

Abstract
MitoNEET (mNT) (CDGSH iron-sulfur domain-containing protein 1 or CISD1) is an outer mitochondrial membrane protein that donates 2Fe-2S clusters to apo-acceptor proteins. In the present study, using a global mNT knock-out (mNTKO) mouse model, we investigated the in vivo functional role of mNT in the development of alcoholic steatohepatitis. Experimental alcoholic steatohepatitis was achieved by pair feeding wild-type (WT) and mNTKO mice with Lieber-DeCarli ethanol-containing diets for 4 weeks. Strikingly, chronically ethanol-fed mNTKO mice were completely resistant to ethanol-induced steatohepatitis as revealed by dramatically reduced hepatic triglycerides, decreased hepatic cholesterol level, diminished liver inflammatory response, and normalized serum ALT levels. Mechanistic studies demonstrated that ethanol administration to mNTKO mice induced two pivotal endocrine hormones, namely, adipose-derived adiponectin and gut-derived fibroblast growth factor 15 (Fgf15). The elevation in circulating levels of adiponectin and Fgf15 led to normalized hepatic and serum levels of bile acids, limited hepatic accumulation of toxic bile, attenuated inflammation, and amelioration of liver injury in the ethanol-fed mNTKO mice. Other potential mechanisms such as reduced oxidative stress, activated Sirt1 signaling, and diminished NF-κB activity also contribute to hepatic improvement in the ethanol-fed mNTKO mice. In conclusion, the present study identified adiponectin and Fgf15 as pivotal adipose-gut-liver metabolic coordinators in mediating the protective action of mNT deficiency against development of alcoholic steatohepatitis in mice. Our findings may help to establish mNT as a novel therapeutic target and pharmacological inhibition of mNT may be beneficial for the prevention and treatment of human alcoholic steatohepatitis.
Keywords: adiponectin, alcohol, intestine, iron, liver injury
Introduction
NEET proteins (mitoNEET (CISD1, mNT),3 nutrient-deprivation autophagy factor-1 (NAF-1; CISD2), and Miner 2 (CISD3)) are a class of redox active iron-sulfur (2Fe-2S) proteins (1, 2). Among the three NEET family members, mNT encoded by the CISD1 gene is the founding member of the NEET family, and it is localized in the outer mitochondrial membrane and regulates the maturation and shuttling of 2Fe-2S clusters. mNT has been identified as a mitochondrial target of the anti-diabetic thiazolidinedione drugs (3, 4). Thiazolidinediones such as pioglitazone are capable of binding and stabilizing the mNT protein against 2Fe-2S cluster release, and thus, protecting tissue from mitochondrial injury (4). NAF-1 encoded by CISD2 is closely related to mNT, sharing 44% overall sequence identity and highly similar structure and functions (1, 5). However, the functions of CISD3 are not well understood.
Increasing evidences have revealed that mNT is an important regulator of diverse biological processes, including mitochondrial function, iron metabolism, reactive oxygen species (ROS) homeostasis, lipid metabolism, inflammation process, and autophagy (1–4, 6–15). Utilizing gain and loss of function mouse models, studies have demonstrated that overexpression of mNT in adipose or in liver is associated with a lower mitochondrial membrane potential, reduced rate of β-oxidation, lower levels of ROS-induced damage, and attenuated inflammation in those organs. Conversely, a systemic reduction in mNT expression heightens ROS in adipose and liver and leads to insulin resistance in mice (11). Additionally, mNT overexpression in adipose of mice also enhances subcutaneous adipose tissue browning (12).
Adiponectin is a hormone secreted largely from adipocytes that exerts multiple beneficial effects including lipid lowering and anti-inflammatory properties (16, 17). Adiponectin circulates as multimers of high (HMW), middle, and low molecular weight. The HMW form is the active form of adiponectin. Interestingly, mNT is known to involve in regulation of adipose adiponectin production (11). Adipocyte-specific mNT overexpression increases adiponectin production, transcriptionally and post-transcriptionally, and eventually increases circulating levels of total and HMW adiponectin in mice (11). The ability of mNT to stimulate adiponectin production and release is partially mediated by a selective modulation of the mitochondrial electron transport activity and regulation of intracellular iron metabolism in adipose tissues (11).
Fibroblast growth factor (Fgf) 15 (human homolog, FGF19) is a terminal small intestine (ileum)-derived hormone that functions as a central regulator of glucose, lipid, and bile acid metabolism (18). In liver, circulating Fgf15 signals through binding and activating receptor complex comprised of fibroblast growth factor receptor R4 (Fgfr4) and β-Klotho (β-Kl) (19, 20). Physiological regulation of bile acid synthesis is maintained through a feedback mechanism mediated by Fgf15 via inhibition of cholesterol 7α-hydroxylase 1 (Cyp7a1), the rate-limiting enzyme for hepatic bile acid synthesis (21).
Growing evidences suggest an association and cross-talk between adipocyte-derived adiponectin and ileum Fgf15 in the regulation of hepatic bile acid homeostasis, lipid metabolism, and inflammation (22–27). For instance, circulating adiponectin and Fgf15 were concomitantly elevated in Fgfr4 knock-out mice leading to improvement in glucose metabolism, insulin sensitivity, and reduction in body weight under high fat diet feeding (22). Emerging evidence also demonstrates a role of adiponectin in regulating bile acid homeostasis. Bile acid synthesis and serum bile acid levels are correlated with disease severity in liver diseases such as non-alcoholic fatty liver disease, whereas adiponectin is reversely correlated (28). Nonetheless, whether and how adiponectin and Fgf15 regulate each other and the underlying mechanisms of their concerted action in the liver are unknown.
Clinically, alcoholic steatosis/steatohepatitis is considered as the initial stages of alcoholic liver disease (ALD), which can progress to more severe forms of liver injury, including fibrosis, cirrhosis, and liver failure (29). Considerable evidences suggest that functional alterations of hepatic mitochondria, compromised iron metabolism, and aberrant adiponectin signaling are major anomalies in ALD in humans (29–31). However, the exact pathogenic mechanisms of ethanol-induced dysregulation of liver mitochondria function and adiponectin, ethanol-caused aberrant iron metabolism, and their contributions to the development and progression of alcoholic steatosis/steatohepatitis remain elusive.
The involvements of mNT in mitochondrial homeostasis, iron and lipid metabolism, and adiponectin production prompted us to test a hypothesis that mNT might play a pivotal role in the development of alcoholic steatosis/steatohepatitis. To test this hypothesis, we induced alcoholic fatty liver injury by pair feeding wild-type (WT) and mNT knock-out (mNTKO) mice with Lieber-DeCarli ethanol-containing diets for 4 weeks. Remarkably, following chronic ethanol feeding, mNTKO mice were completely resistant to steatosis, inflammation, and liver injury compared with ethanol-fed WT or mNTKO mice on a control diet. Mechanistically, the protective action of mNT deficiency against alcoholic steatohepatitis in mice was mediated, at least in part, through coordinated regulation of adipose-derived adiponectin and ileum Fgf15, and their concerted hepatic signaling.
Results
mNT Deficiency Alleviated Experimental Alcoholic Fatty Liver Injury in Mice
The in vivo function of mNT in the development of alcoholic stestosis/steatohepatitis was investigated by pair feeding WT and mNTKO mice using Lieber-DeCarli liquid diet for 4 weeks (32, 33).
During the 4-week feeding period, ethanol consumption had no apparent effect on the health status of the mice except inducing fatty liver injury. As shown in Table 1, the liver weight was significantly increased in ethanol-fed WT mice compared with WT control mice, it was unchanged in mNTKO mice fed with or without ethanol. The liver/body weight ratio was significantly increased in ethanol-fed WT and ethanol-fed mNTKO mice compared with WT control mice (Table 1). However, in comparison with ethanol-fed WT mice, the liver/body weight ratio in ethanol-fed mNTKO mice was significantly attenuated (Table 1).
TABLE 1.
Selected parameters in WT and mNTKO mice following ethanol feeding
10–12-Week-old male WT and mNTKO mice were divided into 4 groups as follows: 1) pair-fed WT control (WT); 2) ethanol-fed WT ethanol-containing diet; 3) mNTKO control; 4) ethanol-fed mNTKO. The animals were sacrificed after 4-weeks. Results are expressed as mean ± S.E. of 4–11 animals. Means without a common letter differ, p < 0.05.
| Parameters | WT | WT + ethanol | mNTKO | mNTKO + ethanol |
|---|---|---|---|---|
| Starting body (g) | 27.6 ± 0.5a | 27.4 ± 0.5a | 26.9 ± 0.6a | 27.6 ± 0.5a |
| Final body (g) | 32 ± 2.0a | 30 ± 2.0a | 27 ± 2b | 31 ± 2.5a |
| Liver (g) | 0.7 ± 0.03b | 0.8 ± 0.03a | 0.6 ± 0.02b | 0.6 ± 0.03b |
| Liver/body (%) | 1.9 ± 0.08c | 2.5 ± 0.07a | 1.6 ± 0.07c | 2.18 ± 0.06b |
| Plasma | ||||
| Insulin (ng/ml) | 1.0 ± 0.09a | 0.3 ± 0.03b | 1.0 ± 0.06a | 1.2 ± 0.2a |
| Glucose (mg/dl) | 126 ± 10b | 105 ± 7b | 189 ± 12a | 159 ± 2b |
The hepatic mRNA and protein expression levels of mNT were not significantly altered in the ethanol-fed WT mice compared with the WT pair-fed control mice (Fig. 1). However, mNT was completely removed in the livers of mNTKO mice fed either with or without ethanol (Fig. 1). Hepatic mRNA expression levels of Cisd2 and Cisd3 were similar in the WT mice fed either with or without ethanol (Fig. 1A). Ablation of mNT led to significant increases in the mRNA levels of Cisd2 and Cisd3 in the livers of mNTKO mice fed with a control diet compared with the WT controls (Fig. 1A). However, ethanol administration to mNTKO mice suppressed mRNAs of Cisd2 and Cisd3 compared with WT controls (Fig. 1A).
FIGURE 1.

Deletion of mNT alleviates experimental alcoholic steatohepatitis in mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, relative liver mRNA levels of Cisd1, Cisd2, and Cisd3. B, representative Western analysis of liver Cisd1. C, relative liver protein levels of Cisd1. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
As expected, ethanol feeding significantly increased liver triglyceride (TG) and cholesterol contents compared with the pair-fed WT control mice (Fig. 2, A and B). Strikingly, mNTKO mice fed with or without ethanol displayed significantly lower TG contents than the WT mice (Fig. 2B). Consistently, mNTKO mice fed either with or without ethanol also displayed significantly lower cholesterol contents than their corresponding WT mice (Fig. 2C). The magnitudes of TGs and cholesterol reduction in the mNTKO mice were similar with or without ethanol feeding (Fig. 2, B and C). Concordantly, H&E staining of livers revealed that mNTKO mice fed with or without ethanol exhibited less accumulation of hepatic lipid droplets, whereas there was prominent accumulation of lipid droplets in livers of ethanol-fed WT mice (Fig. 3A). Furthermore, serum alanine aminotransferase (ALT) was significantly elevated in the ethanol-fed WT and control diet-fed mNTKO mice but were normalized to WT control levels in the ethanol-fed mNTKO mice (Fig. 2D).
FIGURE 2.

mNT deficiency alleviates experimental alcoholic steatohepatitis in mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, pictures of mouse livers. B, liver TG levels. C, liver cholesterol (CHOL) levels. D, serum ALT. E, serum TG levels. F, serum cholesterol levels. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
FIGURE 3.

mNT deficiency alleviates experimental alcoholic steatohepatitis in mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, immunohistochemical staining for hematoxylin and eosin (H&E), F48/80+, and MPO (original magnification ×40) of liver sections. B, relative liver mRNA levels of F4/80+, Cd68, and Ly6G. C, liver MPO activity. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
Serum TG levels were not altered by ethanol feeding in the WT mice (Fig. 2E). In comparison with the WT controls, serum TG levels were reduced by ∼40% in the pair-fed mNTKO control mice and by ∼20% in the ethanol-fed mNTKO mice (Fig. 2E). Moreover, ethanol administration to WT mice significantly reduced serum cholesterol levels by ∼40% compared with WT controls, whereas the cholesterol levels in the mNTKO mice fed with or without ethanol were normalized to the WT controls (Fig. 2F). Collectively, these data demonstrated that mNT deficiency ameliorated alcoholic fatty liver injury in mice.
mNT Deficiency Ameliorated Ethanol-induced Hepatic Inflammation in Mice
In comparison with WT mice fed with or without ethanol, mNT deficiency provoked inflammation response as revealed by increased F4/80+ staining, and elevated gene expression of inflammation markers, F4/80+ and Cd68, in livers of the mice fed a control diet (Fig. 3, A and B). Remarkably, ethanol administration to the mNTKO mice diminished hepatic inflammation by suppressing those inflammation markers (Fig. 3, A and B). Note that ethanol feeding to the WT mice did not alter those inflammation markers (Fig. 3, A and B).
Neutrophil infiltration is a hallmark of alcoholic hepatitis (34). Immunohistochemical staining for myeloperoxidase (MPO) revealed that infiltration of neutrophils was higher in livers from the ethanol-fed WT mice or the control diet-fed mNTKO mice compared with the WT controls (Fig. 3A). However, fewer MPO+ neutrophils infiltrated livers of the ethanol-fed mNTKO mice (Fig. 3A). Accordingly, hepatic MPO activity and mRNA levels of the neutrophil marker Ly6G were markedly increased in ethanol-fed WT mice and control diet-fed mNTKO mice compared with WT controls (Fig. 3, B and C). However, both MPO activity and Ly6G gene expression were drastically reduced in ethanol-fed mNTKO mice to the levels of WT controls (Fig. 3, B and C).
Analysis of mRNAs from livers of control diet-fed mNTKO mice further revealed a substantial increase in the mRNAs of several important pro-inflammatory cytokines, including Inf-γ, Emr1, Mip-1α, Tgf-β, and E-selectin compared with the WT control mice (Fig. 4). Strikingly, mRNA expressions of these cytokines were greatly diminished in the mNTKO mice after ethanol administration (Fig. 4). Additionally, in comparison with the control-fed WT mice, the mRNAs of Tnf-α (Tnf-α1 and Tnf-α2) were modestly increased in the ethanol-fed WT mice, and the induction was augmented in the mNTKO mice fed a control diet (Fig. 4). However, ethanol administration to mNTKO mice completely abolished the ability of mNT deficiency to induce hepatic Tnf-α gene expression (Fig. 4).
FIGURE 4.

mNT deficiency ameliorates ethanol (E)-induced hepatic inflammation in mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol-containing diet for 4 weeks. Liver mRNA levels of Tnf-α1, Tnf-α2, Tgf-β, Mip-1α, Emr1, Inf-γ, and E-selection are shown. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
Collectively, our observations indicated that mNT deficiency provoked hepatic inflammation despite displaying lower lipid accumulation in the mice on a control diet. Remarkably, liver inflammation was ameliorated only when ethanol was administrated to mNTKO mice.
mNT Deficiency Elevated Circulating Levels of Lipocalin-2 (Lcn2) But Not Serum Amyloid A-1 (Saa1) in the Ethanol-fed Mice
To understand the underlying mechanism of the attenuated hepatic inflammation in the ethanol-fed mNTKO mice, we further profiled expressions of Lcn2 and Saa1, two major acute-phase proteins known to be involved in the development and progression of ALD (34–37).
In comparison with the WT controls, hepatic Lcn2 mRNA expression levels was modestly elevated in the ethanol-fed WT mice, and Lcn2 gene expression was augmented in the ethanol-fed mNTKO mice (Fig. 5A). Accordingly, the hepatic Lcn2 protein levels were mildly but significantly increased in ethanol-fed mNTKO mice compared with the other three groups (Fig. 5, B and C). In adipose, whereas Lcn2 gene expression was similar in the WT mice fed with or without ethanol, the Lcn2 mRNA levels were drastically increased in ethanol-fed mNTKO mice compared with all other groups (Fig. 5D). Serum levels of Lcn2 were slightly but significantly elevated in the ethanol-fed WT mice or control diet-fed mNTKO mice in comparison with the WT control mice (Fig. 5E). Remarkably, circulating Lcn2 was markedly increased up to ∼2-fold in the ethanol-fed mNTKO mice compared with all other groups (Fig. 5E).
FIGURE 5.

mNT deficiency elevated circulating levels of Lcn2 but not Saa1 in the ethanol(E)-fed mice. A, relative hepatic mRNA levels of Lcn2 and Saa1. B, representative Western analysis of liver Lcn2 and Saa1. C, relative hepatic protein levels of Lcn2 and Saa1. D, relative adipose mRNA levels of Lcn2 and Saa1; E, relative serum Lcn2 and Saa1 protein levels. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
Although hepatic Saa1 mRNA and protein levels were markedly elevated in the ethanol-fed WT and control diet-fed mNTKO mice compared with the WT controls, ethanol feeding to the mNTKO mice drastically exacerbated the increases in hepatic Saa1 gene and protein expression (Fig. 5, A–C). On the contrary, whereas adipose Saa1 mRNA levels were significantly elevated in the ethanol-fed WT and mNTKO control mice, ethanol feeding to mNTKO mice normalized adipose Saa1 mRNA to the levels of WT controls (Fig. 5D). Paradoxically, serum Saa1 levels were similar in ethanol-fed WT and WT control mice (Fig. 5E). The circulating Saa1 levels in the ethanol-fed mNTKO mice were also not altered compared with WT controls, whereas there was a slight elevation in serum Saa1 in the control diet-fed mNTKO mice compared with the other three groups (Fig. 5E). Our results demonstrated that mNT deficiency elevated circulating levels of Lcn2 but not Saa1 in the ethanol-fed mice.
mNT Deficiency Led to Induction of Adipose-derived Hormone Adiponectin in the Ethanol-administrated Mice
To dissect the molecular mechanisms underlying the mNT deficiency amelioration of alcoholic liver injury, we examined adiponectin production in adipose tissues (11, 16, 17).
Consistent with our previous findings (38), serum adiponectin protein levels were significantly lower in ethanol-fed WT mice compared with WT controls (Fig. 6A). Remarkably, ethanol administration to the mNTKO mice caused a prominent ∼32% increase in circulating adiponectin concentrations compared with all other groups (Fig. 6A). Accordingly, ethanol-fed mNTKO mice also exhibited the highest circulating levels of HMW adiponectin in comparison with the other groups (Fig. 6A). However, adipose adiponectin mRNA levels in ethanol-fed mNTKO mice were not significantly increased compare with WT controls (data not shown), implying that adiponectin might be regulated post-transcriptionally by mNT deficiency in response to ethanol challenge. Although mRNA expression of fatty acid transporter protein Cd36 were significantly reduced in adipose tissues of ethanol-fed WT mice compared with WT controls, a significant up-regulation in Cd36 mRNA was observed in adipose tissues of the ethanol-fed mNTKO mice compared with all other groups (Fig. 6B).
FIGURE 6.

mNT deficiency leads to induction of adipose-derived hormone adiponectin in ethanol-administrated mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, serum protein levels of HMW and total adiponectin levels. B, relative liver mRNA levels of Ucp1 and Cd36. C, relative mitochondria DNA copy number. D, relative adipose mRNA levels of Cisd1, Cisd2, and Cisd3. Results are expressed as mean ± S.E. (n = 4–10 mice). Means without a common letter differ, p < 0.05.
Compromised adipose mitochondrial function driven by mNT is associated with an increase in adiponectin production (11). The uncoupling protein 1 (Ucp1) is a mitochondrial protein in adipose (39). Indeed, whereas ethanol feeding to WT mice markedly increased Ucp1 gene expression, Ucp1 mRNA levels in the ethanol-fed mNTKO mice dramatically reduced to the WT control levels (Fig. 6B).
Adipose mitochondrial DNA (mtDNA) copy number was significant elevated in ethanol-fed WT and control diet-fed mNTKO mice compared with WT controls (Fig. 6C). Ethanol feeding to mNTKO mice markedly decreased the adipose mtDNA copy number to the WT control levels (Fig. 6C). These results suggest that reduced adipose mitochondrial activity in ethanol-fed mNTKO mice might increase adiponectin secretion and production. The hepatic mtDNA copy number was significantly decreased in ethanol-fed WT mice, control diet-fed mNTKO mice, and ethanol-fed mNTKO mice to the same extent as in WT control mice (Fig. 6C), implying that amelioration of liver mitochondria function might not be the main contributor to the improvements observed in livers of the ethanol-fed mNTKO mice.
Like mNT, NAF-1 (CISD2) primarily localizes in the outer mitochondrial membrane, regulates mitochondria functions, and promotes adiponectin production in adipose tissue (5, 40–42). Remarkably, ethanol administration to mNTKO mice significantly increased adipose Cisd2 mRNA expression levels compared with all other groups (Fig. 6D). Note that adipose Cisd3 mRNA levels were significantly decreased in the ethanol-fed WT mice and in mNTKO mice fed either with or without ethanol compared with the WT controls (Fig. 6D). Taken together, our data revealed that mNT deficiency led to elevation of adipose-derived adiponectin and its HMW form in the serum of ethanol administrated mice.
mNT Deficiency Increased Ileum Fgf15 Synthesis and Ameliorated Perturbation of Bile Acid Homeostasis in the Ethanol-fed Mice
As shown in Fig. 7A, mRNA expression levels of ileum Fgf15 were slightly elevated in the ethanol-fed WT mice and control diet-fed mNTKO mice, but were increased robustly in the ethanol-fed mNTKO mice, up to ∼6-fold of the WT controls.
FIGURE 7.

mNT deficiency increases Fgf15 synthesis and ameliorates perturbation of hepatic bile acid homeostasis in ethanol (E)-fed mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol-containing diet for 4 weeks. A, relative ileum mRNA levels of Fgf15. B, relative liver mRNA levels of Cyp7a1, Cyp27a1, and Cyp8a1. C, serum total of bile acids. D, total liver bile acids. Results are expressed as mean ± S.E. (n = 5–9 mice). Means without a common letter differ, p < 0.05.
Ileum Fgf15 signaling inhibits hepatic Cyp7a1, the rate-limiting enzyme for the “neutral” pathway of bile acid synthesis in the liver (21). As shown in Fig. 7B, mRNA levels of hepatic Cyp7a1 were markedly suppressed by ethanol feeding to WT mice, and the repression was exacerbated in the ethanol-fed mNTKO mice, indicating that the enhanced ileum Fgf15 synthesis in the ethanol-fed mNTKO mice could transduce signals to liver.
Hepatic mRNAs of Cyp27a1, which catalyzes the initial step of the alternative bile acid biosynthetic pathway, were significantly suppressed in both the WT and mNTKO mice to the same extent as in WT control mice (Fig. 7B). The mRNA levels of hepatic Cyp8b1, an enzyme controlling the synthesis of cholic acid in the ethanol-fed mNTKO mice were the lowest among all groups (Fig. 7B).
Ethanol feeding to WT mice significantly increased serum bile acid to ∼2-fold compared with the WT controls, indicating dysregulation of bile acid metabolism (Fig. 7C). Serum bile acid levels in the control diet-fed mNTKO mice were reduced by ∼40% of their WT controls (Fig. 7C). More strikingly, the ethanol feeding elevated serum levels of total bile acids were completely normalized in the ethanol-fed mNTKO mice (Fig. 7C). In line with these observations, the hepatic pool of bile acids was modestly elevated in the ethanol-fed WT and mNTKO control mice (Fig. 7D). The elevated hepatic bile acids were also completely normalized in the ethanol-fed mNTKO mice (Fig. 7D). Collectively, these data demonstrated that mNT deficiency induced ileum Fgf15 synthesis, normalized the ethanol-induced elevation of serum and hepatic bile acids, and limited the hepatic accumulation of toxic bile in mice administrated ethanol.
mNT Deficiency Modulated Hepatic and Adipose Iron Homeostasis in Mice
In liver, ethanol feeding did not alter the total concentrations of iron and ferrous (Fe2+, reduced form) in WT mice (Fig. 8A). However, hepatic iron or Fe2+ concentrations were significantly reduced in the mNTKO mice fed either a control diet or an ethanol-containing diet in comparison with the WT controls (Fig. 8A). Liver ferric (Fe3+, oxidized form) concentrations were substantially increased in the ethanol-fed WT mice up to ∼8-fold compared with the WT controls. mNTKO mice fed either a control diet or an ethanol-containing diet also displayed a mild ∼2- or ∼4-fold increase in Fe3+ concentrations compared with the WT controls (Fig. 8A). However, in comparison with the ethanol-fed WT mice, there were ∼30% decreases in Fe3+ concentrations in the ethanol-fed mNTKO mice, indicating that mNT deficiency partially abolished the ability of ethanol to induce hepatic Fe3+ contents (Fig. 8A).
FIGURE 8.

mNT deficiency modulates hepatic or adipose iron homeostasis in mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, liver total iron (Fe), ferrous (Fe2+), ferric (Fe3+) concentrations. B, adipose total iron (Fe), ferrous (Fe2+), ferric (Fe3+) concentrations. C, liver MDA levels. Results are expressed as mean ± S.E. (n = 6–9 mice). Means without a common letter differ, p < 0.05.
In adipose, the concentrations of iron and Fe2+ were significantly increased in the ethanol-fed WT mice by ∼4- and ∼2.5-fold, respectively, compared with the WT controls (Fig. 8B). Adipose iron and Fe2+ concentrations were significantly reduced in mNTKO mice fed with or without ethanol in comparison with the WT controls (Fig. 8B). In comparison with the WT controls, Fe3+ concentrations in the ethanol-fed WT, control-fed mNTKO, and ethanol-fed mNTKO mice were markedly increased by ∼170-, ∼8-, and ∼5-fold, respectively (Fig. 8C). Strikingly, there were dramatic decreases in Fe3+ concentrations in adipose tissues of the ethanol-fed mNTKO mice in comparison with the ethanol-fed WT mice (Fig. 8B).
Taken together, we observed lower levels of hepatic or adipose iron concentrations and normalized iron homeostasis in mNTKO mice fed with or without ethanol, consistent with a role of mNT as a pivotal factor in the regulation of iron metabolism (1, 11). More importantly, mNT deficiency partially but significantly abolished the ability of ethanol to induce oxidized Fe3+ contents in liver and adipose tissue of mice.
mNT Deficiency Attenuated Hepatic Oxidative Stress in the Ethanol-fed Mice
mNT regulates oxidative stress (9, 11). Interestingly, hepatic oxidative stress was not altered in WT mice fed either with or without ethanol and in the mNTKO control mice, as demonstrated by similar levels of hepatic malondialdehyde (MDA) (Fig. 8C). However, ethanol administration to mNTKO mice markedly decreased hepatic MDA levels by ∼50% compared with all other groups (Fig. 8C). The data demonstrated that mNT deficiency attenuated the generation of hepatic oxidative stress in mice after ethanol feeding.
mNT Deficiency Activated Hepatic Sirtuin 1 (Sirt1) and Attenuated Nuclear Factor κB (NF-κB) Activity in the Ethanol-fed Mice
We dissected the mechanism for mNT deficiency in mediating hepatic inflammatory process in the ethanol-fed mice by investigating the major inflammatory regulator hepatic Sirt1-NF-κB axis (43, 44).
Consistent with our previous findings (44), ethanol administration to WT mice significantly reduced hepatic Sirt1 protein levels compared with the WT control mice (Fig. 9, A and B). When mNTKO mice were administrated ethanol, the protein levels of Sirt1 were markedly increased to significantly higher than the other three groups, suggesting that mNT deficiency stimulated Sirt1 in the ethanol-fed mice (Fig. 9, A and B).
FIGURE 9.

mNT deficiency activates hepatic Sirt1 and attenuates NF-κB activity in ethanol-administered mice. Wild-type (WT) or mNTKO (KO) mice were pair-fed either a control diet or an ethanol (E)-containing diet for 4 weeks. A, representative Western analysis of liver Sirt1, Ac-NF-κB, NF-κB. B, relative liver protein levels of Sirt1, acetylated (Ac)-NF-κB, NF-κB. C, relative liver mRNA levels of Cd36, PPARα, and Mcad. D, relative serum β-hydroxybutyrate levels. Results are expressed as mean ± S.E. (n = 4–9 mice). Means without a common letter differ, p < 0.05.
Ethanol feeding to WT mice led to markedly stimulated NF-κB activity, as revealed by increased acetylated NF-κB and elevated NF-κB protein expression levels compared with WT control mice (Fig. 9, A and B). The levels of acetylated NF-κB and NF-κB were significant elevated in ethanol-fed mNTKO mice compared with WT controls. However, in comparison with ethanol-fed WT mice, hepatic NF-κB activity was substantially attenuated in the ethanol-fed mNTKO mice (Fig. 9, A and B). Collectively, these results demonstrated that mNT deficiency led to activation of Sirt1 and partial repression of NF-κB activity in ethanol-fed mice, suggesting that the amelioration of inflammation in the livers of ethanol-fed mNTKO mice may be regulated by both NF-κB-dependent and -independent mechanisms.
mNT Deficiency Inhibited the Expression of Lipid Uptake Cd36 and Activated the Expressing of Genes Implicated in Lipid Oxidation in Mice, and Prevented Ethanol-induced Ketoacidosis in Mice
mNT induces lipid uptake by signaling via Cd36, and reduces mitochondrial fatty acid oxidation in adipose and liver (11). As shown in Fig. 9C, mRNAs for PPARα and medium chain acyl-CoA dehydrogenase (Mcad) had no significant differences between the ethanol-fed and control diet-fed WT mice, ablation of mNT elevated the mRNAs of Mcad and PPARα in both the ethanol-fed and control-fed mNTKO mice. In addition, whereas ethanol feeding to WT mice slightly reduced hepatic mRNA expression of Cd36, Cd36 gene expression was markedly inhibited in both the ethanol-fed and control-fed mNTKO mice compared with their WT littermates (Fig. 9C).
Ethanol feeding dramatically increased serum β-hydroxybutyrate (β-OHB) by ∼16-fold compared with the WT controls, indicating ketoacidosis in the ethanol-fed WT mice (Fig. 9D). Strikingly, the elevated serum levels of β-OHB in the ethanol-fed WT mice were largely normalized in the ethanol-fed mNTKO mice, suggesting that mNT deficiency prevented the ethanol-induced ketoacidosis in mice (Fig. 9D).
It is worthwhile to note that the hepatic mRNAs of enzymes involved in lipogenesis such as fatty acid synthase and acetyl-CoA carboxylase were not significantly altered in the ethanol-fed mNTKO mice compared with other groups (data not shown), suggesting that mNT deficiency might reduce hepatic lipid accumulation in mice via lipogenesis-independent mechanisms.
Taken together, in line with the published studies (11), we found that mNT deficiency down-regulated CD36 and enhanced expression of the genes involved in lipid oxidation in both the ethanol-fed and control diet-fed mNTKO mice. These results suggested that such effects might suffice to elicit profound hepatic fat reduction in the mNTKO mice regardless of ethanol.
Discussion
mNT is emerging as a pivotal regulator of mitochondrial function, which impacts the dynamics of iron homeostasis, lipid metabolism, and inflammation processes in organs such as adipose and liver (6–15). In the present study, utilizing a mNT knock-out mouse model, we uncovered an intriguing role of mNT in the development of experimental alcoholic steatohepatitis in mice. We found that the pair-fed mNTKO control mice exhibited reduction of steatosis but enhancement of liver inflammation and exacerbation of liver injury. Remarkably, following chronic ethanol feeding, mNTKO mice exhibited a dramatic reduction of hepatic fat accumulation, diminished hepatic inflammation, attenuated hepatic production of ROS, and alleviation of liver injury. Mechanistic studies revealed that ethanol administration to mNTKO mice significantly increased the adipose-derived adiponectin and ileum Fgf15 synthesis. Correlating closely with induction of adiponectin and Fgf15, mNT deficiency normalized the ethanol-induced elevation of hepatic bile acid pool and serum levels of bile acids, diminished hepatic NF-κB signaling, and reduced hepatic oxidative stress, which, in turn, limited hepatic accumulation of toxic bile and led to prevention of hepatic inflammation, and amelioration of liver injury in mice after ethanol administration. Altogether, our data support a model in which adiponectin and Fgf15 function in concert in mediating the protective effect of mNT deficiency against experimental alcoholic steatohepatitis in mice (Fig. 10).
FIGURE 10.
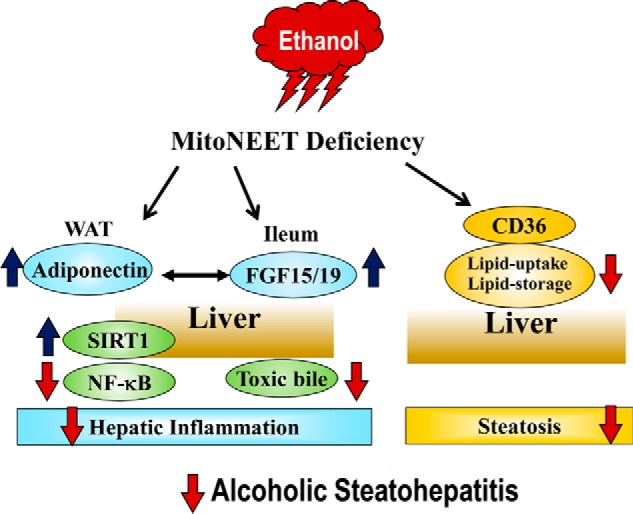
Proposed putative mechanisms that underlie the protective action of mNT deficiency against experimental alcoholic steatohepatitis in mice. Following chronic ethanol administration, mNT deficiency orchestrates induction of adiponectin in adipose and ileum Fgf15 in mice. Elevated adiponectin and Fgf15 subsequently work in concert to signal the liver and act as fine-tuning regulators of hepatic inflammation in an autocrine/paracrine fashion, which, in turn, alleviates experimental alcoholic steatohepatitis in mice.
Our current findings identified a compelling link between the adipocyte-derived adiponectin and ileum Fgf15 synthesis in mNTKO mice after ethanol administration. However, it is not entirely clear whether ethanol primarily induced adipose adiponectin and subsequently (or parallel) led to increases in ileum Fgf15 synthesis in the mNTKO mice. The increased adiponectin in ethanol-fed mNTKO mice might lead to ileum Fgf15 induction. On the other hand, injection of adeno-associated virus-expressing Fgf15 in mice increased adipose adiponectin synthesis (22). Therefore, it is possible that the ileum Fgf15 production might be induced by ethanol feeding to mNTKO mice, which, in turn, increased adipose adiponectin production. The interplay between adipose adiponectin and ileum Fgf15 and how the adiponectin-Fgf15 axis is mediated by mNT deficiency and ethanol warrants future investigation.
The combined increases in serum total and HMW adiponectin levels might be sufficient to elicit profound hepatic improvements in the ethanol-fed mNTKO mice. Given that both adiponectin and Fgf15 exhibited potent anti-inflammatory properties (15, 16, 45–47), elevation of circulating adiponectin and Fgf15 in the ethanol-fed mNTKO mice might also synergistically or additively trigger an anti-inflammation signaling network and protected mNTKO mice from ethanol-induced liver injury.
The diminished inflammatory response in the livers of ethanol-fed mNTKO mice is likely due to decreased liver ferric content, attenuated oxidative stress, and reduced hepatic NF-κB activity. Nevertheless, it is intriguing that the deficiency of mNT anti-inflammatory properties required ethanol in the diet. Despite reduced steatosis, mNTKO mice on a control diet exhibited pronounced inflammation, suggesting that reduction of intrahepatic lipid storage enhanced inflammation and exacerbated liver injury. Hence, it is unlikely that attenuated inflammatory response could just be secondary to reduced hepatic lipid accumulation in ethanol-fed mNTKO mice.
Adiponectin directly up-regulates Sirt1 in cultured hepatic cells (44, 48). The increased hepatic Sirt1 protein levels in ethanol-fed mNTKO mice might be due to their elevated circulating adiponectin levels. Sirt1 exerts anti-inflammatory effects by deacetylation of lysine residues on NF-κB and inhibits NF-κB activity (44). Conceivably, the increased hepatic Sirt1 could contribute to the attenuated acetylated NF-κB levels and diminished NF-κB signaling in the livers of ethanol-fed mNTKO mice.
As it encodes an intestinal endocrine hormone, Fgf15 induction in the ethanol-fed mNTKO mice is of particular interest because Fgf15 is released into portal circulation to negatively regulate hepatic bile acid synthesis (21). Bile acids are increasingly being recognized as the most sensitive markers of inflammation and liver dysfunction and injury (45, 46). Conceivably, the normalized hepatic and serum bile acids in the ethanol-fed mNTKO mice might limit accumulation of toxic bile acids such as deoxycholic acid in liver and ultimately ameliorated ethanol-induced liver damage. Fgf15 also has the ability to directly suppress inflammation via signaling through its receptor Fgfr4 (47). Given that hepatocytes are the only cell type in liver that expresses the Fgf15 receptor complex Fgfr4-βKl, it is logical to speculate that increased Fgf15 in ethanol-fed mNTKO mice might exert anti-inflammatory effects by targeting hepatocytes (49).
It is intriguing that hepatic bile acid synthesis genes such as Cyp7a1 were repressed despite elevated serum and hepatic bile acids in ethanol-fed WT mice. Liver Cyp7a1 gene expression was decreased in chronically ethanol-fed mice (36, 37). Human alcoholic fatty liver samples also had lower hepatic CYP7A1 mRNA expression levels (37). On the other hand, several lines of evidence demonstrate that ethanol consumption alters bile acid synthesis by up-regulating the expression of bile acid synthesis genes, including Cyp7a1, Cyp8b1, and Cyp27a1 (50–52). Therefore, it is likely that ethanol-mediated accumulation of bile acids might be regulated by both bile acid synthesis-dependent and -independent pathways.
Emerging evidences have revealed a potential role of adiponectin in regulating bile acid metabolism and liver function. The serum adiponectin level is inversely correlated with hepatic bile acid synthesis, serum bile acid levels, and hepatocellular injury in patients with nonalcoholic liver disease (28). Interestingly, adiponectin also directly regulates bile acid homeostasis-related genes such as Cyp7a1 (28). Moreover, adiponectin is capable of alleviating toxic bile deoxycholic acid-induced inflammation in esophageal adenocarcinoma cells (53). In this scenario, it is possible that elevated adiponectin in the ethanol-fed mNTKO mice might directly or indirectly via up-regulation of ileum Fgf15 ameliorate the ethanol-mediated perturbation of bile acid homeostasis, and alleviate liver injury.
A surprising discovery was that observed reduction of ferric concentrations, decrease of mitochondria activity, and increase of adiponectin production in adipose tissues of the ethanol-fed mNTKO mice were similar to the phenotypes of adipocyte-specific mNT over-expression mice. More strikingly, mRNA expression of Cisd2 was significantly elevated in adipose but not in liver of the ethanol-fed mNTKO mice. Therefore, it is tempting to speculate that adipose CISD2 induction might act as a compensatory molecule in regulating the effect of ethanol in the mNT-deficient mice (5, 40–42). Performing ethanol feeding studies by using genetically modified mouse models such as adipose-specific Cisd2 null mice or mNT/Cisd2 double knock-out mice in the future could help to provide more definitive answers.
Lcn2 has been identified as a proinflammatory molecule, and plays a key role in the pathogenesis of ALD (34, 37). We have recently found that myeloid cell-specific lipin-1 deficiency reduced serum Lcn2 levels and ameliorated alcoholic hepatitis in mice.4 Paradoxically, mNT deficiency increased circulating Lcn2 in ethanol-fed mice. Emerging evidences suggest that Lcn2 also exerts protective functions against liver injuries by acting as a “help me” signal (54). It is tempting to speculate that mNT deficiency-mediated elevation of Lcn2 may trigger anti-inflammatory signaling and protect the mice from ethanol-induced liver damage. Additional studies are required to further dissect the mechanism underlying this intriguing phenotype.
The mechanisms underlying the disassociation of hepatic Saa1 gene and protein expression and serum levels of Saa1 in ethanol-fed mNTKO mice remain to be elucidated. Liver is the most important organ for expression and secretion of Saa1 during the acute-phase response. However, during the non-acute phase, adipose tissue is the main source of Saa1 in obese subjects (55, 56). Unlike in liver, adipose Saa1 gene expression was not elevated in the ethanol-fed mNTKO mice. Therefore, it is tempting to speculate that serum Saa1 is closely associated with adipose Saa1 but not liver.
Ethanol metabolizes in the liver via alcohol dehydrogenase, aldehyde dehydrogenase 2, or cytochrome P-450 2E1 to produce metabolic byproducts such as acetaldehyde or ROS. Considerable evidence indicates that hepatic ethanol metabolism is associated with alterations in hepatic mitochondria function, and consequently, promotes liver injury (29, 30). For instance, hepatic ethanol metabolism is required for mitochondrial depolarization in mouse liver (56). The role of mNT in mediating effects of hepatic ethanol metabolism on mitochondrial function is currently under investigation in our laboratory.
In summary, our data suggested that the regulations of various endocrine hormones, in particular, the induction of adipocyte-derived adiponectin and ileum Fgf15 synthesis were the main contributors to the observed improvements in livers of the ethanol-fed mNTKO mice. Our findings are consistent with the large body of evidence that ALD is driven by organ cross-talk among adipose, intestine, and liver (29). Furthermore, we have provided new evidence that consuming alcohol could be a potent and efficient way of eliciting anti-inflammatory properties and of alleviating liver injury in mNT-deficient mice. Our novel findings raise the exciting possibility of utilizing mNT antagonists as novel treatments for human alcoholic steatohepatitis.
Experimental Procedures
Reagents
Most chemicals and supplies were purchased from Thermo Fisher Scientific Inc. (Rockford, IL) and Sigma.
Animal Models of Chronic Alcohol Feeding
Male wild-type mice (C57BL/6J) were purchased from Jackson Laboratory or bred in the vivarium associated with our laboratory. Global mNTKO mice were obtained from Taconic Laboratories, and were kindly provided by Dr. Werner J. Geldenhuys (Northeast Ohio Medical University) (57). mNTKO mice on C57BL/6 background were viable and phenotypically normal under a chow diet.
All mice were maintained on a normal chow diet (NIH-31 Open Formula Mouse/Rat Sterilizable Diet 7917, Teklad Diets, Madison, WI) before beginning the ethanol feeding study. Lieber-DeCarli control and ethanol liquid diets were purchased from Bio-Serv (Frenchtown, NJ) (32, 33, 37).
Ten to 12-week-old male mNTKO and their age-matched WT mice were divided into following four groups: 1) WT control; 2) WT plus ethanol (identical to the control diet but with 5% (w/v) ethanol added); 3) mNTKO control; 4) mNTKO plus ethanol. The dietary and nutritional intake of the control mice were matched to those of the ethanol-fed mice by pair feeding the same volume of isocaloric liquid diet for 4 weeks. Ethanol was introduced gradually into the diet as described previously (32, 33, 37). For mice on an ethanol-containing diet, animal cages were placed on heating pads to maintain body temperature to prevent ethanol-induced hypothermia.
After 4 weeks on the liquid diets, the mice were euthanized, and blood and tissue samples (liver, adipose, and intestine) were collected. All animal experiments were approved by the Institutional Animal Care and Use Committee of Northeast Ohio Medical University. All experiments were performed in accordance with relevant guidelines and regulations.
ELISA from Mouse Serum
The serum level of Lcn2 was measured by using a Lipocalin-2/ab199083 Mouse ELISA Kit (Abcam, Cambridge, MA). The serum level of Saa1 was measured by using a Saa1/ab157723 Mouse Elisa Kit (Abcam). Serum levels of ALT and AST were determined by using the MaxDiscovery ALT and AST Assay Kits (Bioo Scientific, Austin, TX). Plasma or tissue triglyceride and cholesterol were determined by using the Infinity Cholesterol and Triglyceride Reagents (Thermo Scientific). Serum levels of total adiponectin were determined using a commercial ELISA kit from BioVendor (Candler, NC). Serum levels of HMW forms of adiponectin were determined by using a Mouse Adiponectin Assay Kit from ALPCO (Salem, NH). Plasma β-OHB was measured by using a β-Hydroxybutyrate Assay Kit (Cayman Chemicals, Ann Arbor, MI). The serum level of insulin was measured by using a Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, IL). The serum level of glucose was measured by using a Glucose Assay Kit (Abcam). Analyses were performed with a SpectraMax i3x microplate reader (Molecular Devices, Sunnyvale, CA).
Measurement of Liver Lipid
Liver triglyceride and cholesterol contents were measured as described previously (37).
Staining for H&E, F4/80+, and MPO
Liver tissues were fixed in 10% formalin and embedded in paraffin. Embedded liver tissues were sectioned and stained with hematoxylin and eosin (H&E) (37). Embedded liver tissues were also subjected to immunohistochemical staining for F4/80+ or MPO by using allophycocyanin-Alexa Fluor 750-conjugated rat anti-mouse F4/80+ monoclonal antibody (Thermo Fisher Scientific) or rabbit anti-MPO polyclonal antibody (Abcam) (37). Sections were incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (Thermo Fisher Scientific) for a MPO antibody. Positive cells for F4/80+ or MPO were randomly photographed using a fluorescence microscope, and positive density was quantified using the software Image-Pro Plus (Media Cybernetics, Bethesda, MD).
MPO Activity Assay
To determine MPO activity, the liver tissues were weighed and homogenized on ice in 1 ml of PBS with 0.1% Nonidet P-40. The homogenates were centrifuged at 12,500 rpm for 15 min and the supernatant was transferred. The MPO activity was measured using assay kits (Abcam, ab105136). One unit of MPO activity was defined as the amount of myeloperoxidase to reduce 1 μmol of hydrogen peroxide per minute. The results of MPO activity were expressed as units of MPO activity per g of liver weight (milliunits/g of liver).
Lipid Peroxidation
The lipid peroxidation product in liver was measured using methods based on the formation of the thiobarbituric reactive substances and expressed as the extent of MDA production, as described previously (36).
Hepatic Bile Acid Pool Size and Serum Bile Acid Concentrations
Liver samples were homogenized in 75% ethanol and incubated at 50 °C for 2 h (46). After centrifugation at 6000 × g for 10 min, the pellets were extracted again with 50% ethanol. Supernatants from the two extraction steps were pooled, evaporated, and reconstituted in 50% ethanol. Concentrations of total bile acids in liver extracts or serum were determined by utilizing Total Bile Acid Kit STA-631 (Cell Biolabs, San Diego, CA).
Iron Analyses
Liver and adipose iron concentrations were determined using the Iron Assay Kit (Abcam ab83366) according to the manufacturer's protocol.
RNA Isolation and mRNA Levels Analysis
Total RNA isolation and mRNA levels were measured as previously described (35–37). Briefly, cDNA synthesis was executed using a Verso cDNA Synthesis Kit (Thermo Scientific). Real-time quantitative polymerase chain reaction amplification was performed using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with FastStart Universal SYBR Green Master Mix with ROX (Roche Diagnostics). Gene-specific primer sets were designed or purchased from Qiagen SuperArray Bioscience (Frederick, MD) (Table 2). The relative amount of target mRNA was calculated by using the comparative cycle threshold (ΔΔCt) method (by normalizing target mRNA Ct values to those for GAPDH (ΔCt) with the control group (ΔΔCt)). Fold-changes were calculated by 2∧(−ΔΔCt).
TABLE 2.
Primers used in this study
| Direction | Sequence (5′ → 3′) | |
|---|---|---|
| 16s | Forward | TGATCAACGGACCAAGTTACC |
| Reverse | TCCGGTCTGAACTCAGATCAC | |
| 18s | Forward | ACGGACCAGAGCGAAAGCAT |
| Reverse | GGACATCTAAGGGCATCACAG | |
| AOX(Acox1) | Forward | TGGTATGGTGTCGTACTTGAATGAC |
| Reverse | AATTTCTACCAATCTGGCTGAAC | |
| β-Kl | Forward | TGTTCTGCTGCGAGCTGTTAC |
| Reverse | CCGGACTCACGTACTGTTTTT | |
| Cd36 | Forward | ATGGGCTGTGATCGGAACTG |
| Reverse | GTCTTCCCAATAAGCATGTCTCC | |
| Cd68 | Forward | TGTCTGATCTTGCTAGGACCG |
| Reverse | GAGAGTAACGGCCTTTTTGTGA | |
| Cisd1 | Forward | GCTGTGCGAGTTGAGTGGAT |
| Reverse | TGGTGCGATTCTCTTTAGCGTA | |
| Cisd2 | Forward | GACAGCATCACCGGGTTCG |
| Reverse | CTCATTCACCACCTTGGGATTTT | |
| Cisd3 | Forward | TATCAGAGGCGGGAAATCTCTT |
| Reverse | ATTCTTGCTTCGGCCACATAC | |
| Cyp27a1 | Forward | CCAGGCACAGGAGAGTACG |
| Reverse | GGGCAAGTGCAGCACATAG | |
| Cyp27b1 | Forward | TCCTGGCTGAACTCTTCTGC |
| Reverse | GGCAACGTAAACTGTGCGAA | |
| Cyp7a1 | Forward | TGTGCTTCAAGCTCTCAGCCAGC |
| Reverse | ACTAGTGGCGGGCGTCCCTA | |
| Cyp8b1 | Forward | CCTCTGGACAAGGGTTTTGTG |
| Reverse | GCACCGTGAAGACATCCCC | |
| Emr1 | Forward | TGACTCACCTTGTGGTCCTAA |
| Reverse | CTTCCCAGAATCCAGTCTTTCC | |
| Fgf15 | Forward | GAAGACGATTGCCATCAAGGA |
| Reverse | CGAATCAGCCCGTATATCTTGC | |
| Fgfr4 | Forward | GCTCGGAGGTAGAGGTCTTGT |
| Reverse | CCACGCTGACTGGTAGGAA | |
| Gapdh | Forward | TGACCTCAACTACATGGTCTACA |
| Reverse | CTTCCCATTCTCGGCCTTG | |
| Ifn-γ | Forward | ATGAACGCTACACACTGCATC |
| Reverse | CCATCCTTTTGCCAGTTCCTC | |
| Il-1β | Forward | TGTAATGAAAGACGGCACACC |
| Reverse | TCTTCTTTGGGTATTGCTTGG | |
| Il-6 | Forward | TAGTCCTTCCTACCCCAATTTCC |
| Reverse | TTGGTCCTTAGCCACTCCTTC | |
| Lcn2 | Forward | TGGCCCTGAGTGTCATGTG |
| Reverse | CTCTTGTAGCTCATAGATGGTGC | |
| Ly6g | Forward | TGCGTTGCTCTGGAGATAGA |
| Reverse | CAGAGTAGTGGGGCAGATGG | |
| Mcad | Forward | GCTCGTGAGCACATTGAAAA |
| Reverse | CATTGTCCAAAAGCCAAACC | |
| Mip-1á | Forward | TTCTCTGTACCATGACACTCTGC |
| Reverse | CGTGGAATCTTCCGGCTGTAG | |
| Saa-1 | Forward | TTAGCTCAGTAGGTTGTGCTGG |
| Reverse | ACAATGTTTCCCCAGAGAGCA | |
| Sele | Forward | AGCAGAGTTTCACGTTGCAGG |
| Reverse | TGGCGCAGATAAGGCTTCA | |
| Sirt1 | Forward | ATCGGCTACCGAGACAAC |
| Reverse | GTCACTAGAGCTGGCGTGT | |
| Tgf-β | Forward | CTCCCGTGGCTTCTAGTGC |
| Reverse | GCCTTAGTTTGGACAGGATCTG | |
| Tnf-α1 | Forward | CCCTCACACTCAGATCATCTTCT |
| Reverse | GCTACGACGTGGGCTACAG | |
| Tnf-α2 | Forward | ACCCTCACACTCACAAACCA |
| Reverse | ACAAGGTACAACCCATCGGC | |
| Ucp1 | Forward | GTGAAGGTCAGAATGCAAGC |
| Reverse | AGGGCCCCCTTCATGAGGTC |
Genomic DNA Isolation and mtDNA Copy Number Determination by Quantitative Real-time PCR
Genomic DNA from tissues was isolated by utilizing a DNAzol Genomic DNA Isolation Reagent Kit (catalog number DN 127, Molecular Research Center, Inc., Cincinnati, OH) according to the manufacturer's instructions. The 16S of the mtDNA gene and the 18S of the nDNA gene were amplified by quantitative PCR (Bio-Rad CFX 96 Real-time PCR System) (58). For PCR sample preparation, 4 μl of genomic DNA (50 ng/μl) was mixed with 1 μl of each primer (10 μm), 4 μl of forward and reverse primer mix (5 μm), 2 μl of nuclease-free water, and 10 μl of iTaq Universal SYBR Green Supermix (Bio-Rad). Each sample was run in triplicate or quadruplicate. The reaction was initiated at 95 °C for 10 min, followed by 50 cycles through 95 °C × 15 s and 60 °C × 60 s. Melt curve analysis was performed from 65 to 93 °C by raising temperature increments of 0.5 °C to verify the specificity and identity of the PCR product. Amplification curves and melt-curve analysis were analyzed using Bio-Rad CFX Manager Software and these curves were used to determine the mtDNA:nDNA ratio. Primers sequences used for DNA amplification are listed in Table 2.
Western Blot Analyses
Western blot analyses were performed by utilizing tissue extracts (35–37). Nuclear and tissue extracts were then separated by electrophoresis in 10% and/or AnyKd Mini-Protean TGX gels using Bio-Rad Mini-PROTEAN Tetra Cell System and transferred to PVDF membranes via Bio-Rad Blotting Module. Multiple protein band quantification and analysis were performed using AlphaView software version 3.4 (ProteinSimple). Lcn2 and Saa1 antibodies were purchased from Abcam. Sirt1, NF-κB, acetylated NF-κB and GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA). CISD1 antibody was purchased from Proteintech Group, Inc. (Rosemont, IL).
Statistical Analysis
All results are provided as mean ± S.E. Statistical analysis was performed using GraphPad Prism (San Diego, CA). The significance of differences between two groups was determined by unpaired two-tailed Student's t test. For comparison of multiple groups, one-way analysis of variance test followed by the post hoc Bonferroni test was applied. Significance was accepted at a p value of <0.05.
Author Contributions
X. H., A. J., J. W., C. K., Y. H., and H. S. contributed to generating the data presented, making the figures and tables, and drafting the procedures. J. W. and M. Y. contributed to designing the experiments, coordinating the project, drafting, and the completion of the manuscript through supervision of others, and approving the final version of manuscript.
Acknowledgments
We thank Dr. Werner J. Geldenhuys (Northeast Ohio Medical University, NEOMED) for providing mNTKO mice. We are grateful to Drs. James P. Hardwick, Kwangwon Lee, and Prabodh Sadana (NEOMED) for excellent technical assistance. We thank Paula Rote and Li Lin (NEOMED) for excellent technical assistance. We appreciate the input of Dr. Junchang Zhang (Guangdong Hospital of Traditional Chinese Medicine in Zhuhai, China) regarding discussions related to the mechanisms of clinical alcoholic and nonalcoholic liver diseases.
This work was supported in part by National Institute on Alcoholism and Alcohol Abuse Grants AA013623 and AA015951 (to M. Y.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M. You, unpublished data.
- mNT
- mitoNEET
- mNTKO
- mNT knock-out
- ALT
- alanine aminotransferase
- Cyp7a1
- cholesterol 7α-hydroxylase
- Fgfr4
- fibroblast growth factor receptor 4
- Kl
- Klotho
- Lcn2
- lipocalin 2
- Mcad
- medium chain acyl-CoA dehydrogenase
- Mip
- macrophage inflammatory protein
- NAF-1
- nutrient-deprivation autophagy factor 1
- NF-κB
- nuclear factor κB
- PPAR
- peroxisome proliferator-activated receptor
- Saa1
- serum amyloid A-1
- Sirt1
- sirtuin 1
- Tnf-α
- tumor necrosis factor-α
- TG
- triacylglyceride
- MPO
- myeloperoxidase
- Ucp-1
- uncoupling protein 1
- MDA
- malondialdehyde
- β-OHB
- β-hydroxybutyrate
- ROS
- reactive oxygen species
- HMW
- high molecular weight.
References
- 1. Tamir S., Paddock M. L., Darash-Yahana-Baram M., Holt S. H., Sohn Y. S., Agranat L., Michaeli D., Stofleth J. T., Lipper C. H., Morcos F., Cabantchik I. Z., Onuchic J. N., Jennings P. A., Mittler R., and Nechushtai R. (2015) Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 1853, 1294–1315 [DOI] [PubMed] [Google Scholar]
- 2. Geldenhuys W. J., Leeper T. C., and Carroll R. T. (2014) mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov. Today 19, 1601–1606 [DOI] [PubMed] [Google Scholar]
- 3. Wright M. B., Bortolini M., Tadayyon M., and Bopst M. (2014) Minireview: challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 28, 1756–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paddock M. L., Wiley S. E., Axelrod H. L., Cohen A. E., Roy M., Abresch E. C., Capraro D., Murphy A. N., Nechushtai R., Dixon J. E., and Jennings P. A. (2007) MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. U.S.A. 104, 14342–14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sohn Y. S., Tamir S., Song L., Michaeli D., Matouk I., Conlan A. R., Harir Y., Holt S. H., Shulaev V., Paddock M. L., Hochberg A., Cabanchick I. Z., Onuchic J. N., Jennings P. A., Nechushtai R., and Mittler R. (2013) NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. U.S.A. 110, 14676–14681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tan G., Landry A. P., Dai R., Wang L., Lu J., and Ding H. (2012) Competition of zinc ion for the [2Fe-2S] cluster binding site in the diabetes drug target protein mitoNEET. Biometals 25, 1177–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferecatu I., Gonçalves S., Golinelli-Cohen M. P., Clémancey M., Martelli A., Riquier S., Guittet E., Latour J. M., Puccio H., Drapier J. C., Lescop E., and Bouton C. (2014) The diabetes drug target MitoNEET governs a novel trafficking pathway to rebuild an Fe-S cluster into cytosolic aconitase/iron regulatory protein 1. J. Biol. Chem. 289, 28070–28086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zuris J. A., Ali S. S., Yeh H., Nguyen T. A., Nechushtai R., Paddock M. L., and Jennings P. A. (2012) NADPH inhibits [2Fe-2S] cluster protein transfer from diabetes drug target MitoNEET to an apo-acceptor protein. J. Biol. Chem. 287, 11649–11655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landry A. P., and Ding H. (2014) Redox control of human mitochondrial outer membrane protein MitoNEET [2Fe-2S] clusters by biological thiols and hydrogen peroxide. J. Biol. Chem. 289, 4307–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Colca J. R., McDonald W. G., Waldon D. J., Leone J. W., Lull J. M., Bannow C. A., Lund E. T., and Mathews W. R. (2004) Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am. J. Physiol. Endocrinol. Metab. 286, E252–E260 [DOI] [PubMed] [Google Scholar]
- 11. Kusminski C. M., Holland W. L., Sun K., Park J., Spurgin S. B., Lin Y., Askew G. R., Simcox J. A., McClain D. A., Li C., and Scherer P. E. (2012) MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 18, 1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kusminski C. M., Park J., and Scherer P. E. (2014) MitoNEET-mediated effects on browning of white adipose tissue. Nat. Commun. 5, 3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moreno-Navarrete J. M., Moreno M., Ortega F., Sabater M., Xifra G., Ricart W., and Fernández-Real J. M. (2016) CISD1 in association with obesity-associated dysfunctional adipogenesis in human visceral adipose tissue. Obesity 24, 139–147 [DOI] [PubMed] [Google Scholar]
- 14. Hunter R. L., Choi D. Y., Ross S. A., and Bing G. (2008) Protective properties afforded by pioglitazone against intrastriatal LPS in Sprague-Dawley rats. Neurosci. Lett. 432, 198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salem A. F., Whitaker-Menezes D., Howell A., Sotgia F., and Lisanti M. P. (2012) Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle 11, 4174–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park P. H., Sanz-Garcia C., and Nagy L. E. (2015) Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr. Pathobiol. Rep. 3, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. You M., and Rogers C. Q. (2009) Adiponectin: a key adipokine in alcoholic fatty liver. Exp. Biol. Med. (Maywood) 234, 850–859 [DOI] [PubMed] [Google Scholar]
- 18. Jahn D., Rau M., Hermanns H. M., and Geier A. (2015) Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 26, 625–635 [DOI] [PubMed] [Google Scholar]
- 19. Lin B. C., Wang M., Blackmore C., and Desnoyers L. R. (2007) Liver-specific activities of FGF19 require Klotho β. J. Biol. Chem. 282, 27277–27284 [DOI] [PubMed] [Google Scholar]
- 20. Kurosu H., Choi M., Ogawa Y., Dickson A. S., Goetz R., Eliseenkova A. V., Mohammadi M., Rosenblatt K. P., Kliewer S. A., and Kuro-o M.(2007) Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 282, 26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., Gerard R. D., Repa J. J., Mangelsdorf D. J., and Kliewer S. A. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225 [DOI] [PubMed] [Google Scholar]
- 22. Ge H., Zhang J., Gong Y., Gupte J., Ye J., Weiszmann J., Samayoa K., Coberly S., Gardner J., Wang H., Corbin T., Chui D., Baribault H., and Li Y. (2014) Fibroblast growth factor receptor 4 (FGFR4) deficiency improves insulin resistance and glucose metabolism under diet-induced obesity conditions. J. Biol. Chem. 289, 30470–30480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo Y., Yang C., Ye M., Jin C., Abbruzzese J. L., Lee M. H., Yeung S. C., and McKeehan W. L. (2013) Deficiency of metabolic regulator FGFR4 delays breast cancer progression through systemic and microenvironmental metabolic alterations. Cancer Metab. 1, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang D., Zhu W., Li J., An C., and Wang Z. (2013) Serum concentrations of fibroblast growth factors 19 and 21 in women with gestational diabetes mellitus: association with insulin resistance, adiponectin, and polycystic ovary syndrome history. PLoS ONE. 8, e81190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolumam G., Chen M. Z., Tong R., Zavala-Solorio J., Kates L., van Bruggen N., Ross J., Wyatt S. K., Gandham V. D., Carano R. A., Dunshee D. R., Wu A. L., Haley B., Anderson K., Warming S., et al. (2015) Sustained brown fat stimulation and insulin sensitization by a humanized bispecific antibody agonist for fibroblast growth factor receptor 1/βKlotho complex. EBioMedicine 2, 730–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sánchez-Infantes D., Gallego-Escuredo J. M., Díaz M., Aragonés G., Sebastiani G., López-Bermejo A., de Zegher F., Domingo P., Villarroya F., and Ibáñez L. (2015) Circulating FGF19 and FGF21 surge in early infancy from infra- to supra-adult concentrations. Int. J. Obes. (Lond.) 39, 742–746 [DOI] [PubMed] [Google Scholar]
- 27. Hao Y., Zhou J., Zhou M., Ma X., Lu Z., Gao M., Pan X., Tang J., Bao Y., and Jia W. (2013) Serum levels of fibroblast growth factor 19 are inversely associated with coronary artery disease in chinese individuals. PLoS ONE 8, e72345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bechmann L. P., Kocabayoglu P., Sowa J. P., Sydor S., Best J., Schlattjan M., Beilfuss A., Schmitt J., Hannivoort R. A., Kilicarslan A., Rust C., Berr F., Tschopp O., Gerken G., Friedman S. L., Geier A., and Canbay A. (2013) Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 57, 1394–1406 [DOI] [PubMed] [Google Scholar]
- 29. Gao B., and Bataller R. (2011) Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song B. J., Akbar M., Abdelmegeed M. A., Byun K., Lee B., Yoon S. K., and Hardwick J. P. (2014) Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 3, 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kohgo Y., Ohtake T., Ikuta K., Suzuki Y., Torimoto Y., and Kato J. (2008) Dysregulation of systemic iron metabolism in alcoholic liver diseases. J. Gastroenterol. Hepatol. 23, S78–S81 [DOI] [PubMed] [Google Scholar]
- 32. Bertola A., Mathews S., Ki S. H., Wang H., and Gao B. (2013) Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertola A., Park O., and Gao B. (2013) Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology 58, 1814–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wieser V., Tymoszuk P., Adolph T. E., Grander C., Grabherr F., Enrich B., Pfister A., Lichtmanegger L., Gerner R., Drach M., Moser P., Zoller H., Weiss G., Moschen A. R., Theurl I., and Tilg H. (2016) Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J. Hepatol. 64, 872–880 [DOI] [PubMed] [Google Scholar]
- 35. Yin H., Hu M., Liang X., Ajmo J. M., Li X., Bataller R., Odena G., Stevens S. M. Jr, and You M. (2014) Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu M., Yin H., Mitra M. S., Liang X., Ajmo J. M., Nadra K., Chrast R., Finck B. N., and You M. (2013) Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology 58, 1953–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cai Y., Jogasuria A., Yin H., Xu M. J., Hu X., Wang J., Kim C., Wu J., Lee K., Gao B., and You M. (2016) The detrimental role played by lipocalin-2 in alcoholic fatty liver in mice. Am. J. Pathol. 186, 2417–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. You M., Considine R. V., Leone T. C., Kelly D. P., and Crabb D. W. (2005) Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 42, 568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dong M., Yang X., Lim S., Cao Z., Honek J., Lu H., Zhang C., Seki T., Hosaka K., Wahlberg E., Yang J., Zhang L., Länne T., Sun B., Li X., Liu Y., Zhang Y., and Cao Y. (2013) Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 18, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Y. F., Kao C. H., Chen Y. T., Wang C. H., Wu C. Y., Tsai C. Y., Liu F. C., Yang C. W., Wei Y. H., Hsu M. T., Tsai S. F., and Tsai T. F. (2009) Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 23, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang C. H., Chen Y. F., Wu C. Y., Wu P. C., Huang Y. L., Kao C. H., Lin C. H., Kao L. S., Tsai T. F., and Wei Y. H. (2014) Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 23, 4770–4785 [DOI] [PubMed] [Google Scholar]
- 42. Tsai P. H., Chien Y., Chuang J. H., Chou S. J., Chien C. H., Lai Y. H., Li H. Y., Ko Y. L., Chang Y. L., Wang C. Y., Liu Y. Y., Lee H. C., Yang C. H., Tsai T. F., Lee Y. Y., and Chiou S. H. (2015) Dysregulation of mitochondrial functions and osteogenic differentiation in Cisd2-deficient murine induced pluripotent stem cells. Stem Cells Dev. 24, 2561–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. You M., Liang X., Ajmo J. M., and Ness G. C. (2008) Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G892–G898 [DOI] [PubMed] [Google Scholar]
- 44. Shen Z., Ajmo J. M., Rogers C. Q., Liang X., Le L., Murr M. M., Peng Y., and You M. (2009) Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-α production in cultured macrophage cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1047–G1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mutanen A., Lohi J., Heikkilä P., Jalanko H., and Pakarinen M. P. (2015) Loss of ileum decreases serum fibroblast growth factor 19 in relation to liver inflammation and fibrosis in pediatric onset intestinal failure. J. Hepatol. 62, 1391–1397 [DOI] [PubMed] [Google Scholar]
- 46. Zhou M., Learned R. M., Rossi S. J., DePaoli A. M., Tian H., and Ling L. (2016) Engineered fibroblast growth factor 19 reduces liver injury and resolves sclerosing cholangitis in Mdr2-deficient mice. Hepatology 63, 914–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Drafahl K. A., McAndrew C. W., Meyer A. N., Haas M., and Donoghue D. J. (2010) The receptor tyrosine kinase FGFR4 negatively regulates NF-κB signaling. PLoS ONE 5, e14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shen Z., Liang X., Rogers C. Q., Rideout D., and You M. (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G364–G374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao Y., Meng C., Wang Y., Huang H., Liu W., Zhang J. F., Zhao H., Feng B., Leung P. S., and Xia Y. (2016) IL-1β inhibits β-Klotho expression and FGF19 signaling in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 310, E289–E300 [DOI] [PubMed] [Google Scholar]
- 50. Wu W., Zhu B., Peng X., Zhou M., Jia D., and Gu J. (2014) Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem. Biophys. Res. Commun. 443, 68–73 [DOI] [PubMed] [Google Scholar]
- 51. Xie G., Zhong W., Li H., Li Q., Qiu Y., Zheng X., Chen H., Zhao X., Zhang S., Zhou Z., Zeisel S. H., and Jia W. (2013) Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 27, 3583–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chanda D., Kim Y. H., Li T., Misra J., Kim D. K., Kim J. R., Kwon J., Jeong W. I., Ahn S. H., Park T. S., Koo S. H., Chiang J. Y., Lee C. H., and Choi H. S. (2013) Hepatic cannabinoid receptor type 1 mediates alcohol-induced regulation of bile acid enzyme genes expression via CREBH. PLoS ONE 8, e68845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang R., Yin X., Shi H., Wu J., Shakya P., Liu D., and Zhang J. (2014) Adiponectin modulates DCA-induced inflammation via the ROS/NF-κB signaling pathway in esophageal adenocarcinoma cells. Dig. Dis. Sci. 59, 89–97 [DOI] [PubMed] [Google Scholar]
- 54. Asimakopoulou A., Borkham-Kamphorst E., Tacke F., and Weiskirchen R. (2016) Lipocalin-2 (NGAL/LCN2), a “help-me” signal in organ inflammation. Hepatology 63, 669–671 [DOI] [PubMed] [Google Scholar]
- 55. Sjöholm K., Palming J., Olofsson L. E., Gummesson A., Svensson P. A., Lystig T. C., Jennische E., Brandberg J., Torgerson J. S., Carlsson B., and Carlsson L. M. (2005) A microarray search for genes predominantly expressed in human omental adipocytes: adipose tissue as a major production site of serum amyloid A. J. Clin. Endocrinol. Metab. 90, 2233–2239 [DOI] [PubMed] [Google Scholar]
- 56. Zhong Z., Ramshesh V. K., Rehman H., Liu Q., Theruvath T. P., Krishnasamy Y., and Lemasters J. J. (2014) Acute ethanol causes hepatic mitochondrial depolarization in mice: role of ethanol metabolism. PLoS ONE 9, e91308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Logan S. J., Yin L., Geldenhuys W. J., Enrick M. K., Stevanov K. M., Carroll R. T., Ohanyan V. A., Kolz C. L., and Chilian W. M. (2015) Novel thiazolidinedione mitoNEET ligand-1 acutely improves cardiac stem cell survival under oxidative stress. Basic Res. Cardiol. 110, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rooney J. P., Ryde I. T., Sanders L. H., Howlett E. H., Colton M. D., Germ K. E., Mayer G. D., Greenamyre J. T., and Meyer J. N. (2015) PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 1241, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]