

Abstract
Disease-causing mutations in G protein-coupled receptor (GPCR) genes, including the V2 vasopressin receptor (V2R) gene, often cause misfolded receptors, leading to a defect in plasma membrane trafficking. A novel V2R mutation, T273M, identified in a boy with partial nephrogenic diabetes insipidus (NDI), shows intracellular localization and partial defects similar to the two mutants we described previously (10). Although non-peptide V2R antagonists have been shown to rescue the membrane localization of V2R mutants, their level of functional rescue is weak. Interestingly, it has been reported that a non-peptide agonist, OPC51803, activates misfolded V2R mutants intracellularly without degradation, thus potentially serving as a therapeutic agent against NDI (14). In our current experiments, however, a peptide antagonist blocked arginine vasopressin (AVP)- or OPC51803-stimulated cAMP accumulation both in COS-7 and MDCK cells, suggesting that OPC51803 mainly stimulates cell surface V2R mutants. In addition, our analyses revealed that OPC51803 works not only as a non-peptide agonist that causes activation/β-arrestin-dependent desensitization of V2R mutants expressed at the plasma membrane but also as a pharmacochaperone that promotes the endoplasmic reticulum-retained mutant maturation and trafficking to the plasma membrane. The ratio of the pharmacochaperone effect to the desensitization effect likely correlates negatively with the residual function of the tested mutants, suggesting that OPC5 has a more favorable effect on the V2R mutants with a less residual function. We speculated that the canceling of the desensitization effect of OPC51803 by the pharmacochaperone effect after long-term treatment may produce sustainable signaling, and thus pharmacochaperone agonists such as OPC51803 may serve as promising therapeutics for NDI caused by misfolded V2R mutants.
Keywords: G protein-coupled receptor (GPCR), membrane trafficking, molecular chaperone, receptor regulation, signal transduction
Introduction
Loss-of-function mutations of the V2 vasopressin receptor (V2R)3 cause X-linked congenital nephrogenic diabetes insipidus (NDI), resulting in renal resistance to the antidiuretic hormone arginine vasopressin (AVP) (1–4). Most of these mutations cause intracellular retention of V2R in the endoplasmic reticulum and a failure to interact with AVP. To date, several kinds of pharmacological chaperones have been described (5–9), and we have also recently reported that OPC31260 (OPC3) and OPC41061 (OPC4) show unique action against wild-type and two mutant V2R variants including a novel Ser-333del mutant, V2R, that we had identified previously (10). These agents augmented basal cAMP accumulation in cells expressing mutant V2R by working as pharmacochaperones but suppressed basal cAMP accumulation in cells expressing wild-type V2R by functioning as inverse agonists. With respect to this target-dependent mode of action, these agents have also been found to show protean agonism (11–13). These findings have helped to further elucidate how GPCRs work, both physiologically and pathophysiologically.
From a clinical point of view, however, neither of these compounds could be used to treat at least our partial NDI patients because they inhibit AVP action. Interestingly, a non-peptide agonist, OPC51803 (OPC5), has been reported to activate the endoplasmic reticulum (ER)-retained V2R mutants intracellularly (14). Considering that most of the NDI-causing mutations in V2R genes lead to misfolded receptors that are recognized and retained by the quality control system of the ER, the intracellular activation of ER-retained mutant GPCRs is one of the currently attractive strategies for developing novel treatments for such disorders (5, 15).
Here, we identified a novel V2R mutation in a 4-year-old boy diagnosed with partial NDI due to his high basal AVP levels. This novel mutant receptor was found to localize mainly in the ER, similar to the two NDI V2R mutants we reported previously (10). Based on another previous report (14), we speculated that OPC5 may be a promising compound for treatment of disorders caused by ER-retained V2R mutants. By virtue of the residual function of V2R mutants that cause partial NDI clinically, we identified a novel mechanism of action of OPC5 in our current study that may enable sustained signaling by these receptor mutants.
Results
Identification of a New V2 Receptor Mutation in a Partial NDI Patient
A 4-year-old male patient showing polydipsia and polyuria was suspected of having NDI based on an increased basal AVP level. Two standard diagnostic tests, the water deprivation and pitressin loading tests, were performed on admission. The stimulation of endogenous AVP secretion by water deprivation did not concentrate the patient's diluted urine (<200 mosm/kg · H2O), which was not unexpected because his AVP level even after water deprivation (66.8 pg/ml) was as high as his basal AVP level (66.7 pg/ml). In contrast, exogenous AVP (pitressin) loading, which is a stronger stimulation, concentrated his urine up to 636 mosm/kg · H2O (>400 mosm/kg · H2O). Based on these results, he was diagnosed with partial NDI (10) (Fig. 1A). We subsequently identified a missense mutation in the V2R gene (AVPR2), T273M, causing a substitution of threonine with methionine at the 273rd residue within the sixth transmembrane domain (Fig. 1, B and C). Whereas most NDIs to date have been reported to show a complete phenotype, this case showed only a partial NDI clinically. These findings suggested that the T273M mutation may cause a partial loss-of-function of V2R, which is very rare. To our knowledge, this is only the 13th case of partial NDI to be described but the first to harbor a T273M mutation in the V2R (3, 10, 16–24).
FIGURE 1.

Clinical testing and V2 receptor gene mutation profile of the partial NDI patient. A, water deprivation and pitressin loading test results of the patient. B, identification of a novel V2R gene mutation (T273→M). C, the structure model of the V2 receptor highlighted with the current mutation (T273M) and two previous mutations (Y128S and S333del) discovered in our partial NDI patients.
V2R-T273M Localizes Mainly within the ER, Similar to Other NDI Mutants
Using immunofluorescence confocal microscopy, we examined whether the T273M V2R mutant was expressed correctly. In COS-7 cells transiently expressing Myc-tagged wild-type V2R (COS-WT), Myc-tagged T273M-V2R (COS-T273M), Myc-tagged Y128S-V2R (COS-Y128S), and Myc-tagged Ser-333del-V2R (COS-S333del), all of the mutant receptors appeared to localize in the cytosol, whereas V2R-WT localized mainly at the plasma membrane. All of the V2R mutants were also found to colocalize predominantly with an ER marker, protein disulfide isomerase (Fig. 2A, arrowheads indicate the representative colocalization particles). Based on the findings of a previous report (14), we speculated that OPC5 might be a useful treatment candidate for our patient, as it might activate the ER-retained mutant V2R intracellularly.
FIGURE 2.
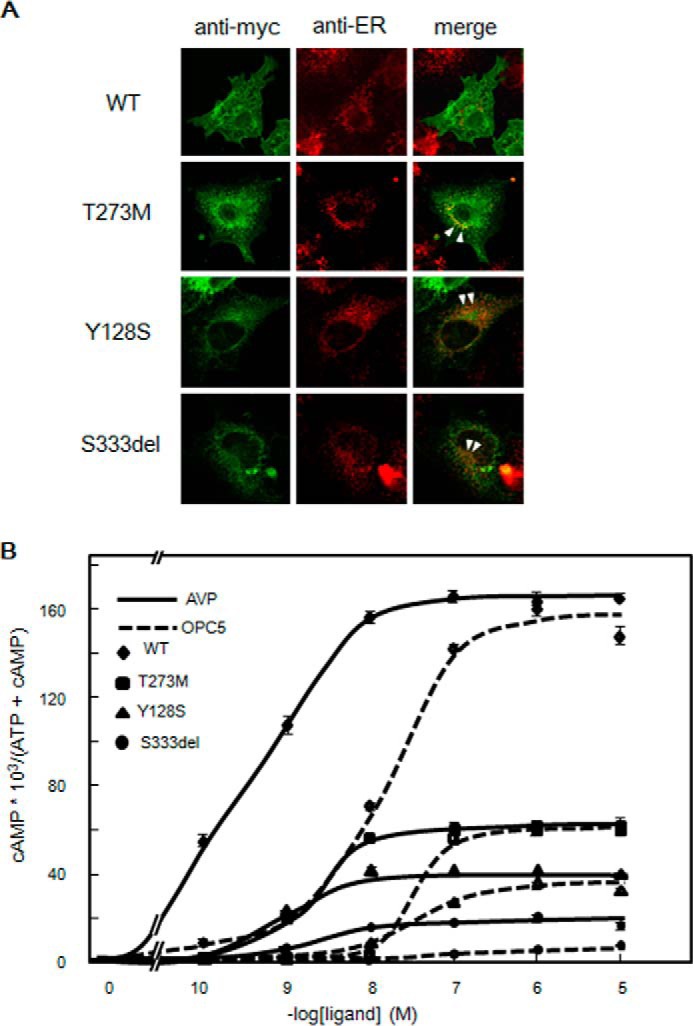
Immunofluorescence microscopy analysis and cAMP accumulation in V2R-WT and V2R mutant-expressing cells. A, colocalization of Myc-tagged V2R-T273M, V2R-Y128S, and V2R-Ser-333del with an ER marker. Immunofluorescence microscopy analysis of COS-7 cells transiently expressing Myc-tagged V2R-WT or the indicated V2R mutant using confocal microscopy is shown. Fluorescent images were developed with anti-Myc or anti-protein disulfide isomerase (ER marker) antibodies as described under “Experimental Procedures.” The arrowheads indicate the representative colocalization particles. B, AVP- or OPC5-stimulated cAMP accumulation associated with V2R-WT and our three partial NDI V2R mutants. COS-7 cells (106 cells) were transiently transfected with V2R-WT (diamond) or partial NDI mutants (V2R-T273M, square; V2R-Y128S, triangle; V2R-Ser-333del, circle); 0.2 μg of pcDNA3 containing each Myc-tagged V2R was used. After 24 h, the cells were reseeded into 24-well plates at 0.75 × 105 cells/well to which [3H]adenine (2 μCi/ml, GE Healthcare) was added. At 48 h later, after washing in assay medium with 1 mm IBMX, the cells were incubated for 30 min at 37 °C in serum-free medium with or without increasing concentrations of AVP (solid line) or OPC5 (dashed line) (from 0.1 nm to 10 μm). cAMP accumulation was measured as described “Experimental Procedures.” Data are the means ± S.D. of three independent experiments. Each set of results is representative of at least two additional experiments. The EC50 (nm) of AVP and OPC5 are 0.3 or 10.8 for WT, 1.8 and 20.0 for T273M, 0.7 and 26.9 for Y128S, and 3.3 and undetermined for S333del, respectively. The Emax values (nm) of AVP and OPC5 are 165 and 150 for WT, 61 and 63 for T273M, 41 and 38 for Y127S, and 18 and undetermined for S333del, respectively. In the case of S333del, EC50 and Emax of OPC5 cannot be calculated because the OPC5 curve does not reach a plateau even at 10 μm OPC5.
cAMP Accumulation Is Associated with V2R-WT and the Three Partial NDI Mutants
To characterize the new V2R mutant in our patient and examine the agonist activity of OPC5 against this variant, we examined AVP- and OPC5-dependent cAMP accumulation in COS-WT, COS-T273M, COS-Y128S, and COS-S333del cells (Fig. 2B). COS-WT cells showed a robust OPC5-dependent cAMP accumulation response in a dose-dependent manner, the peak of which was close to that of the AVP-dependent cAMP accumulation response. In contrast, the dose-response curve of OPC5 was shifted right by about 1 order of magnitude compared with that of AVP. Moreover, COS-T273M, COS-Y128S, and COS-S333del cells showed only partial AVP- and OPC5- dependent cAMP accumulation responses in a dose-dependent manner. In terms of the relative potency (i.e. EC50) and efficacy (i.e. Emax) of AVP and OPC5, COS-T273M cells showed a pattern similar to COS-WT; the dose-response curve for OPC5 was shifted right by about 1 order of magnitude compared with that of AVP. The AVP-dependent partial cAMP accumulation in COS-T273M, COS-Y128S, and COS-S333del cells suggested that at least a portion of each mutant protein might be expressed at the plasma membrane, despite their predominantly intracellular localization as observed by immunofluorescence microscopy. This possibility prompted us to quantify the cell surface expression level of each mutant in detail. By cell surface ELISA, each mutant was indeed found to be expressed, at least in part, at the plasma membrane (Fig. 3A). Based on these results, we speculated that OPC5, similar to peptide AVP, may activate V2R mutants not intracellularly but at the plasma membrane.
FIGURE 3.
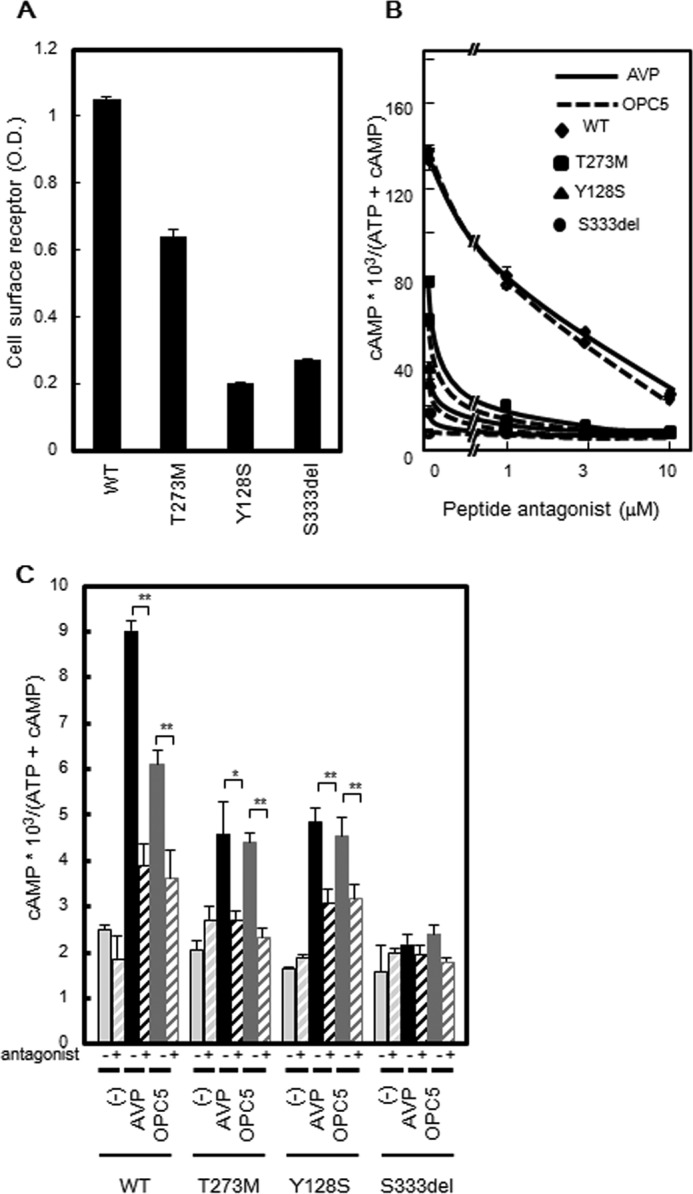
Cell surface expression of WT and mutant V2R proteins and the effect of a peptide antagonist on cAMP accumulation. A, plasma membrane expression of V2R-WT and V2R mutant proteins measured by cell surface ELISA. COS-7 cells (1 × 106 cells) were transiently transfected with V2R-WT and partial NDI mutants (0.2 μg of pcDNA3 containing each Myc-tagged V2R was used). At 24 h later, the cells were reseeded into 24-well plates at 0.75 × 105 cells/well. After a further 24 h, the plasma membrane expression levels of V2R-WT and V2R mutant proteins were measured by cell surface ELISA as described under “Experimental Procedures.” The data shown are the means ± S.D. of three independent experiments. Each set of results is representative of at least two additional experiments. B, effect of the peptide antagonist on AVP- or OPC5-stimulated cAMP accumulation in V2R-WT and V2R mutant-expressing cells. V2R-WT (diamond) and V2R mutant-expressing COS-7 cells (V2R-T273M, square; V2R-Y128S, triangle; and V2R-Ser-333del, circle) were prepared as described in Fig. 2B. At 48 h later, after washing in assay medium with 1 mm IBMX, the cells were incubated for 30 min at 37 °C in serum-free medium containing 10 nm AVP or 100 nm OPC5 with increasing concentrations of peptide antagonist (1 μm to 10 μm). cAMP accumulation was measured as described under “Experimental Procedures.” The data shown are the means ± S.D. of three independent experiments. Each set of results is representative of at least two additional experiments. C, effects of the peptide antagonist on AVP- or OPC5-stimulated cAMP accumulation in MDCK cells transiently expressing V2R-WT and V2R mutant transiently. MDCK cells transiently expressing V2R-WT and V2R mutant (MDCK-WT, MDCK-T273M, and MDCK-Y128S) were prepared as described for Fig. 2B. At 48 h later, after washing in assay medium with 1 mm IBMX, the cells were incubated for 30 min at 37 °C in serum-free medium containing 10 nm AVP or 100 nm OPC5 for MDCK-WT and MDCK-T283M cells and 100 nm AVP or 1 μm OPC5 for MDCK-Y128S with 10 μm peptidic antagonist. cAMP accumulation was measured as described under “Experimental Procedures.” The data shown are the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01. Each set of results is representative of at least two additional experiments.
A Peptide Antagonist Almost Completely Inhibits OPC5-stimulated cAMP Accumulation
To investigate where OPC5-induced cAMP accumulation occurs, we examined cAMP accumulation with or without exposure to a peptide antagonist, [adamantaneacetyl1, O-Et-d-Tyr2, Val4, aminobutyryl6, Arg8,9]-vasopressin. This antagonist showed an inhibitory trend against 10 nm AVP- or 100 nm OPC5- stimulated cAMP accumulation in a dose-dependent manner. At a concentration of 10 μm, it almost completely inhibited AVP- or OPC5- stimulated cAMP accumulation in COS-WT, T273M, Y128S, and S333del cells (Fig. 3B). Similar results were observed in MDCK cells expressing Myc-tagged V2R-WT, V2R-T273M, and V2R-Y128S (Fig. 3C). In MDCK cells expressing Myc-tagged V2R-S333del, however, this effect was not detected because neither AVP nor OPC5 increased cAMP accumulation. In contrast, it was reported previously that OPC5 activates the ER-retained mutants even in the presence of a cell-impermeable V2R antagonist, indicating that OPC5 can activate intracellularly retained V2R mutants in NDI (14). However, these data were not shown in that article. It is noteworthy in this regard that the Y128S mutant was analyzed both in our current study and in that previous report, but the findings appear to differ, at least in part. We speculate that this discrepancy might be caused by the higher dose of OPC5 used in the other study. The authors of that earlier report examined the blocking effect of the cell-impermeable peptide V2R antagonist against stimulation by 1 μm OPC5 (14). In our current experiments, a 10 μm dose of peptide V2R antagonist inhibited the stimulation by 100 nm OPC5 (Fig. 2, B and C) but not that by 1 μm OPC5 (data not shown). Taken together, our present findings suggest that OPC5 may activate not only V2R-WT but also V2R mutants expressed at the plasma membrane, which likely contrasts with the earlier findings, at least in part.
OPC5 Operates as an Agonist Pharmacochaperone and Promotes V2R Mutant Maturation
Some cell-permeable non-peptide ligands have been reported to induce the maturation and cell surface trafficking of ER-retained V2R mutants. Recently we also reported that OPC3 and OPC4 induce the maturation of NDI mutants by working as antagonist pharmacochaperones (10). Based on the characteristics of OPC5 as a cell-permeable V2R ligand, we concluded that it was worth examining whether OPC5 also functions as an agonist pharmacochaperone. When cells expressing Myc-tagged V2R-WT were analyzed by immunoblotting, a band of ∼50 kDa was detected, corresponding to a matured V2R protein (Fig. 4A). In the case of cells expressing V2R mutants, however, only faint bands were detected at that size. However, treatment with OPC4 drastically increased the intensity of the 50-kDa band; treatment with OPC5 also increased this band intensity, although less potently (Fig. 4A, upper left). The 50-kDa band was confirmed to react specifically against Myc-tagged V2R (Fig. 4A, upper right). (We also observed many specific bands other than the 50-kDa band. We speculated that these other bands may have occurred partly because V2R proteins were easily aggregated, either in the cells or during sample preparation, and partly because GPCR proteins are not easily separable on SDS-PAGE. We thus speculated that the relative band density of the 50-kDa protein might not necessarily reflect the correct percentage of mature V2R protein.) Coomassie Blue staining was performed as a loading control (Fig. 4A, lower panel). These results support the possibility that OPC5 may promote the maturation of the three V2R mutants that cause partial NDI.
FIGURE 4.

Effects of V2R ligands on the maturation and localization of V2R mutants. A, effect of OPC4 and OPC5 treatment on the maturation of V2R mutants. HEK293 cells were transfected with pcDNA 3.1-Myc-tagged V2R-WT, V2R mutant, or mock DNA (pcDNA 3.1 alone) and treated for 16 h at 37 °C with 10 μm OPC4, 10 μm OPC5, or vehicle only (0.1% DMSO). Cell lysates were analyzed by immunoblotting with anti-Myc antibodies as described under “Experimental Procedures” (upper panel). Cell lysates were separated in SDS-PAGE and stained by Coomassie Blue (lower panel). B, immunofluorescence microscopy analysis of V2R-WT or V2R mutant-expressing cells. Immunofluorescence microscopy analysis of COS-7 cells that had been transfected with Myc-tagged V2R-WT, V2R mutants, or mock DNA (pcDNA 3.1 alone) was performed. Sixteen h before the collection of fluorescence images, 10 μm OPC4 or OPC5 or vehicle was added. The images were collected with anti-Myc antibody. Each set of results is representative of at least two additional experiments. C, cell surface expression of V2R-WT or V2R mutants after long-time treatment with AVP or OPC5. COS-7 cells expressing V2R-WT or V2R mutants were prepared as described for Fig. 2A, and at 32 h later preincubation with the indicated concentration of AVP or OPC5 was started. At 48 h later, the expression levels of V2R-WT or V2R mutant proteins at the plasma membrane were measured by cell surface ELISA as described under “Experimental Procedures.” Data are the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01. Each set of results is representative of at least two additional experiments.
Effects of OPC5 on the Cell Surface Expression of V2R Mutants
We speculated that treatment with OPC5 might increase the cell surface expression of the V2R mutants as a result of inducing plasma membrane trafficking. However, we could not detect this effect clearly upon treatment with OPC5, unlike OPC4, in immunofluorescence microscopy studies (Fig. 4B). Using quantitative cell surface ELISA, we investigated whether this was due to a weaker pharmacochaperone effect of OPC5. These results revealed an interesting difference between AVP and OPC5, although both are V2R agonists (Fig. 4C). In the V2R-WT cells, incubation with AVP as well as with OPC5 prominently decreased the cell surface V2R levels, suggesting that both molecules working as agonists might have switched on a desensitization pathway and thereby induced V2R internalization. Although incubation with AVP decreased the cell surface expression of V2R-T273M, as it did for V2R-WT, OPC5 incubation caused only a weak effect in this regard. Notably, however, although AVP incubation decreased the cell surface expression of V2R-Y128S and V2R-S333del, OPC5 incubation increased these levels slightly but significantly. These results led us to speculate that OPC5 may work not only as a V2R agonist that induces activation and desensitization but also as a V2R pharmacochaperone.
OPC5 and AVP Both Recruit β-Arrestin to the Plasma Membrane
To investigate whether OPC5 indeed activates a desensitization pathway, we focused on the recruitment of β-arrestin to the plasma membrane as an initial step of this pathway that is followed by receptor internalization. The β-arrestin levels in the membrane fraction increased in COS-WT or COS-T273M cells treated with either OPC5 or AVP (Fig. 5A, upper panel). In the membrane fraction of COS-Y128S or COS-S333del cells, a similar phenomenon appeared to occur but less prominently for both OPC5 and AVP (Fig. 5A, upper panel). Gβ was immunoblotted as a loading control for the membrane fraction (Fig. 5A, lower panel). A total internal reflection fluorescence (TIRF) assay revealed that β-arrestin was significantly recruited to the plasma membrane when OPC5 or AVP was added to HKE293 cells expressing HA-tagged V2R-WT (HEK293-WT) or each V2R mutant with EGFP-tagged β-arrestin 2 (Fig. 5B). Unlike the results of treatment with AVP or OPC5 for 4 h (Fig. 5A), the β-arrestin levels were far higher at the plasma membrane in AVP-stimulated HEK293-WT cells than in the OPC5-stimulated HEK293-WT cells after being treated for 10 min (Fig. 5B). These results suggested that the time course of β-arrestin recruitment to the plasma membrane after treatment of OPC5 may differ from that of AVP. In fact, β-arrestin increased more prominently in the membrane fraction of AVP-stimulated COS-WT cells than in that of OPC5-stimulated COS-WT cells after a 10-min treatment (Fig. 5B, inset). These results support not only the notion that OPC5 switches on the desensitization pathway of V2R mutants but also that it acts on V2R mutants at the plasma membrane and not in the ER.
FIGURE 5.

The effects of AVP or OPC5 on β-arrestin recruitment and ERK phosphorylation. A, AVP- or OPC5-stimulated translocation of β-arrestin to the membrane fractions. COS-7 cells (106 cells) were transiently transfected with V2R-WT or V2R mutants (0.2 μg of pcDNA3 containing each Myc-tagged V2R was used). At 48 h later, the cells were stimulated with 1 μm AVP or 10 μm OPC5 for 4 h and harvested as described under “Experimental Procedures.” Each set of results is representative of at least two additional experiments. B, investigation of AVP- or OPC5-stimulated translocation of β-arrestin to the plasma membranes investigated using TIRF microscopy. HEK293 cells expressing HA-tagged V2R-WT or partial NDI V2R mutants with GFP-tagged β-arrestin 2 were stimulated with 1 μm AVP or 10 μm OPC5 for 10 min. The TIRF images were then analyzed as described under “Experimental Procedures.” The differences in fluorescence intensity at 10 min from that at time zero were analyzed. Inset, the cells as described in A were stimulated with 1 μm AVP or 10 μm OPC5 for 10 min and harvested. The data are the means ± S.D. of three independent experiments. Each set of results is representative of at least two additional experiments. C, AVP- or OPC5-stimulated ERK1/2 phosphorylation associated with the partial NDI V2R mutants. COS-7 cells (106 cells) were transiently transfected with partial NDI V2R mutants (0.2 μg of pcDNA3 containing each Myc-tagged V2R was used). At 32 h later, cells were serum-starved, and at 48 h later the cells were stimulated with the indicated concentrations of AVP or OPC5 for 8 min. Samples were collected and analyzed by immunoblotting as described under “Experimental Procedures.” Each set of results is representative of at least two additional experiments.
OPC5 and AVP Both Stimulate ERK1/2 Phosphorylation
We were interested in other signaling pathways that might be affected by OPC5 treatment. We found that not only AVP but also OPC5 stimulated ERK1/2 phosphorylation in COS-WT, COS-T273M, COS-Y128S, and COS-S333del cells (Fig. 5C, upper panel). ERK1 and ERK2 were immunoblotted as loading controls (Fig. 5C, lower panel). These results also indicated that OPC5 acts at the plasma membrane. It has been reported that GPCR-mediated ERK1/2 phosphorylation is followed by receptor desensitization and internalization via β-arrestin recruitment to the plasma membrane (25). Our present findings (Fig. 5) appear to be consistent with this mechanism.
OPC5 Likely Induces Desensitization and Internalization of Not Only V2R-WT but Also V2R Mutants
Based on our aforementioned data, we speculated that OPC5 acting as an agonist might induce desensitization and internalization of not only V2R-WT but also mutant V2R proteins. To test this possibility, we attempted to block agonist-induced receptor internalization by co-expression of dominant-negative K44A-dynamin with V2R-WT or V2R mutants in COS-7 cells. As anticipated, the co-expression of K44A-dynamin significantly recovered or increased the cell surface expression of V2R-WT and the V2R-T273M and V2R-Y128S mutants following AVP or OPC5 treatment (Fig. 6A). In the case of V2R-S333del, however, this change was not evident. We speculated that this may be because the degree of AVP- or OPC5-stimulated β-arrestin-dependent endocytosis of V2R-S333del is lower than that of the other mutants. These results support our hypothesis that OPC5 induces the internalization of both WT and mutant V2R.
FIGURE 6.

Effects of V2R ligands and dominant-negative dynamin on cell surface expression of V2R-WT and V2R mutants and cAMP accumulation after long-term incubation with AVP and OPC5. A, cell surface expression of V2R mutants after long-term incubation of 1 μm AVP or 10 μm OPC5, OPC3, or OPC4 and effects of dominant-negative K44A-dynamin. COS-7 cells expressing V2R mutants with or without K44A-dynamin were prepared as described for Fig. 3A and preincubated 32 h later with the indicated concentrations of AVP or OPC5. At 48 h later, the plasma membrane expression levels of V2R mutant proteins were measured by cell surface ELISA as described under “Experimental Procedures.” The data are the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01. Each set of results is representative of at least two additional experiments. B, AVP- or OPC5-stimulated cAMP accumulation associated with V2R-WT or V2R mutants with or without AVP or OCP5 pretreatment. COS-7 cells were transiently transfected with V2R-WT and V2R mutant (0.2 μg of pcDNA3 containing each Myc-tagged V2R was used). At 24 h later, the cells were reseeded into 24-well plates at 0.75 × 105 cells/well to which [3H]adenine (2 μCi/ml, GE Healthcare) was added. At 32 h later, cells were treated with 1 μm AVP, 10 μm OPC5, or vehicle. At 48 h later, after washing in assay medium with 1 mm IBMX, the cells were incubated for 30 min at 37 °C in serum-free medium with or without 1 μm AVP or 10 μm OPC5. cAMP accumulation was measured as described under “Experimental Procedures.” Data are the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01. Each set of results is representative of at least two additional experiments.
OPC5 but Not AVP Produces Sustainable cAMP Signaling over a Long-term Treatment in V2R Mutant Cells
Based on our findings thus far, we surmised that the effect of OPC5 treatment in cells expressing V2R mutants likely reflected a balance between its pharmacochaperone and desensitization effects. When V2R mutants, expressed in the basolateral membrane of renal collecting duct principal cells, are activated by AVP binding, the Gs-cAMP-PKA signaling cascade is activated. This results in the phosphorylation and translocation of aquaporin-2 water channels from cytosolic vesicles to the luminal membrane of the collecting duct followed by water reabsorption. From a possible therapeutic standpoint in relation to our NDI patients, we focused on cAMP accumulation after a long-term treatment with OPC5, which we regarded from a clinical perspective to be a determinant of the trafficking of water channels to the luminal membrane. In COS-WT cells, both AVP and OPC5 significantly decreased cAMP accumulation via a second challenge after long-term incubation. In COS-T273M cells, however, OPC5 treatment produced a milder decrease in cAMP accumulation compared with AVP. In COS-Y128S and COS-S333del cells, however, OPC5 treatment did not decrease cAMP accumulation (Fig. 6B). In COS-S333del cells, OPC5 treatment weakly but significantly increased cAMP accumulation (Fig. 6B).
These results suggest that OPC5, but not AVP, enables sustained cAMP signaling after long-term treatment in cells expressing a V2R mutant. This compound may therefore represent a promising new treatment option at least in some patients with partial NDI. The likely mechanism by which OPC5 acts on V2R mutants misfolded in the ER is presented in Fig. 7.
FIGURE 7.
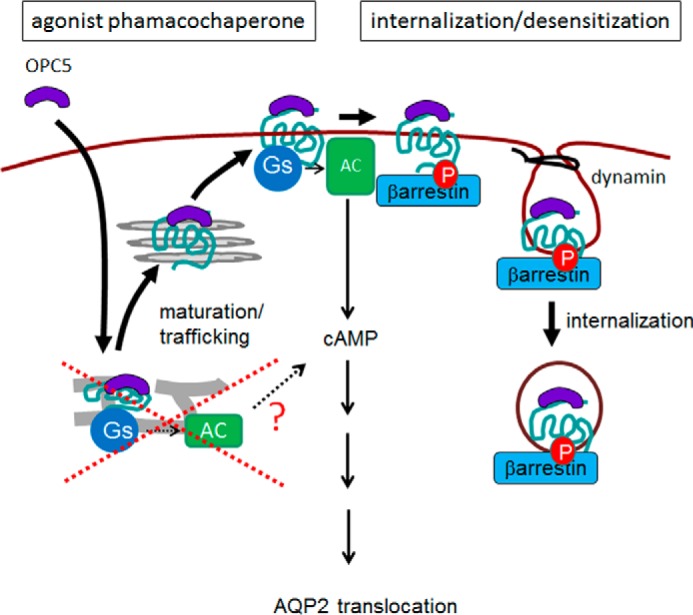
Model of the OPC5 action on partial NDI-causing V2R mutants. OPC5, a non-peptide V2R agonist, passes through the plasma membrane and interacts with ER-retained V2R mutant. By working as a pharmacochaperone, OPC5 promotes the misfolded V2R mutant maturation and trafficking to the plasma membrane. OPC5 then activates the V2R mutant-Gs-adenylyl cyclase (AC)-cAMP cascade, resulting in water channel AQP2 translocation. OPC5 also causes β-arrestin-dependent desensitization/internalization of V2R mutants. Cancelation of the desensitization effect of OPC5 by the pharmacochaperone effect after long-term treatment may enable sustained signaling, which may be beneficial for the treatment of NDI patients. This model likely contrasts, at least in part, with the previous model, in which OPC5 was reported to activate NDI-causing V2R mutants at the intracellular location (i.e. retained in the ER) without their maturation/trafficking to the plasma membrane.
Discussion
We have identified a novel mutation in the V2R gene in a patient diagnosed with partial NDI, evidenced by high basal AVP levels, at the age of 4 years. In the process of analyzing the function of this new V2R mutant, we revealed a new aspect of the V2 receptor ligand, OPC5, as an agonist pharmacochaperone (Fig. 7). Originally, OPC5 was reported to activate NDI-causing V2R mutants at their intracellular location (i.e. retained in the ER) without changing their maturation state (14). We thus speculated that OPC5 would be a highly promising agent for mitigating the effects of our novel V2R mutant, which localized at the ER in a manner similar to the other two mutants we had reported previously (10). We found that OPC5 induced cAMP accumulation in cells expressing these three partial NDI-causing V2R mutants. At the same time, however, peptide AVP was also found to stimulate cAMP accumulation in those cells. We thus could not rule out the possibility that OPC5 might activate the partial NDI mutants at the plasma membrane. Our data showing that a peptide antagonist inhibited OPC5-stimulated cAMP accumulation in cells expressing each of the three V2R mutants in a dose-dependent manner strongly supported this possibility. In addition, OPC5 was found to operate not only as a non-peptide agonist that also causes desensitization of V2R mutants at the plasma membrane but also as a pharmacochaperone that helps ER-retained V2R mutants to mature and traffic to the plasma membrane. From a clinical and therapeutic point of view, we speculated from these findings that the pharmacochaperone effect of OPC5, which cancels the desensitization effect of V2R mutants, may enable sustained signaling. It is noteworthy that the Y128S mutant was analyzed in both our current study and previously and the findings appear to differ, at least in part. We thus speculate that the mechanism presented here (Fig. 7) may enable sustained signaling not only in partial NDI mutants but also in weaker mutants, causing complete NDI clinically.
The Pharmacochaperone Effect of OPC5 Is Likely to Be Underestimated
To elucidate the pharmacochaperone effect of OPC5, we used a dominant-negative dynamin, K44A-dynamin, which blocks V2R internalization and desensitization. However, based on our findings that overexpression of K44A-dynamin did not either completely cancel the AVP-induced internalization of V2R-WT or the V2R mutants or increase OPC5-induced cell surface expression of V2R-S333del, the dominant-negative dynamin appeared not to completely block receptor internalization. These results suggest that the pharmacochaperone effect of OPC5 may be greater than could be detected in our experiments.
OPC5 or Its Related Compounds May Be Feasible Therapeutics for NDI Caused by V2R Mutants
From the perspective of its capacity as a pharmacochaperone, OPC5 was found to show less potency than either OPC3 or OPC4 (10). However, in terms of the agonist functions of OPC5 this compound showed properties that may be advantageous for the treatment of NDI patients. Because of their high affinity, neither OPC3 nor OPC4 as antagonist pharmacochaperones can be easily replaced by AVP after they promote V2R mutant maturation and trafficking to the plasma membrane (8).
Although OPC5 caused desensitization of the V2R mutants as an agonist, it was also found to induce maturation and trafficking as a pharmacochaperone. It may therefore be important to think about the balance between these pharmacochaperone and desensitization effects of OPC5. Considering that the maturation effect of OPC5 upon the three V2R mutants appeared to be similar, as shown by immunoblotting (Fig. 4A), and that the desensitization effect (Fig. 5A) appeared to positively correlate with the residual function of each mutant (Fig. 6B), it may be speculated that OPC5 has a greater pharmacochaperone effect on weaker mutants than on the more potent mutants. In support of this possibility in our current analysis, whereas V2R-T273M was the most potent variant in terms of cAMP accumulation, it showed the most desensitization after long-term treatment with OPC5, thereby having a relatively lower pharmacochaperone effect. In contrast, although V2R-Y128S showed weaker function, it was the least desensitized (Fig. 6B) by OPC5, thus having a relatively greater pharmacochaperone effect (Fig. 6A). Although the balance between the pharmacochaperone and desensitization effects of OPC5 remains to be investigated in other NDI mutants, the balance, at least in our three V2R mutants, seems to favor the weaker V2R mutants. Hence, the pharmacochaperone effect of OPC5, which cancels its desensitization effect, may enable sustained signaling from V2R mutants.
OPC5 Has Protean Agonistic Effects
We have shown by our current analyses that OPC5 produces different pharmacological actions against V2R-WT and V2R mutants; this is known as protean agonism. In COS-S333del cells, OPC5 increased cAMP accumulation because of a relatively high pharmacochaperone effect. In COS-Y128S cells, however, OPC5 apparently showed activity as a biased agonist that stimulated the cAMP signaling pathway but not the V2R desensitization pathway. In COS-T273M cells, OPC5 functioned as an agonist that caused far less desensitization than AVP. In COS-WT cells, OPC5 caused V2R desensitization as potently as AVP. These data showing that OPC5 has different effects dependent on the targeting receptors are a good example of protean agonism. We speculate that this phenomenon will help us to develop adequate ligands in the future that produce more desirable signaling effects depending on the biological context.
Conclusion
A V2R ligand, OPC5, induces the maturation and trafficking of V2R mutants as a pharmacochaperone and desensitizes these receptors as an agonist. In addition, the balance between the pharmacochaperone and desensitization effects of OPC5 seems to favor the weaker V2R mutants. The canceling of the desensitization effect by the pharmacochaperone effect of OPC5 after long-term treatment may enable sustained signaling, which may be beneficial for the treatment of at least some NDI patients.
Experimental Procedures
Pharmacological Chaperones and Ligands
The V2R agonists OPC31260 (OPC3), OPC41061 (OPC4), and OPC51803 (OPC5) were kindly provided by Otsuka Pharmaceutical Co. (Tokyo). Vasopressin (AVP) and the peptide antagonist [adamantaneacetyl1, O-Et-d-Tyr2, Val4, aminobutyryl6, Arg8,9]-vasopressin were purchased from Sigma. The principles of the V2R ligands used in the experiments are follows: AVP, Cys-Tyr-Phe-Gln-Asn-Cys-Pro-Arg-Gly-NH2; OPC3, (+/−)-5-dimethylamino-l-(4-[2-methylbenzoylamino]benzoyl)-2,3,4,5-tetrahydro-1H-benzazepin hydrochloride; OPC4, 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methyl-benzoyl-amino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine; OPC5, (5R)-2-[1-(2-chloro-4-(1-pyrrolidinyl)benzoyl)-2,3,4,5-tetrahydro-1H-1-benzazepin-5-yl]-N-isopropylacetamide.
Antibodies
Anti-Myc monoclonal antibody (M047-3) and anti-GFP monoclonal antibody (clone 1E4, M048-3) were purchased from MBL (Nagoya, Japan). Anti-phospho-p44/42 MAPK (Erk1/2) mouse monoclonal antibody (clone E10) and anti-PDI rabbit monoclonal antibody (clone C81H6) were purchased from Cell Signaling (Danvers, MA). Anti-MAPK rabbit polyclonal antibody (ERK1 + ERK2) (catalog No. 61-7400) was purchased from Zymed Laboratories (San Francisco). Anti-Gβ rabbit polyclonal antibody (sc-378) was purchased from Santa Cruz Biotechnology. Goat anti-mouse HRP-conjugated secondary antibody (M15245) was purchased from Transduction Laboratories (Lexington, KY). Alexa Fluor 488 goat anti-mouse IgG (H+L) (A11029) and Alexa Fluor 594 goat anti-rabbit IgG (H+L) (A11036) were purchased from Thermo Fisher Scientific.
Expression Constructs, Cell Culture, and Transfection
An expression construct encoding a Myc-tagged wild-type V2R (V2R-WT) was kindly provided by M. Bouvier (Université de Montreal, Canada), and each mutant causative for diabetes insipidus was generated using a manual procedure (Stratagene, La Jolla, CA). COS-7 cells, maintained in DMEM containing 10% (v/v) fetal bovine serum, were transfected with constructs encoding Myc-tagged V2R-WT or Myc-tagged V2R mutant using Lipofectamine 2000 (Invitrogen).
cAMP Assay
cAMP accumulation in COS-7 cells transiently expressing V2R-WT and each V2R mutant was assayed as described previously (10, 26–29). Briefly, 1 day after transfection, cells were reseeded into 24-well plates to which [3H]adenine (2 μCi/ml) was added. In the case of examining the effect of long-term treatment, the cells were pretreated with AVP or OPC5 at the indicated concentrations 16 h before performing the assay. 24 h later, the cells were washed and incubated in 0.5 ml of assay medium containing 1 mm isobutylmethylxanthine (IBMX) with or without the indicated concentrations of vasopressin or OPC5. Reactions were terminated after 30 min by removing the medium and lysing the cells in 5% trichloroacetic acid containing ATP and cAMP (1 mm each). [3H]cAMP and [3H]ATP were separated on AG 50W-X4 Dowex and alumina columns, and the data are presented as the ratio of [3H]cAMP to [3H]cAMP plus [3H]ATP, as described previously (10, 26–29).
Cell Surface ELISA
The cell surface expression of exogenous receptors in COS-7 cells transiently transfected with Myc-tagged V2R-WT or each V2R mutant was quantified by ELISA (6, 10, 30, 31). Briefly, 1 day after transfection, cells were reseeded into 24-well plates. 16 h before performing the assay, as needed, cells were pretreated with OPC3, OPC4, or OPC5 at the indicated concentrations. After 24 h, the cells were then washed twice with ice-cold 1% BSA/PBS, placed on ice for 5 min, and incubated in an anti-Myc antibody solution (1:4000) diluted in 1% BSA/PBS for 1 h at 4 °C. This was followed by two further washes in 1% BSA/PBS. The cells were next fixed at 4 °C for 15 min in 4% paraformaldehyde/PBS and again washed twice with 1% BSA/PBS and incubated in anti-mouse HRP-conjugated secondary antibody solution (1:12000) diluted in 1% BSA/PBS at room temperature for 1 h. This was followed by two washes in 1% BSA/PBS for 20 min and a final wash in PBS. Finally, the cells were treated with substrate (o-phenylenediamine dihydrochloride, Sigma) for 5 min at room temperature. This reaction was stopped by the addition of an equivalent volume of 2.5 N HCl. The absorption levels were then read at 492 nm using a plate reader (Bio-Rad).
Immunofluorescence Microscopy
Immunofluorescence staining was performed as described previously (6, 10, 32). Briefly, transiently transfected COS-7 cells were placed onto glass coverslips. At 16 h before performing the assay, cells were pretreated with OPC4 or OPC5 at the indicated concentrations. Cells were then washed twice with PBS, fixed with ice-cold 100% methanol, and incubated at −20 °C for 2 min to evaluate whole cell expression (with permeabilization). After two further washes in PBS, the cells were blocked using Block Ace at room temperature for 20 min and then sequentially incubated with anti-Myc antibody (1:4,000) or anti PDI (an ER marker) antibody (1:4,000) diluted in 20% Block Ace/PBS at 4 °C overnight. The cells were then further washed twice in 20% Block Ace/PBS and incubated with anti-mouse antibody 488 (for anti-Myc antibody) or anti-rabbit antibody 594 (for anti-PDI antibody) diluted in 20% Block Ace at room temperature for 1 h. After two additional washes in 0.1% Tween 20/PBS and one wash in PBS alone, the cells were mounted on glass slides for analysis by inverted microscope or confocal microscope (Nikon, Tokyo).
TIRF Assay
HEK293 cells stably expressing HA-tagged V2R-WT or each V2R mutant were transiently transfected with GFP-tagged β-arrestin 2 and replaced on a glass-bottom 35-mm dish for analysis using a TIRF microscope (Nikon Instruments, Eclipse-Ti-E, Tokyo). The cells on each coverslip were stimulated with 1 μm AVP or 10 μm OPC5 for 10 min. For each TIRF recording, the images were captured every 3 s with a 488 nm excitation laser. Image sequences were recorded using NIS-Elements imaging software (Nikon Instruments) and saved as AVI files. Ten arbitrary individual regions of interest were then selected. The differences in fluorescence intensity at 10 min from time zero were analyzed.
Membrane Localization Assay
The membrane localization of β-arrestin 2 was assayed as described previously (29, 33). Briefly, COS-7 cells were seeded onto 60-mm dishes and transiently transfected with GFP-tagged β-arrestin 2 and Myc-tagged V2R-WT or each V2R mutant. At 48 h later, cells were harvested in ice-cold PBS after incubation with 1 μm AVP or 10 μm OPC5 for 4 h. After centrifugation (400 × g for 5 min at 4 °C), the cells were lysed in hypotonic buffer (20 mm Tris-HCl, pH 7.5, 20 mm NaCl, 2 mm MgCl2, 1 mm EGTA, and proteinase inhibitors) for 10 min and then sheared using a Dounce homogenizer (15 times). The nuclear fractions and unbroken cells were removed by centrifugation (800 × g for 5 min at 4 °C). The supernatants were ultracentrifuged (160,000 × g for 30 min at 4 °C), and the pellet containing the membrane fraction was treated with lysis buffer (40 mm Tris-HCl, pH 7.5, 100 mm NaCl, 2 mm MgCl2, 1 mm EGTA, 1% Triton X-100, and proteinase inhibitors) for 1 h at 4 °C and then centrifuged (200 × g for 5 min at 4 °C). The resulting supernatant was then mixed with the same volume of Laemmli sample buffer and boiled for 5 min. These samples were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with a monoclonal anti-GFP antibody (1:10,000 dilution).
Determination of AVP- or OPC5-stimulated ERK1/2 Activity
Phosphorylation of ERK1/2-stimulated AVP or OPC5 was assayed as described previously (32). Briefly, COS-7 cells seeded in 12-well plates were transiently transfected with Myc-tagged V2R-WT or each V2R mutant and starved for 12–16 h. The cells were then stimulated with 1 μm AVP or 10 μm OPC5 for 5 min. Proteins extracts were prepared and then separated by SDS-PAGE and transferred to PVDF membranes. The membranes were then incubated with monoclonal anti-phospho-MAPK antibody (Cell Signaling).
Genetic Analysis of the V2 Receptor Gene (AVPR2) in Two Patients
Genetic analysis of the V2 receptor gene was performed with approval from the Institutional Review Board of The University of Tokyo. After written informed consent was obtained from the parents of the patients, genomic DNA was prepared from the peripheral blood leukocytes with the use of a DNA isolation kit (Qiagen Japan, Tokyo), and all exons and exon-intron boundaries of AVPR2 were analyzed using a PCR direct sequencing method.
Structure Modeling of the V2 Receptor
The structure model of the human V2 receptor was constructed based on the structure of the human β2 adrenergic receptor (34) as a template using MODELLER 9.16 (35). Energy minimization was performed using Amber 14, and molecular visualization was performed using PyMOL.
Statistical Methods and Analysis
Dunnett's multiple comparison procedures were used for statistical analysis. To evaluate the dose response, the Williams' multiple comparison was used. Averaged data from three independent experiments are shown, and error bars represent S.D. These analyses were performed using Microsoft Excel software.
Author Contributions
N. Makita conducted the experiments, analyzed the results, and wrote the paper. T. S. identified the patient and fulfilled the loaded tests for diagnosis. Y. Y., J. S., K. M., and M. E. performed the experiments. M. O., N. Matsumoto, and M. N. analyzed the data. T. I. conceived the idea for the project and wrote the paper with N. Makita.
Acknowledgments
We thank M. Bouvier for generously donating the Myc-tagged V2R construct. We also thank Otsuka Pharmaceutical Co. for providing a non-peptideV2 receptor ligand.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Science, Sports, and Culture, Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- V2R
- V2 vasopressin receptor
- NDI
- nephrogenic diabetes insipidus
- GPCR
- G protein-coupled receptor
- AVP
- arginine vasopressin
- ER
- endoplasmic reticulum
- MDCK
- Madin-Darby canine kidney
- TIRF
- total internal reflection fluorescence
- IBMX
- isobutylmethylxanthine
- PDI
- protein disulfide isomerase.
References
- 1. Bichet D. G. (2006) Nephrogenic diabetes insipidus. Adv. Chronic Kidney Dis. 13, 96–104 [DOI] [PubMed] [Google Scholar]
- 2. Robben J. H., Knoers N. V., and Deen P. M. (2006) Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am. J. Physiol. Renal Physiol. 291, F257–F270 [DOI] [PubMed] [Google Scholar]
- 3. Neocleous V., Skordis N., Shammas C., Efstathiou E., Mastroyiannopoulos N. P., and Phylactou L. A. (2012) Identification and characterization of a novel X-linked AVPR2 mutation causing partial nephrogenic diabetes insipidus: a case report and review of the literature. Metabolism 61, 922–930 [DOI] [PubMed] [Google Scholar]
- 4. Bockenhauer D., and Bichet D. G. (2015) Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat. Rev. Nephrol. 11, 576–588 [DOI] [PubMed] [Google Scholar]
- 5. Morello J. P., Salahpour A., Laperrière A., Bernier V., Arthus M. F., Lonergan M., Petäjä-Repo U., Angers S., Morin D., Bichet D. G., and Bouvier M. (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest. 105, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernier V., Morello J. P., Zarruk A., Debrand N., Salahpour A., Lonergan M., Arthus M. F., Laperrière A., Brouard R., Bouvier M., and Bichet D. G. (2006) Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 17, 232–243 [DOI] [PubMed] [Google Scholar]
- 7. Macion-Dazard R., Callahan N., Xu Z., Wu N., Thibonnier M., and Shoham M. (2006) Mapping the binding site of six nonpeptide antagonists to the human V2-renal vasopressin receptor. J. Pharmacol. Exp. Ther. 316, 564–571 [DOI] [PubMed] [Google Scholar]
- 8. Robben J. H., Sze M., Knoers N. V., and Deen P. M. (2007) Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: relevance to therapy of nephrogenic diabetes insipidus. Am. J. Physiol. Renal Physiol. 292, F253–F260 [DOI] [PubMed] [Google Scholar]
- 9. Jean-Alphonse F., Perkovska S., Frantz M. C., Durroux T., Méjean C., Morin D., Loison S., Bonnet D., Hibert M., Mouillac B., and Mendre C. (2009) Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 20, 2190–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahashi K., Makita N., Manaka K., Hisano M., Akioka Y., Miura K., Takubo N., Iida A., Ueda N., Hashimoto M., Fujita T., Igarashi T., Sekine T., and Iiri T. (2012) V2 vasopressin receptor (V2R) mutations in partial nephrogenic diabetes insipidus highlight protean agonism of V2R antagonists. J. Biol. Chem. 287, 2099–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kenakin T. (2007) Functional selectivity through protean and biased agonism: who steers the ship? Mol. Pharmacol. 72, 1393–1401 [DOI] [PubMed] [Google Scholar]
- 12. Hill S. J. (2006) G-protein-coupled receptors: past, present and future. Br J. Pharmacol. 147, Suppl. 1, S27–S37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makita N., and Iiri T. (2014) Biased agonism: a novel paradigm in G protein-coupled receptor signaling observed in acquired hypocalciuric hypercalcemia. Endocr. J. 61, 303–309 [DOI] [PubMed] [Google Scholar]
- 14. Robben J. H., Kortenoeven M. L., Sze M., Yae C., Milligan G., Oorschot V. M., Klumperman J., Knoers N. V., and Deen P. M. (2009) Intracellular activation of vasopressin V2 receptor mutants in nephrogenic diabetes insipidus by nonpeptide agonists. Proc. Natl. Acad. Sci. U.S.A. 106, 12195–12200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bichet D. G., and Bockenhauer D. (2016) Genetic forms of nephrogenic diabetes insipidus (NDI): Vasopressin receptor defect (X-linked) and aquaporin defect (autosomal recessive and dominant). Best Pract. Res. Clin. Endocrinol. Metab. 30, 263–276 [DOI] [PubMed] [Google Scholar]
- 16. Sadeghi H., Robertson G. L., Bichet D. G., Innamorati G., and Birnbaumer M. (1997) Biochemical basis of partial nephrogenic diabetes insipidus phenotypes. Mol. Endocrinol. 11, 1806–1813 [DOI] [PubMed] [Google Scholar]
- 17. Ala Y., Morin D., Mouillac B., Sabatier N., Vargas R., Cotte N., Dechaux M., Antignac C., Arthus M. F., Lonergan M., Turner M. S., Balestre M. N., Alonso G., Hibert M., Barberis C., et al. (1998) Functional studies of twelve mutant V2 vasopressin receptors related to nephrogenic diabetes insipidus: molecular basis of a mild clinical phenotype. J. Am. Soc. Nephrol. 9, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 18. Pasel K., Schulz A., Timmermann K., Linnemann K., Hoeltzenbein M., Jääskeläinen J., Grüters A., Filler G., and Schöneberg T. (2000) Functional characterization of the molecular defects causing nephrogenic diabetes insipidus in eight families. J. Clin. Endocrinol. Metab. 85, 1703–1710 [DOI] [PubMed] [Google Scholar]
- 19. Inaba S., Hatakeyama H., Taniguchi N., and Miyamori I. (2001) The property of a novel v2 receptor mutant in a patient with nephrogenic diabetes insipidus. J. Clin. Endocrinol. Metab. 86, 381–385 [DOI] [PubMed] [Google Scholar]
- 20. Chen C. H., Chen W. Y., Liu H. L., Liu T. T., Tsou A. P., Lin C. Y., Chao T., Qi Y., and Hsiao K. J. (2002) Identification of mutations in the arginine vasopressin receptor 2 gene causing nephrogenic diabetes insipidus in Chinese patients. J. Hum. Genet. 47, 66–73 [DOI] [PubMed] [Google Scholar]
- 21. Faerch M., Christensen J. H., Corydon T. J., Kamperis K., de Zegher F., Gregersen N., Robertson G. L., and Rittig S. (2008) Partial nephrogenic diabetes insipidus caused by a novel mutation in the AVPR2 gene. Clin. Endocrinol. (Oxf.) 68, 395–403 [DOI] [PubMed] [Google Scholar]
- 22. Faerch M., Christensen J. H., Rittig S., Johansson J. O., Gregersen N., de Zegher F., and Corydon T. J. (2009) Diverse vasopressin V2 receptor functionality underlying partial congenital nephrogenic diabetes insipidus. Am. J. Physiol. Renal Physiol. 297, F1518–F1525 [DOI] [PubMed] [Google Scholar]
- 23. Bockenhauer D., Carpentier E., Rochdi D., van't Hoff W., Breton B., Bernier V., Bouvier M., and Bichet D. G. (2010) Vasopressin type 2 receptor V88M mutation: molecular basis of partial and complete nephrogenic diabetes insipidus. Nephron Physiol. 114, p1–p10 [DOI] [PubMed] [Google Scholar]
- 24. Sahakitrungruang T., Tee M. K., Rattanachartnarong N., Shotelersuk V., Suphapeetiporn K., and Miller W. L. (2010) Functional characterization of vasopressin receptor 2 mutations causing partial and complete congenital nephrogenic diabetes insipidus in Thai families. Horm. Res. Paediatr. 73, 349–354 [DOI] [PubMed] [Google Scholar]
- 25. McDonald P. H., Chow C. W., Miller W. E., Laporte S. A., Field M. E., Lin F. T., Davis R. J., and Lefkowitz R. J. (2000) β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 26. Iiri T., Herzmark P., Nakamoto J. M., van Dop C., and Bourne H. R. (1994) Rapid GDP release from Gsα in patients with gain and loss of endocrine function. Nature 371, 164–168 [DOI] [PubMed] [Google Scholar]
- 27. Iiri T., Farfel Z., and Bourne H. R. (1997) Conditional activation defect of a human Gsα mutant. Proc. Natl. Acad. Sci. U.S.A. 94, 5656–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iiri T., Bell S. M., Baranski T. J., Fujita T., and Bourne H. R. (1999) A Gsα mutant designed to inhibit receptor signaling through Gs. Proc. Natl. Acad. Sci. U.S.A. 96, 499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Makita N., Sato J., Rondard P., Fukamachi H., Yuasa Y., Aldred M. A., Hashimoto M., Fujita T., and Iiri T. (2007) Human G(sα) mutant causes pseudohypoparathyroidism type Ia/neonatal diarrhea, a potential cell-specific role of the palmitoylation cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 17424–17429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernier V., Lagace M., Lonergan M., Arthus M. F., Bichet D. G., and Bouvier M. (2004) Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol. Endocrinol. 18, 2074–2084 [DOI] [PubMed] [Google Scholar]
- 31. Makita N., Kabasawa Y., Otani Y., Firman, Sato J., Hashimoto M., Nakaya M., Nishihara H., Nangaku M., Kurose H., Ohwada T., and Iiri T. (2013) Attenuated desensitization of β-adrenergic receptor by water-soluble N-nitrosamines that induce S-nitrosylation without NO release. Circ. Res. 112, 327–334 [DOI] [PubMed] [Google Scholar]
- 32. Makita N., Sato J., Manaka K., Shoji Y., Oishi A., Hashimoto M., Fujita T., and Iiri T. (2007) An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc. Natl. Acad. Sci. U.S.A. 104, 5443–5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oishi A., Makita N., Sato J., and Iiri T. (2012) Regulation of RhoA signaling by the cAMP-dependent phosphorylation of RhoGDIα. J. Biol. Chem. 287, 38705–38715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang C. Y., Olieric V., Ma P., Howe N., Vogeley L., Liu X., Warshamanage R., Weinert T., Panepucci E., Kobilka B., Diederichs K., Wang M., and Caffrey M. (2016) In meso in situ serial X-ray crystallography of soluble and membrane proteins at cryogenic temperatures. Acta Crystallogr. D Struct. Biol. 72, 93–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.32 [DOI] [PubMed] [Google Scholar]