

Abstract
A thorough understanding of the signaling pathways involved in the regulation of β cell proliferation is an important initial step in restoring β cell mass in the diabetic patient. Here, we show that epidermal growth factor receptor 1 (EGFR) was significantly up-regulated in the islets of C57BL/6 mice after 50% partial pancreatectomy (PPx), a model for workload-induced β cell proliferation. Specific deletion of EGFR in the β cells of adult mice impaired β cell proliferation at baseline and after 50% PPx, suggesting that the EGFR signaling pathway plays an essential role in adult β cell proliferation. Further analyses showed that β cell-specific depletion of EGFR resulted in impaired expression of cyclin D1 and impaired suppression of p27 after PPx, both of which enhance β cell proliferation. These data highlight the importance of EGFR signaling and its downstream signaling cascade in postnatal β cell growth.
Keywords: beta cell (β cell), cell proliferation, cyclin D1, diabetes, epidermal growth factor receptor (EGFR)
Introduction
Inducing β cell proliferation is a promising pathway to restore β cell mass in the diabetic patient (1–5). Recent studies have led to a better understanding of the signaling pathways regulating β cell proliferation (6–9). Since therapeutic attempts at β cell mass restoration would most often be performed in the adult population, it is increasingly important to completely understand the proliferative signaling pathways in a mature β cell (9, 10).
Epithelial growth factor receptor 1 (EGFR)5 is a member of the ErbB receptor family, which consists of four transmembrane tyrosine kinase receptors (11). Phosphorylation of EGFR tyrosine residues subsequent to ligand binding can initiate a number of downstream signaling cascades leading to different physiological processes (12). In the pancreas, EGFR is predominantly expressed within the islets (13), and it has been suggested to be essential in β cell development, function, and proliferation (13–19). One study found that global EGFR knock-out resulted in delayed β cell differentiation and impaired β cell proliferation during organogenesis (16, 20). Moreover, inhibition of EGFR signaling in the pancreas led to impaired β cell proliferation postnatally (13), during pregnancy (21), and in response to high-fat diet (19, 21). Here, in these studies the β cell phenotype could result from secondary effects due to either a global EGFR knock-out or from a pancreas-specific EGFR knock-out affecting non-β cells. Hence, a β cell-specific EGFR knock-out model seemed attractive.
Recently, we showed that transforming growth factor β (TGFβ) receptor signaling plays differential roles in workload-induced β cell proliferation and in inflammation-induced β cell proliferation (22). β cell proliferation can result from either a direct insult (e.g. local inflammation) to β cells, or from a metabolic need for more β cells, such as after surgical removal of part of the pancreas (PPx) or in a situation of increased physiologic demand (e.g. pregnancy). For the latter, we have defined this type of β cell proliferation as workload-induced β cell proliferation. For inflammation-induced β cell proliferation, activation of EGFR appears to regulate the outcome of the TGFβ receptor signaling on β cell proliferation through blocking the nuclear translocation of SMAD2, a component of the TGFβ receptor signaling pathway and a potential inhibitory factor against β cell proliferation (23). SMAD signaling is an important component of the EGFR pathway as well, and β cell proliferation likely responds to multiple different stimuli (22, 24). Thus, we investigated how the EGFR signaling pathway may be involved in workload-induced β cell proliferation.
Here, EGFR was specifically ablated in pancreatic β cells in order to investigate the role of EGFR signaling in β cell proliferation. Workload-induced β cell proliferation was stimulated by 50% partial pancreatectomy (PPx) (22). We found that EGFR signaling is required for workload-induced β cell proliferation, mediated by enhancing cyclin D1 and suppressing p27.
Results
EGFR Was Up-regulated in Islets after 50% Partial Pancreatectomy
Although we have previously shown that EGFR regulates inflammation-induced pancreatic β cell proliferation (23), the role of EGFR signaling in the regulation of workload-induced β cell proliferation remains unclear. To address this question, a well-documented non-hyperglycemic workload-induced β cell proliferation model, 50% PPx, was utilized to analyze the role of EGFR signaling in β cell proliferation.
One week after PPx, C57BL/6 mice showed the expected robust β cell proliferation (Fig. 1A). To analyze the role of EGFR in this context, the mRNA and protein levels of EGFR in the islets of C57BL/6 mice were measured after either PPx or sham surgery. RT-qPCR showed a more than 4-fold increase in EGFR mRNA (Fig. 1B, p < 0.05). Consistent with this transcriptional up-regulation, the protein level of EGFR was also similarly increased (Fig. 1C, p < 0.05), suggesting that the EGFR signaling is altered after PPx. Next, we examined the levels of the major cell-cycle regulators for β cells. We found that the protein levels of cyclin D2 and Cdk4 were both significantly up-regulated, but their mRNA levels were unchanged (Fig. 1, D–F). The changes in cyclin D1 levels did not reach statistical significance (Fig. 1, D–F). Moreover, p27, a cyclin-dependent kinase inhibitor, was significantly down-regulated at both the mRNA and protein levels (Fig. 1, D–F). These changes in cell-cycle regulators appear to be the bases for the increased β cell proliferation after PPx.
FIGURE 1.

EGFR is up-regulated in islets after 50% partial pancreatectomy. A, β cell proliferation increased 7 days after PPx, compared with sham-operated C57BL/6 mice, shown by representative immunostaining images for BrdU, insulin, and Hoechst (HO). B and C, EGFR levels in islets by RT-qPCR (B), and by Western blotting analysis (C). D, RT-qPCR analyses of mRNA of major cell-cycle regulators. E and F, Western blotting analyses of major cell-cycle regulators, shown by quantification (E), and by representative blots (F). n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance. Scale bars are 100 μm.
Generation of Pdx1-CreERT; EGFRfx/fx Mice
EGFR−/− global null mutant mice are not viable after 2 weeks postpartum, due to epithelial immaturity and abnormalities in organ development (14). There have been some studies in which EGFR signaling specific to the pancreas was partially disrupted through expression of a dominant-negative EGFR under the Pdx1 promoter (13, 21), but there are some drawbacks to these mice. First, expression of a dominant-negative EGFR in Pdx1+ cells likely does not completely disrupt the EGFR signaling pathway. Secondly, the incurred disruption of EGFR signaling in the embryonic pancreas may affect the development and maturation of β cells, thus creating effects that are not related to EGFR signaling in adult β cells.
To circumvent these shortcomings, and to better determine whether EGFR signaling indeed regulates workload-induced β cell proliferation, EGFR was ablated predominantly in pancreatic β cells by generating Pdx1-CreERT; EGFRfx/fx transgenic mice (Fig. 2A). Although EGFR may also be depleted in delta cells in these mice, its contribution to any phenotype in the current experiments should be minimal. In these mice, the expression of CreERT is driven by the Pdx1 promoter, but the CreERT recombinase stays in the cytoplasm. In the presence of tamoxifen, CreERT translocates into the nucleus and deletes exon 3 of the EGFR gene, leading to early termination of translation. Furthermore, the deletion of exon 3 disables the formation of the L1 subdomain, part of the extracellular domain of EGFR, which is required for ligand binding (25). Recent studies have highlighted the caveats of Cre expression in β cells (26, 27). In light of these findings, although here we have used a different mouse strain from the examined ones using hGF mini-enhancer (26), and although we examined 8-week-old mice rather than 24-week-old mice in Brouwers' study (26), we included Pdx1-CreERT mice as another control group in addition to the control EGFRfx/fx mice. We did not find any differences between EGFRfx/fx and Pdx1-CreERT in terms of EGFR expression levels and β cell proliferation. Two weeks after tamoxifen injection, the Pdx1-CreERT; EGFRfx/fx mice showed an ∼97% decrease in islet EGFR mRNA by RT-qPCR (Fig. 2B, p < 0.05), and an ∼80% decrease in islet EGFR protein by Western blot (Fig. 2C, p < 0.05), compared with their littermate control EGFRfx/fx mice, and PDX1-CreERT control mice. No difference was detected in body weight among all 3 groups (Fig. 2D).
FIGURE 2.

Generation of Pdx1-CreERT; EGFRfx/fx mice. A, EGFR was specifically targeted and depleted in pancreatic β cells in Pdx1-CreERT; EGFRfx/fx transgenic mice. In these mice, the expression of CreERT is driven by the Pdx1 promoter, but the recombinase stays in the cytoplasm. In the presence of tamoxifen, CreERT translocates into the nucleus and deletes exon 3 of the EGFR gene, leading to early termination of translation. B and C, EGFR levels in mouse islets were determined 2 weeks after tamoxifen injection in Pdx1-CreERT; EGFRfx/fx mice, and control Pdx1-CreERT and control EGFRfx/fx mice, by RT-qPCR (B, p < 0.05), and by Western blotting analysis (C, p < 0.05). D, mouse body weight. n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance.
EGFR Is Required for Baseline and Workload-induced β Cell Proliferation
We analyzed the effects of EGFR signaling on baseline and workload-induced β cell proliferation. Male Pdx1-CreERT; EGFRfx/fx, control Pdx1-CreERT mice, and control EGFRfx/fx mice received PPx or sham operation at 8 weeks of age, 2 weeks after tamoxifen treatment. These mice were fed with BrdU drinking water for 1 week to aid in immunohistochemical analysis of β cell replication. Ki-67 expression in β cells from these mice was also analyzed at the same time (Fig. 3A). Blood glucose (Fig. 3B) and glucose tolerance (Fig. 3C) were measured in these mice at 1 week after PPx/Sham treatment, showing no difference among the different experimental groups.
FIGURE 3.

EGFR is required for baseline and workload-induced β cell proliferation. A, male Pdx1-CreERT; EGFRfx/fx, control Pdx1-CreERT mice and control EGFRfx/fx mice received PPx or sham operated at 8 weeks of age, 2 weeks after tamoxifen treatment. These mice were fed with BrdU drinking water for 1 week to aid in immunohistochemical analysis of β cell replication. Ki-67 expression in β cells from these mice was also analyzed at the same time. B and C, blood glucose (B) and glucose tolerance (C) were measured in these mice at 1 week after PPx/Sham treatment. D and E, BrdU+/insulin+ cells were quantified, shown by quantification (D), and by representative images (E). n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance. Scale bars are 50 μm.
In sham-treated mice, the ratio of BrdU+/Insulin+ cells to all Insulin+ cells was 1.4 ± 0.2% in EGFRfx/fx mice, 1.5 ± 0.2% in Pdx1-CreERT mice (no difference between the 2 control groups), but was only 0.9 ± 0.1% in Pdx1-CreERT; EGFRfx/fx mice (Fig. 3, D and E, p < 0.05). In PPx-treated mice, the ratio of BrdU+/insulin+ cells to all insulin+ cells was 13.1 ± 1.0% in EGFRfx/fx mice, 13.6 ± 1.1% in Pdx1-CreERT mice (no difference between the 2 control groups), but was only 3.3 ± 0.40% in Pdx1-CreERT; EGFRfx/fx mice (Fig. 3, D and E, p < 0.05). Importantly, the percent reduction in proliferation in Pdx1-CreERT; EGFRfx/fx mice after PPx was significantly greater than the percent reduction in proliferation observed under basal conditions (36% reduction versus 76% reduction; p < 0.05 by ANOVA), suggesting that EGFR signaling is required for workload-induced β cell proliferation.
BrdU should label all proliferating β cells during the 7-day period, while Ki-67 labels cells within the G1 to M phase of the cell cycle at the time the pancreas is harvested (22). We obtained similar results on Ki-67+/insulin+ cells (Fig. 4, A and B, p < 0.05). Moreover, β cell-specific ablation of EGFR resulted in a relative decrease in residual β cell mass, compared with controls 1 week after PPx (Fig. 4C).
FIGURE 4.

Ki-67+/insulin+ cell quantification and β cell mass. A and B, Ki-67+/insulin+ cells were quantified, shown by representative images (A), and by quantification (B). C, β cell mass in the head portion of the pancreas was analyzed at 1 week after PPx/sham treatment. n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance. Scale bars are 50 μm.
Cyclin D1 and p27 May Be Involved in the Regulation of Baseline β Cell Proliferation by EGFR Signaling
Next, the expression of cyclin D1, cyclin D2, Cdk4, and p27 was analyzed in purified islets (28–30). Messenger RNA for all these genes was not altered by EGFR deletion in β cells, except for p27, where a slight but significant increase was detected (Fig. 5A). However, the cyclin D1 protein was significantly decreased in the islets of Pdx1-CreERT; EGFRfx/fx mice (Fig. 5, B and C), suggesting that EGFR may play a role in the post-transcriptional control of cyclin D1 in β cells at baseline.
FIGURE 5.
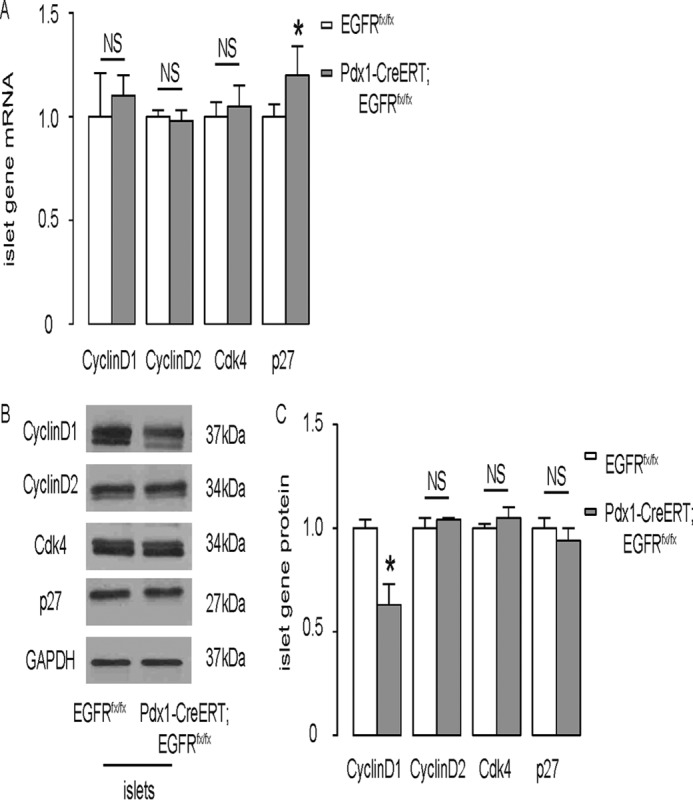
Cyclin D1 and p27 may be involved in the regulation of baseline β cell proliferation by EGFR signaling. A, mRNA of cyclin D1, cyclin D2, Cdk4, and p27 was analyzed in purified islets by RT-qPCR. B and C, protein of cyclin D1, cyclin D2, Cdk4, and p27 was analyzed in purified islets by Western blotting analysis, shown by representative blots (B), and by quantification (C). n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance.
Cyclin D1 and p27 Regulation in β Cells Is Impaired in Pdx1-CreERT; EGFRfx/fx Mice after PPx
To further understand how EGFR signaling regulates β cell proliferation, islets were isolated from both Pdx1-CreERT; EGFRfx/fx mice and control EGFRfx/fx mice after either PPx or sham surgery for analysis of the expression of cell cycle regulators. Consistent with the results obtained with wild-type C57BL/6 mice, for the control EGFRfx/fx mice, the EGFR protein level in the islets was significantly up-regulated after PPx (Fig. 6, A and B, p < 0.05). Cyclin D1, cyclin D2, and p27 protein levels in these mice were unchanged after PPx when compared with the levels of the sham-treated EGFRfx/fx mice, but Cdk4 levels were significantly increased after PPx (Fig. 6A, C–F). In Pdx1-CreERT; EGFRfx/fx mice, however, the protein level of cyclin D1 decreased greatly after PPx in comparison to the sham-treated mice (Fig. 6, A and C, p < 0.05). In addition, cyclin D1 levels were lower in the islets of sham-treated Pdx1-CreERT; EGFRfx/fx mice than in the islets of sham-treated EGFRfx/fx mice (Fig. 6, A and C, p < 0.05), consistent with previous results that EGFR is required for baseline cyclin D1 expression. Moreover, the levels of Cdk4 protein similarly increased after PPx in Pdx1-CreERT; EGFRfx/fx mice (Fig. 6, A and E, p < 0.05), suggesting that the EGFR does not regulate Cdk4, or that PPx increases Cdk4 in β cells in an EGFR signaling-independent manner. Furthermore, islet p27 protein levels were dramatically increased in Pdx1-CreERT; EGFRfx/fx mice after PPx, compared with sham-treatment, while baseline islet p27 levels did not alter after EGFR depletion (Fig. 6, A and F, p < 0.05). Thus, p27 levels in β cells may be regulated in an EGFR signaling-dependent manner upon increased β cell workload.
FIGURE 6.

Cyclin D1 and p27 regulation in β cells may be impaired in Pdx1-CreERT; EGFRfx/fx mice after PPx. Islets were isolated from both Pdx1-CreERT; EGFRfx/fx mice and control EGFRfx/fx mice after either PPx or sham surgery for analysis of the expression of cell cycle regulators. A, representative Western blots for EGFR and cell cycle regulators. B–F, protein quantification of EGFR (B), cyclin D1 (C), cyclin D2 (D), Cdk4 (E), and p27 (F). n = 5. All results were mean ± S.E. *, p < 0.05. NS, no significance.
Discussion
The indispensable nature of EGFR signaling in embryonic β cell development has been convincingly illustrated (13, 14, 16, 20). However, the role for the EGFR signaling pathway in adult β cell proliferation remains ill-defined (13, 18, 19, 21, 23). Two studies showed that combined treatment with EGF and gastrin promotes β cell proliferation and rescues hyperglycemia in alloxan-treated C57BL/6 mice and diabetic NOD mice (31, 32). Another study showed that GLP-1-induced β cell proliferation also requires the activation of EGFR signaling (17). These studies highlight an essential role for the EGFR signaling pathway in the regulation of adult β cell proliferation.
However, there are also reports that suggest a less important role of EGFR signaling in adult β cell proliferation. For example, one study showed that overexpression of EGFR in pancreatic β cells in adult mice did not affect β cell replication, while overexpression during the embryonic period resulted in significantly increased β cell proliferation and β cell mass (18). In another study using transgenic mice with EGFR dominant-negative expression under the control of the Pdx1 promoter (Pdx1-EGFR-DN mice), the authors indicated that inflammation-induced β cell proliferation was not affected by the disruption of EGFR signaling (21). In the homozygous Pdx1-EGFR-DN transgenic mice, phosphorylation of EGFR and activation of its downstream components was measured to be ∼40% of controls (13, 21). In these studies, heterozygous mice were used, and therefore the disruption of the EGFR signaling pathway would be only 20%, which may explain the conflicting results with other studies (17, 23, 31, 32). Additionally, the fact that in this model the EGFR signaling pathway in the pancreas is disrupted beginning with embryonic development raises the concern that early disruption of an important signaling pathway in the pancreas may cause permanent changes that prevent interpretation of a role for EGFR in adult β cells (33). Hence, a better mouse model which circumvents these shortcomings is needed to unveil the true role of the EGFR signaling pathway in adult β cell proliferation.
Here, we were able to address this issue by generating a transgenic mouse model with β cell specific deletion of EGFR only in adulthood. After 50% PPx, adult C57BL/6 mice showed significant up-regulation of islet EGFR. After 5 consecutive daily injections of tamoxifen and a 2-week waiting period for protein degradation, the majority of EGFR in β cell-specific EGFR knock-out mice was gone. Since there are non-β cells within the islets that might express low levels of EGFR, we expect that most of EGFR in β cells was depleted in this model.
We found that baseline β cell proliferation was impaired in β cell-specific EGFR knock-out mice, possibly resulting from the decrease in islet cyclin D1 protein levels. Since cyclin D1 mRNA was not affected by the deletion of EGFR, EGFR may play a role in the post-transcriptional regulation of cyclin D1, e.g. enhancing cyclin D1 protein translation, or suppressing its degradation.
In addition to its effects on baseline β cell proliferation, disruption of the EGFR signaling pathway also impaired workload-induced β cell proliferation. The protein level of cyclin D1 significantly decreased in the islets of PPx-treated β cell-specific EGFR knock-out mice. Moreover, EGFR signaling increased after PPx. These data suggest that EGFR may be required for sustaining the cyclin D1 levels, especially when a higher workload is demanded from β cells. The levels of cyclin D2 seemed to not be significantly regulated by changes in β cell workload, nor did EGFR appear to play a role in its regulation. Interestingly, the levels of Cdk4 similarly increased after PPx in mice, regardless of the presence of β cell-specific EGFR ablation, suggesting that EGFR may not be a regulator of Cdk4, but PPx may induce Cdk4 in β cells through signaling pathways other than EGFR. Finally, islet p27 levels were dramatically increased after PPx only in β cell-specific EGFR knock-out mice, suggesting that EGFR may be necessary for suppressing the up-regulation of p27 upon increased β cell workload. Together, our data suggest that EGFR may be required for sustaining cyclin D1 and suppressing p27 after PPx, which are both needed for an increase in β cell proliferation. Future experiments may be applied to study whether insulin receptor signaling or other signaling pathways may be responsible for activation of Cdk4 during increases in β cell workload. Previous studies have demonstrated a direct regulation of cyclin D1 and p27 by EGFR signaling. For example, Tao et al. showed that nuclear EGFR binds the cyclin D1 promoter to induce its gene transcription (34). Narita et al. showed that EGFR signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway (35). These studies suggest a regulatory relationship between EGFR signaling and cyclin D1/p27.
Overexpression of EGFR in the pancreatic β cells of adult mice did not promote β cell proliferation (18), suggesting that the EGFR signaling pathway is fundamental in maintaining baseline and workload-induced β cell proliferation. Activation of the EGFR signaling pathway alone may be insufficient to stimulate β cell proliferation, but it appears to play an indispensable role in modulating or co-operating with other signaling pathways to induce β cell expansion in certain circumstances (17, 23, 31).
Here small differences in the expression of some genes in β cells after PPx between C57/BL6 and the experimental strains may be due to the relatively small changes in the expression levels, the variability among samples, and the differences between these mouse strains in response to PPx.
Nevertheless, a more detailed dissection of the crosstalk among different signaling pathways and a complete illustration of the network of components that regulate β cell proliferation are required to fully understand adult β cell proliferation (9).
Experimental Procedures
Mouse Strain
All animal experiments were approved by the Animal Research and Care Committee at Children's Hospital of Pittsburgh and the University of Pittsburgh Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from the Jackson Labs (Bar Harbor, ME). Transgenic mice expressing EGFRfx/fx have been described before (25). EGFRfx/fx mice were crossed with Pdx1-CreERT mice, which also have been previously described, to get Pdx1-CreERT; EGFRfx/fx/EGFRfx/fx mice (Pdx1-CreERT; EGFRfx/fx) (36, 37). The CreERT-negative littermates and Pdx1-CreERT mice of same age were used as controls, which showed no phenotypic difference from wild-type C57/BL6 mice. All the mouse strains in this study have a C57/BL6 background. All the mice received surgery at 8 weeks of age.
Partial Pancreatectomy
PPx (50%) was performed as previously described (5, 22). Sham operated mice were used as controls.
Measurement of Blood Glucose, Glucose Tolerance, and β Cell Mass
Random blood glucose, glucose tolerance, and β cell mass were measured at previously described (5, 22, 23, 38). The beta cell mass in PPx-mice (the head portion of the pancreas due to surgical removal of the tail portion) was compared with the same part (the head portion) of the pancreas in the sham-operated mice.
Tamoxifen Injection
Tamoxifen (Sigma-Aldrich) was dissolved in 10 mg/ml corn oil (Sigma-Aldrich) and 2 mg/40 g body weight per day of tamoxifen was administered into the backs of 6-week-old mice subcutaneously for 5 consecutive days in order to induce nuclear translocation of CreERT to induce Cre recombination. Control mice also received injection of Tamoxifen at same dose and frequency. Surgery (sham or PPx) was performed 2 weeks after tamoxifen injection.
Pancreatic Digestion and Islet Isolation
Pancreas digestion and islet isolation procedures were performed as described before (39–42). The pancreas was infused with 0.25 mg/ml collagenase (Sigma-Aldrich) for 40 min to obtain a single cell population. Islets were handpicked at least three times to avoid contamination from non-islet fractions. Total RNA was extracted from isolated islets to confirm the purity of the islets by the absence of amylase and CK19 at the transcriptional level.
Western Blotting Analysis
Western blotting analysis was performed as previously described (23, 38). Primary antibodies for Western blotting analysis are rabbit polyclonal anti-GAPDH (Cell Signaling, San Jose, CA), rabbit anti-EGFR, rabbit anti-cyclin D1, rabbit anti-cyclin D2, rabbit anti-Cdk4, rabbit anti-p27 (all from Santa Cruz Biotechnology). Secondary antibody is HRP-conjugated anti-rabbit (Dako, Carpinteria, CA).
Isolation of RNA and RT-qPCR
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, CA), and then quantified with the Nanodrop 1000 (Thermo Fisher Scientific, Inc., Waltham, MA), followed by cDNA synthesis (Qiagen) and RT-qPCR. RT-qPCR was done as described before (39–41). The primers used were cyclophilin A (QT00247709), EGFR (QT00101584), cyclin D1 (QT00154595), cyclin D2 (QT00170618), Cdk4 (QT00103292), and p27 (QT01058708). All primers were obtained from Qiagen. Results were first normalized against the housekeeping gene cyclophilin A, which is stable across all samples, and then against the experimental controls.
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde overnight at 4 °C, followed by cryoprotection in 30% sucrose overnight, and finally snap-freezing. All samples were sectioned at the thickness of 6 μm. For antigen retrieval, BrdU immunostaining slides were incubated in 2 mmol/liter HCl for 40 min. All slides were incubated with primary antibodies at 4 °C overnight, then incubated with secondary antibodies for 2 h at room temperature. Primary antibodies used in this study were: guinea pig polyclonal insulin (Dako), rat BrdU (Abcam, Cambridge, MA), rabbit Ki-67 (Dako), goat Pdx1 (Abcam). The secondary antibodies used for indirect fluorescence staining were: Cy2, Cy3, or Cy5 conjugated donkey streptavidin, anti-rabbit, anti-guinea pig, anti-rat, and anti-goat. Nucleus staining was performed with Hoechst staining (Becton-Dickinson Biosciences, San Jose, CA). Cryosection imaging was performed with the AxioImager Z.1 microscope (Zeiss) and Volocity software.
BrdU Incorporation, Quantification of Proliferating β Cells, and β Cell Mass
Mice were given BrdU drinking water for 1 week after they received 50% PPx or sham surgery, as described before (22, 23). BrdU water was replaced every 3 days. Quantification of β cell proliferation was based on the percentage of BrdU+ β cells or Ki-67+ β cells. At least 2,000 β cells were counted for each mouse. If the percentage of BrdU+ or Ki-67+ β cells was low, more than 2,000 cells were counted until at least 50 BrdU+ β cells or 20 Ki-67+ β cells were counted, as previously described (23). β cell mass was analyzed as described before (22, 23, 38, 41).
Data Analysis
GraphPad Prism 6.0 (GraphPad Software, Inc. La Jolla, CA) was used for statistical analyses. All values are depicted as mean ± S.E. Five repeats were analyzed in each condition. All data were statistically analyzed using one-way ANOVA with a Bonferroni correction, followed by Fisher's Exact Test to compare two groups. Significance was considered when p < 0.05.
Author Contributions
The study was conceived and designed by X. X. and G. K. G. Acquisition of data was by Z. S., J. F., R. Z., S. F., C. C., D. M. R., K. P., C. S., and X. X. Analysis of data was carried out by Z. S., J. F., R. Z., and X. X., Z. S., X. X., and G. K. G. interpreted the data. Z. S., J. F., and X. X. drafted the article, and all authors revised the article and approved the final version to be published. X. X. and G. K. G. are guarantors of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health (to G. K. G, Grant R01 DK098196) and the Children's Hospital of Pittsburgh. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EGFR
- epidermal growth factor receptor
- PPx
- partial pancreatectomy
- TGFβ
- transforming growth factor β
- BrdU
- bromodeoxyuridine.
References
- 1. Dor Y., Brown J., Martinez O. I., and Melton D. A. (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 [DOI] [PubMed] [Google Scholar]
- 2. Teta M., Rankin M. M., Long S. Y., Stein G. M., and Kushner J. A. (2007) Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev. Cell 12, 817–826 [DOI] [PubMed] [Google Scholar]
- 3. Meier J. J., Butler A. E., Saisho Y., Monchamp T., Galasso R., Bhushan A., Rizza R. A., and Butler P. C. (2008) Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57, 1584–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Georgia S., and Bhushan A. (2004) Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 114, 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xiao X., Chen Z., Shiota C., Prasadan K., Guo P., El-Gohary Y., Paredes J., Welsh C., Wiersch J., and Gittes G. K. (2013) No evidence for beta cell neogenesis in murine adult pancreas. J. Clin. Invest. 123, 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao X., and Gittes G. K. (2015) Concise Review: New Insights Into the Role of Macrophages in beta-Cell Proliferation. Stem Cells Translat. Med. 4, 655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saunders D., and Powers A. C. (2016) Replicative capacity of beta-cells and type 1 diabetes. J. Autoimmun. 71, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunasekaran U., and Gannon M. (2011) Type 2 diabetes and the aging pancreatic beta cell. Aging 3, 565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stewart A. F., Hussain M. A., García-Ocaña A., Vasavada R. C., Bhushan A., Bernal-Mizrachi E., and Kulkarni R. N. (2015) Human beta-cell proliferation and intracellular signaling: part 3. Diabetes 64, 1872–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kulkarni R. N., Mizrachi E. B., Ocana A. G., and Stewart A. F. (2012) Human beta-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 61, 2205–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herbst R. S. (2004) Review of epidermal growth factor receptor biology. Int. J. Rad. Oncol. Biol. Phys. 59, 21–26 [DOI] [PubMed] [Google Scholar]
- 12. Yarden Y., and Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nature Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 13. Miettinen P. J., Ustinov J., Ormio P., Gao R., Palgi J., Hakonen E., Juntti-Berggren L., Berggren P. O., and Otonkoski T. (2006) Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes 55, 3299–3308 [DOI] [PubMed] [Google Scholar]
- 14. Miettinen P. J., Berger J. E., Meneses J., Phung Y., Pedersen R. A., Werb Z., and Derynck R. (1995) Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376, 337–341 [DOI] [PubMed] [Google Scholar]
- 15. Oh Y. S., Shin S., Lee Y. J., Kim E. H., and Jun H. S. (2011) Betacellulin-induced beta cell proliferation and regeneration is mediated by activation of ErbB-1 and ErbB-2 receptors. PloS one 6, e23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huotari M. A., Miettinen P. J., Palgi J., Koivisto T., Ustinov J., Harari D., Yarden Y., and Otonkoski T. (2002) ErbB signaling regulates lineage determination of developing pancreatic islet cells in embryonic organ culture. Endocrinology 143, 4437–4446 [DOI] [PubMed] [Google Scholar]
- 17. Buteau J., Foisy S., Joly E., and Prentki M. (2003) Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 52, 124–132 [DOI] [PubMed] [Google Scholar]
- 18. Hakonen E., Ustinov J., Eizirik D. L., Sariola H., Miettinen P. J., and Otonkoski T. (2014) In vivo activation of the PI3K-Akt pathway in mouse beta cells by the EGFR mutation L858R protects against diabetes. Diabetologia 57, 970–979 [DOI] [PubMed] [Google Scholar]
- 19. Zarrouki B., Benterki I., Fontés G., Peyot M. L., Seda O., Prentki M., and Poitout V. (2014) Epidermal growth factor receptor signaling promotes pancreatic beta-cell proliferation in response to nutrient excess in rats through mTOR and FOXM1. Diabetes 63, 982–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miettinen P. J., Huotari M., Koivisto T., Ustinov J., Palgi J., Rasilainen S., Lehtonen E., Keski-Oja J., and Otonkoski T. (2000) Impaired migration and delayed differentiation of pancreatic islet cells in mice lacking EGF-receptors. Development 127, 2617–2627 [DOI] [PubMed] [Google Scholar]
- 21. Hakonen E., Ustinov J., Mathijs I., Palgi J., Bouwens L., Miettinen P. J., and Otonkoski T. (2011) Epidermal growth factor (EGF)-receptor signalling is needed for murine beta cell mass expansion in response to high-fat diet and pregnancy but not after pancreatic duct ligation. Diabetologia 54, 1735–1743 [DOI] [PubMed] [Google Scholar]
- 22. Xiao X., Wiersch J., El-Gohary Y., Guo P., Prasadan K., Paredes J., Welsh C., Shiota C., and Gittes G. K. (2013) TGFβ Receptor Signaling Is Essential for Inflammation-Induced but Not β-Cell Workload-Induced β-Cell Proliferation. Diabetes 62, 1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiao X., Gaffar I., Guo P., Wiersch J., Fischbach S., Peirish L., Song Z., El-Gohary Y., Prasadan K., Shiota C., and Gittes G. K. (2014) M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc. Natl. Acad. Sci. U.S.A. 111, E1211–E1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rieck S., White P., Schug J., Fox A. J., Smirnova O., Gao N., Gupta R. K., Wang Z. V., Scherer P. E., Keller M. P., Attie A. D., and Kaestner K. H. (2009) The transcriptional response of the islet to pregnancy in mice. Mol. Endocrinol. 23, 1702–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee T. C., and Threadgill D. W. (2009) Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47, 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brouwers B., de Faudeur G., Osipovich A. B., Goyvaerts L., Lemaire K., Boesmans L., Cauwelier E. J., Granvik M., Pruniau V. P., Van Lommel L., Van Schoors J., Stancill J. S., Smolders I., Goffin V., Binart N., in't Veld P., Declercq J., Magnuson M. A., Creemers J. W., Schuit F., and Schraenen A. (2014) Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 20, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Magnuson M. A., and Osipovich A. B. (2013) Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab. 18, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gunasekaran U., Hudgens C. W., Wright B. T., Maulis M. F., and Gannon M. (2012) Differential regulation of embryonic and adult beta cell replication. Cell Cycle 11, 2431–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He L. M., Sartori D. J., Teta M., Opare-Addo L. M., Rankin M. M., Long S. Y., Diehl J. A., and Kushner J. A. (2009) Cyclin D2 protein stability is regulated in pancreatic beta-cells. Mol. Endocrinol. 23, 1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dai C., Brissova M., Hang Y., Thompson C., Poffenberger G., Shostak A., Chen Z., Stein R., and Powers A. C. (2012) Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia 55, 707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suarez-Pinzon W. L., Yan Y., Power R., Brand S. J., and Rabinovitch A. (2005) Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes 54, 2596–2601 [DOI] [PubMed] [Google Scholar]
- 32. Rooman I., and Bouwens L. (2004) Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia 47, 259–265 [DOI] [PubMed] [Google Scholar]
- 33. Brown M. L., and Schneyer A. L. (2010) Emerging roles for the TGFbeta family in pancreatic beta-cell homeostasis. Trends Endocrinol. Metab. 21, 441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tao Y., Song X., Deng X., Xie D., Lee L. M., Liu Y., Li W., Li L., Deng L., Wu Q., Gong J., and Cao Y. (2005) Nuclear accumulation of epidermal growth factor receptor and acceleration of G1/S stage by Epstein-Barr-encoded oncoprotein latent membrane protein 1. Exp. Cell Res. 303, 240–251 [DOI] [PubMed] [Google Scholar]
- 35. Narita Y., Nagane M., Mishima K., Huang H. J., Furnari F. B., and Cavenee W. K. (2002) Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 62, 6764–6769 [PubMed] [Google Scholar]
- 36. El-Gohary Y., Tulachan S., Wiersch J., Guo P., Welsh C., Prasadan K., Paredes J., Shiota C., Xiao X., Wada Y., Diaz M., and Gittes G. (2014) A smad signaling network regulates islet cell proliferation. Diabetes 63, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gu G., Dubauskaite J., and Melton D. A. (2002) Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129, 2447–2457 [DOI] [PubMed] [Google Scholar]
- 38. Xiao X., Fischbach S., Song Z., Gaffar I., Zimmerman R., Wiersch J., Prasadan K., Shiota C., Guo P., Ramachandran S., Witkowski P., and Gittes G. K. (2016) Transient Suppression of TGFβ Receptor Signaling Facilitates Human Islet Transplantation. Endocrinology 157, 1348–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiao X., Prasadan K., Guo P., El-Gohary Y., Fischbach S., Wiersch J., Gaffar I., Shiota C., and Gittes G. K. (2014) Pancreatic duct cells as a source of VEGF in mice. Diabetologia 57, 991–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiao X., Guo P., Shiota C., Prasadan K., El-Gohary Y., Wiersch J., Gaffar I., and Gittes G. K. (2013) Neurogenin3 activation is not sufficient to direct duct-to-beta cell transdifferentiation in the adult pancreas. J. Biol. Chem. 288, 25297–25308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao X., Guo P., Chen Z., El-Gohary Y., Wiersch J., Gaffar I., Prasadan K., Shiota C., and Gittes G. K. (2013) Hypoglycemia reduces vascular endothelial growth factor a production by pancreatic beta cells as a regulator of beta cell mass. J. Biol. Chem. 288, 8636–8646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li D. S., Yuan Y. H., Tu H. J., Liang Q. L., and Dai L. J. (2009) A protocol for islet isolation from mouse pancreas. Nat. Protoc. 4, 1649–1652 [DOI] [PubMed] [Google Scholar]