

Abstract
Rho family small GTPases are important regulators of neuronal development. Defective Rho regulation causes nervous system dysfunctions including mental retardation and Alzheimer's disease. Rac1, a member of the Rho family, regulates dendritic spines and excitatory synapses, but relatively little is known about how synaptic Rac1 is negatively regulated. Breakpoint cluster region (BCR) is a Rac GTPase-activating protein known to form a fusion protein with the c-Abl tyrosine kinase in Philadelphia chromosome-positive chronic myelogenous leukemia. Despite the fact that BCR mRNAs are abundantly expressed in the brain, the neural functions of BCR protein have remained obscure. We report here that BCR and its close relative active BCR-related (ABR) localize at excitatory synapses and directly interact with PSD-95, an abundant postsynaptic scaffolding protein. Mice deficient for BCR or ABR show enhanced basal Rac1 activity but only a small increase in spine density. Importantly, mice lacking BCR or ABR exhibit a marked decrease in the maintenance, but not induction, of long-term potentiation, and show impaired spatial and object recognition memory. These results suggest that BCR and ABR have novel roles in the regulation of synaptic Rac1 signaling, synaptic plasticity, and learning and memory, and that excessive Rac1 activity negatively affects synaptic and cognitive functions.
Introduction
Neuronal development involves diverse molecular and cellular events including the morphogenesis of dendrites and dendritic spines. Rho family small GTPases regulate dendritic and spine development (Carlisle and Kennedy, 2005; Ethell and Pasquale, 2005; Govek et al., 2005; Calabrese et al., 2006; Tada and Sheng, 2006), and defective Rho signaling is associated with mental retardation, Alzheimer's disease, and amyotrophic lateral sclerosis (Blanpied and Ehlers, 2004; Newey et al., 2005; van Galen and Ramakers, 2005; Boda et al., 2006; Zhao et al., 2006; Nadif Kasri and Van Aelst, 2008). Rac1, a well known Rho GTPase, regulates spine/synapse structure (Luo et al., 1996; Nakayama et al., 2000; Tashiro et al., 2000; Penzes et al., 2001; Zhang et al., 2003; Tashiro and Yuste, 2004; Carlisle and Kennedy, 2005; Tada and Sheng, 2006) and synaptic function (Lamprecht and LeDoux, 2004; Wiens et al., 2005; Haditsch et al., 2009; Rex et al., 2009).
Rac1 at excitatory synapses acts in concert with upstream regulators and downstream effectors. Known downstream effectors of synaptic Rac1 include p21-activated kinase (PAK) (Boda et al., 2006), IRSp53 (Soltau et al., 2002; Choi et al., 2005; Kim et al., 2009; Sawallisch et al., 2009), and WAVE (Wiskott–Aldrich syndrome protein family verprolin-homologous protein) (Choi et al., 2005; Pilpel and Segal, 2005; Kim et al., 2006). Activators of synaptic Rac1 that possess guanine nucleotide exchange factor (GEF) activity include Kalirin-7 (Penzes et al., 2003; Xie et al., 2007), βPix (Park et al., 2003; Zhang et al., 2005; Saneyoshi et al., 2008), and Tiam1 (Tolias et al., 2005, 2007; Penzes et al., 2008). However, relatively little is known about inhibitors of synaptic Rac1 with GTPase-activating protein (GAP) activity. Recently, α1-chimaerin, a diacylglycerol-binding protein with Rac1 GAP activity, has been shown to inhibit dendritic spines in a diacylglycerol-dependent manner (Van de Ven et al., 2005; Buttery et al., 2006).
Breakpoint cluster region (BCR) was originally identified as a protein that forms a fusion protein with c-Abl tyrosine kinase via Philadelphia chromosomal translocation, and thereby induces chronic myeloid leukemia (Groffen et al., 1984; Heisterkamp et al., 1985). BCR and its close relative active BCR-related (ABR) (Heisterkamp et al., 1989) show Rac GAP activities both in vitro and in vivo (Diekmann et al., 1991; Heisterkamp et al., 1993; Tan et al., 1993; Chuang et al., 1995; Voncken et al., 1995; Kaartinen et al., 2001; Cho et al., 2007). Importantly, BCR and ABR mRNAs are abundantly expressed in the brain (Heisterkamp et al., 1993). In addition, BCR and ABR double-null mice exhibit hyperactive astroglia and vestibular dysgenesis (Kaartinen et al., 2001, 2002), suggesting that BCR and ABR regulate neural development. However, their involvement in synaptic function/plasticity and higher brain functions has not been explored.
Here, we report that BCR and ABR proteins are mainly expressed in the brain, localize to excitatory synapses, and directly interact with PSD-95, an abundant postsynaptic scaffolding protein (Sheng and Hoogenraad, 2007). Our data from mice lacking BCR or ABR suggest novel involvements of these proteins in the regulation of neuronal Rac1 activity, long-term potentiation (LTP) maintenance, and learning and memory.
Materials and Methods
Constructs.
BCR and ABR expression constructs were generated by subcloning full-length human BCR (amino acids 1–1271) and ABR (1–859) into pEGFP-C1 (Clontech). For pull down, the C-terminal regions of BCR (1059–1271) and ABR (665–859) were subcloned into pGEX4T-1. For yeast two-hybrid, fragments of BCR (1072–1271) and ABR (665–859) were subcloned into pBHA.
Antibodies.
H6-BCR (amino acids 1059–1271) and GST-ABR (650–859) were used to generate polyclonal BCR (1359 rabbit) and ABR (1388 rabbit) antibodies. The following antibodies were purchased: BCR N-20, BCR C-20, synaptophysin, c-Jun N-terminal kinase (JNK), phospho-JNK (Santa Cruz), ABR, pan-extracellular signal-regulated kinase (ERK) (BD Biosciences Transduction Laboratories), synapsin I (Millipore Bioscience Research Reagents), PAK1, PAK3, phospho-PAK1/3, phospho-ERK1/2 (p44/42), p38 mitogen-activated protein kinase (MAPK), and phospho-p38 MAPK (Cell Signaling).
Animals.
Male Sprague Dawley rats were used for experiments including dissociated hippocampal culture, immunoblot analysis, in vivo coimmunoprecipitation, and electron microscopy. BCR−/− and ABR−/− mice in genetic backgrounds of 50% 129Sv plus 50% C57BL/6 and 50% 129Sv plus 50% Black Swiss, respectively (Voncken et al., 1995; Kaartinen et al., 2001), were backcrossed with C57BL/6 females. Littermates were used as controls for spine analysis, LTP/long-term depression (LTD) analysis, and all behavioral assays.
In situ hybridization.
Brain sections (12 μm thick) from postnatal day 14 (P14) rats were used. BCR- and ABR-specific hybridization probes encompassing the C terminus and the 3′-untranslated region were prepared from the following constructs: pGEM-7Zf containing nucleotides 3965–4284 of rat BCR (coding region, nucleotides 131–3943) and nucleotides 3965–4284 of rat ABR (coding region, 251–2866). Antisense riboprobes were prepared using Riboprobe System (Promega) and [α-35S]UTP.
Electron microscopy.
Hippocampal sections (60 μm) from 10-week-old rats were incubated overnight with BCR (C-20) or ABR (BD) antibodies and biotinylated secondary antibodies for 2 h. The sections were incubated with ExtrAvidin peroxidase (Sigma-Aldrich), and the immunoperoxidase was revealed by nickel intensified 3,30-diaminobenzidine tetrahydrochloride. Sections were osmicated, dehydrated, embedded with Durcupan ACM (Fluka), and cured for 48 h at 60°C. The area containing the pyramidal cell layer and stratum radiatum of the CA1 region was trimmed. Thin sections were cut, mounted on nickel grids, and stained with uranyl acetate and lead citrate. Grids were examined on a Hitachi H 7500 electron microscope (Hitachi) at 80 kV accelerating voltage. Images were captured with Digital Montage software driving a MultiScan cooled CCD camera (ES1000W; Gatan) and saved as TIFF files.
Neuron culture, transfection, and immunocytochemistry.
Cultured hippocampal neurons were prepared from embryonic day 18 rat hippocampi. Neurons were transfected by calcium phosphate transfection kit. For immunofluorescence staining, cultured neurons were fixed with 4% formaldehyde and 4% sucrose in PBS, and permeabilized with 0.2% Triton X-100 in PBS. Mouse brain sections (50 μm) for immunostaining were perfused with 4% formaldehyde and permeabilized by 0.5% Triton X-100. ABR signals, but not BCR signals, were amplified by tyramide signal amplification (PerkinElmer Life Sciences).
Pull down of active Rac1.
Amount of active Rac1 was determined using the EZ-Detect Rac1 activation kit (Pierce). Mouse brains (3∼4 weeks) were homogenized in the lysis/binding/washing buffer containing 320 mm sucrose, 10 mm Tris-HCl, pH 7.4, 5 mm EDTA, and protease inhibitors, followed by centrifugation. The supernatant was incubated with GST-PAK1-PBD (p21 binding domain) fusion proteins immobilized on glutathione-agarose bead at 4°C for 1 h, followed by precipitation, washing, and immunoblotting with Rac1 monoclonal antibodies.
DiOlistic spine labeling.
Hippocampal slices from 6-week-old mice were labeled by gene gun-mediated ballistic delivery of lipophilic dye DiI as described previously (Gan et al., 2000). Z-stacked images from the proximal stratum radiatum region of hippocampus were used for blind image analysis. Spines were defined by protrusions with a bulbous head wider than the neck and with length longer than 0.4 μm. Spine density was measured from 40 to 80 μm segments of secondary dendrites on approximately one to two dendrites per neuron. The narrowest point in the spine neck was defined as the border between spine head and neck. MetaMorph software (Molecular Devices) was used for quantitative analysis.
Electrophysiology.
LTP was induced in hippocampal slices (P21–P28) by four episodes of theta-burst stimulation with 10 s intervals. Theta-burst stimulation consisted of 10 stimulus trains delivered at 5 Hz; each train consisted of four pulses at 100 Hz. LTD was induced in hippocampal slices (P21–P28) by low-frequency stimulation (900 pulses at 1 Hz for 900 s). Average responses (±SEM) were expressed as percentage of baseline response (at least 10 min of stable responses). The experimenters were blind to mouse genotypes. Data were analyzed by using custom macros written in Igor (Wavemetrics). For whole-cell recordings, CA1 pyramidal cells (P21) were held at −70 mV using a MultiClamp 700B amplifier (Molecular Devices). Pipette solutions for miniature EPSCs (mEPSCs) contained the following (in mm): 110 Cs-gluconate, 30 CsCl, 20 HEPES, 4 MgATP, 0.3 NaGTP, 4 NaVitC, 0.5 EGTA (10 EGTA for AMPA vs NMDA receptor ratio). TTX (0.5 μm) and bicuculline (20 μm) were added to artificial CSF (ACSF) for mEPSC measurements. To obtain AMPA receptor-mediated EPSCs, neurons in ACSF with 100 μm picrotoxin were held at −70 mV and stimulated at 0.066 Hz. NMDA receptor-mediated EPSCs were isolated by changing the holding potential to +40 mV and adding 10 μm CNQX to ACSF. The 25∼30 EPSC samples were averaged to obtain the AMPA/NMDA EPSC ratio.
Morris water maze.
In the water maze test, BCR−/− male littermates (2–6 months of age) were trained to find the hidden platform in a 100-cm-diameter apparatus using three trials per day with intertrial intervals of 1 h; ABR−/− male littermates were trained using two successive trials per day. Probe tests were performed 24 h after the last training session. Target quadrant occupancy and exact platform crossings were determined using Ethovision software (Noldus).
Novel object recognition.
Novel object recognition tests were conducted in an open-field box on 4 successive days, with 10 min daily trials. For the first 2 d, mice were allowed to explore an empty box for adaptation. On the third day, mice were presented with two different objects: A and B (sample phase). After 24 h, the mice were exposed to two objects: A and C, where C is a novel object. Object exploration was defined as each instance in which a mouse's nose touched the object or was oriented toward and within 2 cm to the object.
Open-field, elevated plus-maze, and rotarod assays.
The open-field assay was performed in a white open-square chamber. The elevated plus-maze consisted of two open arms, two closed arms, and a center area, and was elevated to a height of 50 cm above the floor. Mice were placed in the center area and allowed to explore the space for 5 min. For the accelerating rotarod test, mice were placed on a rotarod rotating at a slow speed (∼4 rpm) for 30 s. The speed was then gradually increased from 4 to 40 rpm over 5 min (one trial per day), and the latencies to falling were measured.
Results
PSD-95 interacts with BCR and ABR but does not promote their synaptic localization
To better understand the role of PSD-95 in excitatory synaptic regulation, we screened PSD-95-interacting proteins using yeast two-hybrid assays, identifying BCR and ABR Rac GAPs as novel PSD-95-binding partners (Fig. 1A). The C termini of BCR and ABR interacted with the first two PDZ domains of PSD-95 and PSD-95 relatives (PSD-93/chapsyn-110, SAP97, and SAP102) (Fig. 1B). Mutant BCR and ABR with V-to-A point mutation of the C-terminal residue failed to bind PSD-95 family proteins. These interactions were further confirmed by glutathione S-transferase (GST) pull down and in vitro coimmunoprecipitation assays (Fig. 1C,D). In rat brains at postnatal days 7, 21, and 42, BCR formed a complex with PSD-95 (Fig. 1E). ABR was also in complex with PSD-95 (Fig. 1F). ABR, but not BCR, weakly associated with SAP97, contrary to the in vitro results.
Figure 1.

PSD-95 interacts with BCR and ABR but does not promote their synaptic localization. A, Domain structure of BCR and ABR. CC, Coiled coil; S/T kinase, serine-threonine kinase; DH, Dbl homology; PH, pleckstrin homology; C2, protein kinase C conserved region 2; PB, PDZ-binding motif that ends with STEV* or STDV*. B, The C termini of BCR and ABR interact with the first two PDZ domains of PSD-95 family proteins (PSD-95, PSD-93/chapsyn-110, SAP97, and SAP102) in yeast two-hybrid assays. GRIP1 PDZ4-6, A negative control. V1271A/V859A, A C-terminal point mutation. C, Pull down of PSD-95 and SAP97 expressed in HEK293T cells by GST-BCR/ABR. GRIP2, A negative control PDZ protein. D, PSD-95 forms a complex with BCR and ABR in HEK293T cells. Cells doubly transfected (Trf) with EGFP-BCR/ABR and PSD-95 were immunoprecipitated (IP) with EGFP antibodies and immunoblotted with PSD-95 antibodies. Input (Inp), 5%. E, F, PSD-95 forms a complex with BCR and ABR in the brain. The crude synaptosomal fraction of rat brain at indicated developmental stages were immunoprecipitated with BCR (1359) and ABR (1388) antibodies and immunoblotted with the indicated antibodies. S-SCAM, A negative control PDZ protein; IgG, control rabbit antibodies. Input (inp), 5%. G, H, BCR and ABR do not depend on PSD-95 binding for synaptic localization. Cultured hippocampal neurons were transfected (DIV 17–19) with WT or mutant BCR/ABR and monitored of the spine localization of the proteins, as determined by the spine/dendrite ratio. Data represent mean ± SEM. n = 13–20 neurons (20–25 ratios per neuron); *p < 0.05 (ANOVA for BCR, and Student's t test for ABR). Scale bars, 10 μm.
We next tested whether PSD-95 promotes synaptic localization of BCR and ABR. In cultured hippocampal neurons, the spine localization of a mutant BCR lacking PSD-95 binding (VA) was comparable with that of wild-type (WT) BCR (Fig. 1G). This suggests that BCR does not require PSD-95 binding for synaptic localization. Interestingly, deletion variants of BCR that lack the N-terminal coiled coil (CC) domain showed significantly reduced spine localization. In addition, a BCR variant containing the CC + S/T kinase domains showed spine localization comparable with that of WT BCR, suggesting that the CC domain is important. ABR, which lacks the CC and S/T kinase domains, showed much lower levels of spine localization compared with BCR (Fig. 1H). As expected, WT and VA ABR showed similar levels of spine localization. These results suggest that BCR and ABR do not require PSD-95 binding for synaptic localization, and that the CC domain of BCR promotes spine localization.
Expression patterns of BCR and ABR mRNAs and proteins in the brain
In situ hybridization revealed that BCR and ABR mRNAs are widely, but distinctly, expressed in various rat brain regions (Fig. 2A). For instance, BCR mRNAs were more abundant in the striatum and cerebellum than in other regions, whereas ABR signals were stronger in forebrain regions, including the olfactory bulb, cortex, and hippocampus. In the hippocampus, BCR transcripts were weaker in the CA3 region, whereas ABR mRNAs were similarly expressed in all hippocampal subregions.
Figure 2.

Expression patterns of BCR and ABR mRNAs and proteins. A, Widespread, but distinct, distribution patterns of BCR and ABR mRNAs in rat brains (P14), revealed by in situ hybridization. Ctx, Cortex; Cb, cerebellum; Hc, hippocampus; OB, olfactory bulb; St, striatum. Scale bar, 1 mm. B, BCR and ABR proteins are mainly expressed in the brain. Br, Brain; Sp, spleen; Lu, lung; Lv, liver; Sm, skeletal muscle; Kd, kidney; Ts, testis. C, Expression of BCR and ABR proteins in various brain regions. Stem, Brainstem; SC, spinal cord. D, Expression of BCR and ABR proteins during rat brain development. W, Week; M, month. E, Widespread distribution of BCR and ABR in subcellular fractions of adult rat brain (6 weeks). H, Homogenates; P2, crude synaptosomes; S2, supernatant after P2 precipitation; S3, cytosol; P3, light membranes; LP1, synaptosomal membranes; LS2, synaptosomal cytosol; LP2, synaptic vesicle-enriched fraction. F, Greater enrichment of BCR (relative to ABR) in PSD fractions, prepared by detergent extraction of the synaptosomal fraction, once with Triton X-100 (PSD I), twice with Triton X-100 (PSD II), once with Triton X-100 plus once with sarcosyl (PSD III). G, Abundant expression of BCR in the CA1 region of hippocampus, with successively lesser expression in the dentate gyrus (DG) and CA3 regions. WT and BCR−/− sections were doubly stained for BCR and NeuN (a neuron-specific protein). Scale bar, 250 μm. H, Similar expression levels of ABR in DG and CA1 regions, with lower expression in CA3. Brain sections were doubly stained for BCR and vGlut1 (an excitatory presynaptic protein; NeuN was not used for antibody incompatibility). Scale bar, 250 μm. I, Ultrastructural localization of BCR and ABR mainly in spines and dendrites of CA1 pyramidal neurons. Signals for BCR (I1–I3) and ABR (I4–I6), shown as dark DAB precipitates (red arrowheads), are mainly detected in dendritic spines (I1, I4; arrows) and dendritic trunks (I2, I5; arrows), although occasionally observed in axon terminals (I3, I6; arrows). Scale bar, 0.2 μm. J, Normal gross morphologies of BCR−/− and ABR−/− brains, revealed by Nissl staining. Scale bars, 1 and 0.25 mm.
BCR and ABR protein expression was mostly restricted to the brain, not or barely being detected in other tissues (Fig. 2B). BCR and ABR proteins were detected in multiple brain regions, including the cortex, hippocampus, cerebellum, and brainstem, as well as the spinal cord (Fig. 2C). During rat brain development, BCR expression levels were initially high at a late embryonic (E18) stage and maintained at slightly decreased levels throughout postnatal brain development, whereas ABR expression gradually increased, similar to the expression pattern of PSD-95 (Fig. 2D).
Both BCR and ABR proteins were widely distributed in subcellular fractions of rat brain extracts (Fig. 2E), although BCR was more abundant in synaptic fractions, such as crude synaptosome (P2) and synaptic membrane (LP1) fractions, than was ABR. Consistently, BCR was highly enriched in PSD fractions, including the most detergent-resistant PSD III fraction, similar to PSD-95 (Fig. 2F). In contrast, a relatively small portion of ABR was detected in PSD fractions, although it was present in PSD III.
Immunohistochemical analyses of brain slices from WT and knock-out (KO) mice revealed a differential expression of BCR and ABR proteins in hippocampal subregions. BCR protein was most abundant in the CA1 region and was expressed at successively lower levels in the dentate gyrus and the CA3 region (Fig. 2G), partly similar to the mRNA expression patterns (Fig. 2A). Substantial BCR signals were observed in the dendritic field of the CA1 region as well as the pyramidal cell body area. ABR proteins were expressed at similarly high levels in the dentate gyrus and CA1 regions, but were lower in the CA3 region (Fig. 2H). The weak ABR signals in the dendritic field may be because ABR, but not BCR, signals were amplified by tyramide.
Electron-microscopic analyses revealed that BCR proteins were detected in dendritic spines and trunks (Fig. 2I1,2), although signals at axon terminals were occasionally observed (Fig. 2I3). Overall, ABR distribution was similar to that of BCR, although ABR was less frequently found in dendritic spines than was BCR (Fig. 2I4–6). BCR−/− and ABR−/− mice had normal gross morphology of the brain (Fig. 2J). Therefore, BCR and ABR mRNAs and proteins have overlapping as well as distinct cellular, subcellular, and ultrastructural distribution patterns in the brain.
Deficiency of BCR or ABR in mice enhances basal Rac1 activity
BCR and ABR show Rac GAP activities in vitro and in vivo; for instance, monocytes derived from BCR and ABR double-null mice show enhanced basal Rac1 activity in assays pulling down GTP-bound Rac1 (Kaartinen et al., 2001). In BCR−/− and ABR−/− brain, basal Rac1 activities were significantly increased by ∼60%, relative to WT brain (Fig. 3A). In addition, phosphorylation levels of PAK1 and PAK3, which correlate with Rac1 activity (Edwards et al., 1999; Bokoch, 2003), were significantly increased in BCR−/− and ABR−/− brain, whereas the total protein levels were unchanged (Fig. 3A). The phosphorylation levels of ERK1/2 (p42/44), which act downstream of PAK1 (Stockton et al., 2007), were also significantly increased, without a corresponding change in total ERK1/2. Phosphorylation levels of JNK or p38 MAPK were unchanged. In addition, expression of other synaptic proteins, including PSD-95 and glutamate receptors, in BCR−/− and ABR−/− mice, was unchanged (Fig. 3B). These results indicate that a deficiency of BCR or ABR in mice leads to enhanced basal Rac1 activity in the brain, and suggest that BCR and ABR function as important negative regulators of neuronal Rac1 activity.
Figure 3.
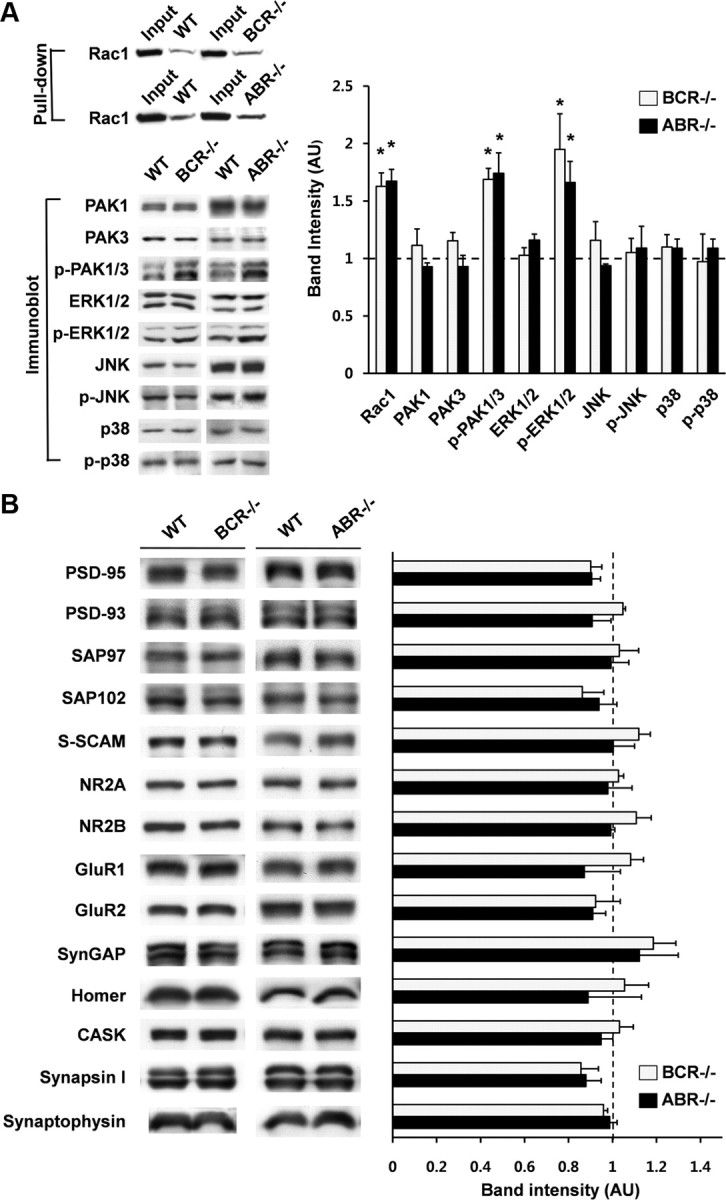
BCR−/− and ABR−/− mice show enhanced basal Rac1 activity. A, Enhanced basal Rac1 activity and phosphorylation levels of PAK1/3 and ERK1/2, but not JNK and p38 MAPKs, in BCR−/− and ABR−/− brains. Protein expression levels in KO mice were normalized to those in WT mice. n = 3–4 (3–4 weeks); *p < 0.05, **p < 0.01; Student's t test. B, Normal expression levels of other synaptic proteins in BCR−/− and ABR−/− mice. BCR and ABR, n = 3. Error bars indicate SEM.
BCR−/− and ABR−/− mice show a small increase in dendritic spine density but unaffected synaptic transmission
Because Rac1 positively regulates dendritic spines, we tested whether the enhanced basal Rac1 activity in BCR−/− and ABR−/− mice exerted a positive effect on dendritic spines. We visualized dendritic spines on the apical dendrites of CA1 pyramidal neurons by ballistic DiI delivery (Gan et al., 2000). BCR−/− and ABR−/− mice showed small, but significant, increases in spine density compared with WT animals (Fig. 4A,B), whereas spine head area was unchanged (Fig. 4C), suggesting that BCR and ABR negatively regulate dendritic spine density.
Figure 4.

BCR−/− and ABR−/− mice show a small increase in spine density but normal synaptic transmission. A–C, Increased spine densities in apical dendrites of BCR−/− and ABR−/− CA1 pyramidal neurons, visualized by ballistic DiI delivery. Scale bar, 5 μm. Spine density, n = 21 frames for BCR and 13 for ABR; spine head area, n = 18 for BCR and 15 for ABR; 3–5 brains; BCR, 6 weeks; ABR, 3 weeks; *p < 0.05, Student's t test. D–F, Normal frequencies and amplitudes of mEPSCs in BCR−/− and ABR−/− CA1 pyramidal neurons. mEPSC traces (D) and average values and cumulative distributions of mEPSC frequencies and amplitudes (E, F; black line, KO; gray line, WT) from WT and KO neurons. BCR, WT, n = 24 cells, 5 mice; KO, n = 19, 4; ABR, WT, n = 21, 3; KO, n = 18, 3. G, Normal AMPA/NMDA ratios of evoked EPSCs at BCR−/− and ABR−/− SC–CA1 synapses. BCR, WT, n = 6 cells, 4 mice; KO, n = 7, 3; ABR, WT, n = 9, 6; KO, n = 8, 6. Error bars indicate SEM.
We next tested whether these structural changes induce changes in synaptic transmission, by measuring mEPSCs. Both the frequencies and amplitudes of mEPSCs in BCR−/− and ABR−/− CA1 pyramidal neurons were comparable with those of WT animals (Fig. 4D–F). In addition, the ratio of evoked AMPA receptor-mediated currents to NMDA receptor-mediated currents at Schaffer collateral (SC)–CA1 pyramidal neuron (CA1) synapses was not changed in BCR−/− or ABR−/− mice (Fig. 4G). Together with the absence of changes in evoked AMPA receptor-mediated EPSCs (see below), these results suggest that both AMPA receptor- and NMDA receptor-mediated excitatory synaptic transmission are normal in BCR−/− and ABR−/− mice. This suggests that genetic ablation of BCR or ABR induces a small increase in spine density without affecting excitatory synaptic transmission.
BCR−/− and ABR−/− mice show reduced maintenance of LTP
Filamentous actin is a major cytoskeletal element of dendritic spines (Matus et al., 1982; Nimchinsky et al., 2002; Hayashi and Majewska, 2005; Matus, 2005; Tada and Sheng, 2006; Alvarez and Sabatini, 2007; Schubert and Dotti, 2007; Sekino et al., 2007; Bourne and Harris, 2008; Cingolani and Goda, 2008). Actin filaments lie in the downstream of Rac1 and have been associated with various forms of synaptic plasticity (Tada and Sheng, 2006; Cingolani and Goda, 2008). Thus, we measured LTP and LTD at SC–CA1 synapses of BCR−/− and ABR−/− hippocampus, using extracellular recordings. The input–output ratio, determined by plotting the slopes of field EPSPs (fEPSPs) against fiber volley amplitudes, was unchanged at BCR−/− and ABR−/− SC–CA1 synapses compared with that in WT mice (Fig. 5A,B), suggesting that AMPA receptor-mediated basal synaptic transmission is normal in both KO mice. In addition, paired-pulse facilitation at SC–CA1 synapses was normal in BCR−/− and ABR−/− mice (Fig. 5C,D), suggesting that the probability of neurotransmitter release was unchanged.
Figure 5.

BCR−/− and ABR−/− mice show reduced maintenance of LTP but normal LTD. A, B, Normal AMPA receptor-mediated basal synaptic transmission at BCR−/− and ABR−/− SC–CA1 synapses. BCR, n = 14 slices, 8 mice for both WT and KO; ABR, n = 10, 8. C, D, Normal paired-pulse facilitation (PPF) at BCR−/− and ABR−/− SC–CA1 synapses. BCR, WT, n = 9, 4; KO, n = 16, 6; ABR, WT, n = 13, 6; KO, n = 12, 6. E, F, Reduced maintenance of LTP induced by theta-burst stimulation at BCR−/− and ABR−/− SC–CA1 synapses. The gray and black traces were taken at 0 min and at the end of recording, respectively. The asterisks indicate statistical significances of the differences in fEPSP slopes between the indicated 10 min windows and the baseline before stimulation (for the bottom two rows) and between WT and BCR−/− or ABR−/− synapses (the top row). ***p < 0.001, **p < 0.01, *p < 0.05, and n.s., not significant, Student's t test. G, H, Normal LTD, induced by low-frequency stimulation, at BCR−/− and ABR−/− SC–CA1 synapses. BCR, WT, n = 15, 8; KO, n = 10, 5; ABR, WT, n = 10, 4; KO, n = 17, 8. Error bars indicate SEM.
Interestingly, LTP induced by theta-burst stimulation was reduced at both BCR−/− and ABR−/− SC–CA1 synapses compared with that at WT synapses, as determined by comparing fEPSP slopes at baseline and during the last 10 min of recordings (Fig. 5E,F) (BCR, 143.9 ± 9.6 and 116.9 ± 7.4% of baseline in WT and KO, respectively; p < 0.05; n = 11 slices from 6 mice for WT and 20, 10 for KO; ABR, 154.0 ± 8.1 and 112.5 ± 9.5% of baseline in WT and KO, respectively; p < 0.01; n = 12, 7 for WT and 17, 8 for KO). In contrast, LTP magnitudes during the first 10 min after LTP at BCR−/− and ABR−/− SC–CA1 synapses were not significantly different from those at WT synapses, although a trend toward decreased LTP magnitudes was apparent, especially at ABR−/− synapses (Fig. 5E,F). These results suggest that a deficiency of BCR or ABR leads to a strong reduction in the maintenance, but not induction, of LTP. In contrast to LTP, LTD induced by low-frequency stimulation (1 Hz, 900 pulses) was comparable in WT and KO (BCR−/− and ABR−/−) animals (Fig. 5G,H). These results suggest that BCR or ABR deficiency selectively suppresses LTP maintenance.
Impaired spatial and object recognition memory in BCR−/− and ABR−/− mice
Because LTP maintenance has been implicated in sustaining memory (Pastalkova et al., 2006; Sacktor, 2008), we tested spatial learning and memory in these animals using the Morris water maze (Morris, 1984). During the learning phase of the maze, BCR−/− and ABR−/− mice showed reduced learning relative to WT mice, exhibiting longer escape latencies at several data points (Fig. 6A,E). In the probe test, performed in a pool without the platform, ABR−/− mice spent a reduced amount of time in the target quadrant compared with WT mice, whereas the time spent by BCR−/− mice was not different from WT mice (Fig. 6B,F). Importantly, the number of exact crossings over the former location of the platform was substantially reduced in both BCR−/− and ABR−/− mice (Fig. 6C,G). No significant changes were observed in swimming speeds (Fig. 6D,H). These results suggest that BCR/ABR deficiency impairs spatial learning and memory.
Figure 6.

Impaired spatial learning and object recognition memory in BCR−/− and ABR−/− mice. A, E, Impaired spatial learning of BCR−/− and ABR−/− mice in Morris water maze. BCR, n = 6 for WT and 7 for KO; ABR, n = 7 for WT and KO; *p < 0.05, Student's t test. B, F, ABR−/−, but not BCR−/−, mice spend reduced amount of time in the target quadrant in the probe test. T, Target; L, left; R, right; O, opposite. *p < 0.05. C, G, BCR−/− and ABR−/− mice show reduced numbers of exact crossings over the former platform area in the probe test. *p < 0.05. D, H, Normal swimming speeds in BCR−/− and ABR−/− mice. I, J, BCR−/− and ABR−/− mice spend equal amounts of time on the two objects (A, B) during the sample phase of object recognition experiments. BCR, n = 11 for WT and 12 for KO; ABR, n = 10 for WT and 9 for KO. K, L, BCR−/− and ABR−/− mice spend equal amounts of time on the two object (A, C), whereas WT mice prefer the novel object (C) in the test phase of object recognition experiments. BCR, n = 11 for WT and 12 for KO; ABR, n = 10 for WT and 9 for KO; ***p < 0.001, *p < 0.05, n.s., not significant, Student's t test. Error bars indicate SEM.
We next tested object recognition, which measures hippocampus-dependent nonspatial visual-discrimination memory (Vnek and Rothblat, 1996). In the sample phase, during which BCR−/− and ABR−/− mice were exposed to two different objects (A and B), the mice spent equal amounts of time on each (Fig. 6I,J), indicating that the two objects were equally preferred. In addition, the total amount of time spent exploring the objects (A + B) was similar in KO and WT mice (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), suggesting that these mice have comparable levels of attention and motivation for the objects. When one of the two objects (B) was replaced with a new one (C) 24 h after the sample phase, WT mice spent more time in the novel object, whereas BCR−/− and ABR−/− mice show an equal preference for the two objects (A and C) (Fig. 6K,L). The total amounts of time spent exploring the objects (A + C) were not different between KO and WT mice (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). These results suggest that BCR/ABR deficiency impairs object recognition memory.
In the open-field test, KO and WT mice showed similar levels of total distance moved and time spent in the center zone, indicating that their motor activities and anxiety levels are similar (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In an elevated plus-maze, KO and WT mice spent comparable amounts of time in open arms and entered the open arms with similar frequencies (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), suggesting normal levels of anxiety in these mice. In the accelerating rotarod assay, the initial latency to fall and the extent of daily motor learning were not different between KO and WT mice (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), suggesting that BCR/ABR deficiency does not affect motor coordination.
Discussion
Previous studies of BCR have mainly focused on BCR involvement in immune regulation and leukemia development. Our study identifies a novel synaptic function of BCR and its close relative ABR, showing that these proteins are mainly expressed in the brain, where they are distributed to excitatory synapses and interact with PSD-95. Moreover, our data provide strong genetic evidence that BCR and ABR regulate synaptic Rac1 signaling, LTP maintenance, and spatial and object recognition memory. These results suggest the novel involvement of BCR and ABR in the regulation of synaptic signaling and plasticity, and related cognitive functions.
Interaction of PSD-95 with BCR and ABR Rac GAPs
Our study identifies a direct interaction of BCR and ABR with PSD-95, a key scaffolding protein of excitatory synapses. One known function of PSD-95 is that it promotes synaptic localization of interacting proteins. Our results, however, indicate that BCR and ABR do not require PSD-95 interaction for their synaptic localization. In this respect, BCR and ABR are similar to SPAR (spine-associated RapGAP), a regulator of dendritic spine morphology (Pak et al., 2001; Pak and Sheng, 2003) that directly interacts with PSD-95 but does not depend on PSD-95 interaction for spine targeting. An alternative function of PSD-95 is to couple interacting proteins with functionally related proteins. For instance, PSD-95 interacts with both NMDA receptors and nNOS (neuronal nitric oxide synthase) and promotes their functional coupling (Sattler et al., 1999).
In this context, it should be noted that PSD-95 interacts with other Rac1-related signaling proteins, including Kalirin-7 (a Rac GEF) and IRSp53 (a Rac effector) (Penzes et al., 2001; Choi et al., 2005). It is thus conceivable that PSD-95 could bring two proteins with opposing functions (e.g., Kalirin-7 and BCR/ABR) into close proximity to promote tight local regulation of Rac1 activity. Alternatively, PSD-95 could link upstream regulators of Rac1, such as Kalirin-7 and BCR/ABR, with downstream Rac1 effectors, including IRSp53.
In addition to direct interactions, PSD-95 indirectly associates with various Rac1 signaling proteins; PSD-95 associates with βPix (a Rac GEF) and the Arp2/3 complex (a Rac effector) via GKAP (guanylate kinase-associated protein), Shank, and cortactin scaffolding proteins (Hering and Sheng, 2003; Park et al., 2003). In addition, PSD-95 is linked to Tiam1 (a Rac GEF), via NMDA receptors (Tolias et al., 2005). Thus, PSD-95 might function to organize synaptic Rac1 signaling in a multiprotein complex in which BCR and ABR function as negative regulators.
Regulation of synaptic Rac1 signaling and spine density by BCR and ABR
We found that, under basal conditions, a deficiency of BCR or ABR in mice leads to enhanced Rac1 activity. Enhanced basal Rac1 activity, in turn, was accompanied by increased levels of phosphorylated PAK1/3 and ERK1/2, which act downstream of Rac1 (Carlisle and Kennedy, 2005; Kennedy et al., 2005; Boda et al., 2006). These results provide strong genetic evidence that BCR and ABR function as important negative regulators of Rac1 activity.
BCR/ABR-deficient mice showed a small increase in spine density in the CA1 region of hippocampus, suggesting that BCR and ABR negatively regulate spine density. One mechanism that might underlie this regulation is the inhibition of Rac1 activity through BCR and ABR Rac-GAP domains, with subsequent suppression of Rac1-dependent F-actin polymerization and spine morphogenesis or maintenance. This possibility is supported by the observation that mice deficient for BCR or ABR show enhanced activity of Rac1 and PAK1/3, both of which positively regulate dendritic spines (Luo et al., 1996; Nakayama et al., 2000; Tashiro et al., 2000; Penzes et al., 2001; Zhang et al., 2003; Boda et al., 2004, 2006, 2008; Hayashi et al., 2004, 2007; Tashiro and Yuste, 2004; Carlisle and Kennedy, 2005; Zhang et al., 2005; Tada and Sheng, 2006; Kreis et al., 2007).
It should be noted, however, that the increase in spine density in BCR−/− and ABR−/− mice was small, and was not accompanied by a concomitant increase in spine head area or spontaneous/evoked excitatory synaptic transmission. These results suggest that compensatory mechanisms that partially uncouple enhanced Rac1 activity from spine density increases may be induced; thus, BCR or ABR may not be major regulators of spine density and synaptic transmission.
Regulation of LTP maintenance by BCR and ABR
BCR−/− and ABR−/− mice showed a marked decrease in the maintenance, but not induction, of LTP (but not LTD), with no effects on basal transmission or presynaptic release. A clue to the potential underlying mechanisms may come from the well known association between the actin cytoskeleton and LTP (Kim and Lisman, 1999; Krucker et al., 2000; Fukazawa et al., 2003; Matsuzaki et al., 2004; Okamoto et al., 2004; Lin et al., 2005; Kramár et al., 2006; Cingolani and Goda, 2008). Particularly relevant to our results is the report that inhibition of F-actin by low concentrations of actin-depolymerizing compounds such as latrunculin and cytochalasin, which preferentially inhibit the dynamic pool of actin filaments, selectively inhibit the maintenance of LTP (Krucker et al., 2000), whereas higher concentrations of these compounds block LTP induction (Kim and Lisman, 1999). More recent studies have shown that LTP induction increases the levels of phosphorylated PAK and cofilin in dendritic spines (Chen et al., 2007), and that distinct signaling pathways involving RhoA-ROCK-cofilin and Rac-PAK regulate the induction and maintenance phases of LTP, respectively (Rex et al., 2009). These results, together with the positive influence of Rac-PAK signaling on excitatory synaptic transmission (Boda et al., 2004; Wiens et al., 2005), suggest the hypothesis that the heightened Rac1 activity in the BCR−/− or ABR−/− brain may partly saturate signaling mechanisms required for the maintenance of LTP. Alternatively, enhanced basal Rac1 activity may evoke compensatory mechanisms that inhibit Rac1 activity under basal conditions in a homeostatic fashion; this, however, would likely have a negative influence on the Rac1 activity required for the stabilization of actin filaments during LTP maintenance. Possible compensatory mechanisms include enhanced activity or levels of Rac GAPs other than BCR and ABR, or reduced activity/levels of Rac GEFs such as Kalirin-7, βPix, and Tiam1.
Regulation of learning and memory by BCR and ABR
BCR−/− and ABR−/− mice showed impairments in spatial and object-recognition memory tasks. One of the most important questions in the field of memory—Does LTP maintenance sustain memory?—hints at a possible underlying mechanism for this observation. That LTP maintenance may indeed be involved in sustaining memory is supported by a large body of evidence (Reymann and Frey, 2007). For instance, PKMζ, an atypical protein kinase M, has been shown to be both necessary and sufficient for LTP maintenance (Pastalkova et al., 2006; Sacktor, 2008). It is thus conceivable that the reduced LTP maintenance in BCR−/− and ABR−/− mice might underlie the impaired memory in these animals.
It should be noted that excessive Rac1 activity in BCR−/− and ABR−/− mice had negative effects on synaptic plasticity and cognitive function. These consequences are similar to those of the opposite situation in which Rac1 signaling is suppressed, for example, by mutations in genes such as αPix/ARHGEF6 and PAK3 that are known to be associated with mental retardation (Newey et al., 2005; Boda et al., 2006; Nadif Kasri and Van Aelst, 2008). Therefore, an appropriate level of Rac1 activity under basal conditions appears to be important in maintaining normal synaptic plasticity and cognitive functions. This view may also shed light on how defective regulation of Rac1 leads to cognitive dysfunction, an important question in the field of mental retardation.
Notably, a recent study showed that increased Rac1 activity promotes passive memory decay and interference-induced forgetfulness in Drosophila (Davis, 2010; Shuai et al., 2010). This study also showed that the PAK-LIMK-cofilin pathway downstream of Rac1 is important in mediating the effect of Rac1. Interestingly, our mice showed enhanced Rac1 and PAK1/3 activities. Whether the underling molecular/neural mechanisms of the reduced performance of BCR−/− and ABR−/− mice in memory tasks have any shared aspects with those in flies remains to be determined.
Similar phenotypes of BCR−/− and ABR−/− mice
It is possible that the various phenotypes observed in BCR−/− and ABR−/− mice are attributable to domains other than the Rac GAP domain, such as the S/T kinase domain of BCR, which is not present in ABR. However, it should be noted that BCR−/− and ABR−/− mice showed many substantially overlapping phenotypes, including enhanced activation of Rac1, PAK1/3, and ERK1/2; increased spine density; reduced LTP maintenance; and impaired spatial and object recognition memory. These results strongly suggest that the shared phenotypes in BCR−/− and ABR−/− mice stem from their shared domains. BCR and ABR share the DH domain in addition to the Rac GAP domain. However, the DH domains of BCR and ABR do not exhibit Rho GEF activity (Cho et al., 2007), suggesting that this domain is unlikely to account for the phenotypes of BCR−/− and ABR−/− mice.
Another possibility is that the phenotypes observed in BCR−/− and ABR−/− mice are caused by long-term changes that accumulate during embryonic brain development. However, ABR expression is weak at the embryonic day 18 stage and sharply increases during the first few weeks of postnatal brain development (Fig. 2D), suggesting that the effect of ABR deletion is likely to be minimal during embryonic development. BCR expression is stronger at the embryonic day 18 stage than in postnatal stages; however, the overall phenotypic similarity between BCR−/− and ABR−/− mice suggests that BCR deficiencies at adult stages are likely to have a greater influence on the observed phenotypes of BCR−/− mice. In addition, the fact that BCR−/− and ABR−/− mice showed enhanced and persistent Rac1 activation at adult stages during which synaptic plasticity and memory performances were measured further reduces the possibility of embryonic defects.
In conclusion, our study provides strong genetic evidence in support of the idea that BCR and ABR regulate synaptic Rac1 signaling, synaptic plasticity, and learning and memory. A direction for future studies would be to elucidate the detailed molecular mechanisms underlying these regulatory functions.
Footnotes
This work was supported by the National Creative Research Initiative Program of the Korean Ministry of Education, Science and Technology (E.K.), Neuroscience Program Grant 2009-0081468 (S.-Y.C.), 21st Century Frontier R&D Program in Neuroscience Grant 2009K001284 (H.K.), Basic Science Research Program Grant R13-2008-009-01001-0 (Y.C.B.), and United States Public Health Service Grants HL071945 (J.G.) and HL060231 (J.G., N.H.). We thank Dr. Owen Witte (University of California, Los Angeles, Los Angeles, CA) for the human BCR cDNA and Dr. Louis Lim (University College London, London, UK) for the human ABR cDNA.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55:1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Boda B, Alberi S, Nikonenko I, Node-Langlois R, Jourdain P, Moosmayer M, Parisi-Jourdain L, Muller D. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J Neurosci. 2004;24:10816–10825. doi: 10.1523/JNEUROSCI.2931-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boda B, Nikonenko I, Alberi S, Muller D. Central nervous system functions of PAK protein family: from spine morphogenesis to mental retardation. Mol Neurobiol. 2006;34:67–80. doi: 10.1385/mn:34:1:67. [DOI] [PubMed] [Google Scholar]
- Boda B, Jourdain L, Muller D. Distinct, but compensatory roles of PAK1 and PAK3 in spine morphogenesis. Hippocampus. 2008;18:857–861. doi: 10.1002/hipo.20451. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttery P, Beg AA, Chih B, Broder A, Mason CA, Scheiffele P. The diacylglycerol-binding protein alpha1-chimaerin regulates dendritic morphology. Proc Natl Acad Sci U S A. 2006;103:1924–1929. doi: 10.1073/pnas.0510655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Wilson MS, Halpain S. Development and regulation of dendritic spine synapses. Physiology (Bethesda) 2006;21:38–47. doi: 10.1152/physiol.00042.2005. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YJ, Cunnick JM, Yi SJ, Kaartinen V, Groffen J, Heisterkamp N. Abr and Bcr, two homologous Rac GTPase-activating proteins, control multiple cellular functions of murine macrophages. Mol Cell Biol. 2007;27:899–911. doi: 10.1128/MCB.00756-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Ko J, Racz B, Burette A, Lee JR, Kim S, Na M, Lee HW, Kim K, Weinberg RJ, Kim E. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci. 2005;25:869–879. doi: 10.1523/JNEUROSCI.3212-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang TH, Xu X, Kaartinen V, Heisterkamp N, Groffen J, Bokoch GM. Abr and Bcr are multifunctional regulators of the Rho GTP-binding protein family. Proc Natl Acad Sci U S A. 1995;92:10282–10286. doi: 10.1073/pnas.92.22.10282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Davis RL. Rac in the act of forgetting. Cell. 2010;140:456–458. doi: 10.1016/j.cell.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Diekmann D, Brill S, Garrett MD, Totty N, Hsuan J, Monfries C, Hall C, Lim L, Hall A. Bcr encodes a GTPase-activating protein for p21rac. Nature. 1991;351:400–402. doi: 10.1038/351400a0. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93–99. doi: 10.1016/0092-8674(84)90077-1. [DOI] [PubMed] [Google Scholar]
- Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, Palmer TD. A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci. 2009;41:409–419. doi: 10.1016/j.mcn.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Ohshima T, Hashimoto M, Mikoshiba K. Pak1 regulates dendritic branching and spine formation. Dev Neurobiol. 2007;67:655–669. doi: 10.1002/dneu.20363. [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron. 2004;42:773–787. doi: 10.1016/j.neuron.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK. Dendritic spine geometry: functional implication and regulation. Neuron. 2005;46:529–532. doi: 10.1016/j.neuron.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph′ translocation. Nature. 1985;315:758–761. doi: 10.1038/315758a0. [DOI] [PubMed] [Google Scholar]
- Heisterkamp N, Morris C, Groffen J. ABR, an active BCR-related gene. Nucleic Acids Res. 1989;17:8821–8831. doi: 10.1093/nar/17.21.8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisterkamp N, Kaartinen V, van Soest S, Bokoch GM, Groffen J. Human ABR encodes a protein with GAPrac activity and homology to the DBL nucleotide exchange factor domain. J Biol Chem. 1993;268:16903–16906. [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Gonzalez-Gomez I, Voncken JW, Haataja L, Faure E, Nagy A, Groffen J, Heisterkamp N. Abnormal function of astroglia lacking Abr and Bcr RacGAPs. Development. 2001;128:4217–4227. doi: 10.1242/dev.128.21.4217. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Nagy A, Gonzalez-Gomez I, Groffen J, Heisterkamp N. Vestibular dysgenesis in mice lacking Abr and Bcr Cdc42/RacGAPs. Dev Dyn. 2002;223:517–525. doi: 10.1002/dvdy.10071. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Choi J, Yang J, Chung W, Kim JH, Paik SK, Kim K, Han S, Won H, Bae YS, Cho SH, Seo J, Bae YC, Choi SY, Kim E. Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci. 2009;29:1586–1595. doi: 10.1523/JNEUROSCI.4306-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, Greengard P. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis P, Thévenot E, Rousseau V, Boda B, Muller D, Barnier JV. The p21-activated kinase 3 implicated in mental retardation regulates spine morphogenesis through a Cdc42-dependent pathway. J Biol Chem. 2007;282:21497–21506. doi: 10.1074/jbc.M703298200. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Growth of dendritic spines: a continuing story. Curr Opin Neurobiol. 2005;15:67–72. doi: 10.1016/j.conb.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Nadif Kasri N, Van Aelst L. Rho-linked genes and neurological disorders. Pflugers Arch. 2008;455:787–797. doi: 10.1007/s00424-007-0385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Pak DT, Sheng M. Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science. 2003;302:1368–1373. doi: 10.1126/science.1082475. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- Park E, Na M, Choi J, Kim S, Lee JR, Yoon J, Park D, Sheng M, Kim E. The Shank family of postsynaptic density proteins interacts with and promotes synaptic accumulation of the beta PIX guanine nucleotide exchange factor for Rac1 and Cdc42. J Biol Chem. 2003;278:19220–19229. doi: 10.1074/jbc.M301052200. [DOI] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. Storage of spatial information by the maintenance mechanism of LTP. Science. 2006;313:1141–1144. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- Penzes P, Johnson RC, Sattler R, Zhang X, Huganir RL, Kambampati V, Mains RE, Eipper BA. The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron. 2001;29:229–242. doi: 10.1016/s0896-6273(01)00193-3. [DOI] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic EphrinB-EphB receptor activation of the Rho-GEF Kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, Srivastava DP. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008;18:405–413. doi: 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Pilpel Y, Segal M. Rapid WAVE dynamics in dendritic spines of cultured hippocampal neurons is mediated by actin polymerization. J Neurochem. 2005;95:1401–1410. doi: 10.1111/j.1471-4159.2005.03467.x. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymann KG, Frey JU. The late maintenance of hippocampal LTP: requirements, phases, “synaptic tagging,” “late-associativity” and implications. Neuropharmacology. 2007;52:24–40. doi: 10.1016/j.neuropharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Sawallisch C, Berhörster K, Disanza A, Mantoani S, Kintscher M, Stoenica L, Dityatev A, Sieber S, Kindler S, Morellini F, Schweizer M, Boeckers TM, Korte M, Scita G, Kreienkamp HJ. The insulin receptor substrate of 53 kDa (IRSp53) limits hippocampal synaptic plasticity. J Biol Chem. 2009;284:9225–9236. doi: 10.1074/jbc.M808425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Sekino Y, Kojima N, Shirao T. Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem Int. 2007;51:92–104. doi: 10.1016/j.neuint.2007.04.029. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Shuai Y, Lu B, Hu Y, Wang L, Sun K, Zhong Y. Forgetting is regulated through Rac activity in Drosophila. Cell. 2010;140:579–589. doi: 10.1016/j.cell.2009.12.044. [DOI] [PubMed] [Google Scholar]
- Soltau M, Richter D, Kreienkamp HJ. The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol Cell Neurosci. 2002;21:575–583. doi: 10.1006/mcne.2002.1201. [DOI] [PubMed] [Google Scholar]
- Stockton R, Reutershan J, Scott D, Sanders J, Ley K, Schwartz MA. Induction of vascular permeability: beta PIX and GIT1 scaffold the activation of extracellular signal-regulated kinase by PAK. Mol Biol Cell. 2007;18:2346–2355. doi: 10.1091/mbc.E06-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Tan EC, Leung T, Manser E, Lim L. The human active breakpoint cluster region-related gene encodes a brain protein with homology to guanine nucleotide exchange proteins and GTPase-activating proteins. J Biol Chem. 1993;268:27291–27298. [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–538. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Kane CG, Tolias CS, Hu L, Greenberg ME. The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptor-dependent dendritic spine development. Proc Natl Acad Sci U S A. 2007;104:7265–7270. doi: 10.1073/pnas.0702044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Ven TJ, VanDongen HM, VanDongen AM. The nonkinase phorbol ester receptor α1-chimerin binds the NMDA receptor NR2A subunit and regulates dendritic spine density. J Neurosci. 2005;25:9488–9496. doi: 10.1523/JNEUROSCI.2450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen EJ, Ramakers GJ. Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog Brain Res. 2005;147:295–317. doi: 10.1016/S0079-6123(04)47022-8. [DOI] [PubMed] [Google Scholar]
- Vnek N, Rothblat LA. The hippocampus and long-term object memory in the rat. J Neurosci. 1996;16:2780–2787. doi: 10.1523/JNEUROSCI.16-08-02780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken JW, van Schaick H, Kaartinen V, Deemer K, Coates T, Landing B, Pattengale P, Dorseuil O, Bokoch GM, Groffen J. Increased neutrophil respiratory burst in bcr-null mutants. Cell. 1995;80:719–728. doi: 10.1016/0092-8674(95)90350-x. [DOI] [PubMed] [Google Scholar]
- Wiens KM, Lin H, Liao D. Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci. 2005;25:10627–10636. doi: 10.1523/JNEUROSCI.1947-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Srivastava DP, Photowala H, Kai L, Cahill ME, Woolfrey KM, Shum CY, Surmeier DJ, Penzes P. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron. 2007;56:640–656. doi: 10.1016/j.neuron.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Horwitz AF. Synapse formation is regulated by the signaling adaptor GIT1. J Cell Biol. 2003;161:131–142. doi: 10.1083/jcb.200211002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Webb DJ, Asmussen H, Niu S, Horwitz AF. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci. 2005;25:3379–3388. doi: 10.1523/JNEUROSCI.3553-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]