

Abstract
The bacterial transcription elongation factor NusG stimulates the Rho-dependent transcription termination through a direct interaction with Rho. The mechanistic basis of NusG dependence of the Rho function is not known. Here, we describe Rho* mutants I168V, R221C/A, and P235H that do not require NusG for their termination function. These Rho* mutants have acquired new properties, which otherwise would have been imparted by NusG. A detailed analyses revealed that they have more stable interactions at the secondary RNA binding sites of Rho, which reduced the lag in initiating its ATPase as well as the translocase activities. These more stable interactions arose from the significant spatial re-orientations of the P, Q, and R structural loops of the Rho central channel. We propose that NusG imparts similar conformational changes in the central channel of Rho, yielding faster isomerization of the open to the closed hexameric states of the latter during its RNA-loading step. This acceleration stabilizes the Rho-RNA interactions at many terminators having suboptimal rut sites, thus making Rho-NusG interactions so essential in vivo. Finally, identification of the NusG binding sites on the Rho hexamer led us to conclude that the former exerts its effect allosterically.
Keywords: allosteric regulation, bacteria, fluorescence, RNA polymerase, transcription termination, NusG, Rho
Introduction
At the end of a significant number of operons in Escherichia coli, DNA sequences do not code for intrinsic termination signals, characterized by a hairpin and a U-stretch (1). Transcription termination factor Rho, a homo-hexameric RNA-dependent ATPase, induces termination in these operons. Rho loads onto unstructured C-rich RNA sequences (rut sites, the Rho-utilization sites) and is capable of translocating along the RNA using its RNA-dependent ATPase activity, and eventually dislodges the elongating RNA polymerase (RNAP)3 (1, 2). By virtue of these properties of Rho, long stretches of untranslated RNA sequences are its ideal targets. Transcription on these stretches can face premature termination by Rho, which usually happens in the cases of transcription-translation uncoupling. Recent genomics study has revealed that Rho-dependent termination is widespread in vivo, which could be the main reason for its essentiality (3–6).
NusG, a transcription elongation factor, being bound to the elongation complex (EC) through its N-terminal domain (NTD) helps the latter to overcome the pauses by increasing the elongation rate (5, 7, 8). Its C-terminal domain (CTD) is capable of interacting with the Rho (9). This interaction stimulates the Rho-dependent termination process in vitro, which is manifested as early termination (10, 11). More recent studies indicated that NusG is essential for recruiting Rho on a subset of terminators in vivo (4, 6). Mechanism of stimulation of Rho-dependent termination by NusG in vitro as well as the molecular basis of NusG dependence of Rho to utilize a subset of terminators in vivo is not known, knowledge of which is essential to understand the intricacies of this transcription termination process.
In this study, by screening a library of randomly mutagenized rho, we have isolated Rho mutants, I168V, R221C, R221A, and P235H (the Rho* mutants) that function independent of NusG both in vivo and in vitro, and also suppress the defects of NusG mutants defective for binding to Rho. These Rho mutants have acquired the properties, which otherwise would have been imparted by NusG. A detailed analyses revealed that they have more stable interactions at the secondary RNA binding channel of Rho, which reduced the lag in initiating the ATPase activity as well as increased the rate of hydrolysis. This more stable interaction arose from significant conformational changes in the R- and Q-loops of the Rho central channel. We propose that interaction of NusG with Rho induces faster isomerization of the open to the closed hexameric states of the latter. This in turn stabilizes Rho-RNA interactions at the “weaker” terminators having suboptimal rut sites, thus making Rho-NusG interaction essential in vivo. Finally, we have delineated that a hydrophobic patch encompassing residues 203–205 of Rho, located away from the central channel is the functional interaction site(s) for the NusG-CTD. Hence, we concluded that NusG exerts its effect allosterically to the secondary RNA binding sites of Rho.
Results
Rationale
To understand the mechanism of the NusG-mediated stimulation of the Rho-dependent termination process, it is important to understand the NusG-induced conformational changes in the Rho molecule as well as the step(s) of the termination process influenced by it. Direct measurements of these conformational changes in vitro may not be simple because of the following reasons. NusG-CTD has six binding sites on a Rho hexamer, one on each protomers. However, the stoichiometry of this binding is, Rho hexamer::NusG monomer (12), which means that at a given time NusG occupies one of six binding sites of the Rho. This poses a problem for in vitro detection of any NusG-mediated conformational changes in the Rho, as the majority of the sites remain unoccupied in a Rho-NusG complex. Moreover, the NusG effect on Rho should be studied in the context of a transcription EC, where NusG-NTD remains bound to RNAP and the CTD interacts with the nascent RNA-bound Rho. Reconstitution of this set-up in vitro is technically quite challenging. To overcome these problems, we decided to genetically screen Rho mutants, which are capable of functioning independent of NusG. It is expected that such Rho mutants will acquire certain properties as well as conformational changes that otherwise would have been imparted by NusG. Characterization of these Rho mutants for their functions (both in vivo and in vitro) and their conformational changes would elucidate the mechanism of the NusG action.
Screening and Isolation of Rho Mutants That Function Independently of NusG
Overexpression of NusG-NTD fragments in vivo titrates out the full-length NusG (Fig. 1A) (13) and occupies the majority of the ECs. Under this condition, due to the absence of the NusG-CTD domain, Rho-NusG interaction does not occur and the termination process becomes inefficient, which leads to toxicity (Fig. 1A, 0.2% arabinose panel). This toxicity may also arise from the NusG-NTD induced alteration in the transcription-translation coupling, if any. An MG1655Δrac (RS862) strain, having a plasmid overexpressing NusG-NTD from the arabinose-controlled PBAD promoter, was electroporated with a mutagenized plasmid library carrying the rho (pHYD567). The transformants that grew in the presence of 0.2% arabinose (an overexpressing condition of NusG-NTD fragment) contain the Rho mutants, which are capable of functioning efficiently in the absence of bona fide Rho-NusG-CTD interactions. These mutations were confirmed by sequencing. We obtained two Rho mutants: I168V and R221C. We also constructed R221A to see the effect of the amino acid change. Previously, some Rho mutants were reported that displayed enhanced as well as early termination activities (14, 15). From that group, we chose the mutants P103L and P235H as their altered properties were most prominent. We tested them in our assays as we envisioned that these may possess some properties that are imparted by NusG. We termed all these Rho mutants as Rho*.
FIGURE 1.

Growth characteristics of the Rho* mutants. A, screening strategies for isolating Rho mutants that can function in the absence of NusG. The Rho-dependent transcription termination requires the interaction of Rho and NusG-CTD (9), absence of which leads to termination read-through and cell death. The NTD of NusG has more affinity for the RNAP than the full-length NusG (13). So, its overexpression replaces the latter from the elongation complex leading to conditional toxicity of the cells. Schematics show the representation of the toxicity of the overexpressed NusG-NTD in the presence of WT Rho. It is expected that the Rho mutants, which can overcome this NusG-NTD mediated toxicity, do not require interaction with NusG implying that they can terminate in the absence of NusG. In the screening protocol, NTD of NusG was cloned under a pBAD promoter and was overexpressed with 0.2% arabinose (see the picture of the growth plate). B, transductants obtained following the deletion of chromosomal nusG by P1 transduction of a MG1655rac−rho− strain. In these strains, WT and Rho* mutants (R221C, I168V, P235H) are expressed from pCL1920 plasmids as indicated. Effect of Rho P103L is shown for comparison. Plates were incubated for ∼20 h at 37 °C. C, homology model of E. coli NusG depicting the Rho binding and termination defective mutations. D, transductants obtained after removing the chromosomal nusG by P1 transduction of MG1655rac−rho− strains that express different combinations of Rho and NusG derivatives expressed from pCL1920 and pHYD3011 plasmids, respectively, as indicated. These NusG mutants are defective for Rho binding (9).
In Vivo Functions of the Rho* Mutants
Deletion of chromosomal nusG by P1 transduction (Fig. 1B, left most panel) or the expression of NusG-CTD mutants defective for Rho-binding (G146D and L158Q expressed from pHYD3011 plasmid; Fig. 1C; see Ref. 9; Fig. 1B, top most panel), cause severe growth defects to MG1655 strains expressing WT Rho from a low-copy pCL1920 plasmid (RS862, chromosomal rho is deleted). At first, we checked whether these Rho* mutants were capable of overcoming the growth defects caused by the deletion of nusG (Fig. 1B) or by its mutations in the CTD (Fig. 1D). When the strains (lacks the chromosomal copy of rho) expressing Rho* mutants from a pCL1920 plasmid were made ΔnusG by P1 transduction, healthy transductants were obtained (Fig. 1, B and D, Rho* mutant panels) in all the cases except for the P103L Rho mutant (Fig. 1B, right most panel). This indicates that all the Rho* mutants, except P103L, are capable of suppressing the growth defects caused either due to the deletion of nusG or by the presence of NusG mutants, G146D and L158Q. We did not include P103L mutant in our subsequent experiments.
We have earlier identified and characterized a Rho-dependent terminator, trac, in the rac prophage region that is highly dependent on NusG in vivo (Fig. 2A) (6). When this terminator was fused upstream of the lacZ reporter, a huge enhancement of the β-galactosidase activities was observed in the presence of NusG-CTD mutants compared with that obtained for the canonical λTR1 terminator (λTR1-lacZ) (6), which proved the NusG dependence of this terminator. We used the trac-lacZ reporter to assess the Rho* mutants-mediated suppression of the termination read-through due to the presence of NusG-CTD mutants. We used strain RS1430 having the trac-lacZ reporter inserted into the chromosome. Each of the Rho* and the NusG mutants were supplied from pCL1920 and pHYD3011 plasmids, respectively. Compared with WT Rho, the β-galactosidase activities were significantly reduced in the presence of different Rho* mutants. I168V exerted the best effect and R221A was least effective (Fig. 2B). Therefore, the Rho* mutants exerted moderate to high levels of suppression of the termination defects caused by NusG-CTD mutants. The above results strongly indicated that these Rho* mutants are capable of suppressing the growth and transcription termination defects that occurred due to the lack of Rho-NusG interaction, and hence these Rho mutants are less dependent on or function independently of NusG in vivo.
FIGURE 2.

In vivo termination assays with the Rho* mutants. A, sequence of the PRM-trac-racR region of the rac prophage fused upstream of a lacZ cassette. Different regions along with primers used for making the DNA templates are indicated. The transcription start site G, is shown as a small arrow. Promoter and racR regions are highlighted. B, bar diagrams showing the β-galactosidase activities indicating the termination efficiencies of WT and Rho* mutant proteins at the NusG-dependent terminator, trac (6), in the presence of different NusG derivatives. The strain RS1430 (see Table 1) was used in the assays. Rho and NusG mutants together with the reporter construct are indicated. Error bars are obtained from the measurements of 5 independent colonies.
In Vitro Properties of the Rho Mutants
Next we assayed different properties of the purified Rho* mutants to understand the step(s) in Rho-dependent termination affected by them. As they efficiently suppressed the Rho binding as well as Rho-dependent termination defects caused by the NusG mutants or its deletion, we hypothesized that these Rho mutants might have acquired unique properties. These properties would have been imparted by NusG upon binding to Rho during the process of transcription elongation.
At first, we performed in vitro transcription termination assays using templates having either the H-19B tR1 terminator (16) or the trac (6) terminator. The latter is more NusG-dependent (6). Transcription was initiated from the PRM promoter that naturally occurs before the trac termination region (Figs. 2A and 3A), whereas the initiation was from the strong T7A1 promoter for the template having the H-19B tR1 terminator. When the T7A1 promoter was present, we performed single-round transcription assays in the presence of rifampicin, whereas for the weak promoter, PRM, multiple round transcription was employed. The termination events were initiated by adding either WT or Rho* mutants, both in the absence and presence of WT NusG.
FIGURE 3.

In vitro transcription assays with the Rho* mutants. Autoradiograms showing the in vitro Rho-dependent transcription termination assays of the WT and Rho* mutants using the templates having (A) H-19B TR1 and (B) trac terminators. Termination assays were performed both in the presence and absence of NusG. In A, termination zones are marked by double sided arrows. In B, an immobilized DNA template having the trac terminator was used to measure the terminated RNA. The termination zones are indicated by dashed lines. The reactions were fractionated into S (half of the supernatant) and P (half supernatant + pellet) fractions to identify the terminated RNA that appeared as released product. In both the experiments, concentrations of RNAP, Rho, and NusG were 25, 50, and 200 nm, respectively. C, schematic showing the formation of a stalled elongation complex (RB) downstream of the rut sites of the trac terminator using the lac repressor. The elongation complex is immobilized via a 5′-biotin-avidin linkage of the template with magnetic beads. Different components of this stalled complex are indicated. D, autoradiograms showing the time course of RNA release from the EC stalled downstream of the trac terminator sequence as induced by WT and Rho* mutants. RNA release by the WT Rho has been shown both in the presence and absence of NusG. S denotes half of the supernatant and P denotes rest of the sample. RB represents the RNA corresponding to the position of the stalled EC on this template. The 0 min time points are the samples that do not contain Rho. E–G, plots showing the fractions of released RNA against time from the RB, in the presence of WT and Rho* mutants, P235H and R221C both in the presence and absence of NusG as indicated. Average rates of RNA release (k) are indicated inside each of the plots. The experimental data points are fitted as described under ”Materials and Methods.“ Error bars were obtained from three independent measurements.
WT Rho produced two distinct termination zones both in the presence and absence of NusG at the H-19B TR1 terminator that is consistent with the previous report (16). Unlike WT Rho, the Rho* mutants tested in these assays exhibited early termination even in the absence of NusG (Fig. 3A). I168V induced less early termination compared with the others. The presence of more inactive protein fractions (data not shown) could be the reason for the less robust termination property of I168V Rho. However, more transcripts were observed in the early termination zone when NusG was present. In case of the PRM-trac template, reactions were performed using a linearized DNA template immobilized to streptavidin-coated magnetic beads so that the amounts of released RNA could be measured in the solution (Fig. 3B). Following the transcription reactions, supernatant (S) and pellet (S + P) fractions were loaded separately. We observed that WT Rho induced two distinct termination zones in the absence (∼400–700 nt) and presence (∼300–400 nt) of NusG. This early termination in the presence of NusG is similar to some other Rho-dependent terminators (10, 11) (Fig. 3A). In this case also, unlike the WT Rho, early termination was observed by the Rho* mutants even in the absence of NusG. In these cases, many early terminated transcripts were observed in the 300–400-nt zone similar to that observed when NusG was present together with the WT Rho. However, in the presence of NusG more transcripts were observed in the 300–400-nt termination zones of these Rho* mutants. Therefore, Rho* mutants mimic the presence of NusG in the above reactions. However, they are still capable of responding to NusG.
Earlier, induction of early termination by NusG was interpreted as the rate of increase in RNA release by Rho (10), and it was later confirmed by us by measuring the release of RNA from stalled EC (11). We hypothesized that the Rho* mutants would accelerate the rate of RNA release in the absence of NusG. We have used a DNA template having a trac terminator, and a lac-operator sequence that is fused downstream of the terminator (Fig. 3C). Earlier we have shown that at this terminator, the rate and the efficiency of RNA release is highly dependent on the presence of NusG (6), which was interpreted as slow and less efficient formation of the translocase competent Rho molecule due to the presence of suboptimal rut sequences. Transcription was initiated from the PRM promoter. At first, a stalled EC was formed at the lac operator site in the presence of the lac repressor, following which WT and Rho* mutants, R221C and P235H, were added to induce the RNA release. Time course of RNA release in the supernatant fractions were estimated from the supernatant (S) and pellet (P) fractions (Fig. 3D), and the amount of fractions of RNA released were plotted against time (Fig. 3, E–G). We observed that in the absence of NusG, the time course of RNA release induced by the WT Rho was slow and the rate of RNA release increased in the presence of NusG (Fig. 3E). The RNA release rate was observed to be increased by ∼2.5-fold by the Rho* mutants in the absence of NusG, and ∼4.5-fold increase was observed when NusG was added (compare Fig. 3, E with F and G). This indicates that compared with WT Rho, these Rho* mutants are capable of inducing significantly faster RNA release both in the presence and absence of NusG. This faster RNA release could imply that these mutants have altered interactions with RNA as well as a faster rate of ATP hydrolysis, which in turn might have increased their rates of helicase activities.
To probe this, next we followed Rho-RNA interactions. Rho has two RNA binding sites: the primary RNA binding site (PBS) at the N-terminal and the secondary RNA binding site (SBS) at the central channel (1, 2). We monitored the interactions at the PBS by following the fluorescence quenching of the fluorscein-poly(dC)34 DNA (a 34-mer oligonucleotide) upon interaction with WT and Rho* mutants (Table 2). This single-stranded DNA selectively binds to the PBS (11). We observed that the dissociation constants of poly(dC)34 did not change drastically due to the Rho* mutations. We also tested the binding of WT and Rho* mutants with other DNA oligos of similar size either having a random sequence (non-rut oligo) or the rut sequence of the λTR1 terminator (rut oligo) (see “Experimental Procedures”) using gel-shift assays. We estimated that WT and Rho* mutants have comparable affinity for the rut oligo and all of them showed insignificant binding to the non-rut oligo (data not shown). These results suggest that interaction patterns in the PBS did not change significantly in these mutants.
TABLE 2.
Properties of the primary and secondary RNA binding sites of the Rho* mutants
| WT and Rho* mutants | Dissociation constants for poly(dC)34 (Kd)a | Km for rC25 oligob | ATPase activity on λtR1 RNAc |
|---|---|---|---|
| μm | nmol of ATP/min/μg of Rho | ||
| WT Rho | 9.7 ± 2.0 | 467.6 ± 13 | 1.2 ± 0.06 |
| P235H Rho | 6.1 ± 0.3 | 26.7 ± 3 | 4.2 ± 0.2 |
| R221C Rho | 8.4 ± 2 | 32.8 ± 5 | 5.3 ± 0.5 |
| I168V Rho | 11.7 ± 1.5 | 52.3 ± 19 | 2.7 ± 0.4 |
| R221A Rho | 11 ± 0.7 | 62.5 ± 6 | 9.2 ± 0.8 |
a Dissociation constants (Kd) were calculated by fitting the binding isotherms to hyperbolic equation that is obtained from the plots of fluorescence intensities of fluorescein-(dC)34 oligonucleotide at 520 nm upon addition of WT and Rho* mutants. Error bars were obtained from three independent measurements.
b Km was calculated from Lineweaver-Burk plot of the form, (1/V) = (Km + [S])/(Vmax[S]), where, V and S are reaction velocity (nanomoles of ATP hydrolyzed/μg of Rho/min) and substrate concentrations, respectively. Intercept on the x axis gave the measure of Km. Errors were obtained from three measurements.
c Rates of ATP hydrolysis were calculated from the time-course of ATP hydrolysis by WT and different Rho* mutant using λTR1 RNA as an inducer. Release of Pi from [γ−32P]ATP was analyzed on the TLC plates. Error bars were obtained from three measurements.
Next we estimated the affinity constants of the SBS of the WT and the Rho* mutants for an RNA oligo, rC25, by measuring the Km of this RNA oligo from the RNA-induced ATPase activity of the Rho proteins (Table 2). This method, instead of a direct binding assay, was used because this RNA oligo induces enzymatic activities to the Rho protein. It should be noted that the Km is a composite parameter that includes both binding and catalytic steps. The Km values obtained with the Rho* mutants were about 7–17-fold lower than what was obtained with the WT Rho (Table 2). This strongly indicates that the Rho* mutants have significantly higher affinity constants for the RNA at the SBS.
It is likely that the tighter RNA binding at the secondary channel would induce faster RNA-dependent ATP hydrolysis. We measured the amounts of ATP hydrolyzed at different time points (0 to 20 min) in the presence of a fixed concentration of the inducer, λTR1 RNA (the rates of ATP hydrolysis were calculated from the dynamic ranges of the plots; Table 2). We observed that the Rho* mutants have ∼2.5–7.5-fold higher rates of ATP hydrolysis compared with that of the WT Rho, which is consistent with their tighter RNA-binding properties at the SBS. The tighter RNA binding at the SBS and the faster ATP hydrolysis properties of the Rho* mutants are suggestive of the presence of distinctive conformational changes in their central channel. Interestingly, the positions of the Rho* mutations on the Rho structure are not at the central channel, rather they are located far away from the SBS (Fig. 4, A and B). This indicates that the mutations caused conformational changes allosterically. In subsequent sections, we probed the nature of these conformational changes at the SBS of the Rho* mutants.
FIGURE 4.

Locations of the Rho* mutants on the structure of hexameric Rho. A, schematic showing the locations of Rho* mutants on the Rho hexamer structure (PDB 3ICE) in a closed conformation (18). B, distances of the positions of the Rho* mutants from the center of the RNA bound to the SBS.
Conformational Changes in the Central Channel of the Rho Mutants
We employed partial proteolytic digestion using trypsin and V8 proteases to map the changes in the surface accessibility of specific regions of the Rho* mutants. We used end-labeled Rho proteins to identify the proteolytically cleaved sites. In Fig. 5A, partial cleavage of WT and Rho* mutants were induced with trypsin. P235H Rho showed hypersensitivity around amino acids 181–184, compared with that observed with the WT Rho. The hypersensitivity of the same region was moderate in the case of R221C Rho. Due to the R221C change, the cleavage was very weak at this position, and also the mutation caused abnormal mobility of the cleaved fragments around the 181–184 amino acid region. In Fig. 5B, the same experiments were repeated with V8 protease. The major cleavage products in all the cases were observed at the 211–215 glutamic acid stretch. This region of R221C Rho showed the maximum sensitivity toward V8. As R221C mutation is located near the region of 211–215, this hypersensitivity could be a specific effect of this mutation. So we probed the same V8 hypersensitivity using the R221A Rho* mutant, which also has the similar properties as R221C (see Figs. 1 and 2). We observed the same pattern of V8 hypersensitivity for R221A Rho (Fig. 5C). Hence, this hypersensitivity of the glutamic acid stretch defines a specific conformational changes of the Rho* mutants. We mapped these two hypersensitive regions (181–184 and 211–215) on the outer wall of the central channel along the dimeric interfaces (Fig. 5D), which suggests significant changes in local conformations in the channel of the Rho* mutants. It should be noted that amino acids 181–184 are part of the ATP binding pocket of the central channel (18), and hence, conformation of this pocket has also been altered especially in the P235H mutant.
FIGURE 5.

Probing the conformational changes in the Rho* mutants. Autoradiograms showing the cleavage patterns of the radiolabeled WT and Rho* mutants in the presence of (A) trypsin and (B) V8 proteases under the indicated conditions. Profiles of the cleavage intensities are aligned with the patterns shown in the autoradiograms. In A for the ease of comparison, cleavage profiles of WT (—) and Rho* mutants (- - -) and in B for WT (—), R221C (- - -), and P235H (- - -) profiles are overlapped. The cleavage positions were identified from the proteolytic digestions with CNBr (at Met), Lys-C (at Lys), and Arg-C (at Arg) as indicated on the side of the autoradiograms. C, autoradiograms showing the V8 cleavage patterns of WT and R221A Rho mutant under the same conditions as above. The V8 cleavage sites were identified in similar ways as described above. Profile plots of the cleavage intensities are shown adjacent to the autoradiogram. D, the trypsin and V8 hypersensitive regions on the amino acid sequence of Rho are shown. These regions are also indicated on the structure of the Rho dimer (V8, blue; trypsin, red). Locations of the Rho* mutations are also indicated.
From the enhanced RNA-binding affinity at the SBS (Table 2) and the faster ATPase activity (Table 2) results, we reasoned that the P-, Q-, and R-loops structural elements in the central channel (Fig. 6A) have undergone significant conformational changes in the Rho* mutants, because the P-loop constitutes a part of the ATP-binding pocket and the Q- and R-loops directly interact with the RNA in the SBS (2) (Fig. 6A). The flexibility of these loops (17, 18) makes them susceptible to conformational changes. We probed these putative conformational changes using fluorescence spectroscopy by following the accessibility of the site-specifically labeled fluorescent probes at these three important structural elements, P-, Q-, and R-loops.
FIGURE 6.

P-, Q-, and R-loops of Rho hexamer and TbCl3 fluorescence. A, crystal structure of Rho hexamer (PDB ID 3ICE) highlighting the functionally important loops and the RNA at the central channel by the indicated colors. B, fluorescence emission spectra of TbCl3 under different conditions as indicated. Two distinct peaks of the TbCl3 at 488 and 547 nm are visible. C, modified Stern-Volmer plots to determine the quenching constant (KSV) and the nature of quenching by a neutral quencher, acrylamide. F0 and F are the fluorescence intensities of Tb(III)-GTP bound to the ATP-binding site of the WT and Rho* mutants, in the absence and presence of different concentrations of acrylamide, respectively. Average KSV values are calculated from the initial slopes of the curves. Errors were calculated from two to three measurements.
At first, we have used a fluorescent probe, Tb(III)-GTP, which binds to the ATP binding pockets (19), to monitor the changes in the spatial orientations of the P-loop. The free Tb(III) becomes fluorescent upon complexation with GTP (Fig. 6B). Fluorescence intensity of this complex further increased upon binding to the ATP binding pockets of WT and Rho* mutants (Fig. 6B) characterized by the two peaks at 488 and 547 nm. We assessed the accessibility of this pocket by measuring the extent of fluorescence quenching with a neutral quencher, acrylamide (20). The quenching constant, KSV, is directly proportional to the accessibility of the fluorescent probe to the quencher. We observed gradual quenching of the fluorescence intensity with increasing concentrations of acrylamide in all the cases characterized by upward curvatures in the Stern-Volmer plots (Fig. 6C). However, the extent of quenching varied for each of the proteins (Fig. 6C). The quenching constant (KSV) was observed to increase to 2–3.5-fold for the Rho* mutants, P235H and R221C, indicating the presence of more accessible P-loop as well as the ATP binding pockets in the mutants allowing better access of ATP molecules to this region. This observation is consistent with the hypersensitivity of this region to the trypsin cleavage (Fig. 5A). This altered conformation might have also affected the rates of the ATPase activities (kcat of ADP/Pi release) of these mutants indirectly (see Table 2).
Next to probe the state(s) of the Q- and R-loops, we introduced single cysteine residues in these two loops that were subsequently labeled with fluorescein malemide. Rho has a single cysteine at 202 position. We at first made a “zero cysteine” Rho (C202S change in the WT Rho) and introduced single cysteines via the following changes, T276C (Q-loop), S281C (Q-loop), T286C (Q-loop), S323C (R-loop), and S325C (R-loop) (Fig. 7A, see also the monomeric structures in Fig. 8). These single cysteine derivatives were constructed both on the WT and the Rho* mutant, P235H. It should be noted that as Rho is a hexamer, one cysteine residue per monomer will be fluorescent labeled. The hexameric structure of Rho revealed (Fig. 7A) that except for the R-loop cysteine derivatives all the others are surface exposed. The R-loop derivatives are partially buried. We tested the ATPase as well as the RNA release activities from the ECs (Fig. 7, B and C) of these single cysteine derivatives. Except the T286C mutation, all the others were either fully or partially active in both assays. The activities of S323C and S325C markedly improved when they were made on the Rho* mutant, P235H. This reduction in activity is expected as the changes were made in the very important functional elements of Rho. In general, P235H Rho is more efficient in the in vitro experiments compared with the WT one (see also Fig. 3 and Table 2), so the cysteine derivatives made on this Rho mutant were also more efficient upon site-specific labeling with fluorescein, each of the cysteines would function as conformational reporters of the Q- and R-loops. About 80% of the protein molecules were labeled under our experimental conditions.
FIGURE 7.

Single Cys derivatives of Rho. A, location of the single cys substitutions on the hexameric structure of Rho (PDB ID 3ICE). The space-filled models were prepared using PyMOL. Positions of the mutations are indicated. The color codes are as follows: T276C, red; S281C, green; T286C, cyan; T323C, magenta; S325C, blue. In first crystal structure the view is from the side of the PBS, in the second is from the side of the SBS, and the third is a side view. The RNA in the central hole of the hexamer is indicated in schematic form as red spirals. B, autoradiograms showing the in vitro Rho-dependent transcription termination assays of the WT and Rho* mutants with single cysteines at different places of the loop structures on the tR1 terminator. The run-off (RO) products and the termination zones of each of the Rho derivatives are indicated. Concentrations of RNAP, DNA, and Rho were 25, 10, and 50 nm, respectively. Template for the in vitro transcription assay was made by PCR amplification on the pRS22 (T7A1-H-19B TR1) using oligo pairs RS83/RK23B. C, bar diagram showing the rates of ATPase activities of all the single cysteine Rho derivatives induced by the λtR1 RNA. Concentrations of Rho and RNA were 25 nm and 20 μm, respectively.
FIGURE 8.
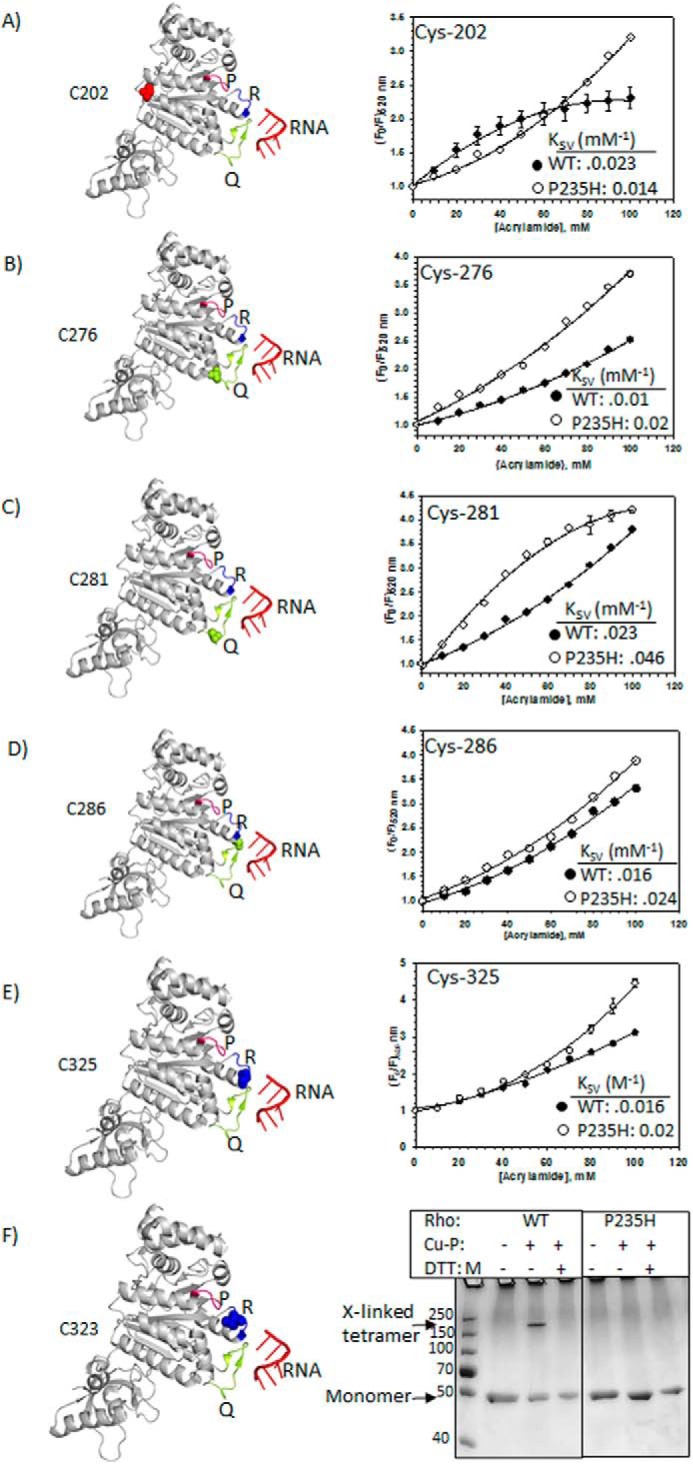
Probing the conformational changes at the central channel of the Rho mutants. Locations of different single cysteine derivatives (left panels) and corresponding modified Stern-Volmer plots obtained from the acrylamide quenching of the fluorescein molecule attached to each of the cysteine derivatives are shown in A–E. Due to non-linear nature of the plots, we used the modified Stern-Volmer equations to fit the curves as described under ”Materials and Methods.“ We calculated the KSV from the initial slopes of the curves, which approximately equals to the respective dynamic quenching constants. The P-, Q-, R-loops are indicated in the structure with the single cysteines as spheres. F, 12% (w/v) SDS-PAGE showing the formation of inter-molecular disulfide bonds between the cysteines of neighboring molecules using copper phenanthroline (Cu-P) as a reducing agent.
We used a fluorescence quenching technique with the neutral quencher, acrylamide, to probe the microenvironment of the Q- and R-loops. Upon excitation at 470 nm, fluorescein emission was monitored in the 500 to 600-nm range (spectra are not shown). Increasing concentrations of the acrylamide induced gradual quenching, and the fractions of quenching (F0/F) were plotted against the concentrations of the quencher (Fig. 8, A–E). The nature of the curves as well as the quenching constants varied from the positions of the cysteine residues, indicating the effectiveness of the probes and the quenching technique to monitor the changes in the microenvironments. From these Stern-Volmer plots (Fig. 8, A–E), we made the following observations.
C202
These cysteines are the natural ones of Rho and are located at the interface of the protomers. The downward bend in the WT Rho curve indicates the presence of more than one population of cysteines with variable accessibility. This might have arisen from the asymmetrical open conformation of the Rho hexamer (17). However, instead of a bended curve, an upward curvature was observed for the P235H Rho. This means cysteines of this Rho are present in more homogeneous environment, but are less accessible (has ∼3-fold less KSV value) than their WT counterparts.
Q-loop Cysteines (Cys-276, Cys-281, Cys-286)
Cys-276 and Cys-281 are more accessible in P235H Rho characterized by their higher KSV values. Cys-281 residues in P235H exhibited heterogeneity, indicating that Q-loops in some of its subunits are relatively buried. This trend was not observed for the Q-loops of the WT Rho. Accessibility of Cys-286 in WT and the mutant was similar.
R-loop Cysteines Cys-325 and Cys-323
Cys-325 in WT and P235H Rho were quenched in a comparable fashion. Interestingly, unlike WT Rho, Cys-323 of P235H Rho could not be labeled by 5′-fluorescein malemide (data not shown), which could be due to its complete burial inside the structure. To further support this conclusion, we assessed the ability of this cysteine residue to form disulfide bridges with the nearby cysteines. We used a reducing agent copper phenanthroline (Cu-P) (9) to induce the Cys-Cys bond (Fig. 8F). We observed that partially exposed Cys-323 (see Fig. 7A also) of the WT Rho could form disulfide linkage with neighboring cysteine from another molecule of Rho as intra-molecular bonding is not possible due to the distance constraints (18). Interestingly, these bonds were not at all formed by Cys-323 of P235H Rho that further confirmed that in this Rho* mutant this R-loop cysteine is totally buried in the structure (Fig. 8F). This same assay with other cysteines of both the Rho proteins revealed that all of them are surface accessible to form the disulfide bridges (data not shown). It should be noted that the cross-linked product migrates closer to the position of a tetramer. However, theoretically an inter-molecular dimer is likely to be the only product. The abnormal migration in SDS-PAGE could have arisen from an unusual conformation of the cross-linked product.
Taken together all the fluorescence experiments (Tb-GTP and fluorescein), we concluded that significant conformational re-orientations of the P-, Q-, and R-loops have occurred in the Rho* mutants, P235H, characterized by more surface accessible P- and Q-loops together with a buried R-loop. We predict similar type of changes in the other Rho* mutants as well. These changes may lead to the formation of more “open” central channel, which enables the mRNA to chaperone into the channel more easily. These structural alterations in the channel might be the main reason for the faster ATPase activity (Table 2) and the tighter binding to RNA at the SBS (Table 2) of the Rho* mutants.
However, it can be argued that the structural changes in different single cysteine derivatives arose due to the fluorescence labeling itself, and it is difficult to rule out that possibility. However, we like to incline to aforementioned interpretations, because we are emphasizing the relative changes between the WT and mutant Rho proteins.
Functional NusG Binding Site(s) on Rho and the Allosteric Effect
The aforementioned detailed characterizations of the Rho* mutants strongly suggested that NusG stimulates the Rho-termination process by exerting its effects at the central channel of the latter. The next task was to identify the functional interaction site(s) of NusG on the Rho to understand whether the site (s) of action coincides with the site(s) of binding. In our earlier study (9) (Fig. 9B), we reported that Rho mutants V203L, M205A, V227N, and V228N had caused partial defects in Rho-NusG CTD binary complex formations in vitro. In the same study, Lys-224 of Rho was also shown to be proximal to the NusG-CTD in this binary complex. All these in vitro assays were performed outside the EC. So to confirm whether the region encompassed by these mutations constitutes the functional Rho-NusG interaction sites, we employed various in vivo assays with these mutants (Fig. 9, C and D). In the present analyses, we also added Q193A mutant due to its proximity to this region of Rho.
FIGURE 9.
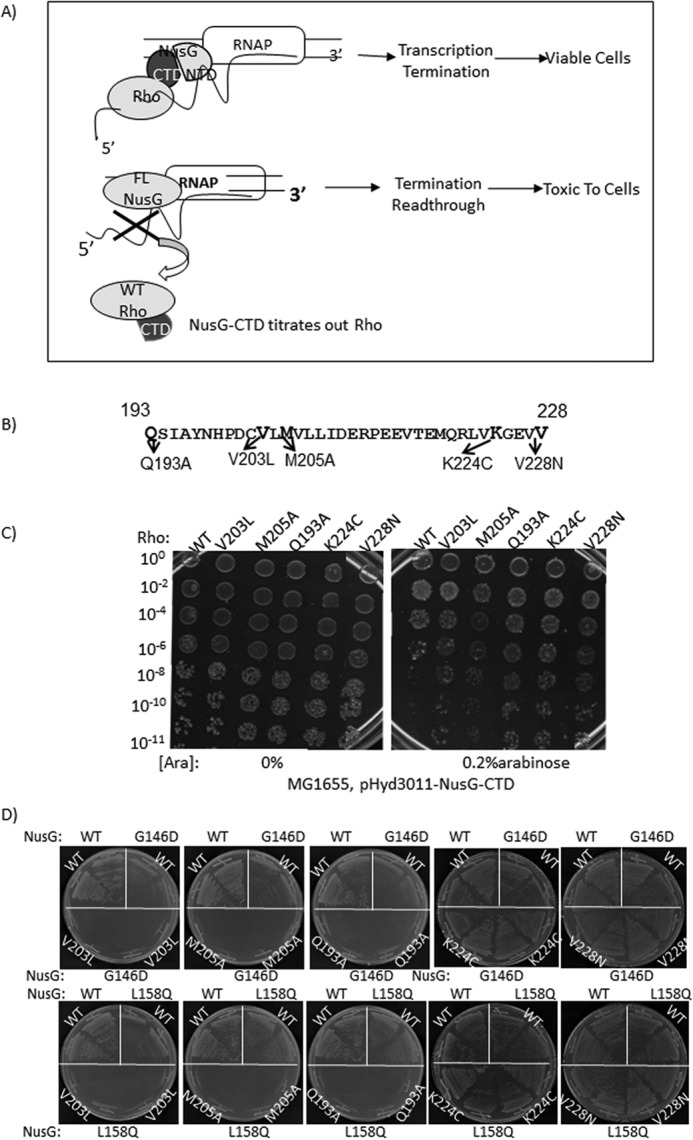
Functional interaction sites of NusG on the Rho hexamer. A, the scheme describing the CTD toxicity assay. The CTD of NusG has more affinity for the Rho than the full-length NusG, and therefore, its overexpression titrates out the latter from the elongation complex leading to conditional toxicity of the cells. Schematics show the representation of the toxicity of the overexpressed NusG-CTD in the presence of WT Rho. B, amino acid sequences of the region of Rho that cross-links to the NusG-CTD (9). The positions of the point mutations in this region are defective in Rho binding in vitro. C, MG1655Δrho strains (RS1305) expressing WT and the indicated Rho mutants from the pCL1920 plasmids were transformed with arabinose-inducible pHYD3011 (a modified pBAD plasmid) plasmid expressing the NusG-CTD fragment. Different dilutions of the overnight culture of the strains were spotted on LB plates either in the absence (0%) or presence of 0.2% arabinose to check the growth defects by the overexpression of the NusG-CTD fragment. Dilutions are indicated on the left side of the figures, which indicate the platting efficiencies. D, growth characteristics of MC4100ΔracΔrho strains having either WT or G146D or L158Q nusG mutants in the chromosome in the presence of different Rho derivatives, WT, V203L, M205A, K224C, and V228N expressed from low-copy pCL1920 plasmids. After transforming with the Rho plasmids, the chromosomal rho was removed by P1 transduction and the transductants were re-streaked on the LB plates to check the synthetic lethality.
Overexpression of NusG-CTD fragment causes toxicity to the cells because it titrates away Rho from the solution and thereby prevents it from interacting with the EC-bound NusG (Fig. 9A). We hypothesized that if the above mentioned Rho mutants have weaker interaction with the NusG-CTD, this toxicity would be alleviated. We transformed MG1655 (RS1305) strains, expressing one of the Rho mutants from a low-copy plasmid, pCL1920, with the arabinose-inducible plasmid pHYD3011-NusG-CTD. Serial dilutions of the overnight cultures were spotted on the LB plates. Overexpression of the NusG-CTD fragment was achieved by adding 0.2% arabinose in the plates. We observed that high level expressions of the CTD fragment caused toxicity only to the strains expressing WT Rho (Fig. 9C, right panel). Strains expressing mutant Rho proteins did not show significant growth defects. This suggests that these Rho mutants have weaker interactions with NusG under in vivo conditions also.
If these Rho mutants are defective in NusG binding, then they might exhibit synthetic defect with NusG-CTD mutants, G146D and L158Q, defective for Rho binding (9) (Fig. 1C). In other words, the partial defects exhibited by these Rho mutants with the WT NusG would be enhanced in the presence of NusG-CTD mutants. Presence of both Rho and NusG mutants might cause synthetic lethality as the Rho-NusG interaction in vivo is essential.
To test the synthetic lethality, MC4100 strains having chromosomal copies of the NusG mutants, G146D (RS1514) and L158Q (RS1523), were used. Different Rho mutants were expressed from a low-copy plasmid (Fig. 9D). We observed that the Rho mutants, Q193A, V203L, and M205A, showed extreme growth defects when they attempted to grow in the presence of these NusG mutants, indicating the presence of strong synthetic lethality for these combinations. Two other Rho mutants, K224C and V228N, did not show a appreciable growth defect when expressed in the presence of the NusG mutants.
Based on these in vivo assays, we concluded that the Rho mutants Q193A, V203L, and M205A are the most defective in interacting with NusG, and thereby the region encompassed by them defines the functional interaction site(s) for NusG on the Rho hexamer. The region defined by these mutants partially overlaps with the region that was defined from the in vitro pull-down or cross-linking assays (9) (Fig. 10A). Most interestingly, however, is that the NusG-binding site(s) on Rho is located far away from the central channel (∼40 Å) and also the locations of Rho* mutants do not overlap with these sites (Fig. 10A). We concluded that the NusG exerts its effect at the Rho central channel allosterically by binding at away site(s).
FIGURE 10.

A, locations of the (i) amino acids important for NusG-binding, (ii) Rho* mutants, and (iii) hypersensitive amino acids are indicated on a Rho-dimer structure (PDB code 2HT1). The NusG-binding site is circled, where amino acids in yellow are K224C, V227N, V228N, and those in green are V203L, Q193A, and M205A. B, schematic showing the kinetic/equilibrium steps during the conversion of OC to CC of the Rho hexamer. Putative step(s) those are targeted by NusG are indicated. ATP-binding and hydrolysis steps are also shown. Hexameric structures were based on the coordinates using PDB codes 3ICE and 1PVO, respectively.
Discussion
In this present study, we have attempted to understand the mechanism of NusG-mediated stimulation of the Rho-dependent transcription termination in E. coli. We isolated Rho mutants (Rho*) that are capable of making the cells viable in the absence of NusG as well as suppressed the defects of NusG-CTD mutants defective for Rho binding (Figs. 1–3). These results led us to reason that the Rho* mutants had acquired properties that NusG may impart to Rho. Analyses of the in vitro properties of these Rho* mutants revealed that they have acquired distinct conformations in the central channel characterized by the spatial re-orientations of the structural elements P-, Q-, and R-loops (Figs. 5, 6, and 8), which enabled them to have tighter RNA binding at the SBS as well as faster ATPase activities (Table 2). We propose that EC-bound NusG induces similar properties to the SBS as well as the central channel of the hexameric Rho. Finally, from the delineations of the functional interaction sites of NusG, we concluded that it exerts its effect at the SBS of Rho allosterically from a distance (Figs. 9 and 10A).
Two kinds of crystal structures of the Rho hexamer have been reported: the open (17) and the closed ring structures (18). In the asymmetrical open ring structure, the PBS is occupied by oligonucleotides, whereas in the more symmetrical closed ring structure both the PBS and the SBS are occupied with DNA and RNA oligonucleotides, respectively. Based on these sequential occupations of the PBS and SBS, we have described open (OC) to close complex (CC) conversion leading to Rho activation via multiple equilibria involving isomerization step(s) as described in Fig. 10 (6, 21). These isomerization step(s) involve major conformational changes in the central channel including RNA and ATP binding at the P-, Q-, and R-loops. In the present report, we provided strong evidence that NusG exerts its stimulatory effect on Rho by altering the interactions at the SBS through reorientations of those loops. We propose that NusG targets the open (OC) to close (CC) isomerization step(s) allosterically, accelerating the forward reactions (k2) to form the irreversible translocation competent state of Rho. This acceleration is more important when Rho-RNA binding at the PBS is relatively unstable. By increasing the k2, the equilibria shifts toward the CC form, which in turn stabilizes the interaction. The overall stimulation of the Rho-dependent termination process by NusG is the direct outcome of the aforementioned acceleration of the rate-limiting steps. This proposed NusG-induced alterations of interactions in the SBS is likely to influence the translocation as well as the helicase properties of the Rho, which might have also contributed in the faster rate of RNA release and the early termination events of the Rho* mutants (Fig. 3).
Rho has six NusG-binding sites, one on each subunit. However, the stoichiometry in the Rho-NusG complex was measured to be 1:1 Rho hexamer:NusG monomer (12), which indicates that at a given time point NusG occupies one of the six sites on the Rho. From the aforementioned model, it is likely that NusG binds to the OC state of the Rho (Fig. 10B), and due to the intrinsic asymmetry of this state, it might prefer one particular subunit of Rho for binding. It is not clear to us how such major conformational changes are induced by NusG at the central channel of Rho by occupying only one of the six sites. If Rho directly binds to the EC-bound NusG, there is a possibility that sequential occupancy of subsequent sites on each of the Rho subunits could occur during the elongation process due to the changes in the orientations of the NusG-CTD along with the moving EC.
The binding affinity for different rut sites having different RNA-sequence compositions vary widely, and therefore many of these sites are likely to be suboptimal rut sites characterized by C-poor sequences. To catch-up a rapidly transcribing EC, it is important that Rho undergoes fast OC to CC conversion at the rut sites via the multiple isomerization steps (Fig. 10B), which is likely to be slower at the suboptimal rut sites due to the weaker binding at PBS (because of high k−1). We propose that on those rut sites, NusG-mediated acceleration at the recruitment steps is essential, and thereby makes NusG an important component of Rho-dependent termination, especially under the in vivo conditions. Consistent with this proposal, genomic studies (4)4 revealed that about one-third of the Rho-dependent termination events are NusG-dependent. Earlier we have shown that a bona fide NusG-dependent terminator, trac of the rac prophage (6), has a weaker rut site characterized by slow formation of the translocation-competent Rho (CC). Hence, the silencing of this prophage through the Rho-dependent termination at trac is actually regulated by the Rho-NusG interaction (3).
Does Rho-NusG CTD Interaction Modulate NusG NTD-RNAP Interactions?
Interaction of NusG-NTD with the two structural elements, β′-clamp helix (CH) and β-gate loop (GL) near the non-template DNA strands (7) of the EC causes anti-pausing and increase in the rate of transcription elongation. It is possible that upon interaction with the NusG-CTD, Rho could alter the NusG NTD-RNAP interaction via the NusG-linker region (Fig. 1C), which might induce pausing instead of anti-pausing to the EC. RfaH, a NusG family protein, has similar NTD conformations as NusG and interacts at the similar site(s) of the EC, but induces pausing at specific sites called the ops sites (7). It is alluring to suggest that Rho-NusG CTD interactions allosterically exert shuttle conformational changes to the NusG-NTD so that the latter behaves functionally similar to RfaH-NTD, and induces enhanced pauses at the Rho-termination sites. This enhanced pausing could act as another way of stimulating Rho-dependent termination by NusG. NusG-induced pause has been documented for Bacillus subtilis NusG (22).
Experimental Procedures
Materials
NTPs were purchased from GE Healthcare. [γ-32P]ATP (3000 Ci/mmol) and [α-32P]CTP (3000 Ci/mmol) were obtained from Jonaki, BRIT (Hyderabad, India). Antibiotics, IPTG, lysozyme, DTT and bovine serum albumin (BSA) were obtained from U.S. Biochemical Corp. Restriction endonucleases, polynucleotide kinase, and T4 DNA ligase were from New England Biolab. E. coli RNA polymerase holoenzyme was purchased from Epicenter Biotechnologies. Streptavidin-coated magnetic beads were from Promega. Taq DNA polymerase was obtained from Roche Applied Science. Site-directed mutagenesis kit was purchased from Stratagene. Ni-NTA-agarose beads were from Qiagen and Sigma. Details of the bacterial strains and plasmids are described in Table 1.
TABLE 1.
Strains and plasmids used in the study
HMK, heart muscle kinase.
| Genotype | Ref. | |
|---|---|---|
| Strains | ||
| RS 257 | MC4100 galEP3 Δrac (GJ3161) | |
| RS330 | GJ3161Δrho:: KanR , with pHYD1201 shelter plasmid, AmpR | 23 |
| RS 734 | RS257 with λRS45 lysogen carrying Plac-nutR-LacZ | |
| RS862 | MG1655 WT rac− strain | 6 |
| RS1017 | RS257 with λRS45 lysogen carrying Plac-H19BnutR-trpt′-LacZ | |
| RS1263 | MG1655 K12 WT strain | |
| RS1305 | MG1655Δrac carrying marker less Δrho (Δrho::FRT) with pHYD1201 AmpR (AI) | This study |
| RS1428 | RS257 with λRS45 lysogen carrying PRM-racR-trac-lacZYA | 6 |
| RS1430 | RS257 with λRS45 lysogen carrying PRM-racR-trac-lacZYA with marker less Δrho::FRT with pHYD1201, AmpR | 6 |
| RS1458 | RS1017, flipped out Kan cassette from RS 1017 (Plac-H19B nutR-trpt′-lacZ) | 6 |
| RS1490 | Rs734 with flipped out Kan cassette made Δrho with keio collection Δrho lysate AI (Δrho::FRT) | 6 |
| RS1514 | MC4100 carrying PRM-racR-trac-lacZYA with G146D nusG in chromosome | 6 |
| RS1523 | MC4100 carrying PRM-racR-trac-lacZYA with L158Q nusG in chromosome | 6 |
| Plasmids | ||
| pHYD1201 | 3.3-kb HindIII-SalI fragment carrying rho+ subcloned from pHYD567 into HindIII SalI sites of pAM34 (pMB1; IPTG dependent replicon, AmpR) | 23 |
| pRS22 | pTL61 T with pT7A1-H-19B nutR-TR_T1T2-lacZYA, AmpR | 20 |
| pRS96 | pET21b with WT rho cloned at NdeI/XhoI site, His-tag at C-terminal; AmpR | |
| pRS604 | T7A1-λTR1 fragment cloned at Hind III site of pRS22. | 24 |
| pRS613 | pET21b with R221C rho cloned at NdeI/XhoI site, His-tag at C-terminal; AmpR | This Study |
| pRS630 | pET21b with P235H rho cloned at NdeI/XhoI site, His-tag at C-terminal; AmpR | This study |
| pRS649 | WT rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | 11 |
| pRS695 | pHYD3011 WT nusG, AmpR | 9 |
| pRS698 | pHYD3011 N-terminal domain nusG, AmpR | 9 |
| pRS699 | pHYD3011 C-terminal domain nusG, AmpR | 9 |
| pRS700 | pHYD3011 G146D nusG, AmpR | 9 |
| pRS728 | pHYD3011 L158Q nusG, AmpR | 9 |
| pRS729 | pHYD3011 V160N nusG, AmpR | 9 |
| pRS766 | pcp20 (ts), Flp recombinase, AmpR, CamR | 25 |
| pRS900 | pET 21b with R221A rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS961 | pET 21b with WT with C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS967 | P235H rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1352 | (PRM-racR-timm-lacZYA) cloned at EcoRI-SmaI of pK8628, AmpR | 6 |
| pRS1439 | V203L rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1440 | Q193A rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1444 | M205A rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1493 | R221C rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1768 | R221A rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1492 | I168V rho with its own promoter cloned at HindIII/SacI sites of pCL1920; SpR, SmR | This study |
| pRS1532 | pET 21b with I168V rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1677 | pET 21b with R221C rho cloned at NdeI/XhoI site, HMKHis tag at C-terminal; AmpR | This study |
| pRS1678 | pET 21b with R221A rho cloned at NdeI/XhoI site, HMKHis tag at C-terminal; AmpR | This study |
| pRS1676 | pET 21b with P235H rho cloned at NdeI/XhoI site, HMKHis tag at C-terminal; AmpR | This study |
| pRS1703 | pET 21b with T276C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1704 | pET 21b with S281C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1705 | pET 21b with T286C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This Study |
| pRS1706 | pET 21b with S325C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1707 | pET 21b with P235H C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1709 | pET 21b with P235H T276C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1710 | pET 21b with P235H S281C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1711 | pET 21b with P235H T286C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1712 | pET 21b with P235H T323C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pRS1713 | pET 21b with P235H S325C C202S rho cloned at NdeI/XhoI site, His tag at C-terminal; AmpR | This study |
| pK8628 | pTL61T with Plac-H19BnutR TR1-lacZAY, AmpR | 26 |
Screening and Isolation of Rho Mutants That Function Independently of NusG
RS862 (MG1655Δrac, rac::tet) was at first transformed with a plasmid overexpressing the NTD of NusG (NusG-NTD) from the arabinose-controlled PBAD promoter. The resulting strain was electroporated with a mutagenized plasmid library carrying the rho (pHYD567). The transformants were at first streaked on a LB plate to get a master plate. The colonies were then further streaked on LB plates supplemented with 0.2% arabinose. The presence of arabinose overexpressed the NusG-NTD fragment (data not shown). Healthy colonies on the arabinose plates were selected. Rho mutant plasmids were isolated from these colonies, and the mutations were confirmed by sequencing. The mutagenized plasmid library were prepared by growing the pHYD567 in the mutator strain, XL-Red (Stratagene).
Growth Assays
The strain RS862 was at first transformed with the plasmid pCL1920 carrying either WT or mutant rho, and were plated on the LB plates. To check the growth abilities of these transformed strains in the absence of nusG, the nusG::KanR cassette was moved into them by P1 transduction. These plates were incubated at 37 °C for ∼20 h to record the growth of the colonies (Fig. 1B).
To study the growth-defect suppression of the NusG mutants (G146D and L158Q) by the aforementioned Rho* mutants, strain RS1305 (MG1655Δrho, having the pHYD1201 shelter plasmid expressing rho) was at first transformed with plasmids pCL1920 carrying either WT or mutant rho and were plated on the LB plates in the absence of IPTG to remove the rho shelter plasmid. Resultant strains were then transformed with the pHYD3011 plasmids carrying either WT or the mutant nusG (G146D and L158Q). Finally, the chromosomal nusG (nusG::kanR) was removed by P1 transduction and the transductants were plated to assess the growth defect suppression by the Rho mutants (Fig. 1C). It should be mentioned that the aforementioned procedure of creating the nusG deletion retained the NusG-NTD fragment in the chromosome. However, this fragment does not support NusG function in vivo, and thereby does not interfere in our studies. Whenever required, rho was deleted by P1 transduction using the lysate grown on RS330 that does not have gene duplication (23) as was observed in the original keio collection.
For the NusG-CTD overexpression-mediated toxicity assays, strain RS1305 was at first transformed with plasmids pCL1920 carrying either WT or different mutant rho and were plated on the LB plate in the absence of IPTG to remove the shelter plasmid expressing a WT copy of rho. Once the shelter plasmid was removed, these strains were transformed with the plasmid (pHYD3011, a pBAD derivative) carrying the CTD domain of NusG. Arabinose-induced high-level of expressions (data not shown) of this domain from these plasmids are toxic to the cells. Serial dilutions of the overnight culture of each of these transformants were spotted both in the absence and presence of 0.2% arabinose and were grown overnight at 37 °C (Fig. 9C). For the synthetic lethality assays with the Rho mutants (V203L, Q193A, M205A, K224C, and V228N) in combination with NusG mutants (G146D, L158Q), RS1514 (MC4100 nusGG146D), and RS1523 (MC4100 nusGL158Q) were first transformed with plasmids pCL1920 carrying either WT or mutant rho. Finally, the chromosomal rho (rho::kanR) was removed by P1 transduction and the transductants were plated to assess the synthetic lethality of the NusG mutants together with the Rho mutants (Fig. 9D).
In Vivo Rho-dependent Transcription Termination Assays
For the in vivo termination assays, we have used reporter system, PRM-trac-lacZYA (NusG-dependent terminator trac fused to lacZ (6). Transcription was initiated from either PRM promoters. These experiments were performed in the MC4100 strain RS1430 containing the trac-lacZ reporter cassette. In all these strains chromosomal rho was deleted and this function was supplied from a shelter plasmid. To introduce the Rho* mutants, strain RS1430 was first transformed with pCL1920 plasmids carrying either WT or mutant rho and were plated on the LB plate in the absence of IPTG to remove the Rho shelter plasmid. Resultant strains were then transformed with pHYD3011 plasmids carrying WT and mutant nusG, following which chromosomal nusG was deleted by P1 transduction (Fig. 2B). In vivo termination efficiencies at different Rho-dependent terminators were estimated by measuring the β-galactosidase activities the following the standard protocols using microtiter plates and the Spectramax Plus plate reader (11).
Preparations of Rho* Mutants
Rho* mutants obtained from genetic screening were cloned in pET21b vectors, and purified using Ni-NTA beads (Qiagen) as per the manufacturer's protocol. The proteins eluted from the Ni-NTA columns were further purified by passing through a heparin-Sepharose column (Amersham Biosciences). To avoid the contamination of the WT Rho expressed from chromosome, the purification was performed in the presence of 1 m NaCl. As the dissociation of the subunits occurred at high salt, non-His-tagged WT protomers, if present, were removed in the wash fractions. CD spectra of different mutants were similar to that of the WT Rho (data not shown), indicating that the overall structural integrity of the mutants was intact. P103L, P235H, and R221A were constructed by site-directed mutagenesis.
Preparations of Single Cys Derivatives of Rho
All the single Cys derivatives of Rho were constructed by site-directed mutagenesis using the Stratagene kit. First, the only Cys residue of the WT Rho (at 202; pRS96) was removed by introducing C202S mutations that yielded a zero Cys Rho (pRS961). The single Cys derivatives, T276C (Cys-276), S281C (Cys-281), T286C (Cys-286), T323C (Cys-323), and S325C (Cys-325) were made using pRS961 as a template for WT Rho. Similarly single Cys derivatives for Rho P235H were made on the zero Cys P235H-C202S (pRS1707) as a template. All these derivatives were His-tagged and the same purification procedures were followed, except that 20 mm β-mercaptoethanol was used in the buffers using Ni-NTA columns and was replaced with 50 mm DTT in the subsequent steps to avoid disulfide formations. The serines (Ser) and threonines (Thr) where selected based on their locations in the central channel of Rho and their proximity to the secondary RNA binding site.
Templates for in Vitro Transcriptions
Linear DNA templates for in vitro transcription assays were made by PCR amplification from the plasmids, pRS22 (PT7A1-H19-BtR1) and pRS1352 (PRM-racR/trac) using the oligo pairs RS83/RS845 and RS83/RS956. To form a road block (RB) on the template, a 22-bp lac operator sequence was inserted after the trac terminator using a downstream primer having the lac operator sequence. A transcription elongation road block is formed at this sequence in the presence of lac repressor. To immobilize the DNA templates to the streptavidin-coated magnetic beads (Promega), a biotin group at the 5′-end of the templates was incorporated by using a biotinylated primer RS83. In the aforementioned templates, transcription was initiated from a PRM promoter.
In Vitro Transcription Assays
We performed single and multiple rounds of in vitro transcription assays for the PT7A1 and PRM promoters, respectively. Since the latter promoter is weak, to improve the yield of RNA, we omitted rifampicin from the reaction mixtures. Single round transcriptions were performed on PT7A1-H19-BtR1 template in the presence of rifampicin in the transcription buffer (T-buffer; 25 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 50 mm KCl, 1 mm DTT, and 0.1 mg/ml of BSA). At first, a 23-mer elongation complex, EC23, was formed by mixing 5 nm DNA template, 25 nm RNA polymerase, 175 mm adenylyl (3′-5′)-uridine, 5 μm each of ATP and GTP and 2.5 μm CTP. [α-32P]CTP (3000Ci/mmol, BRIT, Hyderabad) was used for labeling the transcripts. This EC was then chased with 250 μm NTPs for 15 min. For the PRM-racR/trac template, it was immobilized to the streptavidin-coated beads to assess the terminated products. Reactions were carried out in the transcription buffer (T-buffer) at 37 °C for 10 min. Concentrations of all the NTPs were 250 μm, except that CTP was 50 μm. [γ-32P]CTP (3000 Ci/mmol, BRIT, Hyderabad) was used for labeling the transcripts. Concentrations of DNA, RNAP, Rho, and NusG were 5, 25, 50, and 200 nm, respectively. At the end of the reactions, tubes were held against a magnetic stand and half of the supernatant (S) was removed and directly mixed with formamide loading dye. The rest of the mixture (P; half of the supernatant and pellet) was phenol extracted and mixed with the loading dye. Samples were run onto 6% sequencing gel and analyzed by FLA-9000 PhosphorImager (Fuji) (Fig. 3A).
For the RNA release assays from the stalled ECs, the PRM-trac-lacO template immobilized on streptavidin-coated magnetic beads was used. 100 nm lac repressor was added to the DNA templates to form a road-blocked EC. On this template, at first a 23-mer EC (EC23) was formed by initiating the reactions with 175 μm adenylyl (3′-5′)-uridine, 5 mm GTP, 5 mm ATP, 2.5 mm CTP, and [γ-32P]CTP (3000 Ci/mmol). The EC23 was then chased with 250 μm each of the NTPs for 2 min to form the stalled EC, following which excess NTPs were removed by washing the beads thoroughly. 50 nm Rho and 200 nm NusG (when required) were added to the stalled EC and 10 μl of samples were removed at the indicated time points, separated into S (half of the supernatant) and P (other half of supernatant + pellet) on the magnetic stand. The rest of the procedures were same as described above (Fig. 3D). The fractions of released RNA ((2S)/(S) + (S+P)) were plotted against time and the points were fitted to a curve following exponential rise equations either of the forms: y = a (1-exp−bx) or y = y0 + a(1-exp−bx), where “b” denotes the rate, “a” is amplitude of the RNA release process, and “y0” is minimum value of y at 0 time point. If a lag is observed in the RNA release kinetics, it was fitted to a sigmoidal equation of the form: y = y0 + (a)/[1 + (x/x0)b], where “x0” is the inflection point of the curve and b is the slope (Fig. 3, E–G).
Binding Assays of Rho with Poly(dC)34 and Other Oligonucleotides
The primary RNA-binding property of the WT and mutant Rho proteins was measured by using a 5′-fluorescein labeled 34-mer DNA oligo (dC34) (27). Oligo(dC)34 specifically binds at the primary PBS (11) of Rho. The fluorescence experiments were performed in the buffer containing 25 mm Tris-HCl (pH 8.0), 5 mm MgCl2, 50 mm KCl, 1 mm DTT, and 0.1 mg/ml of BSA at 25 °C in a Hitachi F7000 fluorescence spectrophotometer, under CAT mode, with a PMT voltage of 700 V and scan speed of 240 nm. Excitation was at 470 nm, whereas the emission range was from 500 to 600 nm. Quenching of fluorescence intensity of 10 nm fluorescein-(dC)34 upon addition of increasing concentrations of Rho was used as a measure for determining the primary RNA binding. The ratio of the initial fluorescence intensity at 520 nm (F0) and that obtained upon addition of each concentrations of Rho were plotted against concentrations of Rho. The plots were fitted to a hyperbolic equation of the form, y = (a × b)/(b + x), to obtain the binding isotherms. The b values gave the measure of dissociation constant (Kd) (Table 2).
Binding of other DNA sequences to the PBS of the WT and different Rho mutants were measured qualitatively by gel-shift assays using radiolabeled DNA oligos (data not shown). We have used the following oligonucleotides for these assays: (i) oligo with the λtR1 rut site sequence (RS1387) CCCCGCTCTTACACATTCCAGCCCTGAAAAAGGG and (ii) an oligo with a “non-rut” sequence (RS1386) TGCAGGCCGAAAGATTTTTTTAACTATAAA. The gel-shift assays were performed using 10 nm of radiolabeled oligo in the T buffer with different concentrations of WT or mutant Rho (10–50 nm) proteins. The reaction mixtures were supplemented with 10% glycerol and incubated at 37 °C for 10 min before loading onto a 5–15% gradient native acrylamide gel. Gels were run in 0.5× TBE buffer in a cold room and the signals were analyzed by PhosphorImager.
Measurements of the Rate of ATP Hydrolysis
The rates of RNA-dependent ATP hydrolysis of WT and mutant Rho proteins were measured using λtR1 RNA, containing the rut sites of the λtR1 terminator. DNA templates for transcribing this RNA were prepared by PCR amplification from plasmid pRS604 using the RS139/RS341 oligo pair. The λtR1 RNA was made from this template using the Mega transcript kit (Ambion). ATP hydrolysis was assayed by monitoring the release of Pi from ATP that was observed on the polyethylenimine TLC plates using 0.75 m KH2PO4 (pH 3.5) as a mobile phase buffer. The hydrolysis reactions were performed in T buffer (25 mm Tris-HCl, pH 8.0, 50 mm KCl, 5 mm MgCl2, 1 mm DTT, and 0.1 mg/ml of BSA) at 37 °C. The rate of ATP hydrolysis of 100 mm ATP together with [γ-32P]ATP (3500 Ci/mmol; BRIT, India) were measured using 50 nm Rho. The reaction was initiated by the addition of 0.2 μm RNA. Aliquots were removed, and the reactions were stopped with 1.5 m formic acid at various time points. Release of Pi was analyzed by exposing the TLC sheets to a PhosphorImager screen and subsequently by scanning using Typhoon 9200 (GE Healthcare) (Table 2).
Oligo rC25-Rho Binding Assays
Affinity (Km) of rC25 for the WT and mutant Rho proteins was estimated from the extent of the oligo concentration-dependent ATP hydrolysis using the double reciprocal plots (Lineweaver-Burk plot) of the form: (1/V) = (Km + [S])/(Vmax[S]), where, V and S are reaction velocity (nanomoles of ATP hydrolyzed per μg of Rho/min) and substrate concentrations, respectively. Plots were confined within the dynamic range of the ATP hydrolysis. Experiments were performed essentially following the methods measuring the ATP hydrolysis rate as described above, except that oligo-(dC)34 was added in the reactions to block interactions of RNA at the PBS of Rho (Table 2).
Limited Proteolysis by Trypsin and V8 Proteases
To monitor the proteolytic cleavage products, WT and mutant Rho proteins were end-labeled by 32P at their respective heart muscle kinase tag. This tag was introduced at the C terminus of the Rho proteins. The heart muscle kinase-tagged Rho proteins were radiolabeled with [γ-32P]ATP (3000 Ci/mmol) using protein kinase A. 0.3 μm of labeled Rho in T-buffer supplemented with 1 mm ATP was subjected to proteolytic digestion with 0.05 μg of trypsin for the indicated time period at 37 °C. The reactions were stopped by SDS-loading dyes and samples were heated to 95 °C for 3 min before loading onto a 12–16% gradient SDS-PAGE. For V8 digestion, 0.3 μm of labeled Rho were incubated with increasing concentrations of V8 protease in the T-buffer supplemented with 1 mm ATP for 15 s at 37 °C. Molecular weight markers of end-labeled Rho were generated by cyanogen bromide (CNBr, methionine-specific), Lys-C and submaxillary protease (Arg-C) mediated cleavages following standard protocols. Gels were exposed overnight to a phosphorimager screen and were scanned using PhosphorImager Typhoon 9200 and analyzed by ImageQuant software (Fig. 5, A–C).
Fluorescence Spectroscopy
Tb-GTP Quenching
The fluorescence quenching experiments for studying the accessibility of the ATP binding pocket in WT and different Rho mutants were performed in buffer containing 10 mm Tris-HCl (pH 7.0) and 100 mm KCl at 25 °C in a Hitachi F7000 fluorescence spectrophotometer, under CAT mode, with a PMT voltage of 700 V and scan speed of 240 nm. GTP complexed with TbCl3, Tb-GTP (3:1; 300 μm terbium chloride and 100 μm GTP), upon excitation at 295 nm yields two emission peaks at 488 and 547 nm (28, 29). As Rho can also utilize GTP, it can be assumed that Tb-GTP binds to the same ATP binding pocket. The fluorescence intensity at 547 nm was used to monitor the acrylamide quenching pattern. The changes in fluorescence intensity (F0/F)547 nm was plotted against increasing concentrations of acrylamide (Q). Due to the presence of both static (KS) and dynamic quenching (KD), a modified Stern-Volmer equation: (F0/F)547 = 1 + (KD + KS)[Q] +KDKS[Q]2, where, F0 and F are the initial and final fluorescence intensities, respectively and [Q] is the concentration of acrylamide (Fig. 6C). Quenching constant KSV, which approximately equals to the dynamic quenching, were calculated from the initial slopes of the curves.
Fluorscein-Cys Rho Quenching
Different single Cys derivatives of Rho were at first labeled with fluorescein-5-malemide essentially following the procedure described earlier (19). Fluorescence quenching assays of the fluorescein-labeled Rho proteins were performed using the neutral quencher acrylamide. Quenching was done by adding increasing concentrations of acrylamide (10–100 mm) to 100 nm of labeled Rho in buffer containing 20 mm phosphate buffer, 150 mm NaCl, 8 mm EDTA. The quenching of fluorescence intensity was monitored at 520 nm. The change in fluorescence intensity at 520 nm was plotted against increasing concentrations of acrylamide (Fig. 8, A–E). The quenching constant was obtained in a similar manner as described above for the Tb-GTP fluorescence.
Intermolecular Cys-Cys Formation Assays
A 10 mm stock of Cu-P was prepared by adding 1 μl of 1 m CuSO4 and 3 μl of 1 m phenanthroline in 100 μl. In each reaction, 1 mm Cu-P was added to 1 μg of Rho and incubated for 5 min in a buffer containing 25 mm HCl (pH 7), 50 mm KCl, and 100 mm NaCl. To observe the products, reaction mixtures were run on 12% SDS-PAGE and the gels were visualized by Coomassie Blue staining (Fig. 8F).
Author Contributions
V. V. performed all the in vitro and in vivo experiments. S. A. purified various proteins. R. S. conceived the project and designed the experiments. V. V. and R. S. wrote the manuscript.
Acknowledgments
We thank summer student Shreya Kar for participating in the screening of the Rho* mutants. We also thank Dr. Pallabi Mitra for critically reading the manuscript.
This work was supported in part by DBT-Center of Excellence in Microbial Physiology and Center for DNA Fingerprinting and Diagnostics intramural funding. The authors declare that they have no conflicts of interest with the contents of this article.
V. Valabhoju, S. Agrawal, and R. Sen, unpublished data.
- RNAP
- RNA polymerase
- EC
- elongation complex
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- Ni-NTA
- nickel-nitrilotriacetic acid
- PBS
- primary binding site
- OC
- open complex
- CC
- closed complex
- SBS
- secondary RNA binding site
- nt
- nucleotide(s)
- PDB
- Protein Data Bank
- RB
- road block.
References
- 1. Peters J. M., Vangeloff A. D., and Landick R. (2011) Bacterial transcription terminators: the RNA 30-end chronicles. J. Mol. Biol. 412, 793–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banerjee S., Chalissery J., Bandey I., and Sen R. (2006) Rho-dependent transcription termination: more questions than answers. J. Microbiol. 44, 11–22 [PMC free article] [PubMed] [Google Scholar]
- 3. Cardinale C. J., Washburn R. S., Tadigotla V. R., Brown L. M., Gottesman M. E., and Nudler E. (2008) Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science 320, 935–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peters J. M., Mooney R. A., Grass J. A., Jessen E. D., Tran F., and Landick R. (2012) Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 26, 2621–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mooney R. A., Davis S. E., Peters J. M., Rowland J. L., Ansari A. Z., and Landick R. (2009) Regulator trafficking on bacterial transcription units in vivo. Mol. Cell 33, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shashni R., Qayyum M. Z., Vishalini V., Dey D., and Sen R. (2014) Redundancy of primary RNA-binding functions of the bacterial transcription terminator Rho. Nucleic Acids Res. 42, 9677–9690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tomar S. K., and Artsimovitch I. (2013) NusG-Spt5 proteins-universal tools for transcription modification and communication. Chem. Rev. 113, 8604–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sen R., Chalissery J., Qayyum M. Z., Vishalini V., and Muteeb G. (2014) Nus factors of Escherichia coli. 6, EcoSal Plus ESP-0008–2013 [DOI] [PubMed] [Google Scholar]
- 9. Chalissery J., Muteeb G., Kalarickal N. C., Mohan S., Jisha V., and Sen R. (2011) Interaction surface of the transcription terminator Rho required to form a complex with the C-terminal domain of the antiterminator NusG. J. Mol. Biol. 405, 49–64 [DOI] [PubMed] [Google Scholar]
- 10. Burns C. M., Nowatzke W. L., and Richardson J. P. (1999) Activation of Rho-dependent transcription termination by NusG: dependence on terminator location and acceleration of RNA release. J. Biol. Chem. 274, 5245–5251 [DOI] [PubMed] [Google Scholar]
- 11. Chalissery J., Banerjee S., Bandey I., and Sen R. (2007) Transcription termination defective mutants of Rho: role of different functions of Rho in releasing RNA from the elongation complex. J. Mol. Biol. 371, 855–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pasman Z., and von Hippel P. H. (2000) Regulation of rho-dependent transcription termination by NusG is specific to the Escherichia coli elongation complex. Biochemistry 39, 5573–5585 [DOI] [PubMed] [Google Scholar]
- 13. Mooney R. A., Schweimer K., Rösch P., Gottesman M., and Landick R. (2009) Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators. J. Mol. Biol. 391, 341–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Washburn R. S., and Stitt B. L. (1996) In vitro characterization of transcription termination factor Rho from Escherichia coli rho (nusD) mutants. J. Mol. Biol. 260, 332–346 [DOI] [PubMed] [Google Scholar]
- 15. Pereira S., and Platt T. (1995) Analysis of E. coli Rho factor: mutations affecting secondary-site interactions. J. Mol. Biol. 251, 30–40 [DOI] [PubMed] [Google Scholar]
- 16. Cheeran A., Babu Suganthan R., Swapna G., Bandey I., Achary M. S., Nagarajaram H. A., and Sen R. (2005) Escherichia coli RNA polymerase mutations located near the upstream edge of an RNA:DNA hybrid and the beginning of the RNA-exit channel are defective for transcription antitermination by the N protein from lambdoid phage H-19B. J. Mol. Biol. 352, 28–43 [DOI] [PubMed] [Google Scholar]
- 17. Skordalakes E., and Berger J. M. (2003) Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell 114, 135–146 [DOI] [PubMed] [Google Scholar]
- 18. Skordalakes E., and Berger J. M. (2006) Structural insights into RNA-dependent ring closure and ATPase activation by the Rho termination factor. Cell 127, 553–564 [DOI] [PubMed] [Google Scholar]
- 19. Ranjan A., Sharma S., Banerjee R., Sen U., and Sen R. (2013) Structural and mechanistic basis of anti-termination of Rho-dependent transcription termination by bacteriophage P4 capsid protein Psu. Nucleic Acids Res. 41, 6839–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lakowicz J. R. (1999) Principles of fluorescence spectroscopy, 2nd Ed., Kluwer Academic/Plenum Publishers, New York [Google Scholar]
- 21. Qayyum M. Z., Dey D., and Sen R. (2016) Transcription elongation factor NusA is a negative regulator of Rho-dependent termination. J. Biol. Chem. 291, 8090–8108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yakhnin A. V., Murakami K. S., and Babitzke P. (2016) NusG is a sequence-specific RNA polymerase pause factor that binds to the non-template DNA within the paused transcription bubble. J. Biol. Chem. 291, 5299–5308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harinarayanan R., and Gowrishankar J. (2003) Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. J. Mol. Biol. 332, 31–46 [DOI] [PubMed] [Google Scholar]
- 24. Dutta D., Chalissery J., and Sen R. (2008) Transcription termination factor rho prefers catalytically active elongation complexes for releasing RNA. J. Biol. Chem. 283, 20243–20251 [DOI] [PubMed] [Google Scholar]
- 25. Cherepanov P. P., and Wackernagel W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 [DOI] [PubMed] [Google Scholar]
- 26. Neely M. N., and Friedman D. I. (1998) Functional and genetic analysis of regulatory regions of E. coli phage H-19B: location of shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol. Microbiol. 28, 1255–1267 [DOI] [PubMed] [Google Scholar]
- 27. Kalarickal N. K., Ranjan A., Kalyani B. S., Wal M., and Sen R. (2010) A bacterial transcription terminator with inefficient molecular motor action but with a robust transcription termination function. J. Mol. Biol. 395, 966–982 [DOI] [PubMed] [Google Scholar]
- 28. Kumar K. P., and Chatterji D. (1990) Resonance energy transfer study on the proximity relationship between the GTP binding site and the rifampicin binding site of Escherichia coli RNA polymerase. Biochemistry 29, 317–322 [DOI] [PubMed] [Google Scholar]
- 29. Sen R., and Dasgupta D. (2003) Simple fluorescence assays probing conformational changes of Escherichia coli RNA polymerase during transcription initiation. Methods Enzymol. 370, 598–605 [DOI] [PubMed] [Google Scholar]