

Abstract
The aggregation of α-synuclein (α-syn) into amyloid fibrils is associated with neurodegenerative diseases, collectively referred to as the α-synucleinopathies. In vivo, molecular chaperones, such as the small heat-shock proteins (sHsps), normally act to prevent protein aggregation; however, it remains to be determined how aggregation-prone α-syn evades sHsp chaperone action leading to its disease-associated deposition. This work examines the molecular mechanism by which two canonical sHsps, αB-crystallin (αB-c) and Hsp27, interact with aggregation-prone α-syn to prevent its aggregation in vitro. Both sHsps are very effective inhibitors of α-syn aggregation, but no stable complex between the sHsps and α-syn was detected, indicating that the sHsps inhibit α-syn aggregation via transient interactions. Moreover, the ability of these sHsps to prevent α-syn aggregation was dependent on the kinetics of aggregation; the faster the rate of aggregation (shorter the lag phase), the less effective the sHsps were at inhibiting fibril formation of α-syn. Thus, these findings indicate that the rate at which α-syn aggregates in cells may be a significant factor in how it evades sHsp chaperone action in the α-synucleinopathies.
Keywords: α-synuclein (a-synuclein), amyloid, analytical ultracentrifugation, fibril, molecular chaperone, Parkinson disease, protein aggregation, small heat shock protein (sHsp), synuclein
Introduction
α-Synuclein (α-syn)5 is a protein primarily found in neuronal tissue, where it is predominantly localized to presynaptic terminals (1). The definitive function of α-syn is yet to be established, however, it has been implicated in modulating synaptic activity through membrane processes, including membrane biogenesis, vesicular trafficking, and neurotransmitter release (2, 3). Although it has traditionally been considered an intrinsically disordered protein (4, 5), there have been recent studies indicating that it may form an α-helical tetramer in vivo (6, 7). In addition, α-syn aggregates to form amyloid fibrils under physiological conditions. The deposition of fibrillar α-syn into inclusion bodies, known as Lewy bodies or Lewy neurites when they are present in the cell body or processes, respectively, is a pathological hallmark of a number of neurodegenerative disorders, including Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy, collectively referred to as the α-synucleinopathies (8). Moreover, multiplication of SNCA, which encodes for α-syn, or missense mutations (A53T, A30P, and E46K) in this gene, cause familial early-onset Parkinson's disease (9–11). Under conditions of physiological pH and temperature, purified recombinant α-syn forms fibrillar aggregates in vitro via a nucleation-dependent mechanism (12). The lag phase in this process, which is the rate-limiting step, corresponds to the formation of oligomeric nuclei from aggregation-prone intermediates. This is followed by the rapid growth of nuclei into fibrillar species (elongation phase) until thermodynamic equilibrium is reached (plateau phase) between monomeric and aggregated α-syn forms (13).
Although the main component of Lewy bodies and Lewy neurites is α-syn, auxiliary proteins, including small heat-shock proteins (sHsps), have also been identified in these inclusions (14–16). The expression of sHsps is dramatically up-regulated in response to cellular stress (17), aging (18), and in some protein aggregation disorders, including the α-synucleinopathies (15). The sHsps are an integral component of the cellular network that acts to maintain protein homeostasis (proteostasis) (19, 20). This family of molecular chaperones is defined by the relatively small molecular mass of their monomeric subunits and the presence of a central, conserved α-crystallin domain, which is flanked by variable N- and C-terminal regions (21). The sHsps are often referred to as “holdase” chaperones, although this does not fully describe their chaperone action (22). The sHsps prevent protein aggregation by recognizing, interacting with, and stabilizing partially folded (hydrophobic) intermediate states of target proteins that have left the protein on-folding pathway and entered an off-folding pathway (23–25). As their chaperone action is ATP-independent, sHsps are specialized for preventing inappropriate associations that could otherwise lead to aggregation in the energy-depleted cell (26). A defining feature of some mammalian sHsps (there are 11 in humans, HSPB1–11), such as Hsp27 (HspB1) and αB-crystallin (αB-c; HspB5), is that they form large polydisperse oligomers in solution, which undergo rapid subunit exchange (27, 28). The consensus model of sHsp chaperone action is that small, dissociated species are the chaperone active species as these likely contain a higher relative degree of exposed hydrophobicity than the larger oligomers (27). The larger oligomers are therefore regarded as reservoirs of these chaperone-active dissociated forms. Based on this model, the rate of subunit exchange governs the speed of production of chaperone-active species capable of interacting with aggregation-prone states of target proteins. Phosphorylation at three key residues, Ser15, Ser45 and Ser59 in αB-c (29) or Ser15, Ser78, and Ser82 in Hsp27 (30, 31), has been suggested to be an additional regulatory mechanism with regards to chaperone function of sHsps during times of cellular stress (32–34).
The ability of sHsps such as αB-c and Hsp27 to prevent the fibrillar formation of α-syn in vitro is well established (35–37). However, the mechanism by which sHsps achieve this inhibition remains to be definitively established (38). Moreover, given the α-synucleinopathies (and other diseases) are associated with protein aggregation, it is clear that, under certain circumstances, aggregation-prone proteins can evade the chaperone action of the sHsps. This failure of the sHsps to prevent aggregation is often attributed to them being “overwhelmed” in the context of disease (39–41). However, specific factors that lead to sHsp chaperone activity being overwhelmed are yet to be determined. In this study, we have directly addressed this by characterizing the nature of the interaction between the archetypal sHsps, αB-c and Hsp27, and monomeric α-syn. We show that these chaperones prevent α-syn aggregation through transient interactions and that the chaperone efficacy of these sHsps in preventing α-syn fibril formation is highly dependent upon the rate at which α-syn aggregates.
Results
sHsp Inhibition of α-Syn Aggregation Is Concentration Dependent
The amyloid fibrillar aggregation of wild-type α-syn has been well characterized in vitro (35). As expected, there was no increase in thioflavin-T (ThT) fluorescence when Hsp27, bovine serum albumin (BSA), or buffer were incubated alone. However, incubation of α-syn under these experimental conditions resulted in an increase in ThT fluorescence over time indicative of fibril formation (Fig. 1A). This increase in ThT fluorescence was inhibited by the addition of Hsp27 at a 1:1 (α-syn:Hsp27) molar ratio, but was not inhibited by the presence of the non-chaperone control protein BSA. The change in ThT associated with α-syn aggregation can be fitted by a Boltzmann sigmoidal curve. This reveals that when α-syn was incubated alone there was a lag phase of 15 ± 3 h, and the maximum ThT fluorescence occurred after 36 ± 3 h (Fig. 1B). Addition of the sHsps αB-c and Hsp27 prevented the fibrillar aggregation of α-syn in a concentration-dependent manner (Fig. 1B). At a 1:10 molar ratio (sHsp:α-syn), αB-c and Hsp27 inhibited the change in ThT fluorescence associated with α-syn fibril formation by 65 ± 4 and 74 ± 5%, respectively (Fig. 1C).
FIGURE 1.

sHsp inhibit α-syn aggregation in a concentration-dependent manner. Recombinant α-syn was incubated at 300 μm in 50 mm sodium phosphate buffer with 100 mm NaCl and 0.01% NaN3 (pH 7.4), in the absence or presence of sHsps or the non-chaperone control protein BSA. Fibril formation was monitored by the change in ThT fluorescence at 490 nm over time. A, a representative trace of 2 independent experiments is shown for α-syn in the absence or presence of Hsp27 or BSA at a 1:1 molar ratio (Hsp27/BSA:α-syn). Hsp27, BSA, and buffer alone samples are also included for comparison, and the data for these overlay one another along the x axis due to them showing no change in fluorescence over the course of the assay. B, a representative trace of 4 independent experiments is shown for α-syn in the presence of various ratios of wild-type αB-c. Data were fitted with a Boltzmann sigmoidal curve using GraphPad Prism version 4.02. C, values obtained for the maximum change in ThT fluorescence from these fits was used to determine the percent protection afforded by wild-type αB-c or Hsp27 at a range of molar ratios. Results are presented as mean ± S.E. (n ≥ 4).
In Preventing the Fibrillar Aggregation of α-Syn, the sHsps Do Not Form Stable, High Molecular Mass sHsp-Target Protein Complexes
To investigate whether the sHsps inhibit the fibrillar aggregation of α-syn via the formation of high molecular mass complexes, as has been reported for the interaction with amorphously aggregating proteins (22, 42, 43), the samples containing a 1:1 molar ratio of the sHsp and α-syn (i.e. a concentration corresponding to near complete inhibition of fibril formation of α-syn by the sHsps, see Fig. 1B) were collected after the aggregation assay for subsequent analysis by size exclusion chromatography (Fig. 2A). Prior to incubation, in the absence of sHsp, α-syn eluted from the column in a peak centered at 17 ml. Following incubation of α-syn in the absence of sHsp, there was very little soluble α-syn present in the sample, consistent with its aggregation into fibrils. When present alone in solution, αB-c eluted as a well resolved peak at 13 ml, consistent with it being a large polydisperse oligomer of average mass ∼650 kDa under these solution conditions (44, 45). In the sample containing both αB-c and α-syn, there was no detectable shift in the elution volume or size of the individual peaks corresponding to monomeric α-syn and oligomeric αB-c, or the appearance of any additional peaks. Thus, there was no significant difference in the amount of soluble oligomeric αB-c and monomeric α-syn in this sample compared with when each of these (non-aggregated) proteins were analyzed alone. The presence of only αB-c in the peak eluting at 13 ml and only α-syn in the peak eluting at 17 ml in this sample was confirmed by immunoblotting (Fig. 2C) (the detectable limits of the immunoblotting procedure used in this work was ∼30 nm for each protein). Even when a cross-linker, bis(sulfosuccinimydyl) (with a spacer arm length of 8 atoms or 11.4 Å), was added to the sample following incubation and prior to size exclusion chromatography, there was no evidence of co-elution of αB-c and α-syn from the column (data not shown). Thus, under the conditions used in this work, we found no evidence of a stable high molecular mass complex formed between αB-c and aggregation-prone α-syn. Similar results were obtained for samples containing Hsp27 and α-syn (Fig. 2, B and D).
FIGURE 2.

sHsps prevent α-syn aggregation but, in doing so, do not form a stable high molecular mass sHsp-target protein complex. A and B, size exclusion chromatograms of soluble α-syn (300 μm), αB-c (300 μm), Hsp27 (300 μm), or post-aggregation samples of soluble α-syn (300 μm) in the absence and presence of 1:1 molar ratio of αB-c or Hsp27. C and D, immunoblot analysis of the eluate fractions collected from the size exclusion column after loading with the sample from A or C containing α-syn and αB-c or Hsp27. Aliquots from every fraction (1 ml) collected between 8 and 20 ml were loaded on to a SDS-PAGE gel, transferred to nitrocellulose membrane, and blotted with an anti-αB-c, anti-Hsp27, or anti-α-syn antibody. Results shown are representative of two independent experiments.
We sought to further investigate the interaction between these sHsps and aggregation-prone α-syn by analytical ultracentrifugation (AUC). Due to the propensity of the wild-type sHsps to form large, high molecular mass, polydisperse oligomers in solution (see Fig. 2A), variant forms of these sHsps that either mimic phosphorylation (Hsp273D) or consist of only the core α-crystallin domain (αB-ccore) were selected for use in these experiments. This is because these variants exist predominately as either monomers or dimers (46, 47) and therefore their molecular masses more closely match that of α-syn (an important factor in these absorbance-based AUC experiments as it ensures that both species sediment at similar rates). We first established that both variant forms also inhibit the fibrillar aggregation of α-syn at similar levels to the wild-type proteins (Fig. 3A). Notably, the ability of the core domain (i.e. αB-ccore) to inhibit α-syn aggregation indicates that the ability of the sHsps to prevent α-syn fibril formation is inherent to the α-crystallin domain, and does not require the N- or C-terminal regions.
FIGURE 3.
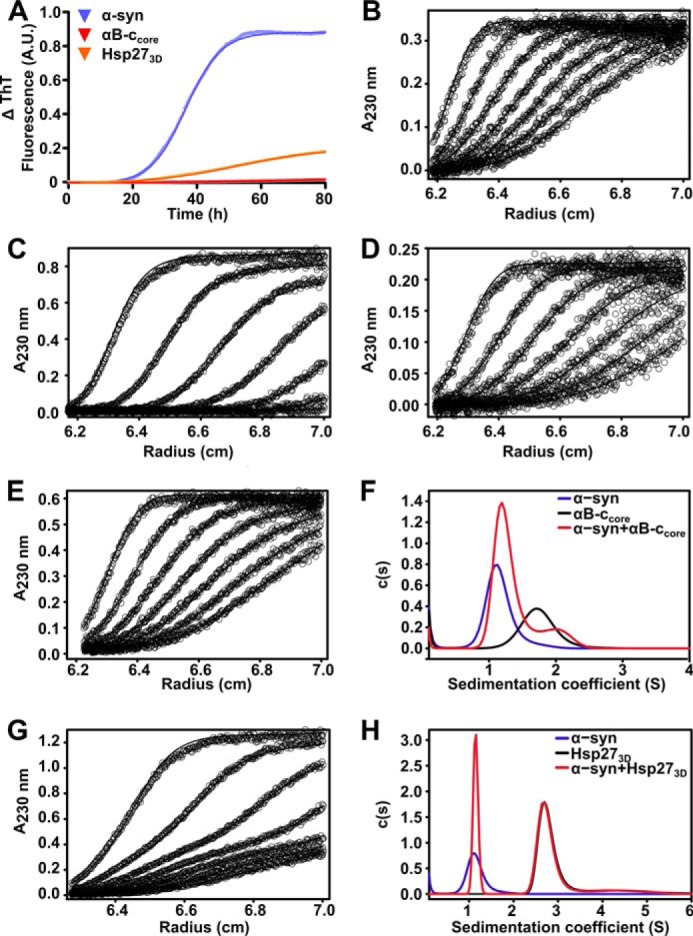
Analysis of the interaction between sHsps and aggregation-prone α-syn by absorbance-based AUC. A, recombinant α-syn was incubated at 300 μm in 50 mm sodium phosphate buffer with 100 mm NaCl and 0.01% NaN3 (pH 7.4), in the absence or presence of sHsps variants (30 μm). Fibril formation was monitored by the change in ThT fluorescence at 490 nm over time, and data were fitted with a Boltzmann sigmoidal curve using GraphPad Prism version 4.02. Sedimentation velocity analysis of 5 μm α-syn (B), αB-ccore (C), or Hsp273D (D) is shown. Data collected at 60-min intervals are presented, overlaid with theoretical fits to the c(s) model generated by SEDFIT. Sedimentation velocity profiles of α-syn co-incubated with αB-ccore (E) or Hsp273D (G) are also shown, along with c(s) distributions for these profiles (F and H, respectively).
Sedimentation velocity (SV) profiles for α-syn alone, and in the presence of either Hsp273D or αB-ccore are shown in Fig. 3. These profiles were fitted using a c(s) model, which considers both sedimentation and diffusion to determine sedimentation coefficients for the species in solution (48). Single peaks for α-syn, αB-ccore, and Hsp273D correspond to sedimentation coefficients of ∼1.1, 1.7, and 2.7 S, respectively. Fitting the SV profile resulting from co-incubation of α-syn with αB-ccore (Fig. 3D) results in a bimodal distribution, with peak positions of ∼1.2 and 1.9 S (Fig. 3E). The small shift of the peak positions may suggest that these proteins interact transiently. Comparison of the predicted weight-average sedimentation coefficient for the two components with the weight-average sedimentation coefficient calculated for the mixture shows a small shift from 1.37 to 1.40 S. Similarly, co-incubation of α-syn with Hsp273D (Fig. 3F) results in a bimodal distribution, with peaks corresponding to sedimentation coefficients of ∼1.1 and 2.7 S (Fig. 3G). In contrast to the mixture of α-syn with αB-ccore, the peak positions for α-syn and Hsp273D do not change significantly. The absence of additional peaks confirms the lack of a detectable stable complex between α-syn and Hsp273D.
The use of fluorescently labeled protein allows specific detection of one component of a complex mixture using fluorescence-detected AUC. As such, CF488A-labeled wild-type Hsp27 was used in fluorescence-based AUC experiments to obtain SV profiles with a range of α-syn concentrations (Fig. 4). As above, these profiles were fitted using the c(s) model and the resulting distribution had three maxima (Fig. 4B). The shoulder centered at 0.9 S results from non-sedimenting fluorescence, and is attributed to residual free CF488A dye in solution from the labeling process. The other two main peaks, centered at ∼2.4 and 10.5 S, reflect the equilibrium characteristic of wild-type Hsp27 in which smaller species (most likely predominately dimers) dissociate from polydisperse oligomers to provide chaperone-active subunits (27). Importantly, with increasing unlabeled α-syn concentration, there is no concentration-dependent increase in the weight-average sedimentation coefficient of Hsp27 that would otherwise be indicative of a stable, high molecular mass complex being formed between Hsp27 and α-syn. Rather, the weight-average sedimentation coefficient decreases as the concentration of α-syn increases (Fig. 4C), indicating that the average size of the Hsp27 species in solution decreases with increasing amounts of α-syn. This trend is supported by a reduction in the signal at 10.5 S and an increase in the signal at 2.4 S with increasing (unlabeled) α-syn concentration. Together, these data suggest that the presence of aggregation-prone α-syn causes the dissociation of Hsp27 polydisperse oligomers in a concentration-dependent manner.
FIGURE 4.

Aggregation-prone α-syn induces the dissociation of oligomeric Hsp27. Sedimentation velocity analysis of 0.5 μm CF488-labeled Hsp27 in the absence or presence of increasing concentrations of α-syn (0–50 μm). A, radial scans are displayed for the highest α-syn concentration overlaid with theoretical fits to the c(s) model generated by SEDFIT. Data are collected at 60-min intervals are presented. B, c(s) distributions for each profile are shown, and C, the weight-average sedimentation coefficient was calculated via integration from 1 to 25 S for each concentration of α-syn. Data were then fitted with a linear regression model.
The Ability of sHsps to Prevent the Aggregation of α-Syn Is Dependent on the Kinetics of the Aggregation Process
We next sought to identify possible mechanism(s) by which aggregation-prone α-syn may “overwhelm” sHsps, leading to the formation of amyloid fibrils associated with disease. To determine whether the rate at which α-syn aggregates affects the ability of the sHsps to prevent fibril formation, we used two methods to alter the kinetics of aggregation of α-syn, which avoid the confounding effect a change in solution conditions (e.g. pH, temperature) may have on the chaperone activity of the sHsps (28, 49–51). First, we exploited the nucleation-dependent mechanism of amyloid fibril formation of α-syn, whereby an increase in the concentration of aggregation-prone monomeric protein increases the rate of aggregation (13). Because the dissociation of small chaperone-active species from larger polydisperse oligomers is proposed to be a key component of the chaperone activity of sHsps (33, 52, 53), we also determined the rate of subunit exchange over the sHsp concentration range to be used in these experiments (i.e. 3–90 μm) using FRET. During subunit exchange, mixing of labeled and unlabeled sHsp subunits resulted in an exponential decrease in the emission fluorescence intensity of the acceptor, which can be used to calculate the rate of subunit exchange (Fig. 5A). Over the concentration range used in this work, the rate of subunit exchange of αB-c reached a maximum at ∼30 μm (Fig. 5B). Importantly, this rate was not significantly affected by the presence of α-syn (Fig. 5B). In contrast to αB-c, the oligomeric state of Hsp27 (46) and its subunit exchange rate (Fig. 5C) are both significantly affected by concentration. Therefore, Hsp27 was unsuitable for use in these experiments due to these confounding effects.
FIGURE 5.

The ability of αB-c to inhibit the fibrillar aggregation of α-syn is dependent on the kinetics of aggregation. A, fluorescently labeled αB-c was incubated for 1 h at 37 °C in PBS (pH 7.4) at concentrations ranging from 3 to 90 μm, consisting of an equimolar mixture of fluorescently labeled protein capable of FRET. Samples were diluted 10-fold into unlabeled αB-c, in the absence or presence of α-syn at a 1:10 (αB-c:α-syn) molar ratio, and the loss of fluorescence in the acceptor fluorescence channel was used to calculate: B, the rate of subunit exchange in the absence and presence of α-syn; C, the rate of subunit exchange was similarly calculated for Hsp27, in the absence and presence of α-syn. D, recombinant A53T α-syn was incubated at concentrations ranging from 150 to 750 μm in 50 mm phosphate buffer containing 100 mm NaCl and 0.01% NaN3 (pH 7.4), in the presence or absence of a 1:10 molar ratio of αB-c. Samples were incubated at 37 °C for 60 h and aggregation was monitored via the change in ThT fluorescence at 490 nm. A representative plot is shown for α-syn in the absence of αB-c with Boltzmann-sigmoidal curves fitted to the data. Values obtained from D were used to calculate the lag phase, elongation rate, and plateau phase for each α-syn concentration. The percent protection afforded by αB-c when present in the sample was calculated and correlated with the (E) duration of the lag phase and (F) rate of elongation. Symbols represent the calculated parameters from each of three independent repeats, with each point corresponding to values calculated from a fit of triplicate samples and shaded according to the concentration of α-syn as indicated in panel D. Data in B, E, and F were fitted with a non-linear regression model and data in C were fitted with a linear regression model. The R2 coefficients of determination are shown.
The disease-associated A53T variant of α-syn was incubated at concentrations from 150 to 750 μm in the absence and presence of αB-c (Fig. 5D). In the absence of the chaperone, increasing the concentration of α-syn led to an increase in the rate and maximum ThT fluorescence associated with α-syn fibril formation, and a decrease in the lag phase (from 9 ± 2 to 3 ± 1 h over this concentration range). When αB-c was present, such that the molar ratio of α-syn:αB-c remained constant (i.e. 1:10 αB-c:α-syn), the ability of αB-c to inhibit α-syn fibril formation was dependent on the kinetics of the aggregation process (Fig. 5, E and F). Thus, when the kinetics of α-syn aggregation were relatively slow (i.e. longer lag phase and slower rate of aggregation), αB-c was a more effective inhibitor of aggregation. When the kinetics of aggregation increased (i.e. shorter lag phase and faster rate of aggregation), αB-c was a less effective chaperone, only decreasing the amount of aggregation of α-syn by ∼20%.
The second method employed to alter the aggregation kinetics of α-syn aggregation was the use of disease-related mutants, which aggregate at different rates (35). Recombinant α-syn mutant proteins were incubated at 300 μm in the absence or presence of either αB-c or Hsp27 (1:10 molar ratio, sHsp:α-syn). As expected, each of the mutant proteins displayed different aggregation kinetics, with α-synA30P having a similar lag phase, elongation rate, and maximal fibril formation to that of α-synWT (Fig. 6A). In contrast, α-synA53T had the shortest lag phase (3 ± 1 h), slowest elongation rate, and lowest maximum increase in ThT fluorescence. The α-synE46K variant was characterized by a slow rate of elongation, high maximal ThT fluorescence, and longest lag phase (42 ± 9 h) of any of the α-syn variants tested. Although both αB-c and Hsp27 were able to inhibit the aggregation of the α-syn proteins (Fig. 6A), their ability to do so was dependent upon the isoform of α-syn. Both chaperones were least effective at inhibiting the aggregation of α-synA53T, which aggregated the fastest (i.e. shortest lag phase), whereas they were most effective at inhibiting α-synE46K, which aggregated the slowest (i.e. longest lag phase) (Fig. 6, B and C).
FIGURE 6.

The ability of the sHsps αB-c and Hsp27 to prevent the aggregation of disease-associated mutant forms of α-syn. Recombinant WT or disease-related mutant forms of α-syn (A30P, A53T, or E46K) were incubated at 300 μm in 50 mm phosphate buffer containing 100 mm NaCl and 0.01% NaN3 (pH 7.4), in the presence or absence of 30 μm αB-c or Hsp27. Samples were incubated at 37 °C for 60 h and aggregation was monitored via the change in ThT fluorescence at 490 nm. A, representative plots of the four independent repeats, each consisting of triplicate samples, are shown for each of the α-syn proteins, including Boltzmann-sigmoidal curves fitted to the data. Values obtained from these fits were used to calculate the lag phase for each protein, which was then correlated with the percent protection when (B) αB-c or (C) Hsp27 was present in the sample. Data shown are the results of four independent repeats, with each point corresponding to values calculated from a fit of triplicate samples. Non-linear regression analysis of the correlation between the lag phase and the percent protection was performed using GraphPad Prism version 4.02.
Discussion
The sHsps play a critical role in maintaining cellular proteostasis by preventing protein aggregation associated with disease. Here, we confirm that the sHsps αB-c and Hsp27 are potent inhibitors of α-syn fibril formation in vitro (37, 54, 55). The phosphomimicking variant of Hsp27 was also an effective chaperone at inhibiting α-syn fibril formation, adding to the work demonstrating that phosphorylation of sHsps acts to increase their chaperone activity (56). Notably, the αB-c core domain inhibited the aggregation of α-syn with similar efficacy to the wild-type (full-length) protein, demonstrating that the sites required to inhibit α-syn aggregation are present in the core domain of these sHsps. The finding that the core domain of αB-c is sufficient to prevent α-syn aggregation is consistent with previous studies showing that this region is capable of preventing other target proteins from forming amorphous or fibrillar aggregates (47, 57).
Although the ability of the sHsps to inhibit the amyloid fibrillar aggregation of a range of target proteins in vitro is well established, the mechanism by which they do so is not clearly defined and may be dependent on the target protein (22, 58–60). We therefore sought to characterize the molecular mechanism by which αB-c and Hsp27 interact with α-syn to prevent its aggregation. Our findings that both Hsp27 and αB-c increase the lag phase of α-syn aggregation and inhibit the elongation phase suggest that they primarily act through stabilizing aggregation-prone monomeric α-syn to prevent it forming fibrils. However, we were unable to detect a high molecular mass complex formed between α-syn and αB-c or Hsp27 under the experimental conditions used in this work. Thus, in the absence of any evidence of a stable interaction, we conclude that these proteins interact transiently and this acts to prevent α-syn aggregation.
Transient interactions between the sHsps and α-syn are difficult to detect. Small shifts in the c(s) distributions and calculated weight-average sedimentation coefficient for the α-syn/αB-ccore mixture may suggest a transiently interacting, rapidly exchanging system. That these shifts were not observed for Hsp273D does not rule out that α-syn and Hsp273D interact transiently. Rather it indicates that this interaction was below the detection limit of the AUC experimental conditions. This may be due to differences in the binding kinetics or the amount of α-syn interacting with Hsp273D compared with αB-ccore. If the fraction of species in this interacting population is too small, or if the interaction occurs too fast, it may not be able to be resolved in the sedimentation experiment (61–63). Transient interactions between αB-c and α-syn (and also of αB-c with other fibril-forming target proteins) are apparent from NMR spectra acquired on mixtures of these proteins due to general broadening of spectra of the target protein with no indication of specific binding site(s) (37, 64–66). We therefore conclude that these sHsps prevent α-syn aggregation via transient interactions in a similar way as has been previously described for apoC-II and reduced and carboxymethylated α-lactalbumin (22). In doing so, the manner by which these sHsps act as chaperones to prevent α-syn aggregation is not through a holdase mechanism, as has been well characterized for sHsp interaction with amorphously aggregating target proteins under stress conditions, particularly elevated temperature (42, 43). Rather, these data support a model in which sHsps can act as protein stabilizers, interacting transiently with relatively ordered protein intermediates that have entered off-folding pathways, a process that facilitates them re-entering the on-folding pathway (38, 58, 67). Considering the cellular implications of this type of interaction, a transient interaction mechanism is favorable as it does not deplete the pool of sHsps available to interact with aggregation-prone proteins in the cells. In contrast, a holdase-type chaperone mechanism, in which stable high molecular mass complexes are formed, likely occurs with more disordered intermediates that expose higher degrees of hydrophobicity on their surface prior to undergoing hydrophobic collapse and aggregation, as occurs under significant stress conditions with the formation of amorphous aggregates (22).
Importantly, it is unclear at this stage whether the interaction between the sHsps and α-syn impacts on the conformation of aggregation-prone monomeric α-syn. However, previous studies have demonstrated that depletion of αB-c from aggregation-inducing conditions containing the amyloidogenic target can allow aggregation to proceed (22). This implies that transient interactions between aggregation-prone species and sHsps may not significantly alter the conformation or aggregation propensity of the target protein.
Our previous work has extensively investigated the interaction of αB-c with a variety of amorphously aggregating target proteins under conditions of elevated temperature, reductive and chemical stress. As a result of these studies, it is apparent that there are similarities between the mechanisms of sHsp chaperone interaction with amorphous and fibrillar aggregating target proteins. As observed in the current study with fibril-forming α-syn, sHsps are more efficient chaperones when interacting with slowly (amorphously) aggregating target proteins (68, 69). They also interact with target proteins early along their aggregation pathway, i.e. monomeric forms that are in a disordered, intermediate state (68, 69). Dynamic, transient interactions between the target protein and the sHsp are also crucial factors in determining chaperone efficacy with target proteins (70).
Given that both αB-c and Hsp27 are highly effective molecular chaperones at inhibiting α-syn fibril formation in vitro, the question remains as to how α-syn intermediates escape the sHsps to form fibrils and plaques in the context of the α-synucleinopathies? A key factor explored in this study was whether the rate of aggregation has a significant effect on the ability of these chaperones to prevent aggregation. Similar to the reported effect of metals (71), pesticides, membrane lipids (72), and molecular crowding (73, 74), increases in the initial monomer concentration of α-syn promotes nucleation during the rate-limiting step, increasing the kinetics of aggregation (73, 75–77). Furthermore, the three most studied disease-related mutations in α-syn exhibit markedly different aggregation kinetics (35), which was also observed in this study. We therefore exploited both increases in α-syn monomer concentration and disease-related mutants of α-syn to alter the kinetics of fibrillar aggregation of α-syn. In both cases, increasing the rate of α-syn aggregation led to a decrease in the chaperone ability of αB-c and Hsp27 to prevent fibril formation. In particular, there was a marked correlation between the efficacy of the chaperone and the lag phase of aggregation, i.e. the longer the time taken to form nuclei, the more efficacious the chaperones were in preventing aggregation. These results also support our previous work in which we increased the rate of α-syn fibril formation by adding the inert crowding agent dextran (37). Under these conditions of αB-c was a much poorer chaperone. However, the presence of crowding agent also reduces the rate of subunit exchange of αA-c (78), which may have contributed to the decrease in chaperone efficacy of αB-c in this case.
The effect of aggregation kinetics on the ability of sHsps to prevent fibril formation is significant given the association of mutations and duplication or triplication of the SCNA gene with early onset Parkinson's disease (9–11, 79, 80). Moreover, the link between increased aggregation rate and a decrease in chaperone efficacy provides a potential mechanism for aggregation-prone α-syn overwhelming the chaperone capability of the sHsps. Although fluorescence-based AUC suggested that the presence of aggregation-prone α-syn causes the dissociation of Hsp27 oligomers, this technique provides a measure of the oligomeric distribution at equilibrium. The real-time availability of chaperone-active subunits, through their dissociation from large polydisperse oligomers, which are able to interact with aggregation-prone proteins, is governed by the subunit exchange rate (81). The rate of subunit exchange for αB-c increased with concentration up to ∼30 μm, after which it remained constant, which supports the notion that subunit exchange is a result of dissociation from sHsp oligomers, as opposed to oligomeric collisions, similar to that demonstrated for αA-crystallin (28). Moreover, factors that increase the rate of α-syn aggregation in cells (such as mutation, gene multiplication, or macromolecular crowding) are likely to overwhelm the protective capacity of the sHsps due to an insufficient supply of chaperone-active subunits capable of interacting with the aggregation-prone α-syn monomer. Such a mechanism is also consistent with the dissociated subunits of sHsps being the chaperone-active species in cells.
This study aimed to characterize the interaction of sHsps with monomeric species. This required the addition of chaperone to monomeric samples before aggregation has commenced, similar to many other studies examining sHsp chaperone action (33, 35, 37, 82). Recently, the interaction of sHsps with preformed amyloid fibrils has also been investigated, and αB-c has been shown to bind along the length of amyloid fibrils with moderate (μm) affinity (36, 83, 84). Addition of monomeric units to fibril ends is essential during the elongation phase of aggregation. In addition, sites of exposed hydrophobicity along the face of the fibril have been identified as possible locations for secondary nucleation (via fragmentation), leading to rapid fibril growth and dominating the overall kinetics of aggregation (85–87). Interaction with these species may contribute to the ability of sHsps to inhibit aggregation, by competing with monomeric units for access to fibril ends or occluding sites of potential secondary nucleation (36). These mechanisms undoubtedly play a role in the sHsp inhibition of α-syn aggregation, and may complement the interaction of the sHsps with monomeric α-syn.
The current study contributes to a greater understanding of the molecular mechanisms by which sHsps interact with disease-related target proteins to prevent their aggregation. We provide further support for the reclassification of the sHsps as protein stabilizers rather than holdase chaperones because the latter does not fully describe the manner by which they can interact with aggregation-prone proteins (22). Thus, although the ability to form complexes with destabilized, particularly amorphously aggregating, proteins is a key element of their chaperone activity (26), this work adds to a growing body of evidence highlighting that this is not the only mechanism by which sHsps inhibit protein aggregation (22, 35, 37, 58, 65). Furthermore, we demonstrate that the rate of aggregation is a significant factor that governs the relative ability of αB-c and Hsp27 to prevent α-syn fibrillar aggregation. This provides a potential rationale for how sHsp chaperone activity is overwhelmed in the context of diseases associated with protein aggregation, i.e. factors that increase the rate at which aggregation occurs also compromise the ability of sHsps to prevent it.
Experimental Procedures
Materials
The pET24d or pET24a bacterial expression vectors, containing the human HSPB1 (Hsp27), HSPB5 (αB-c), or SNCA (α-syn) genes, were used for expression of recombinant wild-type proteins. Disease-related mutants of α-syn and sHsp variants used in this work were produced via site-directed mutagenesis of the wild-type gene (GenScript). In particular, a variant of Hsp27 designed to mimic phosphorylation was generated by mutation of serine residues to aspartic acid at sites that are modified in vivo (i.e. S15AD, S78D, and S82D) to produce Hsp273D. The plasmid for the expression of the core domain of αB-c, i.e. residues 68–153 (αB-ccore), was a kind gift from Professor A. Laganowsky (Texas A&M Health Science Center). All recombinant proteins were expressed in Escherichia coli BL21(DE3) cells transformed with each plasmid, and purified as described previously (12, 88, 89). Dithiothreitol (DTT) was purchased from Amresco (Solon, OH), and protein molecular mass standards used in gel electrophoresis were Protein Precision Plus Dual Color obtained from Bio-Rad Laboratories. Protein mass calibrants for size exclusion chromatography, containing bovine thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.35 kDa), was also purchased from Bio-Rad. All other chemicals, including ThT, were obtained from Sigma, unless otherwise stated. Protein concentrations were determined using a NanoDrop 2000c spectrophotometer (ThermoFisher Scientific, Waltham, MA), based upon A280 nm0.1% values of 0.44 m−1 cm−1 for α-syn calculated using the ExPASy ProtParam tool (90), 0.83 m−1 cm−1 for αB-c, and 1.65 m−1 cm−1 for Hsp27 (91). Mouse monoclonal anti-αB-c antibody (clone 1B6.1–3G4) and mouse monoclonal anti-Hsp27 antibody (clone G3.1) were purchased from Abcam (Cambridge, UK). Both the mouse monoclonal anti-α-syn antibody (clone Syn211) and the peroxidase-conjugated anti-mouse IgG secondary antibody were obtained from Sigma.
Aggregation Assays
Fibrillar aggregation of α-syn in the absence or presence of sHsps was monitored using a ThT fluorescence assay via a previously described method (33) with adaptations. Briefly, α-syn was incubated at 300 μm in 50 mm phosphate buffer containing 100 mm NaCl (pH 7.4) and 0.01% sodium azide unless otherwise indicated. Assays were conducted in triplicate using clear 384-microwell plates (Greiner Bio-One, Frickenhausen, Germany) with each well containing 30 μl of sample. Plates were incubated in a POLARstar OPTIMA plate reader (BMG Labtechnologies, Melbourne, Australia) at 37 °C, with the plate sealed to prevent evaporation. The ThT fluorescence was measured using excitation and emission filters of 440 and 490 nm, respectively. Readings were taken every 625 s for a period of up to 60 h. Plates were subjected to linear shaking at 600 rpm for 540 s after each reading.
At the end of each assay, data for the change in ThT over time were fitted with Boltzmann sigmoidal curves using GraphPad Prism version 4.02 (GraphPad Software Inc., San Diego, CA). Data were only used when the R2 coefficient of determination was >0.8. Parameters from these fits were used to derive the length of the lag phase and rate of α-syn aggregation using equations previously derived (92), with modification (Fig. 7). The equation of the Boltzmann curve is given as the following.
 |
The slope of the line at any point is,
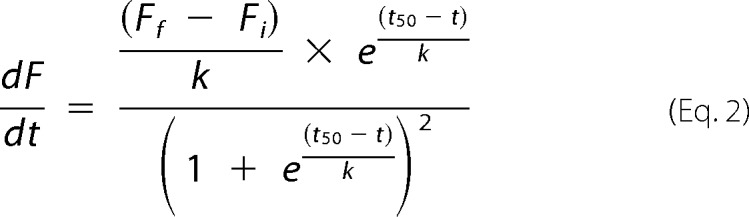 |
when t = t50 (i.e. the inflection point, when the elongation rate is maximal).
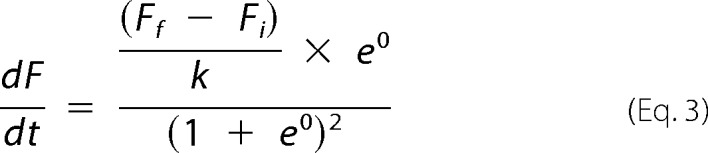 |
Therefore the rate of α-syn aggregation at t50 was calculated according to the following equation,
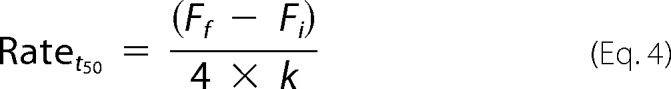 |
where Fi corresponds to the initial fluorescence value, Ff corresponds to the final fluorescence value, t50 refers to the time taken to reach half-maximal fluorescence, and k describes the gradient of the curve.
FIGURE 7.
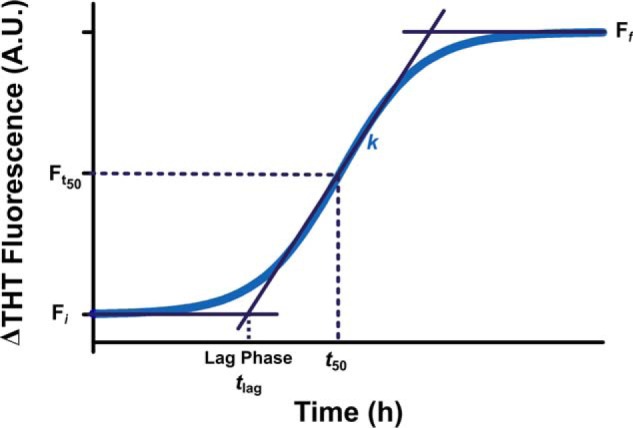
Schematic illustration of the Boltzmann sigmoidal curve used to describe the increase in ThT fluorescence upon α-syn fibril formation. Fi corresponds to the initial fluorescence value, Ff corresponds to the final fluorescence value, t50 refers to the time taken to reach half-maximal fluorescence (Ft50), tlag is the lag time, and k denotes the steepness of the curve.
The lag phase (tlag) is when
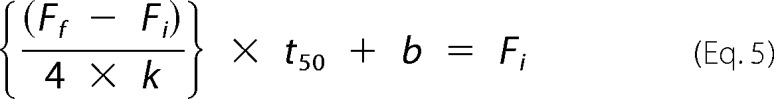 |
To solve for b, at t50 (i.e. the mid-point between Ff and Fi) is shown as,
 |
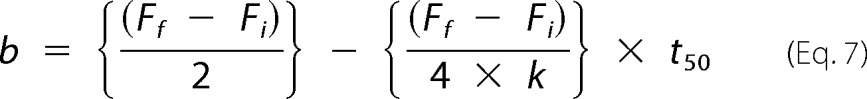 |
So substituting b into Equation 5,
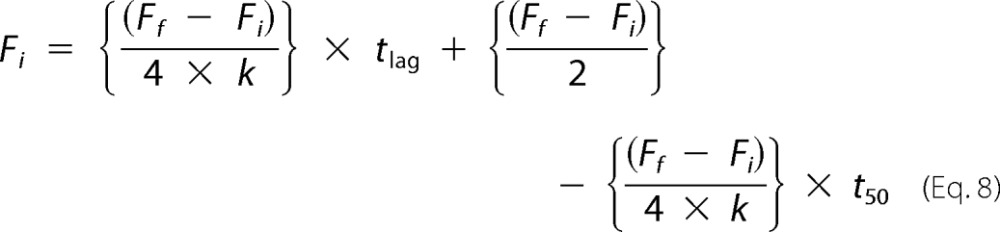 |
Solving for tlag,
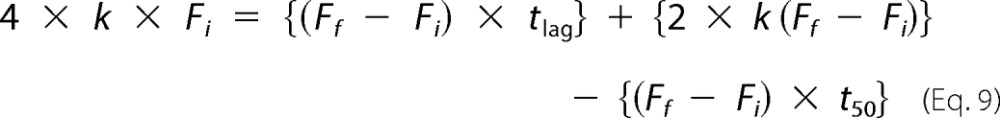 |
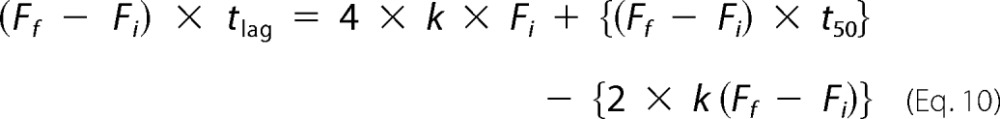 |
Therefore, tlag is determined using the following equation.
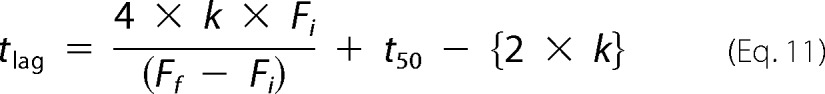 |
The relative efficacy of the sHsps to inhibit α-syn fibril formation was determined by calculating the protection provided by each sHsp at the conclusion of the assay, according to the difference in maximum ThT fluorescence in the absence and presence of the chaperone using the equation,
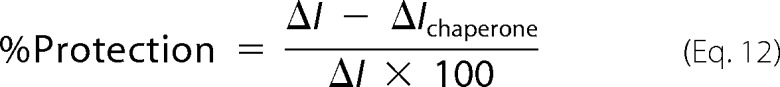 |
where ΔI and ΔIchaperone correspond to the change in ThT fluorescence of α-syn in the absence and presence of the sHsp, respectively. In each case, samples were assayed in triplicate and the percent protection is reported as a mean ± S.E. of at least 3 independent (biological) replicates.
Analytical Size Exclusion Chromatography
The nature of the interaction between the sHsps and α-syn was analyzed via size exclusion chromatography of the samples at the end of the aggregation assays. Samples containing 300 μm α-syn in the absence or presence of 300 μm αB-c or Hsp27 were collected immediately following incubation and centrifuged at 14,000 × g for 10 min at 4 °C to remove any insoluble protein. Supernatants were collected and loaded onto a Superose-6 size exclusion column (GE Healthcare, Uppsala, Sweden), pre-equilibrated with 50 mm phosphate buffer containing 100 mm NaCl (pH 7.4) and 0.01% sodium azide, at a flow rate of 0.5 ml/min. Protein elution was monitored using an in-line UV detector, and concentrations were determined using the peak integration function of PrimeView version 5.0 (GE Healthcare), to monitor any loss of protein following centrifugation. Eluate fractions (1 ml) were collected and analyzed via SDS-PAGE and immunoblotting. Representative results are presented from two independent experiments from two separate aggregation assays.
Immunoblotting
Following SDS-PAGE, in which proteins were resolved on 15% (v/v) gels, proteins were transferred to nitrocellulose membrane at 100 V for 1 h. Membranes were blocked with 5% (w/v) skim milk powder in Tris-buffered saline (TBS; 10 mm Tris-HCl, 150 mm NaCl, pH 7.5) for 1 h at room temperature. Blots were then incubated with primary antibodies directed against αB-c, Hsp27 or α-syn, diluted 1:5000 in 5% (w/v) skim milk powder in TBS containing 0.05% Tween 20 (TBS-T) for 1 h at room temperature. Blots were washed four times (10 min) in TBS-T, and incubated with the peroxidase-conjugated anti-mouse secondary antibody (1:5000 dilution in 5% (w/v) skim milk powder in TBS-T) for 1 h at room temperature. Blots were washed as above and labeled proteins were detected with SuperSignal West Pico Chemiluminescent Substrate according to the manufacturer's instructions (ThermoFisher Scientific).
Fluorescent Labeling of sHsps
Recombinant proteins of interest were fluorescently labeled with either succinimidyl ester or maleimide variants of CF488A or CF647 dyes (Biotium, Hayward, CA) according to the manufacturer's instructions. Briefly, those samples intended for maleimide labeling were preincubated with a 10-fold molar excess of Tris(2-carboxyethyl)phosphine for 30 min at room temperature. Recombinant protein was then loaded onto a PD10 column (GE Healthcare) pre-equilibrated with degassed phosphate-buffered saline (PBS; 2.7 mm KCl, 1.75 mm K2HPO4, 135 mm NaCl, 10 mm NaH2PO4, pH 7.4) and 500-μl fractions were collected. Protein elution was monitored by determining the absorbance of each fraction at 280 nm. Fractions containing protein were pooled and dye conjugates were added in a 1.5 m (succinimidyl ester) or 10 m (maleimide) excess relative to the protein concentration for labeling. Labeling reactions were incubated overnight at 4 °C with agitation, and then labeled protein was separated from unreacted dye using a pre-equilibrated PD10 column as described above. The concentration and degree of labeling of each protein were calculated as per the manufacturer's instructions and the latter was found to be greater than 60% in all cases. Labeled proteins were stored at −20 °C. Protein labeling with the fluorophore was also confirmed via electrospray ionization mass spectrometry.
Analytical Ultracentrifugation
UV absorbance-detected AUC was performed as previously described (83). Briefly, the maximum concentration was calculated for each protein such that the absorbance at 230 or 280 nm was within the optimal detection range of the instrument. Samples (380 μl) were prepared at room temperature and incubated for 1 h before being loaded into a 12-mm double-sector epon-filled centerpiece, alongside the relevant reference solution (400 μl). Samples were centrifuged in an XL-I analytical ultracentrifuge (Beckman Coulter, CA) at 50,000 rpm using either a Ti60 or Ti50 rotor at 20 °C. Radial absorbance scans were collected at 230 or 280 nm at 6-min intervals with a radial step size of 0.003 cm for a total of 10 h. SV profiles were analyzed with SEDFIT software using a continuous c(s) distribution model and regularization by maximum entropy. Using a regularization parameter of p = 0.95 and 200 sedimentation coefficient increments, the data were fitted to produce c(s) distributions. Weight-average sedimentation coefficients of each sample were then calculated by integration of the sedimentation distributions over the given range. The buffer density (ρ), buffer viscosity (η), and partial specific volume (v̄) used for analysis were estimated using SEDNTERP software.
Fluorescence-detected AUC was performed as previously described (83), using Hsp27-WT labeled with CF488A and unlabeled monomeric α-syn. Briefly, samples (350 μl) were prepared at room temperature and incubated for 1 h before being loaded into a 12-mm double-sector epon-filled centerpiece, and covered with 50 μl of FC-43 perfluorotributylamine (Scientific Instrument Services Inc., Ringoes, NJ). Samples were centrifuged in an XL-A analytical ultracentrifuge fitted with a fluorescence detection system (Aviv Biomedical, Lakewood, NJ) at 50,000 rpm using either a Ti60 or Ti50 rotor at 20 °C. Excitation was at 488 nm and fluorescence above 505 nm was measured. Radial fluorescence scans were collected at 3-min intervals with a radial step size of 0.002 cm for a total of 5 h. Sedimentation velocity profiles were analyzed as described above, fitting time-independent noise and according to a fixed sample meniscus position.
Bulk Förster Resonance Energy Transfer (FRET) Analysis of sHsp Subunit Exchange Rate
Subunit exchange rates of the sHsps were determined by monitoring changes in FRET between fluorescently labeled sHsp oligomers. Aliquots of fluorescently labeled sHsp, composed of an equimolar mixture of CF488A- and CF647-labeled protein, were prepared in PBS (pH 7.4) at concentrations ranging from 3 to 90 μm. Samples were incubated at 37 °C with shaking at 60 rpm using a VorTemp incubator (Labnet, Edison, NJ) for 1 h, to allow complete mixing of the two labeled populations (confirmed by monitoring the quenching of donor fluorescence at 495 nm). The labeled sHsps were then diluted 10-fold into a sample of unlabeled sHsp at an equivalent concentration so as to maintain the overall concentration of the sHsp. The loss of FRET due to subunit exchange of labeled sHsps with unlabeled sHsps was determined by the decrease in acceptor fluorescence at 670 nm and monitored using a POLARstar Omega platereader (BMG Labtechnologies, Melbourne, Australia). Experiments were performed in the absence or presence of a 10-fold molar excess of α-synWT, in black polystyrene clear-bottom 384-well plates (Greiner Bio-One, Frickenhausen, Germany). Readings were taken every 120 s over a period of 4 h, with linear shaking at 600 rpm for 90 s after each cycle. The rate constant was determined by fitting the data to a one-phase exponential decay curve using GraphPad Prism version 4.02 (GraphPad Software Inc.). The rate of subunit exchange was derived from the equation,
where k is defined as the rate constant of the reaction, and Y0 and Yt correspond to the fluorescence intensity at time = 0 and t respectively.
Author Contributions
H. E. conceived and coordinated the experiments. H. E., D. C., J. C., and M. D. designed the experiments. D. C., E. S., and M. D. performed the experiments. All authors analyzed and reviewed the data. D. C. and H. E. wrote the paper. All authors read and approved the final manuscript.
Acknowledgment
We thank the Illawarra Health and Medical Research Institute for technical support.
This work was supported in part by grants from the Australian Department of Health and Aging and the University of Wollongong. The authors declare that they have no conflicts of interest with the contents of this article.
- α-syn
- α-synuclein
- αB-c
- αB-crystallin
- AUC
- analytical ultracentrifugation
- sHsp
- small heat-shock protein
- SV
- sedimentation velocity
- ThT
- thioflavin-T.
References
- 1. Maroteaux L., Campanelli J. T., and Scheller R. H. (1988) Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davidson W. S., Jonas A., Clayton D. F., and George J. M. (1998) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449 [DOI] [PubMed] [Google Scholar]
- 3. Jenco J. M., Rawlingson A., Daniels B., and Morris A. J. (1998) Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by α- and β-synucleins. Biochemistry 37, 4901–4909 [DOI] [PubMed] [Google Scholar]
- 4. Weinreb P. H., Zhen W., Poon A. W., Conway K. A., and Lansbury P. T. (1996) NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 35, 13709–13715 [DOI] [PubMed] [Google Scholar]
- 5. Uversky V. N., Gillespie J. R., and Fink A. L. (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins Struct. Funct. Bioinformat. 41, 415–427 [DOI] [PubMed] [Google Scholar]
- 6. Dettmer U., Newman A. J., Luth E. S., Bartels T., and Selkoe D. (2013) In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 288, 6371–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartels T., Choi J. G., and Selkoe D. J. (2011) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martí M. J., Tolosa E., and Campdelacreu J. (2003) Clinical overview of the synucleinopathies. Mov. Disord. 18, S21–S27 [DOI] [PubMed] [Google Scholar]
- 9. Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., et al. (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047 [DOI] [PubMed] [Google Scholar]
- 10. Krüger R., Kuhn W., Müller T., Woitalla D., Graeber M., Kösel S., Przuntek H., Epplen J. T., Schöls L., and Riess O. (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat. Genet. 18, 106–108 [DOI] [PubMed] [Google Scholar]
- 11. Zarranz J. J., Alegre J., Gómez-Esteban J. C., Lezcano E., Ros R., Ampuero I., Vidal L., Hoenicka J., Rodriguez O., Atarés B., Llorens V., Gomez Tortosa E., del Ser T., Muñoz D. G., and de Yebenes J. G. (2004) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 [DOI] [PubMed] [Google Scholar]
- 12. Narhi L., Wood S. J., Steavenson S., Jiang Y., Wu G. M., Anafi D., Kaufman S. A., Martin F., Sitney K., Denis P., Louis J. C., Wypych J., Biere A. L., and Citron M. (1999) Both familial Parkinson's disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 274, 9843–9846 [DOI] [PubMed] [Google Scholar]
- 13. Wood S. J., Wypych J., Steavenson S., Louis J. C., Citron M., and Biere A. L. (1999) α-Synuclein fibrillogenesis is nucleation-dependent. J. Biol. Chem. 274, 19509–19512 [DOI] [PubMed] [Google Scholar]
- 14. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., and Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 15. Outeiro T. F., Klucken J., Strathearn K. E., Liu F., Nguyen P., Rochet J.-C., Hyman B. T., and McLean P. J. (2006) Small heat shock proteins protect against α-synuclein-induced toxicity and aggregation. Biochem. Biophys. Res. Commun. 351, 631–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McLean P. J., Kawamata H., Shariff S., Hewett J., Sharma N., Ueda K., Breakefield X. O., and Hyman B. T. (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of α-synuclein aggregation. J. Neurochem. 83, 846–854 [DOI] [PubMed] [Google Scholar]
- 17. Bartelt-Kirbach B., and Golenhofen N. (2014) Reaction of small heat-shock proteins to different kinds of cellular stress in cultured rat hippocampal neurons. Cell Stress Chaperones 19, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walther D. M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R. I., Dobson C. M., Vendruscolo M., Mann M., and Hartl F. U. (2015) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 20. Treweek T. M., Meehan S., Ecroyd H., and Carver J. A. (2015) Small heat-shock proteins: important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 72, 429–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haslbeck M., Franzmann T., Weinfurtner D., and Buchner J. (2005) Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 12, 842–846 [DOI] [PubMed] [Google Scholar]
- 22. Kulig M., and Ecroyd H. (2012) The small heat-shock protein αB-crystallin uses different mechanisms of chaperone action to prevent the amorphous versus fibrillar aggregation of α-lactalbumin. Biochem. J. 448, 343–352 [DOI] [PubMed] [Google Scholar]
- 23. Carver J. A., Rekas A., Thorn D. C., and Wilson M. R. (2003) Small heat-shock proteins and clusterin: intra- and extracellular molecular chaperones with a common mechanism of action and function? IUBMB Life 55, 661–668 [DOI] [PubMed] [Google Scholar]
- 24. Ehrnsperger M., Gräber S., Gaestel M., and Buchner J. (1997) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 16, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feder M. E., and Hofmann G. E. (1999) Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu. Rev. Physiol. 61, 243–282 [DOI] [PubMed] [Google Scholar]
- 26. Arrigo A.-P., Simon S., Gibert B., Kretz-Remy C., Nivon M., Czekalla A., Guillet D., Moulin M., Diaz-Latoud C., and Vicart P. (2007) Hsp27 (HspB1) and αB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 581, 3665–3674 [DOI] [PubMed] [Google Scholar]
- 27. Van Montfort R., Slingsby C., and Vierling E. (2001) Structure and function of the small heat shock protein/α-crystallin family of molecular chaperones. Adv. Protein Chem. 59, 105–156 [DOI] [PubMed] [Google Scholar]
- 28. Bova M. P., McHaourab H. S., Han Y., and Fung B. K. (2000) Subunit exchange of small heat shock proteins: analysis of oligomer formation of αA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J. Biol. Chem. 275, 1035–1042 [DOI] [PubMed] [Google Scholar]
- 29. Miesbauer L. R., Zhou X., Yang Z., Yang Z., Sun Y., Smith D. L., and Smith J. B. (1994) Post-translational modifications of water-soluble human lens crystallins from young adults. J. Biol. Chem. 269, 12494–12502 [PubMed] [Google Scholar]
- 30. Landry J., Lambert H., Zhou M., Lavoie J. N., Hickey E., Weber L. A., and Anderson C. W. (1992) Human HSP27 is phosphorylated at serines 78 and 82 by heat shock and mitogen-activated kinases that recognize the same amino acid motif as S6 kinase II. J. Biol. Chem. 267, 794–803 [PubMed] [Google Scholar]
- 31. Gaestel M., Schröder W., Benndorf R., Lippmann C., Buchner K., Hucho F., Erdmann V. A., and Bielka H. (1991) Identification of the phosphorylation sites of the murine small heat shock protein hsp25. J. Biol. Chem. 266, 14721–14724 [PubMed] [Google Scholar]
- 32. Kato K., Hasegawa K., Goto S., and Inaguma Y. (1994) Dissociation as a result of phosphorylation of an aggregated form of the small stress protein, Hsp27. J. Biol. Chem. 269, 11274–11278 [PubMed] [Google Scholar]
- 33. Ecroyd H., Meehan S., Horwitz J., Aquilina J. A., Benesch J. L., Robinson C. V., MacPhee C. E., and Carver J. A. (2007) Mimicking phosphorylation of αB-crystallin affects its chaperone activity. Biochem. J. 401, 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ahmad M. F., Raman B., Ramakrishna T., and Rao Ch. M. (2008) Effect of phosphorylation on αB-crystallin: differences in stability, subunit exchange and chaperone activity of homo and mixed oligomers of αB-crystallin and its phosphorylation-mimicking mutant. J. Mol. Biol. 375, 1040–1051 [DOI] [PubMed] [Google Scholar]
- 35. Bruinsma I. B., Bruggink K. A., Kinast K., Versleijen A. A., Segers-Nolten I. M., Subramaniam V., Kuiperij H. B., Boelens W., de Waal R. M., and Verbeek M. M. (2011) Inhibition of α-synuclein aggregation by small heat shock proteins. Proteins 79, 2956–2967 [DOI] [PubMed] [Google Scholar]
- 36. Waudby C. A., Knowles T. P., Devlin G. L., Skepper J. N., Ecroyd H., Carver J. A., Welland M. E., Christodoulou J., Dobson C. M., and Meehan S. (2010) The interaction of αB-crystallin with mature α-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 98, 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rekas A., Adda C. G., Andrew Aquilina J., Barnham K. J., Sunde M., Galatis D., Williamson N. A., Masters C. L., Anders R. F., Robinson C. V., Cappai R., and Carver J. A. (2004) Interaction of the molecular chaperone αB-crystallin with α-synuclein: effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 340, 1167–1183 [DOI] [PubMed] [Google Scholar]
- 38. Cox D., Carver J. A., and Ecroyd H. (2014) Preventing α-synuclein aggregation: the role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta 1842, 1830–1843 [DOI] [PubMed] [Google Scholar]
- 39. Leak R. K. (2014) Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 8, 293–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Healy E. F., Little C., and King P. J. (2014) A model for small heat shock protein inhibition of polyglutamine aggregation. Cell Biochem. Biophys. 69, 275–281 [DOI] [PubMed] [Google Scholar]
- 41. Bakthisaran R., Tangirala R., and Rao Ch. M. (2015) Small heat shock proteins: role in cellular functions and pathology. Biochim. Biophys. Acta 1854, 291–319 [DOI] [PubMed] [Google Scholar]
- 42. Haslbeck M., Walke S., Stromer T., Ehrnsperger M., White H. E., Chen S., Saibil H. R., and Buchner J. (1999) Hsp26: a temperature-regulated chaperone. EMBO J. 18, 6744–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee G. J., Roseman A. M., Saibil H. R., and Vierling E. (1997) A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 16, 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horwitz J. (2005) α-Crystallin: its involvement in suppression of protein aggregation and protein folding. in Protein Folding Handbook II, pp. 858–875, Wiley-VCH Verlag GmbH, Weinheim, Germany [Google Scholar]
- 45. Haley D. A., Horwitz J., and Stewart P. L. (1998) The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 277, 27–35 [DOI] [PubMed] [Google Scholar]
- 46. Jovcevski B., Kelly M. A., Rote A. P., Berg T., Gastall H. Y., Benesch J. L., Aquilina J. A., and Ecroyd H. (2015) Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem. Biol. 22, 186–195 [DOI] [PubMed] [Google Scholar]
- 47. Hochberg G. K., Ecroyd H., Liu C., Cox D., Cascio D., Sawaya M. R., Collier M. P., Stroud J., Carver J. A., Baldwin A. J., Robinson C. V., Eisenberg D. S., Benesch J. L., and Laganowsky A. (2014) The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc. Natl. Acad. Sci. U.S.A. 111, E1562–E1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun Y., and MacRae T. (2005) Small heat shock proteins: molecular structure and chaperone function. Cell. Mol. Life Sci. 62, 2460–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lelj-Garolla B., and Mauk A. G. (2006) Self-association and chaperone activity of Hsp27 are thermally activated. J. Biol. Chem. 281, 8169–8174 [DOI] [PubMed] [Google Scholar]
- 51. Fu X., and Chang Z. (2004) Temperature-dependent subunit exchange and chaperone-like activities of Hsp16.3, a small heat shock protein from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 316, 291–299 [DOI] [PubMed] [Google Scholar]
- 52. Aquilina J. A., Benesch J. L., Ding L. L., Yaron O., Horwitz J., and Robinson C. V. (2004) Phosphorylation of αB-crystallin alters chaperone function through loss of dimeric substructure. J. Biol. Chem. 279, 28675–28680 [DOI] [PubMed] [Google Scholar]
- 53. Benesch J. L., Ayoub M., Robinson C. V., and Aquilina J. A. (2008) Small heat shock protein activity is regulated by variable oligomeric substructure. J. Biol. Chem. 283, 28513–28517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J., Martin E., Gonzales V., Borchelt D. R., and Lee M. K. (2008) Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol. Aging 29, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aquilina J. A., Shrestha S., Morris A. M., and Ecroyd H. (2013) Structural and functional aspects of hetero-oligomers formed by the small heat shock proteins αB-crystallin and HSP27. J. Biol. Chem. 288, 13602–13609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bakthisaran R., Akula K. K., Tangirala R., and Rao Ch. M. (2016) Phosphorylation of αB-crystallin: role in stress, aging and patho-physiological conditions. Biochim. Biophys. Acta 1860, 167–182 [DOI] [PubMed] [Google Scholar]
- 57. Mainz A., Peschek J., Stavropoulou M., Back K. C., Bardiaux B., Asami S., Prade E., Peters C., Weinkauf S., Buchner J., and Reif B. (2015) The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905 [DOI] [PubMed] [Google Scholar]
- 58. Hatters D. M., Lindner R. A., Carver J. A., and Howlett G. J. (2001) The molecular chaperone, α-crystallin, inhibits amyloid formation by apolipoprotein C-II. J. Biol. Chem. 276, 33755–33761 [DOI] [PubMed] [Google Scholar]
- 59. Raman B., Ban T., Sakai M., Pasta S. Y., Ramakrishna T., Naiki H., Goto Y., and Rao Ch. M. (2005) αB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid β-peptide and β2-microglobulin. Biochem. J. 392, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilhelmus M. M., Boelens W. C., Otte-Höller I., Kamps B., de Waal R. M., and Verbeek M. M. (2006) Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 1089, 67–78 [DOI] [PubMed] [Google Scholar]
- 61. Lebowitz J., Lewis M. S., and Schuck P. (2002) Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Balbo A., and Schuck P. (2005) Analytical ultracentrifugation in the study of protein self-association and heterogeneous protein-protein interactions. Protein-Protein Interactions. A Molecular Cloning Manual, Edition 2, Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 63. Howlett G. J., Minton A. P., and Rivas G. (2006) Analytical ultracentrifugation for the study of protein association and assembly. Curr. Opin. Chem. Biol. 10, 430–436 [DOI] [PubMed] [Google Scholar]
- 64. Rekas A., Jankova L., Thorn D. C., Cappai R., and Carver J. A. (2007) Monitoring the prevention of amyloid fibril formation by α-crystallin. FEBS J. 274, 6290–6304 [DOI] [PubMed] [Google Scholar]
- 65. Robertson A. L., Headey S. J., Saunders H. M., Ecroyd H., Scanlon M. J., Carver J. A., and Bottomley S. P. (2010) Small heat-shock proteins interact with a flanking domain to suppress polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 107, 10424–10429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Esposito G., Garvey M., Alverdi V., Pettirossi F., Corazza A., Fogolari F., Polano M., Mangione P. P., Giorgetti S., Stoppini M., Rekas A., Bellotti V., Heck A. J., and Carver J. A. (2013) Monitoring the interaction between β2-microglobulin and the molecular chaperone αB-crystallin by NMR and mass spectrometry: αB-crystallin dissociates β2-microglobulin oligomers. J. Biol. Chem. 288, 17844–17858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jakob U., Gaestel M., Engel K., and Buchner J. (1993) Small heat shock proteins are molecular chaperones. J. Biol. Chem. 268, 1517–1520 [PubMed] [Google Scholar]
- 68. Lindner R. A., Treweek T. M., and Carver J. A. (2001) The molecular chaperone α-crystallin is in kinetic competition with aggregation to stabilize a monomeric molten-globule form of α-lactalbumin. Biochem. J. 354, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carver J. A., Lindner R. A., Lyon C., Canet D., Hernandez H., Dobson C. M., and Redfield C. (2002) The interaction of the molecular chaperone α-crystallin with unfolding α-lactalbumin: a structural and kinetic spectroscopic study. J. Mol. Biol. 318, 815–827 [DOI] [PubMed] [Google Scholar]
- 70. Devlin G. L., Carver J. A., and Bottomley S. P. (2003) The selective inhibition of serpin aggregation by the molecular chaperone, α-crystallin, indicates a nucleation-dependent specificity. J. Biol. Chem. 278, 48644–48650 [DOI] [PubMed] [Google Scholar]
- 71. Uversky V. N., Li J., Bower K., and Fink A. L. (2002) Synergistic effects of pesticides and metals on the fibrillation of α-synuclein: Implications for Parkinson's disease. Neurotoxicology 23, 527–536 [DOI] [PubMed] [Google Scholar]
- 72. Lee H. J., Choi C., and Lee S. J. (2002) Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678 [DOI] [PubMed] [Google Scholar]
- 73. Munishkina L. A., Cooper E. M., Uversky V. N., and Fink A. L. (2004) The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464 [DOI] [PubMed] [Google Scholar]
- 74. Uversky V. N., M Cooper E., Bower K. S., Li J., and Fink A. L. (2002) Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 515, 99–103 [DOI] [PubMed] [Google Scholar]
- 75. Shtilerman M. D., Ding T. T., and Lansbury P. T. (2002) Molecular crowding accelerates fibrillization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry 41, 3855–3860 [DOI] [PubMed] [Google Scholar]
- 76. Uversky V. N. (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Uversky V. N. (2007) Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 103, 17–37 [DOI] [PubMed] [Google Scholar]
- 78. Ghahghaei A., Rekas A., Price W. E., and Carver J. A. (2007) The effect of dextran on subunit exchange of the molecular chaperone αA-crystallin. Biochim. Biophys. Acta 1774, 102–111 [DOI] [PubMed] [Google Scholar]
- 79. Campbell B. C., McLean C. A., Culvenor J. G., Gai W. P., Blumbergs P. C., Jäkälä P., Beyreuther K., Masters C. L., and Li Q.-X. (2001) The solubility of α-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J. Neurochem. 76, 87–96 [DOI] [PubMed] [Google Scholar]
- 80. Baba M., Nakajo S., Tu P. H., Tomita T., Nakaya K., Lee V. M., Trojanowski J. Q., and Iwatsubo T. (1998) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 152, 879–884 [PMC free article] [PubMed] [Google Scholar]
- 81. Vos M. J., Hageman J., Carra S., and Kampinga H. H. (2008) Structural and functional diversities between members of the human HSPB, HSPH, HSPA, and DNAJ chaperone families. Biochemistry 47, 7001–7011 [DOI] [PubMed] [Google Scholar]
- 82. Horwitz J. (1992) α-Crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. U.S.A. 89, 10449–10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Binger K. J., Ecroyd H., Yang S., Carver J. A., Howlett G. J., and Griffin M. D. (2013) Avoiding the oligomeric state: αB-crystallin inhibits fragmentation and induces dissociation of apolipoprotein C-II amyloid fibrils. FASEB J. 27, 1214–1222 [DOI] [PubMed] [Google Scholar]
- 84. Shammas S. L., Waudby C. A., Wang S., Buell A. K., Knowles T. P., Ecroyd H., Welland M. E., Carver J. A., Dobson C. M., and Meehan S. (2011) Binding of the molecular chaperone αB-crystallin to Aβ amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ruschak A. M., and Miranker A. D. (2007) Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 12341–12346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Knowles T. P., Waudby C. A., Devlin G. L., Cohen S. I., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., and Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 [DOI] [PubMed] [Google Scholar]
- 87. Cohen S. I., Vendruscolo M., Welland M. E., Dobson C. M., Terentjev E. M., and Knowles T. P. (2011) Nucleated polymerization with secondary pathways: I. time evolution of the principal moments. J. Chem. Phys. 135, 065105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Horwitz J., Huang Q. L., Ding L., and Bova M. P. (1998) Lens alpha-crystallin: chaperone-like properties. Methods Enzymol. 290, 365–383 [DOI] [PubMed] [Google Scholar]
- 89. Laganowsky A., Benesch J. L., Landau M., Ding L., Sawaya M. R., Cascio D., Huang Q., Robinson C. V., Horwitz J., and Eisenberg D. (2010) Crystal structures of truncated αA and αB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 19, 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Walker J. M. (2005) The proteomics protocols handbook, Springer, New York [Google Scholar]
- 91. Hayes D., Napoli V., Mazurkie A., Stafford W. F., and Graceffa P. (2009) Phosphorylation dependence of Hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 284, 18801–18807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N., and Fink A. L. (2001) Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 [DOI] [PubMed] [Google Scholar]