Compounds were tested for their ability to be LAT1 substrates using both cis-inhibition and trans-stimulation cell assays. As HA-Phe demonstrated weak substrate activity, its structure-activity relationship (SAR) was further explored by synthesis and testing of HA derivatives of other LAT1 amino acid substrates (i.e. Tyr, Leu, Ile, and Met). The potential for a false positive in the trans-stimulation assay caused by parent amino acid was evaluated by conducting compound stability experiments for both HA-Leu and the corresponding methyl ester derivative. We concluded that HA’s are transported by LAT1. In addition, our results lend support to a recent account that amino acid esters are LAT1 substrates, and that hydrogen bonding may be as important as charge for interaction with the transporter binding site.
Keywords: SLC7A5, Amino Acid, Acyl Sulfonamide, Tetrazole, Transporter Substrate, Transporter Inhibitor
Graphical Abstract
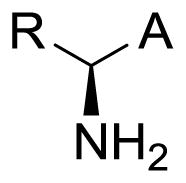
Large neutral amino acids such as tyrosine, tryptophan, leucine, isoleucine, phenylalanine, and methionine are actively transported across cell membranes by LAT1.1–6 Additionally, it transports amino acid-containing drugs such as gabapentin,7, 8 melphalan,9 L-DOPA10, 11 and baclofen12 across the blood-brain barrier (BBB). LAT1 is a sodium-independent heterodimeric membrane protein found mainly in the brain, thymus, testis, placenta, spleen, and skeletal muscle. Much of its appeal as a targeted drug delivery mechanism is due to its relative high abundance at the BBB versus other tissues (>100X BBB selective).2, 13 Besides being an instrument for CNS delivery, it has also been shown that LAT1 is up-regulated in many cancer types, including prostate,14 esophageal,15 colorectal,16 gastric,17 and non-small-cell lung cancer (NSCLC).18 Furthermore, it has been demonstrated that cancer growth can be inhibited by blocking LAT119–23 which is consistent with a cancer cell’s increased nutritional requirements. Thus, drugs that are able to mimic naturally-occurring amino acids (e.g. gabapentin) or prodrugs containing LAT1 recognition elements24–28 may have far-reaching utility for treating CNS diseases and cancer.
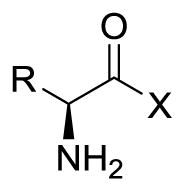
Another advantage favoring LAT1 for drug delivery is that it is relatively tolerant to substrate structural modifications.4, 29 For example, it has been shown that in addition to hydrophobic natural α-amino acids, some β and γ amino acids (e.g. gabapentin) are also transported by LAT1.8, 30–32 Despite some flexibility in the presentation of the amine and carboxylic acid functional groups, it has been maintained that both of these functional groups are essential for transporter recognition.4, 24, 33 The primary evidence supporting a carboxylic acid requirement has been centered on observations that replacement with esters and a closely related sulfonic acid resulted in loss of activity.4, 24, 29 However, many of the traditional carboxylic acid bioisosteres34, 35 have apparently not been explored. Lately, this story has become further convoluted as Nagamori et al. reported that several carboxylic esters and a hydroxamic acid derivative of L-leucine were LAT1 substrates.32 Their conclusions, based in part on a trans-stimulation assay,36 contradicted previous reports that esters do not bind LAT1.4, 24, 37
Our group had been exploring carboxylic acid bioisosteres as LAT1 substrates, including hydroxamic acids38, 39 as part of our ongoing effort to better understand LAT1 SAR40 prior to the recent report by Nagamori.32 We have been focused on modifying the carboxylic acid rather than the amine due to the potential metabolism and toxicity41 liabilities of the former. Besides contributing to the knowledge of LAT1 SAR, replacing the carboxylic acid functional group has the potential for altering the pharmacokinetics of prodrugs34, 35 intended for LAT1 transport. Moreover, learning what functional groups may serve as surrogates for the amino acid carboxyl opens up many possibilities for the design of drugs that might benefit from this delivery mechanism.
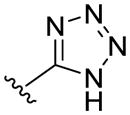
Though numerous carboxylic acid bioisosteres have been described,34, 35, 42, 43 we choose to prioritize the acylsulfonamide, tetrazole, and hydroxamic acid functionalities (Table 1). These groups were selected due to their comparable pKa and/or structural similarity to the carboxylic acid functional group,34 and they have previously demonstrated biological activity in other series.44–46 The acylsulfonamide (or sulfonimide) has a similar geometry and pKa as a carboxylic acid, and it was successfully applied as a cysteinyl leukotriene (LTE4) receptor antagonist that demonstrated greater activity than the parent carboxylic acid.47 Acylsulfonamides were also chosen because they are convenient to synthesize from the corresponding carboxylic acid. Though slightly larger than a carboxylic acid,48 tetrazoles faithfully reproduce their trigonal planar shape and acidity (pKa: 4.5–4.9), as the tetrazole anion is stabilized by delocalization. The tetrazole group, which is present in the orally active angiotensin II receptor antagonist Losartan,46 has the potential to improve oral bioavailability of resulting prodrugs relative to parent carboxylic acid. Hydroxamic acids (pKa: 8–9) are known primarily for their metal-chelating abilities; and though dramatically less acidic than the previous two bioisosteres, they have been reported as MAP/ERK kinase inhibitors where they displayed similar ADME properties to carboxylic acids.44, 49 However, use of hydoxamic acids could be limited by hydrolysis to parent carboxylic acid in vivo.50
Table 1.
Exchange efflux rate and uptake inhibition of [3H]-gabapentin in HEK-hLAT1 cells for carboxylic acid bioisosteres and their parent amino acids tested at 200 μM.
| |||||
|---|---|---|---|---|---|
| Compounda | A | R | Efflux Rateb | % Inhibitionc | pKa1, pKa2d |
| 1a (Phe) | -CO2H | PhCH2- | 3.6 ± 0.7 | 85 ± 0.6 | 1.8, 9.1 |
| 1b (Tyr) | -CO2H | p-HOPhCH2- | 2.6 ± 0.4 | 68 ± 0.5 | - |
| 1h (Gly) | -CO2H | H | 0.78 ± 0.2 | 33 ± 3 | - |
| 1i (Arg) | -CO2H | NH2(NH)CNH(CH2)3- | 0.75 ± 0.1 | 49 ± 2 | - |
|
| |||||
| 10a | -CONHSO2Me | PhCH2- | 0.61 ± 0.06 | 15 ± 0.7 | 1.8, 8.4 |
| 10b | -CONHSO2Me | p-HOPhCH2- | 0.63 ± 0.03 | 12 ± 0.4 | - |
| 11 |
|
PhCH2- | 0.61 ± 0.01 | 11 ± 1 | 2.5, 7.8 |
| 12a (HA-Phe) | -CONHOH | PhCH2- | 1.3 ± 0.1 | 24 ± 0.5 | 6.9, 8.0 |
| 12b (HA-Tyr) | -CONHOH | p-HOPhCH2- | 1.0 ± 0.1 | 39 ± 30 | - |
Cell assay data was obtained at least in triplicate. Amino acids and their corresponding derivatives possess S stereochemistry at the α-carbon.
Compounds were tested at 200 μM for their ability to cause efflux (fmol/min) of [3H]-gabapentin from pre-loaded HEK-hLAT1 cells.
Compounds were tested at 200 μM for their ability to inhibit uptake of [3H]-gabapentin into HEK-hLAT1 cells. Data is presented as % inhibition relative to background signal in the absence of a test compound.
Compounds were evaluated in both cis-inhibition and trans-stimulation assays using HEK cells engineered to overexpress human LAT1.51, 52 Cis-inhibition studies were used to identify LAT1 transport inhibitors, which may be potential substrates. However, to more directly identify substrates, we performed a trans-stimulation experiment36 which exploits LAT1’s alternating access mechanism8, 53 by loading cells with a radiolabeled substrate followed by incubation with extracellular test compound. The exchange efflux rate of the radiolabel in the presence of a test compound is compared with the efflux rate in the absence of the test compound. Compounds that are LAT1 substrates should increase the efflux rate of the radiolabeled amino acid compared with its efflux rate in the absence of test compound. We selected [3H]-gabapentin as a probe substrate due to its selectivity for LAT1 relative to other membrane transporters.8
Acyl sulfonamides 10a–b were prepared from protected amino acids according to methodology described by Drummond.54 Tetrazole bioisostere 11 was synthesized from the primary amide of Cbz-protected Phe in 3 steps using a previously published route,55 and our resulting NMR characterization was consistent with what had previously been reported. After discovering a lack of activity for tetrazole 11, we choose not to pursue additional amino acid analogs. Hydroxamic acids (HA’s) were synthesized using two different routes. Aromatic analogs (i.e. 12a–12d) were prepared using methodology previously described by Ahlford and Adolfsson.56 Due to problems with over-reduction of hydroxamic acids to give primary amide (e.g. 13e) during hydrogenolysis of benzyl protected hydroxamic acid, we used a different route to prepare HA’s of aliphatic amino acids (Scheme 1).
Scheme 1.

Synthesis of compounds 13e and 12e–12g. Reagents and conditions: (a) SOCl2, MeOH, 9e: 56%, 9f: 86%, 9g: 67%; (b) Boc2O, DCM, Boc-9g: 85%; (c) 7N NH3 in MeOH, 50 °C, sealed tube, 13e: 60%; (d) 50% NH2OH in water, MeOH or 1,4-dioxane, 12e: 23%, 12f: 3%, 12g: 20% (2 steps); (e) 4N HCl in 1,4-dioxane. 13e and 12e–g were purified by conversion to their HCl salts and recrystallization to >99% purity by HPLC.
Hydroxamic acids of aliphatic amino acids Leu, Ile and Met (12e–g) were synthesized according to Scheme 1. Nucleophilic acyl substitution with hydroxylamine on methyl esters 9 gave low yields, but avoided having to use an amine protecting group for Leu and Ile analogs. However, reaction of 9g with hydroxylamine to form HA-Met gave a complex mixture that could not be purified by recrystallization. We found that Boc protected 9g gave a cleaner conversion to HA-Met, albeit the recrystallized yield was still relatively poor (20% for steps de). Since our objective was to obtain HA’s of high purity with negligible levels of parent amino acids to avert a false positive result in our cell assay, we were generally unconcerned about isolated yield and the potential losses resulting from multiple recrystallization steps. Moreover, potentially better methods57–60 for preparing HA’s were not pursued, as the current routes provided satisfactory amounts of material for testing in a relatively short time period. In contrast, the yield for substitution with ammonia to generate leucinamide 13e was significantly better (60% recrystallized yield) than for the corresponding HA analogs. Generally HA’s demonstrated poor solubility in both water and organic solvents; however, we found that solubility was dramatically improved by conversion to the hydrochloride salt.
Of these three bioisosteres, only the hydroxamic acid 12a had significant activity in our trans-stimulation assay relative to non-substrates Gly and Arg (Table 1). We were surprised by this result. We had expected the tetrazole 11 and acylsulfonamides 10a to have been better surrogates for the acidic carboxylic acid than 12a given that the measured pKa1 values61 for the former (pKa1 = 2.5 and 1.8, respectively) were much closer to that of parent amino acid Phe 1a (pKa1 = 1.8) than HA-Phe 12a (pKa1 = 6.9) was. It is worth noting that our pKa1 values were considerably lower than those reported for these bioisosteres when they were present as isolated functional groups,34 which demonstrates as might be expected that the α-amino group depresses their pKa as it would for an adjacent carboxylic acid.
To determine whether HA’s of other LAT1 amino acid substrates (e.g. Leu, Ile, Met) were LAT1 ligands, compounds of Table 2 were prepared and tested. All of the HA’s, with the exception of 12h, had diminished activity in both our trans-stimulation and cis-inhibition assays relative to the parent amino acids. Based on their % inhibition of [3H]-gabapentin cell uptake or IC50 values, it is clear that HA’s are weaker ligands of LAT1 than the parent amino acids. And none of the HA’s had IC50 values below 200 μM in our assay. Conversely, all of the HA’s demonstrated greater efflux rates of [3H]-gabapentin from pre-loaded HEK-hLAT1 cells than did the negative controls Arg and Gly. The notable exception to this trend was HA-Gly 12h, which we did not expect to be a LAT1 substrate by analogy to its non-substrate, parent amino acid Gly. Of the HA’s tested, 12a and 12e–g (HA’s of Phe, Leu, Ile, Met) demonstrated significant activity relative to the negative controls. The larger efflux rates measured for HA-Leu 12e and HA-Ile 12f relative to HA-Phe 12a (1.5 vs. 1.3 fmol/min) were juxtaposed with the activity of the parent amino acids, in which Phe 1a demonstrated a superior efflux rate (3.6 fmol/min). It has previously been shown that both Leu and Ile have slightly greater LAT1 transport capacity (Vmax) values than Phe.63, 64 Since the trans-stimulation assay relies upon the kinetics of exchange between intracellular [3H]-gabapentin and extracellular test compound, it is conceivable that a similar trend for Vmax applies to the HA’s as it does to the parent amino acids. However, considering the bounce in our assay relative to the observed efflux rates, we cannot confidently distinguish the substrate activity of the HA’s from each other.
Table 2.
Exchange efflux rate and uptake inhibition of [3H]-gabapentin in HEK-hLAT1 cells for hydroxamic acids, related carboxylic acid derivatives, and their parent amino acids.
| |||||
|---|---|---|---|---|---|
| Compounda | X | R | Efflux Rateb | % Inhibitionc | IC50 (μM)d |
| 1a (Phe) | PhCH2- | 3.6 ± 0.7 | 85 ± 0.6 | - | |
| 1b (Tyr) | p-HOPhCH2- | 2.6 ± 0.4 | 68 ± 0.5 | - | |
| 1d (Trp) |
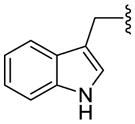
|
1.6 ± 0.3 | 79 ± 0.6 | - | |
| 1e (Leu) | -OH | (CH3)2CHCH2- | 3.2 ± 0.5 | 73 ± 0.7 | 87 ± 10 |
| 1f (Ile) | (S)-CH3CH2(CH3)CH- | 2.5 ± 0.1 | - | 150 ± 40 | |
| 1g (Met) | CH3S(CH2)2- | 2.4 ± 0.1 | - | 180 ± 3 | |
| 1h (Gly) | H | 0.78 ± 0.2 | 33 ± 3 | >200 | |
| 1i (Arg) | NH2(NH)CNH(CH2)3- | 0.75 ± 0.1 | 49 ± 2 | - | |
|
| |||||
| 12a (HA-Phe) | PhCH2- | 1.3 ± 0.1 | 24 ± 0.5 | - | |
| 12b (HA-Tyr) | p-HOPhCH2- | 1.0 ± 0.1 | 39 ± 30 | - | |
| 12d (HA-Trp) |
|
1.0 ± 0.1 | 35 ± 1 | - | |
| 12e (HA-Leu) | -NHOH | (CH3)2CHCH2- | 1.5 ± 0.1 | 50. ± 1 | >200 |
| 12f (HA-Ile) | (S)-CH3CH2(CH3)CH- | 1.5 ± 0.2 | - | >200 | |
| 12g (HA-Met) | CH3S(CH2)2- | 1.1 ± 0.01 | - | >200 | |
| 12h (HA-Gly) | H | 0.70 ± 0.1 | 38 ± 1 | >200 | |
|
| |||||
| 9e (Leu ester) | -OMe | 2.1 ± 0.2 | - | >200 | |
| 13e (Leucinamide) | -NH2 | (CH3)2CHCH2- | 0.69 ± 0.01 | - | >200 |
Cell assay data was obtained at least in triplicate. Amino acids and their corresponding derivatives possess S stereochemistry at the α-carbon.
Compounds were tested at 200 μM for their ability to cause efflux (fmol/min) of [3H]-gabapentin from pre-loaded HEK-hLAT1 cells.
Compounds were tested at 200 μM for their ability to inhibit uptake of [3H]-gabapentin into HEK-hLAT1 cells. Data is presented as % inhibition relative to background signal in the absence of a test compound.
Given the disparity in the literature4, 32, 37 as to whether esters are LAT1 substrates, we also tested the Leu methyl ester 9e, which was an intermediate in the preparation of HA-Leu 12e (Scheme 1). And to further probe the SAR for close-in derivatives of the HA’s (Table 2), we also decided to test the structurally-related primary amide Leucinamide 13e. Though ester 9e was recently reported to be a LAT1 substrate,32 to our knowledge this was the first time that 13e or any amino acid primary amide has been tested for LAT1 activity. Interestingly, 13e did not demonstrate significant substrate activity (efflux rate: 0.69 fmol/min). This result also indicated that 13e was sufficiently stable to the assay conditions so as not to generate adequate parent Leu 1e to cause trans-stimulation. Furthermore, it is apparent that the hydroxamic acid ‘-OH’ group plays an important role in the observed LAT1 activity; whether that be due to its effect on acidity, hydrogen bonding, or some other factor is currently unclear.
A different story unfolded for ester 9e. In a trans-stimulation experiment performed by Nagamori,32 both ester 9e and its parent Leu appeared to have almost identical activity. In our hands, 9e exhibited significantly less activity than parent Leu in both trans-stimulation (efflux rate: 2.1 vs. 3.2 fmol/min) and cis-inhibition assays (IC50: >200 μM vs. 87 μM). One possible explanation for this disparity may be due to the fact that the cells used by Nagamori were different from the cells we used. Nagamori et al. used non-transfected HeLa S3 cells (a cervical cancer cell line) whereas, we used HEK-hLAT1 cells52 that demonstrated 8-fold higher uptake of [3H]-gabapentin relative to the control cell line HEK-EV (supplementary material). LAT1 expression may have been higher in our transfected cells and the contribution of other transporters could differ between the two cell lines. We selected HEK cells due to their having relatively low levels of transporters,65 so we would expect the observed activity in our assays to be due solely to LAT1. Our IC50 value for 9e was consistent with earlier SAR presented by Uchino4 that the methyl ester of phenylalanine poorly inhibited uptake of L-[14C]-Phe into oocytes expressing LAT1. But the ostensible interpretation of the results from our trans-stimulation assay is the same as Nagamori’s—that methyl ester 9e does appear to be a LAT1 substrate, despite lacking an acidic carboxylic acid functional group.
Because of our concerns and those raised by others about the potential for a false positive result in LAT1 cell assays,37 we evaluated how much parent Leu 1e would need to be present as an impurity in test compounds (i.e. 9e, 12e, or 13e), either from the synthesis or formed under the conditions of the cell assay, to result in a significant efflux rate (>1 fmol/min) in our trans-stimulation assay. We tested the efflux rate at concentrations ranging from 4 μM up to 200 μM covering a range of Leu 1e impurity from 2% to 100%, respectively (in relation to previous studies). The background efflux rate (0.7 ± 0.05 fmol/min) was subtracted from total efflux and the net contribution to [3H]-gabapentin efflux rate is depicted in Figure 1. The effect of increasing concentrations of Leu 1e on the net LAT1 efflux rate was fitted to the Michaelis-Menten equation (Km of 36.8 ± 9.8 μM and Vmax of 1.99 ± 0.19 fmol/min). The Km and Vmax were similar to previously reported values.64 A Leu 1e concentration of 4 μM did not increase the LAT1 exchange rate significantly in comparison to background signal (p value=0.146), and higher concentrations were required to facilitate the exchange of [3H]-gabapentin. In fact, more than 10% of a parent Leu 1e impurity would be required to fully account for HA-Leu 12e’s LAT1 activation (1.5 ± 0.1 fmol/min). Nevertheless, contamination of test compounds with parent amino acid may result in increased efflux ratio, and this should be examined carefully when performing trans-stimulation assays.
Figure 1.

Plot of net exchange efflux rate (fmol/min) of [3H]-gabapentin from pre-loaded HEK-hLAT1 cells vs. Leu (1e) concentration (μM). Net efflux rate was calculated by subtracting the exchange rate without Leu (1e) (marked by a dotted line) from the efflux rate at Leu (1e) concentrations of 4, 10, 32, 80 and 200 μM. Solid line represents a non-linear regression fit of the data to Michaelis-Menten kinetics.
Compelled by the implications of Figure 1, we carefully scrutinized the purity of all of our HA’s by NMR and HPLC (supplementary material), in particular checking for the presence of residual parent amino acid. We recrystallized all of the HA’s at least once to improve purity, and the amount of parent amino acid detected by HPLC was less than 0.5%, and in most cases it was below our limit of detection. Thus, we conclude that for the HA’s of Table 2 substrate activity was not due to parent amino acid carried over from the synthesis.
In addition to being vigilant about purity, we also performed a series of simple stability experiments. Thus, we exposed 13C-labeled 9e and 12e (Figure 2; synthesized using similar methods as described for unlabeled 9e and 12e, above) to conditions to mimic our cell assays, including incubation of compounds with ‘buffer only’ or with ‘buffer and cells’ for various periods of time. The resulting mixture was analyzed by 13C NMR (supplementary material).
Figure 2.

Carbon-13 labeled leucine derivatives [13C]-9e (methyl ester) and [13C]-12e (HA-Leu) used in stability experiments to assess the amount of parent leucine that could form under the conditions of the LAT1 cell assays.
Within the time period of our cell assay (~5 mins), 13C-9e hydrolyzed to give 4% parent amino acid, whereas 13C-12e only gave a marginal increase in parent 13C-1e relative to its initial amount (0.8% vs. 0.4%, respectively) that was likely within the variability of NMR peak integration. To take the experiment further, we incubated compounds with cells for an hour at 37 °C. This resulted in a moderate increase in the amount of 13C-1e (12%) from ester 13C-9e, but only a nominal amount (1%) arising from HA-Leu 13C-12e hydrolysis. Even after incubation of HA-Leu 13C-12e with cells for 5h at 37 °C, only 3% 13C-1e was observed in the 13C NMR spectrum.
Due to 13C NMR analysis requirements, these stability experiments had to be performed with 1 mg of compound/well, which was ~25–50X more compound than typically used in our cell assays, done at 200 μM concentration. Consequently, we can’t rule out the possibility of enzyme saturation at this higher concentration, and that either 9e and/or 12e might be undergoing intracellular enzymatic cleavage as the assay is normally conducted. However, it was previously shown68 that leucinamide 13e is more sensitive to enzymatic hydrolysis than the leucine methyl ester 12e. So, if enzyme-catalyzed formation of parent Leu was problematic, it seems likely that leucinamide would also have given a false positive; yet, 13e lacked activity in our trans-stimulation assay.
Upon relating these stability results to Figure 1, it appears that the concentration of parent Leu 1e within the timeframe of our cell assay from either methyl ester 9e or HA-Leu 12e (~8 and 2 μM, respectively) does not explain our trans-stimulation assay results (2.1 and 1.5 fmol/min, respectively). For the observed activity to be solely due to parent amino acid, Figure 1 suggests that Leu would have to be present at ~80 and ~30 μM concentrations for 9e and 12e, respectively, and all within the 3 minutes of the assay. Our stability experiments indicated that ≤10% of these levels were actually present, supporting the notion that the observed exchange efflux of [3H]-gabapentin (Table 2) was mostly caused by the test compounds themselves. Even so, it is probable that a small fraction of the activity was due to Leu, particularly for the less stable methyl ester 9e.
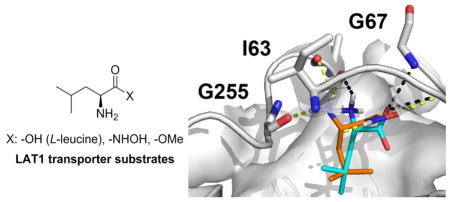
Though the atomic structure of the human LAT1 is not known, we have developed a homology model based on a structure of a related transporter, the arginine-agmatine transporter AdiC from E. coli.51,69 This model has been recently refined using newly characterized ligands and improved LAT1/AdiC alignment.40 The LAT1 model helped rationalize the amino acid selectivity among amino acid transporters, and virtual screening against this model followed by experimental testing identified previously unknown LAT1 ligands.51 Docking of 12e against our LAT1 model40 suggests that hydroxamic acids establish hydrogen bonds with backbone atoms of Ile63 and Gly67 in a manner similar to that of LAT1 amino acid ligands such as leucine (Figure 3A). We postulate that maintaining these hydrogen bonds is important for activity, and that this is the primary reason hydroxamic acids are LAT1 substrates. Interestingly, docking of non-ligand leucinamide 13e (Figure 3B) using two different docking programs (i.e. FRED70 and Glide SP71; supplementary material) does not rule out that 13e is a LAT1 ligand. We therefore estimated the binding energies of Leu (1e), Leu methyl ester (9e), HA-Leu (12e), and leucinamide (13e) to LAT1 using Molecular Mechanism Generalized Born Surface Area (MMGBSA) calculated by Prime (Schrödinger suite).72 Though the predicted ΔGbind values of −79, −64, and −58 kcal/mol for 1e, 12e, and 13e, respectively, correlated with experimental data, the predicted ΔGbind for 9e (−52 kcal/mol) did not. As a large component of the calculated binding energies was due to electrostatic interactions (i.e. ΔGCoulomb of −63, −53 and −47 kcal/mol for 1e, 12e and 13e, respectively), it is possible that our calculations are underestimating other interactions (e.g. dipole-dipole) with LAT1 that exist for ester 9e which account for its observed activity.
Figure 3.

Predicted binding mode of LAT1 with leucine and its analogs. Predicted pose of the known substrate leucine 1e is shown in cyan sticks, and the two leucine analogs including (A) the recently discovered substrate HA-Leu 12e and (B) non-ligand Leucinamide 13e are illustrated with orange and green sticks, respectively. The hydrogen bonds between LAT1 binding site residues and the leucine analogs are shown as dashed lines, including 1e (yellow), 12e (black), and 13e (black).
Based on our results and those recently reported by Nagamori,32 it appears that the previous view that LAT1 substrates must possess an acidic functional group needs to be revised. As most of the earlier conclusions4, 37, 73 were based on inhibition experiments (e.g. cis-inhibition or rat brain perfusion) at fixed concentrations of test compound, it is possible that those assays were not sensitive enough to detect substrates such as esters with weaker interactions with LAT1. The trans-stimulation assay may be more sensitive to identify weak ligands, as it is based on the exchange of pre-loaded substrates (e.g. [3H]-gabapentin) only for test compounds that employ LAT1 to cross a cell membrane, rather than inhibition potency.
Though we do not currently have an explanation for why tetrazoles and acylsulfonamides lacked activity, our data point toward LAT1 binding being less sensitive to the pKa of the carboxylic acid surrogate and more sensitive to its H-bonding capabilities. Thus, our results support the observation made by Nagamori32 that both oxygens of an amino acid carboxylic acid are likely involved in H-bonding with LAT1. We are currently expanding our work to include additional carboxylic acid bioisosteres35 to test this hypothesis.
Supplementary Material
Acknowledgments
E.A., A.F., K.F., L.H., and S.M. all thank the University of Nebraska at Kearney (UNK) University Research Fellows (URF) Program for financial support. E.A. also thanks the UNK Summer Student Research Program (SSRP). This work was supported by the UNK Research Services Council (RSC) and the Nebraska EPSCoR Undergraduate Research Experiences (URE) Program (to A.A.T.). We appreciate OpenEye Scientific Software Inc. for granting us access to its high-performance molecular modeling applications through its academic license program. This work was supported in part by the National Institutes of Health grant R01 GM108911 (to A.S. and C.C.), by the Department of Defense grant W81XWH-15-1-0539 (to A.S. and C.C.), and by the National Institutes of Health’s National Institute of General Medical Sciences grant U01 GM61390 (to A.A.Z., H.C.C., L.L. and K.M.G.).
Footnotes
Full experimental details, compound characterization data, IC50 curves, stability data and model refinement and ligand docking description can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kanai Y, Segawa H, Miyamoto K, Uchino H, Takeda E, Endou H. J Biol Chem. 1998;273:23629. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- 2.Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Proc Natl Acad Sci USA. 1999;96:12079. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kido Y, Tamai I, Uchino H, Suzuki F, Sai Y, Tsuji A. J Pharm Pharmacol. 2001;53:497. doi: 10.1211/0022357011775794. [DOI] [PubMed] [Google Scholar]
- 4.Uchino H, Kanai Y, Kim DK, Wempe MF, Chairoungdua A, Morimoto E, Anders MW, Endou H. Mol Pharmacol. 2002;61:729. doi: 10.1124/mol.61.4.729. [DOI] [PubMed] [Google Scholar]
- 5.del Amo EM, Urtti A, Yliperttula M. Eur J Pharm Sci. 2008;35:161. doi: 10.1016/j.ejps.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT. Curr Pharm Des. 2014;20:1422. doi: 10.2174/13816128113199990463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Welty DF. Pharm Res. 1996;13:398. doi: 10.1023/a:1016092525901. [DOI] [PubMed] [Google Scholar]
- 8.Dickens D, Webb SD, Antonyuk S, Giannoudis A, Owen A, Radisch S, Hasnain SS, Pirmohamed M. Biochem Pharmacol. 2013;85:1672. doi: 10.1016/j.bcp.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Cornford EM, Young D, Paxton JW, Finlay GJ, Wilson WR, Pardridge WM. Cancer Res. 1992;52:138. [PubMed] [Google Scholar]
- 10.Kageyama T, Nakamura M, Matsuo A, Yamasaki Y, Takakura Y, Hashida M, Kanai Y, Naito M, Tsuruo T, Minato N, Shimohama S. Brain Res. 2000;879:115. doi: 10.1016/s0006-8993(00)02758-x. [DOI] [PubMed] [Google Scholar]
- 11.Soares-da-Silva P, Serrao MP. Am J Physiol Renal Physiol. 2004;287:F252. doi: 10.1152/ajprenal.00030.2004. [DOI] [PubMed] [Google Scholar]
- 12.Van Bree JBMM, Audus KL, Borchardt RT. Pharm Res. 1988;5:369. doi: 10.1023/a:1015959628008. [DOI] [PubMed] [Google Scholar]
- 13.Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Yan AT, Cwirla SE, Grindstaff KK. Neuroscience. 2008;155:423. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 14.Yanagisawa N, Satoh T, Hana K, Ichinoe M, Nakada N, Endou H, Okayasu I, Murakumo Y. Cancer Biomark. 2015;15:365. doi: 10.3233/CBM-150486. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi H, Ishii Y, Takayama T. J Surg Oncol. 2005;90:233. doi: 10.1002/jso.20257. [DOI] [PubMed] [Google Scholar]
- 16.Ebara T, Kaira K, Saito J, Shioya M, Asao T, Takahashi T, Sakurai H, Kanai Y, Kuwano H, Nakano T. Anticancer Res. 2010;30:4223. [PubMed] [Google Scholar]
- 17.Ichinoe M, Mikami T, Yoshida T, Igawa I, Tsuruta T, Nakada N, Anzai N, Suzuki Y, Endou H, Okayasu I. Pathol Int. 2011;61:281. doi: 10.1111/j.1440-1827.2011.02650.x. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi K, Ogata S, Nakanishi K, Ozeki Y, Hiroi S, Tominaga S, Aida S, Matsuo H, Sakata T, Kawai T. Lung Cancer. 2010;68:58. doi: 10.1016/j.lungcan.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Yun D-W, Lee SA, Park M-G, Kim J-S, Yu S-K, Park M-R, Kim S-G, Oh J-S, Kim CS, Kim H-J, Kim J-S, Chun HS, Kanai Y, Endou H, Wempe MF, Kim DK. J Pharm Sci. 2014;124:208. doi: 10.1254/jphs.13154fp. [DOI] [PubMed] [Google Scholar]
- 20.Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, Hagenbeek T, Spits H, Reverso J, Ambrosetti D, Michiels JF, Bailly-Maitre B, Endou H, Wempe MF, Peyron JF. Leukemia. 2015;29:1253. doi: 10.1038/leu.2014.338. [DOI] [PubMed] [Google Scholar]
- 21.Ohkawa M, Ohno Y, Masuko K, Takeuchi A, Suda K, Kubo A, Kawahara R, Okazaki S, Tanaka T, Saya H, Seki M, Enomoto T, Yagi H, Hashimoto Y, Masuko T. Biochem Biophys Res Commun. 2011;406:649. doi: 10.1016/j.bbrc.2011.02.135. [DOI] [PubMed] [Google Scholar]
- 22.Shennan DB, Thomson J. Oncol Rep. 2008;20:885. [PubMed] [Google Scholar]
- 23.Wang Q, Holst J. Am J Cancer Res. 2015;5:1281. [PMC free article] [PubMed] [Google Scholar]
- 24.Gynther M, Laine K, Ropponen J, Leppanen J, Mannila A, Nevalainen T, Savolainen J, Jarvinen T, Rautio J. J Med Chem. 2008;51:932. doi: 10.1021/jm701175d. [DOI] [PubMed] [Google Scholar]
- 25.Killian DM, Hermeling S, Chikhale PJ. Drug Deliv. 2007;14:25. doi: 10.1080/10717540600559510. [DOI] [PubMed] [Google Scholar]
- 26.Walker I, Nicholls D, Irwin WJ, Freeman S. Int J Pharm. 1994;104:157. [Google Scholar]
- 27.Peura L, Malmioja K, Huttunen K, Leppänen J, Hämäläinen M, Forsberg MM, Rautio J, Laine K. Pharm Res. 2013;30:2523. doi: 10.1007/s11095-012-0966-3. [DOI] [PubMed] [Google Scholar]
- 28.Rautio J, Gynther M, Laine K. Ther Deliv. 2013;4:281. doi: 10.4155/tde.12.165. [DOI] [PubMed] [Google Scholar]
- 29.Ylikangas H, Malmioja K, Peura L, Gynther M, Nwachukwu EO, Leppanen J, Laine K, Rautio J, Lahtela-Kakkonen M, Huttunen KM, Poso A. ChemMedChem. 2014;9:2699. doi: 10.1002/cmdc.201402281. [DOI] [PubMed] [Google Scholar]
- 30.Jandeleit B, Fischer W-N, Koller KJ. WO2015117147. 2015
- 31.Jandeleit B, Fischer W-N, Koller KJ. WO2015117146. 2015
- 32.Nagamori S, Wiriyasermkul P, Okuda S, Kojima N, Hari Y, Kiyonaka S, Mori Y, Tominaga H, Ohgaki R, Kanai Y. Amino Acids. 2016;48:1045. doi: 10.1007/s00726-015-2158-z. [DOI] [PubMed] [Google Scholar]
- 33.Smith QR. Int Congr Ser. 2005;1277:63. [Google Scholar]
- 34.Ballatore C, Huryn DM, Smith AB., 3rd ChemMedChem. 2013;8:385. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meanwell NA. J Med Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 36.Fraga S, Serrao MP, Soares-da-Silva P. Eur J Pharmacol. 2002;441:127. doi: 10.1016/s0014-2999(02)01416-4. [DOI] [PubMed] [Google Scholar]
- 37.Rautio J, Karkkainen J, Huttunen K, Gynther M. Eur J Pharm Sci. 2014;66C:36. doi: 10.1016/j.ejps.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 38.Augustyn E, Heeren N, Miller S, Zur AA, Lin L, Giacomini K, Thomas AA. Abstract of Papers, 50th Midwest Regional Meeting of the American Chemical Society; St. Joseph, MO. Washington, DC: American Chemical Society; 2015. p. MWRM279. [Google Scholar]
- 39.Thomas AA, Augustyn E, Finke K, Hansen LM, Heeren N, Miller S, Zur AA, Lin L, Giacomini K. Presented at the 50th Midwest Regional Meeting of the American Chemical Society; St. Joseph, MO. October 2015; p. MWRM3. [Google Scholar]
- 40.Augustyn E, Finke K, Zur AA, Hansen L, Heeren N, Chien H-C, Lin L, Giacomini KM, Colas C, Schlessinger A, Thomas AA. Bioorg Med Chem Lett. 2016;26:2616. doi: 10.1016/j.bmcl.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li C, Benet LZ, Grillo MP. Chem Res Toxicol. 2002;15:1309. doi: 10.1021/tx020013l. [DOI] [PubMed] [Google Scholar]
- 42.Poulie CB, Bunch L. ChemMedChem. 2013;8:205. doi: 10.1002/cmdc.201200436. [DOI] [PubMed] [Google Scholar]
- 43.Lassalas P, Gay B, Lasfargeas C, James MJ, Tran V, Vijayendran KG, Brunden KR, Kozlowski MC, Thomas CJ, Smith AB, 3rd, Huryn DM, Ballatore C. J Med Chem. 2016;59:3183. doi: 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, Przybranowski SA, Sebolt-Leopold J, Van Becelaere K, Doherty AM, Kennedy RM, Marston D, Howard WA, Jr, Smith Y, Warmus JS, Tecle H. Bioorg Med Chem Lett. 2008;18:6501. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- 45.Uehling DE, Donaldson KH, Deaton DN, Hyman CE, Sugg EE, Barrett DG, Hughes RG, Reitter B, Adkison KK, Lancaster ME, Lee F, Hart R, Paulik MA, Sherman BW, True T, Cowan C. J Med Chem. 2002;45:567. doi: 10.1021/jm0101500. [DOI] [PubMed] [Google Scholar]
- 46.Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, Timmermans PB. J Med Chem. 1996;39:625. doi: 10.1021/jm9504722. [DOI] [PubMed] [Google Scholar]
- 47.Yee YK, Bernstein PR, Adams EJ, Brown FJ, Cronk LA, Hebbel KC, Vacek EP, Krell RD, Snyder DW. J Med Chem. 1990;33:2437. doi: 10.1021/jm00171a018. [DOI] [PubMed] [Google Scholar]
- 48.Costantino G, Maltoni K, Marinozzi M, Camaioni E, Prezeau L, Pin JP, Pellicciari R. Bioorg Med Chem. 2001;9:221. doi: 10.1016/s0968-0896(00)00270-4. [DOI] [PubMed] [Google Scholar]
- 49.Wallace EM, Lyssikatos J, Blake JF, Seo J, Yang HW, Yeh TC, Perrier M, Jarski H, Marsh V, Poch G, Livingston MG, Otten J, Hingorani G, Woessner R, Lee P, Winkler J, Koch K. J Med Chem. 2006;49:441. doi: 10.1021/jm050834y. [DOI] [PubMed] [Google Scholar]
- 50.Summers JB, Mazdiyasni H, Holms JH, Ratajczyk JD, Dyer RD, Carter GW. J Med Chem. 1987;30:574. doi: 10.1021/jm00386a022. [DOI] [PubMed] [Google Scholar]
- 51.Geier EG, Schlessinger A, Fan H, Gable JE, Irwin JJ, Sali A, Giacomini KM. Proc Natl Acad Sci USA. 2013;110:5480. doi: 10.1073/pnas.1218165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zerangue N. US 7,462,459 B2. 2008
- 53.Forrest LR, Kramer R, Ziegler C. Biochim Biophys Acta. 2011;1807:167. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 54.Drummond JT, Johnson G. Tetrahedron Lett. 1988;29:1653. [Google Scholar]
- 55.Sureshbabu VV, Venkataramanarao R, Naik SA, Chennakrishnareddy G. Tetrahedron Lett. 2007;48:7038. [Google Scholar]
- 56.Ahlford K, Adolfsson H. Catal Commun. 2011;12:1118. [Google Scholar]
- 57.Pirrung MC, Chau JHL. J Org Chem. 1995;60:8084. [Google Scholar]
- 58.Bailen MA, Chinchilla R, Dodsworth DJ, Najera C. Tetrahedron Lett. 2001;42:5013. [Google Scholar]
- 59.Thouin E, Lubell WD. Tetrahedron Lett. 2000;41:457. [Google Scholar]
- 60.Reddy AS, Kumar MS, Reddy GR. Tetrahedron Lett. 2000;41:6285. [Google Scholar]
- 61.Analiza. pKa Determination. 2016 May 8; http://www.analiza.com/physchem/pka.html.
- 62.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5. W.H. Freeman; New York: 2002. [Google Scholar]
- 63.Smith QR. J Nutr. 2000;130:1016S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- 64.Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, Nii T, Cha SH, Matsuo H, Fukushima J, Fukasawa Y, Tani Y, Taketani Y, Uchino H, Kim JY, Inatomi J, Okayasu I, Miyamoto K, Takeda E, Goya T, Endou H. Biochim Biophys Acta. 2001;1514:291. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 65.Mateus A, Matsson P, Artursson P. Mol Pharm. 2013;10:2467. doi: 10.1021/mp4000822. [DOI] [PubMed] [Google Scholar]
- 66.Segawa H, Fukasawa Y, Miyamoto K, Takeda E, Endou H, Kanai Y. J Biol Chem. 1999;274:19745. doi: 10.1074/jbc.274.28.19745. [DOI] [PubMed] [Google Scholar]
- 67.Kim CS, Cho SH, Chun HS, Lee SY, Endou H, Kanai Y, Kim do K. Biol Pharm Bull. 2008;31:1096. doi: 10.1248/bpb.31.1096. [DOI] [PubMed] [Google Scholar]
- 68.Shippey SS, Binkley F. J Biol Chem. 1958;230:699. [PubMed] [Google Scholar]
- 69.Schlessinger A, Khuri N, Giacomini KM, Sali A. Curr Top Med Chem. 2013;13:843. doi: 10.2174/1568026611313070007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGann M. J Chem Inf Model. 2011;51:578. doi: 10.1021/ci100436p. [DOI] [PubMed] [Google Scholar]
- 71.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 72.Sherman W, Beard HS, Farid R. Chem Biol Drug Des. 2006;67:83. doi: 10.1111/j.1747-0285.2005.00327.x. [DOI] [PubMed] [Google Scholar]
- 73.Ylikangas H, Peura L, Malmioja K, Leppanen J, Laine K, Poso A, Lahtela-Kakkonen M, Rautio J. Eur J Pharm Sci. 2013;48:523. doi: 10.1016/j.ejps.2012.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.