

Abstract
TIPE2 (TNF-α-induced protein 8-like 2) is a novel death effector domain protein and is a negative regulator of the innate and adaptive immune response. Although it has been demonstrated that caspase-8 contributes to the negative regulation of TIPE2, the negative regulatory mechanism is not entirely understood. Here, we demonstrate that TIPE2 interacts with TGF-β-activated kinase 1 (TAK1), a crucial regulatory molecule of inflammatory and immune signals, and consequently acts as a powerful negative regulator of TAK1. The interaction between endogenous TIPE2 and TAK1 was observed in RAW264.7 macrophage-like cells and mouse primary cells derived from spleen and thymus. The TIPE2 amino acid 101–140 region interacted with TAK1 by binding to the amino acid 200–291 region of the internal kinase domain of TAK1. TIPE2 interfered with the formation of the TAK1-TAB1-TAB2 complex and subsequently inhibited activation of TAK1 and its downstream molecules. Importantly, silencing TIPE2 through RNA interference attenuated the inhibitory action of TIPE2 on LPS- and TNF-α-stimulated TAK1 activity. Exogenous TIPE2 101–140, the region that interacts with TAK1, also inhibited LPS- and TNF-α-stimulated NF-κB reporter activity. Interestingly, cell-permeable TIPE2 protein maintained its binding ability with TAK1 and exhibited the same inhibitory action of native TIPE2 on TLR4 signaling in vitro. Thus, cell-permeable TIPE2 protein is a potential candidate for intracellular protein therapy for TAK1-related diseases. The present study demonstrates that TIPE2 acts as a novel negative regulator of inflammatory and immune responses through TAK1 signaling.
Keywords: cell-penetrating peptide (CPP), immunology, inflammation, signal transduction, tumor necrosis factor (TNF)
Introduction
The TNFAIP8 (TNF-α-induced protein 8) family is generally considered to be a subfamily of death effector domain proteins (1–4). Despite the structural similarity to this family, several studies have demonstrated that the various members of the TNFAIP8 family exhibit different biological functions (2, 5–8). Recently, Sun et al. (9) identified TIPE2 (TNFAIP8-like 2) as a member of the TNFAIP8 family. TIPE2 protein is preferentially expressed in lymphoid tissues such as the spleen, lymph nodes, and thymus. Interestingly, Sun et al. (9) also demonstrated that TIPE2-deficient mice are hyper-responsive to TLR and TCR signals, leading to inflammation of multiple organs. In addition, it has been demonstrated that TIPE2 inhibits the activation of NF-κB and AP-1, which are involved in inflammatory and antigen-specific immune responses. Therefore, TIPE2 is considered to be a key negative regulator that plays an important role in homeostasis of the immune system. Recent studies (10, 11) have also demonstrated the novel functions of TIPE2 as a potent inhibitor of Ras-mediated oncogenesis and atherosclerosis in relation to the macrophage response to oxidized low density lipoprotein. Thus, because TIPE2 acts as a potent inhibitor of inflammatory diseases and cancers, this negative regulator is a potential candidate for the development of drugs for inflammatory diseases and cancers. Although Sun et al. (9) have suggested interaction with caspase-8 as a possible mechanism through which TIPE2 negatively regulates the immune system, in view of the multipotential actions of TIPE2, our interests were to explore the novel target molecules of TIPE2 and to investigate its novel regulatory mechanism(s). Therefore, in this study we investigated whether TIPE2 is able to interact with several signal molecules that underlie innate and adaptive immune signal systems. Interestingly, we found that TIPE2 interacted with TGF-β-activated kinase 1 (TAK1),2 an important regulatory molecule of inflammatory and immune signals.
TAK1 was originally identified as a key regulator of TGF-β/bone morphogenic protein signals and is a member of the MAPK kinase kinase family that acts in TGF-β-mediated MAPK activation (12–16). Importantly, TAK1 plays a pivotal role in the regulation of cellular responses stimulated by growth factors, proinflammatory cytokines, and TLR ligands. Many studies (17–21) have demonstrated that TAK1-transduced signals from TCR and BCR, as well as antigen stimulation, play an important role in activation and survival of T cells or B cells. These demonstrations suggest that TAK1 is a fundamental molecule in the regulation of cellular events induced by changes in the environment. In this study, we demonstrate that TIPE2 acts as a novel negative regulator of TAK1, and interestingly, that a cell-permeable TIPE2 protein exhibits the ability of a potent inhibitor of the TAK1 signal.
Experimental Procedures
Cells
HEK293T cells were cultured in DMEM (Wako Pure Chemical, Osaka, Japan) supplemented with 10% FBS (Biowest, Nuaillé, France) and 1% penicillin and streptomycin solution (Nacarai Tesque, Kyoto, Japan) at 37 °C under 5% CO2 and 95% air. RAW264.7 cells were cultured in RPMI 1640 medium (Wako Pure Chemical) supplemented with 10% FBS and antibiotics at 37 °C under 5% CO2 and 95% air. FLAG-TAK1-stable RAW264.7 cells were transfected with a FLAG-mouse TAK1/pcDNA3 vector, and the transfected cells were selected in the presence of 1% Geneticin G418 (Calbio Chem, San Diego, CA) in RPMI 1640 medium supplemented with 10% FBS. Individual Geneticin-resistant colonies were cloned and expanded. The expression levels of TAK1 in each clone were monitored by immunoblot analysis using an anti-FLAG antibody, as described below.
Preparation of Mouse Tissue Homogenates
The spleens, thymi, and lungs of C57BL/6 N males (5 weeks old) were removed immediately after cervical dislocation. Extracts of their homogenates were prepared as follows: each tissue was suspended in TNE-N+ buffer solution (60 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.1% Nonidet P-40, and protease inhibitor mixture (Nacarai Tesque, Kyoto, Japan)) and then homogenized with a homogenizer pestle. Their homogenates were clarified by centrifugation at 12,000 × g for 10 min and subsequently used for immunoprecipitation and Western blotting assays as described below. The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Nihon University.
TIPE2 cDNA Cloning, Plasmid Construction, and siRNA
Human TIPE2 cDNA was isolated and amplified from Michigan Cancer Foundation (MCF)-7 mRNA by RT-PCR using specific primers, which were added to each restriction site: 5′-AAAGAATTCCCCATGGAGTCCTTCAGCTCAAAGAG-3′ (sense), 5′-AAAGTCGACCTCTCAGAGCTTCCCTTCGTCTAGCAGC-3′ (antisense-stop), and 5′-AAAGTCGACAGAGCTTCCCTTCGTCTAGCAGCTTCC-3′ (antisense-nonstop). The amplified cDNAs were digested with EcoRI and SalI in high salt-buffered solution. The cDNAs were purified with a QIAEX II DNA purification kit (Qiagen) after separation by agarose gel electrophoresis. The purified TIPE2 fragments were inserted using Ligation High DNA ligase (Toyobo, Osaka, Japan) in a pcDNA3 plasmid (Life Technologies), which was digested with EcoRI and XhoI. Ligation products were transformed into Escherichia coli DH5α competent cells (Takara, Shiga, Japan) and copied. TIPE2 cDNA insertion into pcDNA3 was confirmed by sequencing analysis (Applied Biosystems) and comparison with the National Center for Biotechnology Information database. TIPE2 cDNA was subcloned using EcoRI and XbaI enzymes in 6× Myc-tagged, FLAG-tagged, human influenza HA-tagged, Halo tagged, and GST-tagged fusion plasmids, respectively. All plasmids were prepared and purified by using Qiagen tip 500 (Qiagen). Mouse TAK1, human TAB2 (TAK1-binding protein 2), and human TNF receptor-associated factor 6 (TRAF6) cDNAs were kindly provided by Dr. K. Matsumoto. Human TAB1 was provided by Dr. K. Nishida. IL-1 receptor-associated kinase (IRAK) 4, IRAK-M, and IκB kinase β were generated by PCR, using human or mouse testis cDNA libraries as a template, and inserted into HA-tagged fusion pcDNA3 plasmid, respectively. HA-TAK1 deletion mutants (aa 1–150, 1–200, 1–291, 292–579, and 200–291) were generated by PCR using specific primers and cloned into HA-tagged pcDNA3 or FLAG-tagged pSG5. TIPE2 deletion mutants (aa 1–140, 1–100, 100–184, and 101–140) were also generated by PCR, using HA-TIPE2 wild type as a template and inserted into GST-tagged pGEX4T-1 or 6× Myc-tagged pSG5.
TIPE2-specific siRNA and a nonspecific negative control were purchased from Life Technologies. Transfection into cells with plasmid or siRNA was performed using X-tremeGENE siRNA transfection reagent (Roche Diagnostics) according to the manufacturer's protocols.
Antibodies
The following monoclonal antibodies were used for immunocytochemistry, immunoprecipitation, and Western blotting assays: anti-FLAG (Sigma-Aldrich), anti-His (Santa Cruz), anti-Halo (Promega, Madison, WI), anti-Myc (Santa Cruz), anti-human TIPE2 (Protein Tech), anti-mouse TAK1 (Cell Signaling, Beverly, MA), anti-human pTAK1 (Thr187; Cell Signaling), anti-human IκB (Cell Signaling), anti-human pIκB (Cell Signaling), anti-mouse p38 (Santa Cruz), anti-mouse pp38 (Cell Signaling), anti-mouse ERK (Santa Cruz), anti-mouse pERK (Santa Cruz), anti-human MKK6 (Cell Signaling), anti-human pMKK3/6 (Cell Signaling), anti-β-actin (Abcam), anti-rat Alexa Fluor 594 (Life Technologies), and anti-goat Alexa Fluor 488 (Life Technologies).
Preparation of Recombinant MTM-TIPE2
Jo et al. (22) previously described a membrane-translocating motif (MTM) composed of 12 amino acids from a hydrophobic signal sequence from fibroblast growth factor 4. This motif was attached to C-terminal ends to mediate uptake into cells. We constructed plasmids His-TIPE2-MTM/pET28a or His-TIPE2/pET28a by cloning full-length TIPE2 cDNA into pET28a-MTM vector (MTM encoded oligonucleotides (sense, 5′-GGCCGCGCAGCCGTTCTTCTCCCTGTTCTTCTTGCCGCACCCTAAC-3′, and antisense, 5′-TCGAGTTAGGGTGCGGCAAGAAGAACAGGGAGAAGAACGGCTGCGC-3′) ligated into the NotI-XhoI site of empty pET28a vector). MTM-TIPE2 or TIPE2 was expressed in E. coli BL21 (DE3) transformed with His-TIPE2-MTM/pET28a or His-TIPE2/pET28a plasmid. The cells were grown at 37 °C in lysogeny broth supplemented with kanamycin (30 mg/ml) until cultures reach an A600 of 0.3. In addition, to induce the expression of the recombinant proteins, 0.1 mm isopropyl-8-d-thiogalactopyranoside was added to the cultures. The cells were then cultured for an additional 3 h at 37 °C, harvested, and disrupted in 20 mm Tris-HCl buffer (pH 7.5) with an ultrasonicator (Vibra Cell). The disrupted cell suspensions were centrifuged, and the soluble fraction of the precipitate was prepared with guanidium lysis buffer (6 m guanidine hydrochloride, 20 mm sodium phosphate, pH 7.8, 500 mm NaCl). The recombinant proteins in the soluble fraction were purified using the nickel-nitrilotriacetic acid purification system (Life Technologies) according to the manufacturer's instructions. The purified denatured recombinant proteins were dialyzed with 0.5 m arginine containing buffer (0.5 m l-arginine, 50 mm sodium diphosphate, 2 mm DTT, 0.02% Tween 80) and then with DMEM or RPMI 1640 medium. For the preparation of soluble protein, the aggregations were separated by centrifugation at 13,000 × g for 5 min at 4 °C. The purity and quantity of proteins were assessed by SDS-PAGE.
Immunostaining and Confocal Fluorescence Microscopy
As described previously (23, 24), RAW264.7 cells were cultured on cover glasses in 35-mm culture dishes and then transfected using X-tremeGENE HP (Roche Diagnostics) with the GFP-empty, enhanced GFP (EGFP)-TAK1, and/or FLAG-TIPE2 plasmids. Transfected cells were grown for 24 h at 37 °C in RPMI 1640 medium containing 10% FBS. The cells were then washed three times with PBS and fixed in 4% paraformaldehyde/PBS for 30 min at room temperature. The cells were then washed again three times with PBS. For the detection of FLAG-TIPE2 or EGFP-TAK1, after the wash with PBS, the cells were incubated with anti-FLAG antibody or anti-GFP antibody in blocking solution (5% skim milk; Wako Pure Chemical) for 1 h. The cells were subsequently washed five times with TBST (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) and then incubated for 1 h with anti-rat Alexa Fluor 594 or anti-goat Alexa Fluor 488 in blocking solution. Finally, the cells were washed five times with TBST, and the coverslip was mounted with Vectorshield (Vector Laboratories). Samples were examined using FV-1000 confocal laser imaging microscopy (Olympus, Tokyo, Japan).
For co-localization of TIPE2 and TAK1 at endogenous level in spleen, paraffin-embedded sections of spleen were obtained from Genostaff Co. Ltd. (Tokyo, Japan). The tissue sections were deparaffinized with lemosol and dehydrated with a series of ethanol solutions in PBS. The sections were treated for 5 min with proteinase K (5 μg/ml) and washed with TBST. Then the sections were incubated with anti-mouse TAK1 antibody and/or anti-human TIPE2 antibody in blocking solution (StartingBlock T20) for 1 h. The sections were subsequently washed, incubated with anti-mouse Alexa Fluor 594 or anti-rabbit Alexa Fluor 488 as described above, and then examined using LSM 510 META laser scanning microscopes (Carl Zeiss).
In addition, for cell-transducing experiments of recombinant MTM-TIPE2, RAW264.7 cells cultured on cover glasses were incubated for 30 min with FITC-labeled MTM-TIPE2 (1 μm). Then the cells were fixed in 4% paraformaldehyde/PBS and subsequently were treated for 15 min with proteinase K (5 μg/ml). Thereafter, samples were examined using FV-1000 confocal laser imaging microscopy.
Flow Cytometry
RAW264.7 cells were cultured in 35-mm culture dishes containing RPMI 1640 medium with 10% FBS and treated for 15 min with or without FITC-labeled MTM-TIPE2 or FITC-labeled TIPE2 at 1 μm. The cells were then washed three times with PBS, fixed for 4 h at −20 °C in cold 70% ethanol/PBS, washed a further twice with PBS, and then treated for 15 min with proteinase K. Finally, the cell sample was analyzed using FACS Cant (BD Biosciences).
Immunoprecipitation and Western Blotting Analysis
As described previously (24), cells transfected with plasmids encoding each tag fusion protein were extracted with TNE-N+ buffer solution. Each tagged protein was precipitated from the cell lysates using anti-tag-coated agarose (Sigma-Aldrich). Immunocomplexes were washed five times with TNE buffer, and then each tagged protein was eluted from the anti-tag-coated agarose (3× FLAG peptide). These samples were denatured in SDS buffer for SDS-PAGE.
Western blotting analysis was attempted with the semi-dry method. Denatured proteins from cells were separated by molecular weight using SDS-PAGE. The proteins in the gels were transferred to Immobilon transfer membranes (Millipore) using a Trans-Blot Cell (Bio-Rad). The membrane was blocked from nonspecific recognition of antibody with 5% skim milk dissolved in TBST. The proteins were detected using the indicated antibodies by means of an enhanced chemiluminescence system.
To determine the interaction domain of TIPE2 with TAK1, E. coli was transformed with plasmids encoding GST-TIPE2 deletion mutants. Then cell extracts were obtained to prepare the recombinant proteins. The GST-TIPE2 deletion proteins were purified using glutathione Sepharose 4B beads (Roche Diagnostics). The purified GST-TIPE2 proteins were mixed with FLAG-TAK1 that was overexpressed in HEK293T cells. FLAG-TAK1 was immunoprecipitated and then detected by Western blotting assay using an anti-FLAG antibody.
His Pulldown Assay
For the pulldown assay to examine the interaction of His-MTM-TIPE2 protein with TAK1, RAW246.7 cells were transiently transfected with or without FLAG-TAK1 and were incubated for 1 h with or without MTM-TIPE2 protein or TIPE2 protein at 1 μm. Then cell extracts were prepared and interaction of TIPE2 with TAK1 in cell lysates was detected by His pulldown assay with anti-His tag purification resin beads (Roche Diagnostics) and by Western blotting assay with anti-FLAG antibody.
NF-κB-dependent Luciferase Reporter Assay
RAW264.7 cells, HEK293T cells were plated in 24-well culture plates. On the following day, the cells were transiently transfected with the indicated expression vectors using X-tremeGENE 9 DNA transfection reagent (Roche Diagnostics). The total amount of DNA was kept constant by supplementation with empty vector, pcDNA3 (Life Technologies). Every transfection step included 100 ng of reporter plasmid together with 4 ng of Renilla for normalization of transfection efficiency. We used the NF-κB-luciferase reporter plasmid, provided by Dr. Y. Ohmori. After 24 h, the cells were washed and then incubated for additional 24 h under serum-free culture conditions. Subsequently, the cells were treated with the indicated stimulator (LPS, 01114B, 100 ng/ml (Sigma-Aldrich); poly(I·C), 50 μg/ml (Sigma-Aldrich); CpG DNA, 1 μm (Enzo Life Science, Farmingdale); TNF-α, 10 ng/ml (R&D Systems); and IL-1β 10 ng/ml (Wako Pure Chemical)) for 5 h. Then cells were lysed in a luciferase lysis buffer (Promega), and luciferase activity was measured using a Dual-Luciferase reporter assay system (Promega) with a VeritasTM Microplate luminometer (Promega). All luciferase experiments were performed in triplicate.
Measurement of Inflammatory Cytokines
RAW264.7 cells were transfected using X-tremeGENE siRNA transfection reagent (Roche Diagnostics) with or without FLAG-TIPE2 or TIPE2 siRNA. After 24 h, the cells were incubated for an additional 24 h under serum-free culture conditions. Subsequently, the cells were treated for 8 h with or without LPS at 100 ng/ml in the serum-free medium. The IL-6 level in the culture supernatants was measured by mouse IL-6 ELISA Ready-SET-Go (eBioscience) according to the manufacturer's protocol.
Quantitative RT-PCR Analysis
Quantitative RT-PCR (qRT-PCR) analysis was performed on a Rotor-Gene Q (Qiagen) using a Rotor-Gene SYBR Green PCR kit (Qiagen). Isolation of total RNA and cDNA synthesis were performed following the protocols provided with AccessQuickTM RT-PCR system (Promega). Primers used for RT-PCR were specific for one of the indicated cytokines. QuantiTect Primer Assay (Qiagen) primers were used for qRT-PCR: IL-1β (Mm_Il1b_2_SG, QT01048355), IL-6 (Mm_Il6_1_SG, QT00098875), and actin (Mm_Actb_2_SG, QT01136772). The qRT-PCR reaction was performed in a Rotor-Gene Q. More than 40 cycles of amplification were performed, each consisting of a denaturation (95 °C, 5 s) and an annealing/extension (60 °C, 10 s) step. Data acquisition and the analysis of the qRT-PCR assays were performed using Rotor-Gene Analysis (Qiagen).
Statistical Analysis
The data are expressed as the means ± S.D. in triplicate. A p value < 0.05 was considered to indicate statistical significance.
Results
TIPE2 Interacts with TAK1 by Binding with Its Internal Kinase Domain
We first explored whether TIPE2 is able to interact with major signal molecules, such as IRAK4, IRAK-M, TRAF6, TAK1, and IκB kinase β that lie down the immune signal pathway. To examine these interactions, we co-transfected FLAG-TIPE2 and each HA-tagged signal molecule into HEK293T cells, respectively, and then immunoprecipitated their cell extracts with an anti-FLAG antibody. Interestingly, although the immunoprecipitation-Western blotting assay showed that TIPE2 clearly interacted with TAK1, TIPE2 did not interact with the other signal molecules tested (Fig. 1A). To determine whether the interaction with TAK1 in the overexpressed cells would also occur between endogenous TIPE2 and TAK1, we examined the interaction using RAW264.7 cell extracts. Fig. 1B clearly shows the endogenous interaction between both molecules. In addition, because a previous study (9) demonstrated that TIPE2 is predominantly expressed in lymphoid tissues, we attempted to determine whether TIPE2 is able to interact with TAK1 in mouse primary cells derived from spleen and thymus. Fig. 1C shows that TIPE2 also interacted with TAK1 in cell extracts derived from mouse spleen and thymus, although no such interaction was observed in lung cell extracts. We also used confocal microscopy to determine whether TIPE2 and TAK1 co-localize in the cytoplasm during their interaction. As expected, Fig. 1D shows that the exogenous TIPE2 and TAK1 co-localized in the cytoplasm of RAW264.7 cells. Furthermore, co-localization of the both proteins at endogenous level was also observed in spleen cells (Fig. 1E). These results suggest that TIPE2 is a novel molecule that interacts with TAK1. Based on these results, we experimented to determine the binding region involved in the interaction between TIPE2 and TAK1. We constructed several deletion mutants of TAK1 (Fig. 1F) and prepared their expression vectors. Then we transiently overexpressed FLAG-TIPE2 and HA-TAK1 deletion mutants in HEK293T cells, respectively. As shown in Fig. 1G, although TIPE2 interacted with TAK1 wild type and the TAK1 1–291 mutant, it did not interact with the TAK1 1–150, 1–120, and 292–579 mutants. These observations suggested that TIPE2 interacted with region aa 200–291, located in the internal kinase domain of TAK1. We also generated several GST-TIPE2 fusion proteins (Fig. 1H). Using a His pulldown assay, we determined the binding region of TIPE2 involved in interaction with TAK1. Although TIPE2 wild type and the TIPE2 1–140 and TIPE2 100–184 mutants bound to TAK1, the TIPE2 1–100 mutant was unable to bind to TAK1 (Fig. 1I). These results indicated that the aa 101–140 region of TIPE2 interacted with the TAK1 aa 200–291 region of its internal kinase domain. To confirm the interaction between TIPE2 aa 101–140 and TAK1 aa 200–291, we co-transfected 6× Myc-TIPE2 101–140 and FLAG-TAK1 200–291 into HEK293T cells and then investigated their interaction by immunoprecipitation- Western blotting assay. Fig. 1J shows that the molecules interacted with each other via the determined binding regions. Fig. 1K illustrates the interaction between TIPE2 and TAK1 via their binding regions.
FIGURE 1.

TIPE2 interacts with TAK1 via binding with the TAK1 internal kinase domain. A, HEK293T cells were transfected with FLAG-TIPE2 alone or with both FLAG-TIPE2 and HA-tagged signal molecules, respectively. The cells were extracted 24 h after transfection and immunoprecipitated with anti-FLAG antibody. Interactions were detected by Western blotting assay using an anti-HA antibody. The expression of FLAG-TIPE2 and the indicated HA-tagged signal molecules in the immunoprecipitates and whole cell lysates was also detected by Western blotting assay. B, RAW264.7 cells were extracted and immunoprecipitated with control IgG or anti-TAK1 antibody. The presence of TIPE2 or TAK1 in the immunoprecipitates and whole cell lysates was detected by Western blotting assay with anti-TIPE2 antibody or anti-TAK1 antibody. C, mouse spleen, thymus, and lung cell extracts were prepared and immunoprecipitated with control IgG or anti-TAK1 antibody. The presence of TIPE2 or TAK1 in the immunoprecipitates and whole cell lysates was detected by Western blotting assay with anti-TIPE2 antibody or anti-TAK1 antibody. D, RAW264.7 cells were transfected with FLAG-TIPE2 or EGFP-TAK1 vector. After 24 h, the cells were immunostained with Alexa 594 (red) for FLAG-TIPE2 or with EGFP antibody for EGFP-TAK1. E, the sections of spleen were incubated with anti-TIPE2 and/or with anti-TAK1 antibody for 1 h and then immunostained with Alexa 488 (red) for TIPE2 or Alexa594 (green) for TAK1. The nucleus was stained with TO-PRO-3. Scale bars, 10 μm. F, five truncated mutants of TAK1 (TAK1 1–150, TAK1 1–200, TAK1 1–291, TAK1 292–579, and TAK1 200–291) were generated from the control WT vector. G, HEK293T cells were transfected with FLAG-TIPE2 alone or with both FLAG-TIPE2 and the indicated HA-tagged truncated mutant vectors. After 24 h, the cells were extracted and immunoprecipitated using an anti-FLAG antibody. Interactions were detected by Western blotting assay using an anti-HA antibody. The expression of FLAG-TIPE2 and the indicated HA-TAK1 deletion mutants in the immunoprecipitates and whole cell lysates was detected by Western blotting assay. H, four truncated mutants of TIPE2 (TIPE2 1–100, TIPE2 1–140, TIPE2 100–184, and TIPE2 101–140) were generated from the control WT vector. I, HEK293T cells were transiently transfected with or without FLAG-TAK1 vector. At 24 h after the transfection, the cells were lysed, and the cell lysates were incubated for 60 min with 0.4 nmol of the indicated GST-TIPE2 protein. The mixture was immunoprecipitated using an anti-GST antibody. The bound proteins were detected by Western blotting assay with anti-FLAG antibody. J, HEK293T cells were transfected with TIPE2 mutant 6× Myc-TIPE2 101–140 alone, FLAG-TAK1 200–291 alone, and a combination of both vectors. At 24 h after transfection, the cells were extracted and immunoprecipitated using an anti-FLAG antibody. Interactions were detected by Western blotting assay using an anti-Myc antibody. The expression of FLAG-TAK1 and 6× Myc-TIPE2 in the immunoprecipitates and whole cell lysates was detected by Western blotting assay. Interactions were detected by Western blotting assay using an anti-Myc antibody. K, a model for TIPE2 interaction with TAK1 via the TAK1 internal kinase domain. IB, immunoblot; IP, immunoprecipitation; CBB, Coomassie Brilliant Blue.
TIPE2 Interferes with the Binding of TABs to TAK1 and Inhibits TAB1- and TAB2-dependent TAK1 Activity
As demonstrated in previous studies (25–30), the formation of the TAK1-TAB1-TAB2 complex is an important event in initiating TAK1 activation, and its subsequent phosphorylation and ubiquitination is essential in inducing its activation. Therefore, the based on the molecular steps involved in TAK1 activation, we investigated whether TIPE2 acts as a negative regulator in subsequent molecular events for TAK1 activation. Initially, we investigated whether TIPE2 was able to interfere with the binding between TAK1 and TAB1 or TAB2 in HEK293T cells. We transiently overexpressed the combination of FLAG-TAK1, 6× Myc-TIPE2, and HA-TAB1 or HA-TAB2 in the cells and then examined their bindings by immunoprecipitation-Western blotting assay. As shown in Fig. 2 (A and B), although TAK1 bound with TAB1 or TAB2 in the cells, TIPE2 clearly interfered with the binding between exogenous TAK1 and TAB1 or TAB2. Because the binding interference by TIPE2 suggested the possibility that TIPE2 inhibits TAB-dependent phosphorylation and ubiquitination of TAK1, we examined whether TIPE2 is actually able to inhibit TAB1- or TAB2-dependent phosphorylation of TAK1. FLAG-TAK1 and 6× Myc-TAB1 or 6× Myc-TAB2 were transiently transfected in the presence or absence of HA-TIPE2 into HEK293T cell, respectively. TIPE2 dramatically inhibited TAB1- or TAB2-dependent phosphorylation of TAK1 in the cells (Fig. 2, C and D). Because these results suggested that TIPE2 might also interfere with TAB1-dependent ubiquitination of TAK1, we transfected FLAG-TAK1, 6× Myc-TAB1, and HA-K63-Ub in the presence or absence of Halo-TIPE2 into HEK293T cells and then examined K63 ubiquitination of immunoprecipitated FLAG-TAK1 by Western blotting assay. Fig. 2E shows the inhibitory action of TIPE2 on TAB1-dependent ubiquitination of TAK1 in the cells. Collectively these results suggest that TIPE2 acts as a powerful inhibitor of TAK1 activation through interfering its binding with TABs and subsequent phosphorylation and ubiquitination. Therefore, we investigated whether TIPE2 is actually able to inhibit TAB1-dependent phosphorylation of MKK3/6, a substrate of TAK1. As shown in Fig. 2F, TIPE2 inhibited TAB1-dependent phosphorylation of MKK3/6 in HEK293T cells. These results suggested that TIPE2 plays a crucial role as a negative regulator of TAK1 signaling.
FIGURE 2.
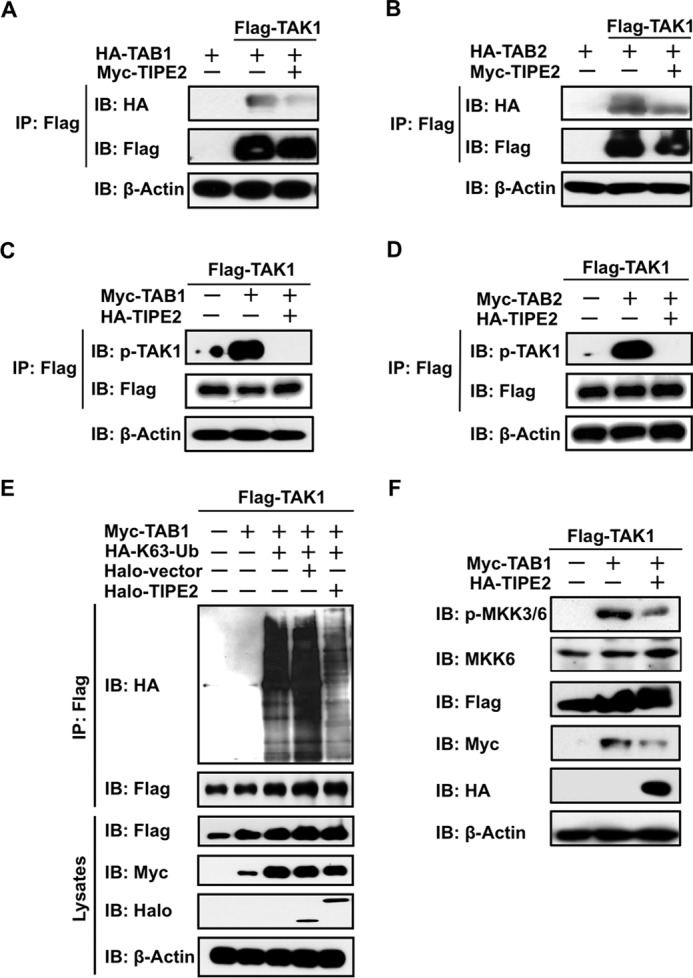
TIPE2 interferes with the binding of TAB1 and TAB2 with TAK1, which inhibits phosphorylation and ubiquitination of TAK1 and its activity. A and B, HEK293T cells were transfected with 6× Myc-TIPE2 alone or the indicated combination of 6× Myc-TIPE2, FLAG-TAK1, and HA-TAB1 (A) or HA-TAB2 (B). After 24 h, the cells were extracted and immunoprecipitated using an anti-FLAG antibody. Binding of TAB1 or TAB2 with TAK1 was detected by Western blotting assay using an anti-HA antibody. Expression of FLAG-TAK1, 6× Myc-TIPE2, HA-TAB1, and HA-TAB2 in the immunoprecipitates was detected by Western blotting assay using the appropriate anti-tag antibody. C and D, HEK293T cells were transfected with HA-TIPE2 alone or the indicated combination of HA-TIPE2, FLAGTAK1, and 6× Myc-TAB1 (C) or 6× Myc-TAB2 (D). After 24 h, the cells were extracted and immunoprecipitated using an anti-FLAG antibody. The phosphorylation of TAK1 was detected by Western blotting assay using an anti-pTAK1 antibody. The expression of FLAG-TAK1, HA-TIPE2, 6× Myc-TAB1, and 6× Myc-TAB2 in the immunoprecipitates was detected by Western blotting assay. E, HEK293T cells were transfected with the indicated combination of FLAG-TAK1, Halo-TIPE2, Halo-vector, HA-K63-Ub, and 6× Myc-TAB1 vectors. After 24 h, whole cell lysates were extracted and immunoprecipitated using an anti-FLAG antibody. The ubiquitination of TAK1 was detected by Western blotting assay using an anti-HA antibody. The expression of FLAG-TAK1, Halo-TIPE2, and 6× Myc-TAB1 in whole cell lysates was detected by Western blotting assay. F, HEK293T cells were transfected with the indicated combination of HA-TIPE2, FLAG-TAK1, and 6× Myc-TAB1 vectors. After 24 h, the cells were extracted, and then TAK1 activity in the whole lysates was monitored by detecting phosphorylation of MKK3/6, a substrate of TAK1, by Western blotting assay using an anti-pMKK3/6 antibody. The expression of FLAG-TAK1, HA-TIPE2, 6× Myc-TAB1, MKK6, and phosphorylation of MKK3/6 in whole lysates was detected by Western blotting assay using the appropriate anti-tag antibody, anti-MKK6, and anti-pMKK3/6 antibody. IB, immunoblot; IP, immunoprecipitation.
TIPE2 Inhibits Phosphorylation of TAK1 and Its Downstream Molecules in Cells Stimulated with LPS and Inflammatory Cytokines
Next, we examined whether TIPE2 inhibits the ligand-stimulated activation of TAK1 by ligands such as LPS, TNF-α, and IL-1β. We developed TAK1-stable RAW264.7 cells and transiently overexpressed HA-TIPE2 in the stable cells. Subsequently, we examined LPS-stimulated phosphorylation of TAK1 in the cells. As shown in Fig. 3A, LPS-stimulated phosphorylation of TAK1 in the stable cells was strongly inhibited by TIPE2. In addition, we also examined the inhibitory action of TIPE2 on TNF-α- or IL-1β-stimulated phosphorylation of TAK1 in HEK293T cells. Fig. 3 (B and C) shows that TIPE2 clearly inhibited TNF-α- and IL-1β-stimulated phosphorylation of TAK1 in the cells. In addition, by silencing TIPE2 with RNA interference, we defined the specificity of TIPE2 inhibition for phosphorylation of TAK1 in TNF-α-stimulated HEK293T cells. Importantly, we observed that TIPE2 silencing attenuated its inhibitory action on TAK1 phosphorylation stimulated by the inflammatory cytokine (Fig. 3D). Collectively, these results defined the inhibitory action of TIPE2 on ligand-stimulated activation of TAK1. To demonstrate the action of TIPE2 as a TAK1 inhibitor, we examined the inhibitory effect of TIPE2 on phosphorylation of MKK3/6 in TNF-α-stimulated cells. As expected, TIPE2 dramatically inhibited TNF-α-stimulated phosphorylation of MKK3/6 in the cells (Fig. 3E). Because these results suggest that TIPE2 might also inhibit phosphorylation of p38, ERK, and IκB, which play crucial roles in cellular events that are driven by TAK1 signaling, we examined this action. We overexpressed HA-TIPE2 in TAK1-stable RAW264.7 cells and examined phosphorylation of IκB, p38, and ERK in the cells by LPS or IL-1β. Fig. 3 (F and G) shows that TIPE2 also inhibits the phosphorylation of p38, ERK, and IκB in the cells at 60 min after LPS or IL-1β treatment.
FIGURE 3.

TIPE2 inhibits phosphorylation of TAK1 and its downstream molecules in ligand-stimulated cells. A, TAK1-stable RAW246.7 cells were transfected with or without HA-TIPE2. The cells were incubated for 24 h and then incubated for an additional 24 h in serum-free culture conditions. Next, the cells were treated with or without LPS at 100 ng/ml. At the indicated times, the cells were extracted, and phosphorylation of TAK1 was detected by Western blotting assay using an anti-pTAK1 antibody. B and C, HEK293T cells were transfected with FLAG-TAK1 alone or FLAG-TAK1 with or without HA-TIPE2. After 24 h, the cells were incubated for an additional 24 h under serum-free culture conditions. The cells were then treated with or without TNF-α at 10 ng/ml (B) or IL-1β at 10 ng/ml (C) and extracted 5 min after the treatment. Then phosphorylation of TAK1 was detected by Western blotting assay using an anti-pTAK1 antibody. D, HEK293T cells were transiently transfected with FLAG-TAK1 alone or FLAG-TAK1 with or without HA-TIPE2, TIPE2 siRNA, and negative control siRNA. After 24 h, the cells were incubated for an additional 24 h in serum-free culture conditions and then treated for 5 min with or without TNF-α at 10 ng/ml. Then cells were extracted, and phosphorylation of TAK1 was detected by Western blotting assay using an anti-pTAK1 antibody. The expression of FLAG-TAK1 and HA-TIPE2 in whole cell lysates was detected by Western blotting assay. E, HEK293T cells were transfected with FLAG-TAK1 alone or FLAG-TAK1 with or without HA-TIPE2 vectors. After 24 h, the cells were incubated for an additional 24 h in serum-free culture conditions and then treated for 15 min with or without TNF-α at 10 ng/ml. TAK1 activity was monitored by detecting phosphorylation of MKK3/6 by Western blotting assay using an anti-pMKK3/6 antibody. The expression of FLAG-TAK1 and HA-TIPE2 in whole cell lysates was detected by Western blotting assay. F and G, TAK1-stable RAW246.7 cells were transfected with or without HA-TIPE2 vectors. After 24 h, the cells were incubated for an additional 24 h in serum-free culture conditions and then treated for the indicated times with or without LPS at 100 ng/ml (F) or IL-1-β at 10 ng/ml (G). Next, the cells were extracted at the indicated times, and phosphorylation of p38, ERK, and IκB was detected by Western blotting assay using anti-pp38, anti-pERK, and anti-pIκB antibodies. The expression of p38, ERK, and IκB in whole cell lysates was determined by Western blotting assay. IB, immunoblot; IP, immunoprecipitation.
TIPE2 Inhibition of Activation of TAK1 Signaling Involves the Binding Region of TAK1
Many studies (12, 28, 31, 32) have demonstrated that TLR ligands and inflammatory cytokines (TNF-α, IL-1β) stimulate NF-κB activity via activation of TAK1. Because we demonstrated that TIPE2 acts as a potent negative regulator of TAK1 activation, we examined the inhibitory effect of TIPE2 on LPS-stimulated NF-κB reporter activity in TIPE2-overexpressed cells and also on production of IL-6 by the stimulated cells. As shown in Fig. 4A, although strong NF-κB reporter activity was observed in RAW264.7 cells stimulated with LPS, TIPE2 significantly inhibited the NF-κB reporter activity in the cells. However, silencing of TIPE2 by RNA interference attenuated its inhibitory action on LPS-stimulated production of IL-6 by the cells (Fig. 4B). In addition, the inhibitory effect of TIPE2 on TNF-α-stimulated NF-κB reporter activity in HEK293T cells was also attenuated by TIPE2-specific siRNA (data not shown). Because we determined that TIPE2 aa 101–140 is the binding region that interacts with TAK1, we investigated whether this binding region contributes to the inhibitory effect of TIPE2 on TNF-α-stimulated NF-κB reporter activity. Importantly, the TIPE2 101–140 mutant markedly inhibited LPS- or TNF-α-stimulated NF-κB reporter activity in RAW264.7 cells (Fig. 4C) and in HEK293T cells (Fig. 4D). Collectively, these results demonstrate that the TIPE2 binding region contributes to TIPE2 inhibition of activation of TAK1 signaling.
FIGURE 4.
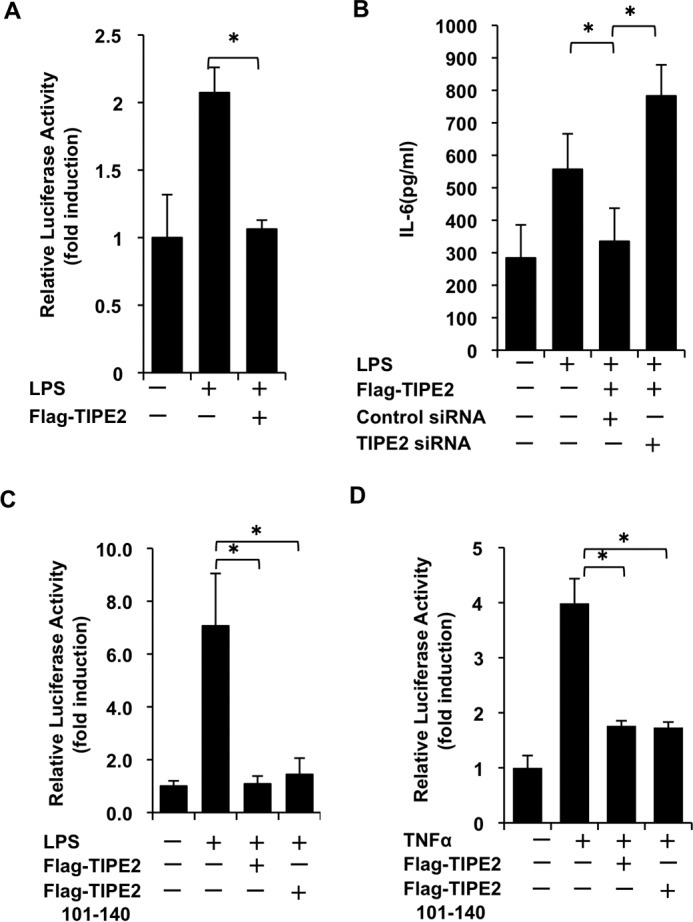
TIPE2 inhibits activation of TAK1 signaling via its binding domain. A, RAW246.7 cells in 24-well plates were transiently transfected with or without FLAG-TIPE2 and NF-κB reporter construct. After 24 h, the cells were incubated for an additional 24 h under serum-free culture conditions. Then cells were treated for 5 h with or without LPS at 100 ng/ml. Next, the cells were lysed, and luciferase activity was measured. All luciferase experiments were performed in triplicate. The results are expressed as means ± standard deviation. *, p < 0.05. B, RAW246.7 cells in 24-well plates were transiently transfected with or without FLAG-TIPE2, TIPE2 siRNA, and control siRNA. After 24 h, the cells were treated for an additional 24 h under serum-free culture conditions. Then the cell culture supernatants were harvested at 8 h after LPS at 100 ng/ml treatment, and IL-6 production in the supernatants was measured using an ELISA system. The results are expressed as means ± standard deviation, in triplicate. *, p < 0.05. C, RAW246.7 cells in 24-well plates were transiently transfected with or without FLAG-TIPE2, FLAG-TIPE2 101–140, and NF-κB reporter construct. After 24 h, the cells were treated for an additional 24 h under serum-free culture conditions. Then cells were treated for 5 h with or without LPS at 100 ng/ml. The luciferase experiments were performed as described above. The results are expressed as means ± standard deviation, in triplicate. *, p < 0.05. D, HEK293T cells in 24-well plates were transiently transfected with or without FLAG-TIPE2 or FLAG-TIPE2 101–140 and NF-κB reporter construct. After 24 h, the cells were incubated for an additional 24 h under serum-free culture conditions. Then cells were treated for 5 h with or without TNF-α at 10 ng/ml. The luciferase experiments were performed as described above. The results are expressed as means ± standard deviation, in triplicate. *, p < 0.05.
Cell-permeable TIPE2 Inhibits LPS-stimulated Gene Expression of Inflammatory Cytokines
Jo et al. (22) suggested that cell-permeable MTM suppressor of cytokine signaling 3 is a useful candidate for intracellular protein therapy for JAK-STAT-mediated inflammatory diseases. Therefore, we speculated that MTM-TIPE2 is a target for the development of intracellular protein therapy for TAK1-related inflammatory diseases. Thus, we designed an MTM-TIPE2 gene construct and developed its recombinant protein. We then explored the inhibitory ability of MTM-TIPE2 on NF-κB reporter activity and gene expression of inflammatory cytokines in LPS-stimulated RAW264.7 cells. First, using confocal laser scanning microscopy, we observed the delivery of MTM-TIPE2 into the cytoplasm of RAW264.7 cells. As shown in Fig. 5A, FITC-labeled MTM-TIPE2 was detected abundantly in cytoplasm of most cells, although some TIPE2 without MTM was observed in the cells. Penetration of MTM-TIPE2 into the cytoplasm was supported by flow cytometry analysis (Fig. 5B). We then used a His pulldown assay to examine whether MTM-TIPE2 sustains its characteristics and interacts with TAK1. MTM-TIPE2 was incubated with cell lysates from FLAG-TAK1-overexpressed RAW264.7 cells, and interaction of MTM-TIPE2 with FLAG-TAK1 was detected by His pulldown and Western blotting assays. Fig. 5C shows that MTM-TIPE2 preserves its ability to interact with TAK1 in the manner of native TIPE2. Therefore, we attempted to determine whether MTM-TIPE2 is able to inhibit NF-κΒ reporter activity and gene expression of inflammatory cytokines in LPS-stimulated cells. Using FLAG-TIPE2-overexpressed cells as a positive control, we observed that MTM-TIPE2 also significantly inhibited LPS-stimulated reporter activity of NF-κB in the cells (Fig. 5D). The inhibitory action of MTM-TIPE2 also was observed in poly(I·C)- and CpG DNA-treated cells (data not shown). Because these results suggest that MTM-TIPE2 might inhibit gene expression of proinflammatory cytokines in cells stimulated with TLR ligands, we used qRT-PCR to examine the inhibitory effect of MTM-TIPE2 on gene expression of proinflammatory cytokines in LPS-stimulated cells. As hypothesized, MTM-TIPE2 significantly inhibited gene expression of proinflammatory cytokines (IL-1β, IL-6) in LPS-stimulated cells (Fig. 5, E and F). MTM-TIPE2 also inhibited gene expression of proinflammatory cytokines in poly(I·C)- and CpG DNA-treated cells (data not shown). Collectively, these results suggest the possibility that cell-permeable MTM-TIPE2 could be a potential tool for intracellular protein therapy for TAK1-related inflammatory diseases.
FIGURE 5.
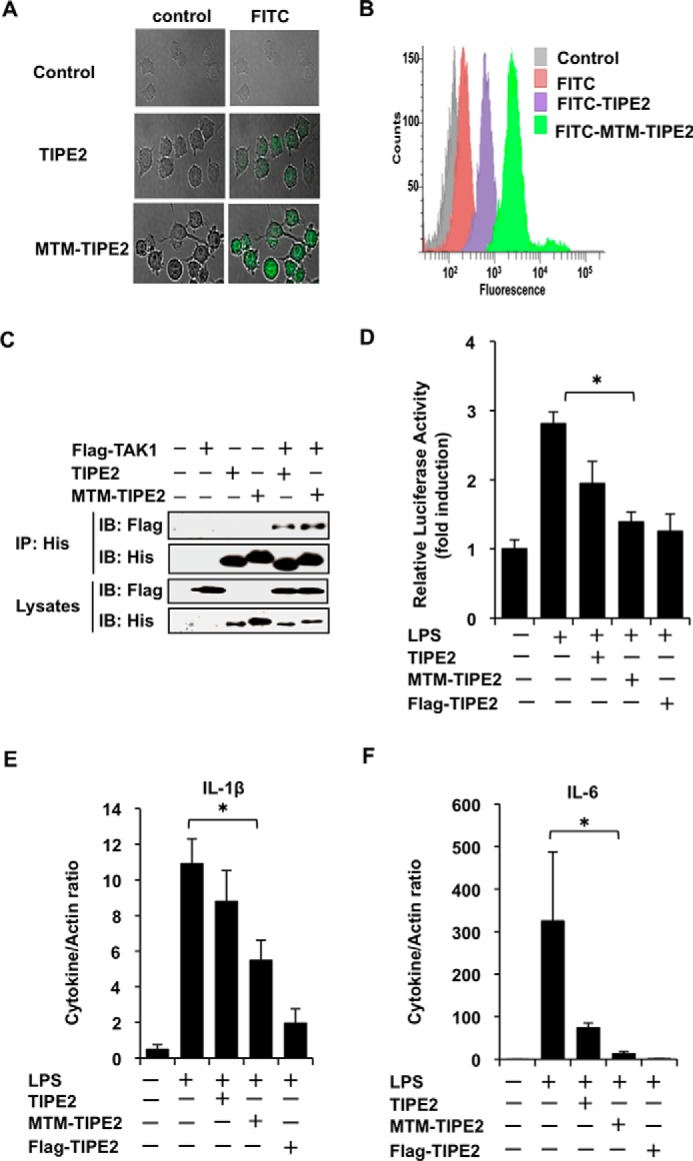
MTM-TIPE2 inhibits LPS-stimulated NF-κB activity and gene expression of inflammatory cytokines in vitro. A and B, RAW246.7 cells were incubated for 30 min in the absence or presence of FITC-labeled MTM-TIPE2 or FITC-labeled TIPE2 at 1 μm, and subsequently the cells were treated for 15 min with proteinase K. Then the cells were observed by confocal microscopy (A) and analyzed by flow cytometry (B). C, the cells were transiently transfected with or without FLAG-TAK1. After 24 h, the cells were incubated for 1 h with or without His-MTM-TIPE2 or His-TIPE2 at 1 μm. The mixtures were used for the His pulldown assay. The bound proteins were detected by Western blotting assay using anti-FLAG and anti-His antibodies. D, cells in 24-well plates were transiently transfected with or without NF-κB reporter construct and then were incubated for an additional 24 h under serum-free culture conditions. Then cells were incubated for 1 h with or without MTM-TIPE2 or TIPE2 at 1 μm. The treated cells were then treated with or without LPS at 100 ng/ml. The cells were lysed after 5 h, and NF-κB reporter activity was measured. FLAG-TIPE2-transfected cells were used as the positive control. The results are expressed as means ± standard deviation in triplicate. *, p < 0.05. E and F, the cells were cultured for 24 h under serum-free culture conditions and then incubated for 1 h with or without MTM-TIPE2 or TIPE2 at 1 μm. Then the cells were treated with LPS at 100 ng/ml. After 2 h, mRNA was prepared, and gene expression of the indicated cytokines was analyzed by qRT-PCR. FLAG-TIPE2-transfected cells were used as the positive control. The results are expressed as means ± standard deviation in triplicate. *, p < 0.05. IB, immunoblot; IP, immunoprecipitation.
Discussion
Although caspase 8 is one of the target molecules of TIPE2 (9), in this study we found that TAK1was another novel target molecule of TIPE2. For the interaction between TIPE2 and TAK1, we demonstrated that the TIPE2 aa 101–140 region interacted via binding with the TAK1 aa 201–291 region in its internal kinase domain. Importantly, TIPE2 inhibited formation of the TAK1-TAB1-TAB2 complex, and subsequently interfered with TAK1 signaling. Silencing of TIPE2 by RNA interference eliminated its inhibitory action on TAK1 signaling. Therefore, the present study is the first to demonstrate that TIPE2 is a novel negative regulator of TAK1-mediated signals. In addition, because TAK1 plays a pivotal role in inflammatory and immune signals, this finding also suggested the possibility that TIPE2 acts as a negative regulator of these signal systems.
Many studies (25, 33–38) have indicated that formation of the TAK1-TAB1-TAB2 complex is an important cellular event in the initiation of TAK1 activation and subsequently induces activation of MAP2Ks, such as MKK4/7 and MKK3/6, and NF-κB. Although TAB1 is constitutively associated with TAK1 in resting cells and TAB2 is bound to the membrane in the absence of TAK1 ligands, with ligand stimulation, the membrane-binding TAB2 migrates to the cytoplasm and is associated with TRAF6 to trigger TAK1 activation. Therefore, TAB1-TAB2-TRAF6 binding to TAK1 is an important event for its activation. Recently, an interesting study (28) demonstrated that O-GlcNAcylation of TAB1 on Ser395 was essential for TAK1 activation. In contrast, the negative regulatory mechanism of TAK1 following its ligand stimulation was largely unknown. Based on the requirement for TAB1-dependent phosphorylation of TAK1 in induction of its activation, Kim et al. (39) demonstrated that PP2A, a Ser/Thr protein phosphatase, functions as a negative regulator in TGF-β1-stimulated TAK1 activation via binding with TAK1 and TAB1. Mochida et al. (40) also demonstrated that apoptosis signal-regulating kinase 1, a member of the MAPK kinase kinase family, inhibits the activation of NF-κB induced by IL-1 through disruption of TRAF6-TAK1 interaction. In addition, most recently, it has demonstrated that S6K1 interacts with the N terminus of TAK1 and negatively regulates the activation of TAK1 through competition with the TAK1-TAB1 interaction (41). Through biochemical and molecular studies, we showed here that exogenous TIPE2 attenuated the binding of TAB1 and TAB2 to TAK1 and inhibited TAB-dependent induction of its phosphorylation and ubiquitination, consequently negatively regulating its activation. Although we do not yet completely understand the molecular mechanism of TIPE2 inhibition of the binding of TAB1 and TAB2 to TAK1, we hypothesize that the inhibitory action of TIPE2 may result from 1) the conformational changes of TAK1 structure through the interaction with TIPE2 and 2) competition of TIPE2 with TAB1 or TAB2 for interaction with TAK1, because the TAB1 aa 437–481 region binds to the internal kinase domain of TAK1, which is the same region (aa 101–140) that interacts with TIPE2. Collectively, as illustrated in Fig. 6, although the TLR ligands, TNF-α and IL-1 activate both the MAPKs and NF-κB via TAK1 activation, TIPE2 inhibited activation of their signal molecules via inhibiting a series of cellular events in TAK1 activation. Thus, we propose that TIPE2 functions as a novel potential negative regulator of TAK1 signaling.
FIGURE 6.
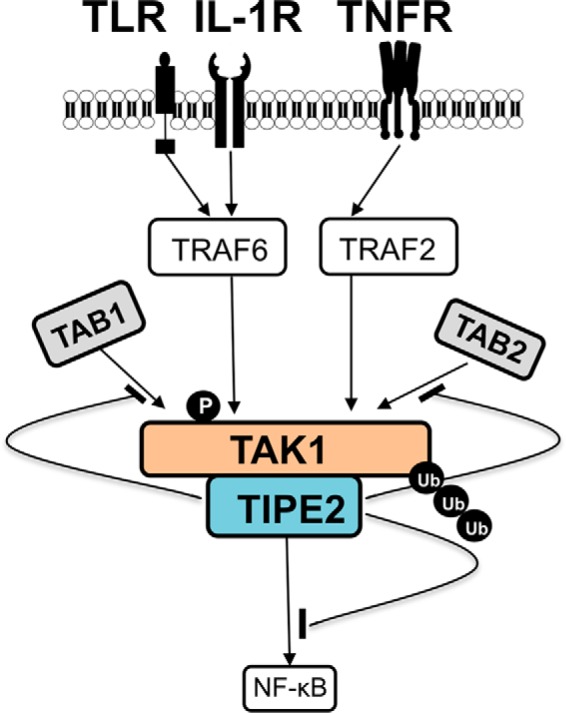
A possible model of TIPE2 inhibition of activation of TAK1 signaling. When TLR, IL-1R, and TNFR are stimulated via binding with their specific ligands, TAK1 is activated through subsequent downstream molecular events in the TAK1 signaling pathway. This study demonstrates that TIPE2 (aa 101–140) interacts with TAK1 via binding to the internal kinase domain (aa 200–291) of TAK1. Consequently, formation of the TAK1-TAB1-TAB2 complex is inhibited, as are the subsequent phosphorylation and ubiquitination of TAK1. Therefore, the negative regulation by TIPE2 of TAK1 activation may contribute to suppression of TAK1-NF-κB-mediated inflammatory responses.
MAPKs (MKK3/6, p38, ERK, and JNK), IκB, and NF-κB in the TAK1 signaling pathway play fundamental roles in cellular events mediated by the TAK1 signal. TIPE2 inhibited TLR ligand-stimulated phosphorylation of the MAPKs and proinflammatory cytokines and also inhibited NF-κB activity via suppressing phosphorylation of IκB. Consequently, TIPE2 inhibited ligand-stimulated gene expression and production of proinflammatory cytokines. Interestingly, although we demonstrated that exogenous TIPE2 101–140, the region that interacts with TAK1, was able to dramatically inhibit LPS- and TNF-α-stimulated reporter activity of NF-κB in RAW264.7 and HEK293T cells, these observations indicated that the TIPE2 binding region was involved in its function as a negative regulator of TAK1 signaling. Because activation of TAK1 is induced by proinflammatory cytokines, TLR ligands, and growth factors, the inhibitory action of TIPE2 on TAK1 signaling suggests that TIPE2 behaves as a potential negative regulator of a large number of cellular responses evoked by changes in the cell environment, such as inflammation, infectious diseases, cancer, apoptosis, and bone metabolism. Based on these findings, TIPE2 will be a pivotal target molecule in the development of novel drugs for diseases mediated by TAK1. In particular, TIPE2 101–140 protein will be a primary candidate during drug development.
The signal peptide-fused membrane transducing technique is a useful method for transduction of signal molecules into the cytoplasm and is a potential therapy for inhibition of inflammatory and autoimmune diseases (22, 42). Interestingly, by transducing cell-permeable Yamanaka factors into MEFs, Zhou et al. (43) developed the generation of inducible pluripotent stem cells. Although methods for transducing genes that encode target proteins using retroviral and lentiviral vectors have been widely investigated, there are several questions concerning transfection efficiency and safety for human trials. These studies have suggested that cell-permeable technology for transducing signal regulatory molecules or transcription factors into many kinds of cells is a useful strategy for developing intracellular protein therapy for TAK1-mediated diseases. Therefore, our interest was in developing cell-permeable TIPE2 (MTM-TIPE2) as an intracellular protein therapy for TAK1-mediated diseases. As described here, MTM-TIPE2 was rapidly delivered into the cytoplasm of most cells and interacted with TAK1 in the cells. MTM-TIPE2 transduction into RAW264.7 cells clearly inhibited NF-κB activity and gene expression of proinflammatory cytokines in cells stimulated by LPS. These observations strongly indicate that MTM-TIPE2 sufficiently maintained the negative regulatory activities of native TIPE2 in vitro. In addition, as described here, because exogenous TIPE2 101–140 sufficiently acted as a potent inhibitor of TAK1 ligand signals in vitro, in further studies, we intend to explore and to determine whether cell-permeable TIPE2 and TIPE2 101–140 protein will be useful as a potential intracellular protein therapy for inflammatory diseases and cancer mediated by TAK1 signaling in vivo.
In conclusion, we demonstrate here that TIPE2 is a novel negative regulator of TAK1 signal. Also we propose that cell-permeable MTM-TIPE2 will be a powerful candidate for intracellular protein therapy in TAK1 signal-mediated diseases.
Author Contributions
S. H. and Y. M. conceived and designed the study. S. H. wrote the paper. M. O., Risa N., Ryutarou N., W. S., and A. M. constructed vectors and analyzed the biochemical experiments by immunoblots, IP Western, qRT-PCR, ELISA, and luciferase assays in Figs. 1–4. M. O. performed immunostaining in Fig. 1. Ryutarou N. performed expression and purification of cell-permeable TIPE2 proteins and analyzed the experiments in Fig. 5. All authors reviewed the results and the final version of the manuscript.
Acknowledgments
We are grateful to Drs. K. Matsumoto (Nagoya University), Y. Ohmori (Meikai University), and K. Nishida (Suzuka University of Medical Science) for providing plasmids. We thank Drs. T. Narita and H. Sugiya (Nihon University) for technical advice. We also thank all Hanazawa lab members for discussion and assistance.
This work was supported in part by a grant-in-aid for science research from the ministry of Education, Culture, Sports, Science and Technology of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- TAK1
- TGF-β-activated kinase 1
- MTM
- membrane translocating motif
- qRT-PCR
- quantitative real-time PCR
- aa
- amino acid(s)
- TRAF
- TNF receptor-associated factor
- IRAK
- IL-1 receptor-associated kinase
- TIPE2
- tumor necrosis factor α-induced protein 8-like 2
- TNFAIP8
- TNF-α-induced protein 8
- TLR
- toll-like receptor
- TCR
- T cell receptor
- TAB
- TAK1-binding protein.
References
- 1. Woodward M. J., de Boer J., Heidorn S., Hubank M., Kioussis D., Williams O., and Brady H. J. (2010) Tnfaip8 is an essential gene for the regulation of glucocorticoid-mediated apoptosis of thymocytes. Cell Death Differ. 17, 316–323 [DOI] [PubMed] [Google Scholar]
- 2. Lou Y., and Liu S. (2011) The TIPE (TNFAIP8) family in inflammation, immunity, and cancer. Mol. Immunol. 49, 4–7 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y., Wang M. Y., He J., Wang J. C., Yang Y. J., Jin L., Chen Z. Y., Ma X. J., Sun M. H., Xia K. Q., Hong X. N., Wei Q. Y., and Zhou X. Y. (2012) Tumor necrosis factor-α induced protein 8 polymorphism and risk of non-Hodgkin's lymphoma in a Chinese population: a case-control study. PLoS One 7, e37846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi T. Y., Cheng X., Yu K. D., Sun M. H., Shao Z. M., Wang M. Y., Zhu M. L., He J., Li Q. X., Chen X. J., Zhou X. Y., Wu X., and Wei Q. (2013) Functional variants in TNFAIP8 associated with cervical cancer susceptibility and clinical outcomes. Carcinogenesis 34, 770–778 [DOI] [PubMed] [Google Scholar]
- 5. Cao X., Zhang L., Shi Y., Sun Y., Dai S., Guo C., Zhu F., Wang Q., Wang J., Wang X., Chen Y. H., and Zhang L. (2013) Human tumor necrosis factor (TNF)-α-induced protein 8-like 2 suppresses hepatocellular carcinoma metastasis through inhibiting Rac1. Mol. Cancer 12, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y., Wei X., Liu L., Liu S., Wang Z., Zhang B., Fan B., Yang F., Huang S., Jiang F., Chen Y. H., and Yi F. (2012) TIPE2, a novel regulator of immunity, protects against experimental stroke. J. Biol. Chem. 287, 32546–32555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ha J. Y., Kim J. S., Kang Y. H., Bok E., Kim Y. S., and Son J. H. (2014) Tnfaip8 l1/Oxi-β binds to FBXW5, increasing autophagy through activation of TSC2 in a Parkinson's disease model. J. Neurochem. 129, 527–538 [DOI] [PubMed] [Google Scholar]
- 8. Zhang S., Zhang Y., Wei X., Zhen J., Wang Z., Li M., Miao W., Ding H., Du P., Zhang W., He M., and Yi F. (2010) Expression and regulation of a novel identified TNFAIP8 family is associated with diabetic nephropathy. Biochim. Biophys. Acta 1802, 1078–1086 [DOI] [PubMed] [Google Scholar]
- 9. Sun H., Gong S., Carmody R. J., Hilliard A., Li L., Sun J., Kong L., Xu L., Hilliard B., Hu S., Shen H., Yang X., and Chen Y. H. (2008) TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell 133, 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gus-Brautbar Y., Johnson D., Zhang L., Sun H., Wang P., Zhang S., Zhang L., and Chen Y. H. (2012) The anti-inflammatory TIPE2 is an inhibitor of the oncogenic Ras. Mol. Cell 45, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lou Y., Liu S., Zhang C., Zhang G., Li J., Ni M., An G., Dong M., Liu X., Zhu F., Zhang W., Gao F., Chen Y. H., and Zhang Y. (2013) Enhanced atherosclerosis in TIPE2-deficient mice is associated with increased macrophage responses to oxidized low-density lipoprotein. J. Immunol. 191, 4849–4857 [DOI] [PubMed] [Google Scholar]
- 12. Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., and Matsumoto K. (1999) The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256 [DOI] [PubMed] [Google Scholar]
- 13. Shim J. H., Greenblatt M. B., Xie M., Schneider M. D., Zou W., Zhai B., Gygi S., and Glimcher L. H. (2009) TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 28, 2028–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goswami M., Uzgare A. R., and Sater A. K. (2001) Regulation of MAP kinase by the BMP-4/TAK1 pathway in Xenopus ectoderm. Dev. Biol. 236, 259–270 [DOI] [PubMed] [Google Scholar]
- 15. Delaney J. R., and Mlodzik M. (2006) TGF-β activated kinase-1: new insights into the diverse roles of TAK1 in development and immunity. Cell Cycle 5, 2852–2855 [DOI] [PubMed] [Google Scholar]
- 16. Sakurai H. (2012) Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharm. Sci 33, 522–530 [DOI] [PubMed] [Google Scholar]
- 17. Mahmud S. A., Manlove L. S., Schmitz H. M., Xing Y., Wang Y., Owen D. L., Schenkel J. M., Boomer J. S., Green J. M., Yagita H., Chi H., Hogquist K. A., and Farrar M. A. (2014) Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 15, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun L., Deng L., Ea C. K., Xia Z. P., and Chen Z. J. (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 [DOI] [PubMed] [Google Scholar]
- 19. Blonska M., Pappu B. P., Matsumoto R., Li H., Su B., Wang D., and Lin X. (2007) The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity 26, 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schuman J., Chen Y., Podd A., Yu M., Liu H. H., Wen R., Chen Z. J., and Wang D. (2009) A critical role of TAK1 in B-cell receptor-mediated nuclear factor κB activation. Blood 113, 4566–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shinohara H., and Kurosaki T. (2009) Comprehending the complex connection between PKCβ, TAK1, and IKK in BCR signaling. Immunol. Rev. 232, 300–318 [DOI] [PubMed] [Google Scholar]
- 22. Jo D., Liu D., Yao S., Collins R. D., and Hawiger J. (2005) Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 11, 892–898 [DOI] [PubMed] [Google Scholar]
- 23. Ishida H., Masuhiro Y., Fukushima A., Argueta J. G., Yamaguchi N., Shiota S., and Hanazawa S. (2005) Identification and characterization of novel isoforms of human DP-1: DP-1α regulates the transcriptional activity of E2F1 as well as cell cycle progression in a dominant-negative manner. J. Biol. Chem. 280, 24642–24648 [DOI] [PubMed] [Google Scholar]
- 24. Masuhiro Y., Kayama K., Fukushima A., Baba K., Soutsu M., Kamiya Y., Gotoh M., Yamaguchi N., and Hanazawa S. (2008) SOCS-3 inhibits E2F/DP-1 transcriptional activity and cell cycle progression via interaction with DP-1. J. Biol. Chem. 283, 31575–31583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sakurai H., Miyoshi H., Mizukami J., and Sugita T. (2000) Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS Lett. 474, 141–145 [DOI] [PubMed] [Google Scholar]
- 26. Ishitani T., Takaesu G., Ninomiya-Tsuji J., Shibuya H., Gaynor R. B., and Matsumoto K. (2003) Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 22, 6277–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walsh M. C., Kim G. K., Maurizio P. L., Molnar E. E., and Choi Y. (2008) TRAF6 autoubiquitination-independent activation of the NFκB and MAPK pathways in response to IL-1 and RANKL. PLoS One 3, e4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pathak S., Borodkin V. S., Albarbarawi O., Campbell D. G., Ibrahim A., and van Aalten D. M. (2012) O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J. 31, 1394–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takaesu G., Kishida S., Hiyama A., Yamaguchi K., Shibuya H., Irie K., Ninomiya-Tsuji J., and Matsumoto K. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 5, 649–658 [DOI] [PubMed] [Google Scholar]
- 30. Shibuya H., Yamaguchi K., Shirakabe K., Tonegawa A., Gotoh Y., Ueno N., Irie K., Nishida E., and Matsumoto K. (1996) TAB1: an activator of the TAK1 MAPKKK in TGF-β signal transduction. Science 272, 1179–1182 [DOI] [PubMed] [Google Scholar]
- 31. Shuto T., Xu H., Wang B., Han J., Kai H., Gu X. X., Murphy T. F., Lim D. J., and Li J. D. (2001) Activation of NF-κB by nontypeable Hemophilus influenzae is mediated by Toll-like receptor 2-TAK1-dependent NIK-IKK α/β-IκBα and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 98, 8774–8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., Hayden M. S., Lee K. Y., Bussey C., Steckel M., Tanaka N., Yamada G., Akira S., Matsumoto K., and Ghosh S. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muñoz-Sanjuán I., Bell E., Altmann C. R., Vonica A., and Brivanlou A. H. (2002) Gene profiling during neural induction in Xenopus laevis: regulation of BMP signaling by post-transcriptional mechanisms and TAB3, a novel TAK1-binding protein. Development 129, 5529–5540 [DOI] [PubMed] [Google Scholar]
- 34. Kishimoto K., Matsumoto K., and Ninomiya-Tsuji J. (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 275, 7359–7364 [DOI] [PubMed] [Google Scholar]
- 35. Kurahashi T., Nomura T., Kanei-Ishii C., Shinkai Y., and Ishii S. (2005) The Wnt-NLK signaling pathway inhibits A-Myb activity by inhibiting the association with coactivator CBP and methylating histone H3. Mol. Biol. Cell 16, 4705–4713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C. H., and Landström M. (2008) The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 37. Ray D. M., Rogers B. A., Sunman J. A., Akiyama S. K., Olden K., and Roberts J. D. (2010) Lysine 63-linked ubiquitination is important for arachidonic acid-induced cellular adhesion and migration. Biochem. Cell Biol. 88, 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fan Y., Shi Y., Liu S., Mao R., An L., Zhao Y., Zhang H., Zhang F., Xu G., Qin J., and Yang J. (2012) Lys48-linked TAK1 polyubiquitination at lysine-72 downregulates TNFα-induced NF-κB activation via mediating TAK1 degradation. Cell Signal. 24, 1381–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S. I., Kwak J. H., Wang L., and Choi M. E. (2008) Protein phosphatase 2A is a negative regulator of transforming growth factor-β1-induced TAK1 activation in mesangial cells. J. Biol. Chem. 283, 10753–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mochida Y., Takeda K., Saitoh M., Nishitoh H., Amagasa T., Ninomiya-Tsuji J., Matsumoto K., and Ichijo H. (2000) ASK1 inhibits interleukin-1-induced NF-κB activity through disruption of TRAF6-TAK1 interaction. J. Biol. Chem. 275, 32747–32752 [DOI] [PubMed] [Google Scholar]
- 41. Kim S. Y., Baik K. H., Baek K. H., Chah K. H., Kim K. A., Moon G., Jung E., Kim S. T., Shim J. H., Greenblatt M. B., Chun E., and Lee K. Y. (2014) S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway. Mol. Cell Biol. 34, 510–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi J. M., Shin J. H., Sohn M. H., Harding M. J., Park J. H., Tobiasova Z., Kim D. Y., Maher S. E., Chae W. J., Park S. H., Lee C. G., Lee S. K., and Bothwell A. L. (2010) Cell-permeable Foxp3 protein alleviates autoimmune disease associated with inflammatory bowel disease and allergic airway inflammation. Proc. Natl. Acad. Sci. U.S.A. 107, 18575–18580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou H., Wu S., Joo J. Y., Zhu S., Han D. W., Lin T., Trauger S., Bien G., Yao S., Zhu Y., Siuzdak G., Schöler H. R., Duan L., and Ding S. (2009) Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]