

Abstract
Neurofibrillar tangles caused by intracellular hyperphosphorylated tau inclusion and extracellular amyloid β peptide deposition are hallmarks of Alzheimer's disease. Tau contains one or two cysteine residues in three or four repeats of the microtubule binding region following alternative splicing of exon 10, and formation of intermolecular cysteine disulfide bonds accelerates tau aggregation. 8-Nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) acts as a novel second messenger of nitric oxide (NO) by covalently binding cGMP to cysteine residues by electrophilic properties, a process termed protein S-guanylation. Here we studied S-guanylation of tau and its effects on tau aggregation. 8-Nitro-cGMP exposure induced S-guanylation of tau both in vitro and in tau-overexpressed HEK293T cells. S-guanylated tau inhibited heparin-induced tau aggregation in a thioflavin T assay. Atomic force microscopy observations indicated that S-guanylated tau could not form tau granules and fibrils. Further biochemical analyses showed that S-guanylated tau was inhibited at the step of tau oligomer formation. In P301L tau-expressing Neuro2A cells, 8-nitro-cGMP treatment significantly reduced the amount of sarcosyl-insoluble tau. NO-linked chemical modification on cysteine residues of tau could block tau aggregation, and therefore, increasing 8-nitro-cGMP levels in the brain could become a potential therapeutic strategy for Alzheimer's disease.
Keywords: Alzheimer disease, chemical modification, oxidative stress, protein aggregation, Tau protein (Tau), tauopathy, 8-nitro-cGMP, S-guanylation, cysteine modification
Introduction
The intracellular accumulation of tau protein that gives rise to neurofibrillary tangles (NFTs),2 accompanied by neuronal loss as well as the deposition of amyloid β (Aβ) peptides in the extracellular space forming senile plaques, are key pathological hallmarks of Alzheimer's disease (AD) (1). Anatomical studies have revealed a strong correlation between a decline in cognitive function and NFTs that have spread from the entorhinal cortex to the limbic system and neocortex (2). Moreover, the fact that the extent of NFT formation correlates with that of neuronal loss (3) suggests that the formation of NFTs is a key factor influencing the occurrence of dementia in AD. The importance of NFT formation is also supported by the discovery of tau gene mutation in cases of frontotemporal dementia with parkinsonism (FTDP)-17 (4). These patients showed evidence of NFTs and neuronal loss without Aβ deposition. Mice expressing this human mutant tau exhibit evidence of NFTs, neuronal loss, and behavioral abnormalities (5). Experiments involving the expression of the human P301L transgene in mice (6) or the human FTDP-17 mutation in fruit flies (7) link neuronal death to tau aggregation rather than to NFT formation. This suggests that tau aggregation is associated with neuronal loss.
Tau aggregation is a three-step process. First, tau molecules bind to each other, forming soluble oligomers. These oligomers of ∼40 tau molecules then grow and precipitate as granular tau oligomers with a β sheet structure. Last, the granular tau oligomers bind to each other and form tau fibrils. Intermolecular tau interaction is thought to involve two distinct mechanisms: cysteine-dependent and cysteine-independent interactions. In polyanion-induced tau aggregation such as by heparin, cysteine-dependent interactions occur before β sheet aggregation or cysteine-independent interactions, suggesting that intermolecular disulfide bonds between cysteine residues trigger tau oligomer formation accompanied by cysteine-independent tau interaction (8–10).
Nitric oxide (NO) produced by NO synthase (NOS) is a well established endogenous gaseous mediator involved in various physiological roles, including vascular regulation (11), neuronal transmission (12), and host defense against microbe invasion (13). NO also modifies cyclic GMP to form nitrated cGMP (8-nitro-cGMP) (14). Electrophilic 8-nitro-cGMP reacts with the thiolate anion of cysteine to form a cysteine-cGMP adduct (14), a process termed S-guanylation, concerning antioxidant-adaptive responses (15) and autophagy (16). Because cysteine residues of tau play a critical role in tau aggregation, we sought to investigate the effect of tau S-guanylation by 8-nitro-cGMP on tau aggregation both in vitro and in vivo.
Results
S-guanylation of Tau in Vitro
To investigate whether tau can be a substrate for 8-nitro-cGMP, we prepared recombinant wild-type 2N3R and 2N4R and their corresponding cysteine-to-alanine substituted mutants. The recombinant tau (10 μm) samples were incubated with 8-nitro-cGMP (100 μm) at 37 °C for 16 h. After removing excess 8-nitro-cGMP from the reaction mixture, S-guanylated tau was subjected to immunoblot analysis and probed using S-guanylated protein-specific antibody (RS-cGMP) or anti-tau monoclonal antibody (HT-7). S-guanylated BSA was used as a positive control for RS-cGMP. RS-cGMP showed a positive signal in 8-nitro-cGMP-treated wild-type 2N3R (Wild-3R) and 2N4R (Wild-4R) tau but not in vehicle-treated 2N3R and 2N4R tau or 8-nitro-cGMP-treated cysteine-to-alanine substitution mutant tau (Mutant-3R, Mutant-4R) (Fig. 1A). To confirm that cGMP binds cysteine residues in tau via 8-nitro-cGMP, a partial cysteine-containing peptide of tau, R3′ (SKVTSKCGSLGN, molecular weight (MW) = 1,180.3), was analyzed by mass spectrometry (Fig. 1B) after incubation with either the vehicle or the 8-nitro-cGMP (MW = 390). Chromatogram of the R3′ peptide incubated with the vehicle exhibited a single peak eluting around 5 min in which mass spectrometry yielded a signal (m/z = 1180.7). During incubation with 8-nitro-cGMP (0.4 mm), the chromatogram showed a distinct peak eluting around 7 min as well as an R3′ peptide peak (elution at 5 min), and the peak height at 7 min was higher, when the R3′ peptide was incubated with a higher concentration of 8-nitro-cGMP (2 mm). Mass spectrometry of the R3′ peptide incubated with 8-nitro-cGMP yielded a signal (m/z = 1523.8). The difference in mass of R3′ peptide by incubation with 8-nitro-cGMP (plus 343.1 of R3′ peptide mass) is due to the cGMP adduct at the cysteine residue of the R3′ peptide (Fig. 1C). These results suggest that 8-nitro-cGMP modulates the cysteine residues of tau and forms S-guanylated tau in vitro.
FIGURE 1.
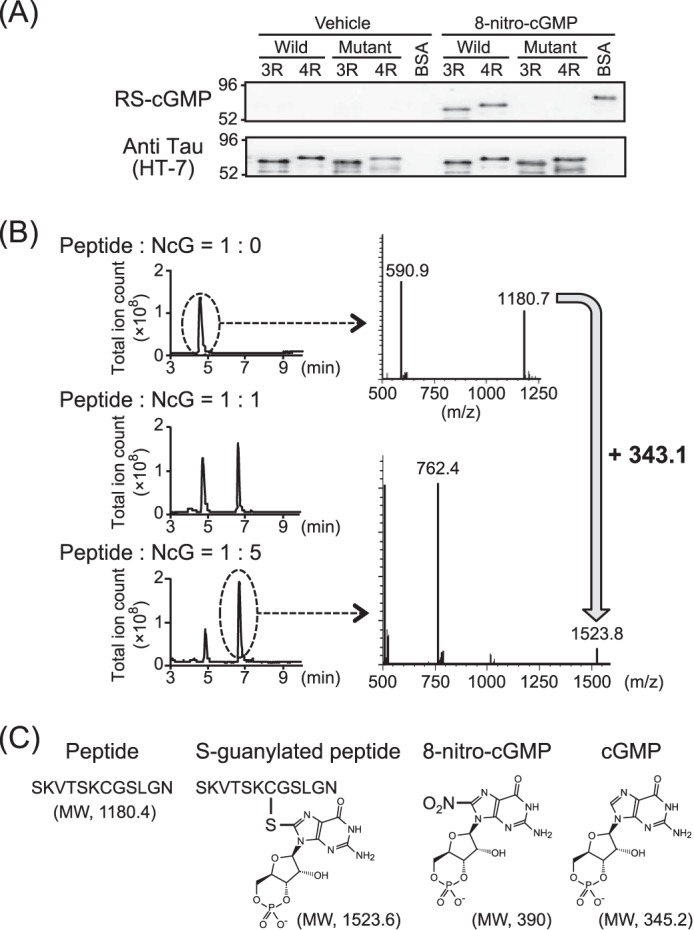
S-guanylation of recombinant tau by 8-nitro-cGMP in vitro. A, recombinant wild-type 2N3R and 2N4R and their corresponding cysteine-to-alanine substituted mutants were treated with 8-nitro-cGMP and subjected to immunoblot analysis with HT-7 or RS-cGMP antibodies. BSA was simultaneously treated with 8-nitro-cGMP as a positive control of S-guanylation. Wild-type tau isomers are indicated as Wild-3R (2N3R) and Wild-4R (2N4R). Cysteine-to-alanine substituted mutants are indicated as Mutant-3R (C291A-2N3R) and Mutant-4R (C291, 322A-2N4R). The experiment was repeated at least three times, and the same results were observed each time. B, the peptide SKVTSKCGSLGN containing the cysteine residue of the tau microtubule binding domain reacted with 0 mm (left panel, top), 0.4 mm (left panel, center), and 2.0 mm (left panel, bottom) 8-nitro-cGMP (NcG). The total ion monitoring chromatogram (positive scan mode (m/z, 500–1600)) of the reaction mixture (left panel) and the mass spectrum of the peak at about 5 and 7 min (right panel) are shown. C, the structure and the molecular weight of related materials using for S-guanylation of peptide are indicated.
S-guanylation of Tau in Tau-expressing HEK293T Cells
Tau can be modulated by S-guanylation. To confirm this chemical modification by 8-nitro-cGMP in HEK293T cells, we overexpressed 2N3R and 2N4R tau in HEK293T cells and treated these cells with 8-nitro-cGMP (200 μm) for 6 h at 37 °C. After treatment, cell lysates were boiled for 15 min, and the heat-stable fraction was used for subsequent immunoprecipitation. Partially purified tau in the heat-stable fraction was immunoprecipitated using RS-cGMP antibody and probed using HT-7 antibody. In 8-nitro-cGMP-treated cells (+), S-guanylated tau was detected at the predicted size but not in vehicle-treated cells (−) (Fig. 2A).
FIGURE 2.

Intracellular S-guanylation of tau and His-4rp by 8-nitro-cGMP. 2N3R and 2N4R tau isomers and the histidine-tagged microtubule binding region (four-repeat) fragment (His-4rp) were transiently expressed in HEK293T cells and cultured in the presence or absence of 8-nitro-cGMP. A, 2N3R and 2N4R tau were S-guanylated under intracellular conditions by 8-nitro-cGMP. S-guanylated tau was immunoprecipitated (IP) with RS-cGMP antibody from the heat-stable cell lysate fraction and then visualized using HT-7 antibody. B, the intracellular microtubule binding region fragment of tau was S-guanylated by 8-nitro-cGMP. S-guanylated His-4rp was trapped by Ni-NTA-agarose from the heat-stable cell lysate fraction and detected by immunoblotting using RS-cGMP antibody. The quantity of total tau or His-4rp was confirmed by immunoblotting the heat-stable fraction using HT-7 and 2B11, respectively (input).
To confirm this result, we expressed a histidine tag-conjugated microtubule binding region of 2N4R tau (His-4rp, residues 252–376 of the longest tau) and treated HEK293T cells with 8-nitro-cGMP (200 μm) for 6 h at 37 °C. His-4rp tau was purified by Ni-NTA-agarose from the heat-stable cell lysate fraction. 8-Nitro-cGMP-treated cells specifically showed an RS-cGMP antibody-positive band of around 18 kDa compared with vehicle treatment. Tau antibody for the microtubule binding region (2B11) showed a positive band at the 18 kDa band, a size similar to that of the RS-cGMP antibody, in both vehicle- and 8-nitro-cGMP-treated cells (Fig. 2B). These results suggest that the cysteine residues of tau are able to undergo S-guanylation in vivo.
The Effect of S-guanylation on Tau Aggregation
Next we investigated whether S-guanylation of tau affects tau aggregation. S-guanylated tau was prepared by 8-nitro-cGMP treatment in recombinant 2N3R or 2N4R tau. After confirming S-guanylation of tau as shown in Fig. 1, 10 μm (2N4R) and 5 μm (2N3R) modified and unmodified tau were incubated with heparin sodium. Tau aggregation was monitored by fluorescence of thioflavin T (ThT). As shown in Fig. 3A, unmodified 3R and 4R tau showed a stark increase in ThT fluorescence upon incubation with heparin sodium and subsequently reached a plateau after 24 h. Conversely, both S-guanylated 3R and 4R tau did not show any increase in ThT fluorescence even after 24 h of incubation, suggesting that cysteine modification by 8-nitro-cGMP inhibits tau aggregation. To confirm this result, the aggregation mixture was visualized using atomic force microscopy (AFM). Wild-type 2N4R tau aggregation showed granular and fibrous tau precipitation, whereas S-guanylated tau showed minimal evidence of precipitates (Fig. 3B). These results indicate that modification of S-guanylation inhibits tau aggregation to form β sheet structures. We next aimed to elucidate the specific tau aggregation process inhibited by S-guanylation. As it has been reported previously that tau oligomer formation occurs before the increase in ThT fluorescence (8), we examined 2N3R and 2N4R tau oligomer formation in the aggregation mixture for 4 h after incubation with heparin sodium. Wild-type tau started oligomer formation after 30 min of incubation and formed multimers after 4 h of incubation, whereas S-guanylated tau formed monomers, dimers, and some trimers even before incubation. This did not change after longer incubation with heparin (Fig. 3, D and E). As a normal tau function, the effect of S-guanylated tau on microtubule-forming ability was investigated. As shown in Fig. 3C, S-guanylated tau showed a significant reduction in tubulin polymerization in vitro. However, the level of tubulin polymerization was maintained at ∼75% of unmodified tau. Taken together, modification of cysteine residues in tau by 8-nitro-cGMP loses the ability of tau/tau interaction in the soluble phase but maintains the ability of microtubule formation.
FIGURE 3.

The effect of S-guanylation on tau aggregation. A, the ThT assay was conducted on unmodified tau (open circles) and S-guanylated tau (closed circles), and the fluorescence intensity was measured at the indicated time points. The ThT assay was performed independently multiple times. Data are mean ± S.D. (2N4R, n = 4; 2N3R, n = 3). B, the reaction mixture from the ThT assay was observed using AFM after 48 h of incubation. AFM observations were repeated using reaction mixtures of independent ThT assays (n = 4), and the same result was obtained each time. C, the microtubule-forming ability of S-guanylated tau. A tubulin polymerization assay was conducted on unmodified tau and S-guanylated tau. Fluorescence intensity was measured after 60 min of incubation. Total amounts of tubulin polymerization by S-guanylated tau were normalized by that of unmodified tau, and result is presented as a percentage of the unmodified tau (mean ± S.D. from independent experiments; n = 3; *, p < 0.01; unpaired t test). D and E, tau oligomer formation was observed at an early point of tau aggregation. Aggregation of tau and S-guanylated tau was triggered by addition of heparin sodium, and the reaction mixture at the early time point indicated was analyzed by immunoblotting with POX-Tau5 (2N4R), HT-7 (2N3R), and RS-cGMP under non-reducing conditions. The experiment was repeated (n = 4), and the same result was obtained each time.
S-guanylation of Cysteine Residues in Tau Is Inhibited in Sarcosyl-insoluble Tau Formation
To apply our in vitro results of S-guanylation of tau to the in vivo context, we treated P301L mutant tau stably expressed in Neuro2A cells (P301L-Neuro2A) with 8-nitro-cGMP (150 μm) for 48 h. Compared with vehicle-treated cells, the amount of tau in the sarcosyl-insoluble fraction was reduced by 8-nitro-cGMP treatment, whereas there was no difference in the RIPA-soluble fraction (Fig. 4A). Quantitatively, 8-nitro-cGMP treatment reduced the amount of insoluble tau by ∼50% of the vehicle control (Fig. 4B). RS-cGMP-positive tau was detected in the RIPA-soluble fraction of 8-nitro-cGMP-treated cells, suggesting that S-guanylated tau could not aggregate and remained in the cytoplasmic soluble fraction.
FIGURE 4.
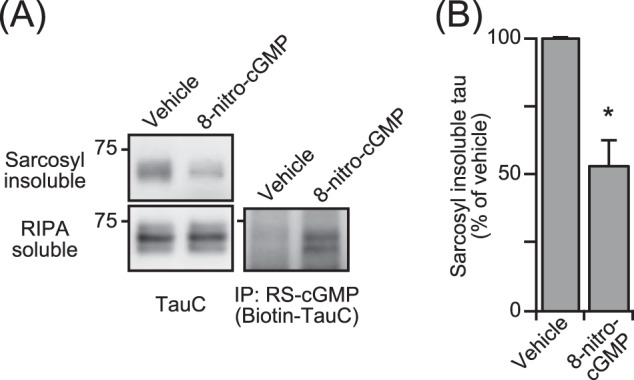
The effect of S-guanylation of tau on intracellular tau aggregation. A, intracellular sarcosyl-insoluble tau from P301L-Neuro2A cells was cultured for 48 h with 8-nitro-cGMP and then detected by immunoblot analysis using TauC antibody. Intracellular tau levels were confirmed by immunoblot analysis against the RIPA-soluble fraction with TauC. Tau S-guanylation was confirmed by immunoblotting with biotin-TauC antibody following immunoprecipitation (IP) from the RIPA-soluble fraction with RS-cGMP antibody. B, quantitative analysis of intracellular sarcosyl-insoluble tau was performed. The intensity of the immunoblot of the sarcosyl insoluble fraction from 8-nitro-cGMP-treated cells was normalized to that of vehicle, and the result is indicated as a percentage of the vehicle (mean ± S.D. from independent experiments, n = 7). *, p < 0.0001 (unpaired t test).
Discussion
This study investigated the effect of cysteine modification of tau by 8-nitro-cGMP on heparin-induced tau aggregation. Cysteine residues in both 2N3R and 2N4R tau were S-guanylated by 8-nitro-cGMP in vitro, causing S-guanylated tau to lose the ability to form tau aggregates in vitro and in vivo. This indicates that NO-linked posttranslational modification of cysteine residues in tau by 8-nitro-cGMP prevent tau oligomer formation. These results were confirmed in the cellular model Neuro2A cell line overexpressing P301L mutant tau.
Cysteine residues in tau are affected by oxidation because of the formation of disulfide bridges between thiol groups of cysteine. In four-repeat Tau containing two cysteine residues (Cys-291 and Cys-322) in the microtubule binding domain, oxidation of tau can either lead to the formation of intramolecular disulfide bonds that prevent tau aggregation or the formation of intermolecular disulfide bonds that accelerate tau aggregation (19, 20). However, following substitution of cysteine to alanine, tau retained the ability to form fibrils (21) despite the delay in the nucleation step, suggesting that other regions within tau and not just cysteine residues are involved in tau aggregation. A local sequence motif of PHF6 (VQIVYK) and of PHF6* (VQIINK) were reported to form β sheet structures without cysteine disulfide formation (22, 23). Also, intermolecular disulfide bridges may bring PHF6 into close proximity, accelerating tau aggregation (22).
We recently found that isoproterenol inhibits neuronal loss by blocking tau aggregation (24). Isoproterenol covalently binds to cysteine residues of tau, completely inhibiting tau oligomer formation as well as 8-nitro-cGMP, suggesting that an adduct of cysteine residues may block interaction of PHF6 in addition to cysteine disulfide bridges.
8-Nitro-cGMP is generated after NO production (14), and NO is involved in various physiological functions, including vascular regulation, neuronal transmission, and inflammation (11–13, 25). 8-Nitro-cGMP acts as a second messenger of NO signaling and modifies a thiolate in cysteine residues of various proteins to regulate their functions (14–16). It is well established that 8-nitro-cGMP-modified KEAP1 plays a role in the defense against oxidative stress (15).
Oxidative stress is believed to be one cause of AD (26). Observations in the AD brain show that Aβ binds to copper-induced reactive oxygen, leading to mitochondrial dysfunction by fragmentation and increased fusion (27, 28). Oxidative stress also affects tau pathology by accelerating tau aggregation through disulfide bridge formation between tau molecules (19, 20, 24). Indeed, vitamin E administration is able to delay the formation of NFTs in a mouse model (29). NO signaling may serve as a defense mechanism against tau aggregation triggered by oxidative stress. In a mouse model expressing a mutated amyloid protein precursor, the genetic ablation of NO synthase 2 resulted in the accumulation of hyperphosphorylated endogenous tau, suggesting that the absence of NO facilitates Aβ-induced tau aggregation signaling (30). Although the level of NO synthase in the AD brain is controversial (31), NO signaling may work as a defense mechanism to protect against AD. As such, activating NOS in the brain could potentially prevent the development of AD. In other words, NO synthase could be an important target for AD therapy.
Experimental Procedures
Materials
Thioflavin T and heparin sodium were purchased from Sigma and Acros Organics, respectively. All other materials used were of the highest commercial grade available and purchased from Nacalai Tesque Inc. 8-Nitro-cGMP and S-guanylated protein-specific rabbit polyclonal antibody, RS-cGMP (14), were kindly provided by Dr. Akaike (Tohoku University). Anti-tau monoclonal antibody, clone Tau5 (AHB0042, Invitrogen); anti-human Tau monoclonal antibody, HT-7 (MN1000, Thermo Scientific); and anti-repeat domain monoclonal antibody, clone 2B11 (10237, Immuno-Biological Laboratories Co., Ltd.) were also purchased. Rabbit polyclonal antibody TauC specific for the C-terminal of tau we reported previously (18) was used in this study. Peroxidase-conjugated Tau5 antibody (POX-Tau5) and biotin-conjugated TauC (biotin-TauC) were prepared using the peroxidase labeling kit NH2 (LK11, Dojindo) and biotin labeling kit NH2 (LK03, Dojindo), respectively, in accordance with the instructions of the manufacturer.
Purification of Recombinant Tau Isoforms
Wild-type three-repeat (2N3R) and four-repeat (2N4R) tau and their corresponding cysteine-to-alanine substituted mutants (C322A-2N3R, C291,322A-2N4R) were cloned into a pRK172 vector and purified by a modified method reported previously (9). Briefly, wild-type and mutant tau in pRK172 vectors were expressed in Escherichia coli strain BL21 (DE3) cells (Takara Bio Inc.). Cells were then sonicated and heated in boiling water to partially purify the heat-stable tau. After centrifugation, the supernatant was loaded onto a phosphocellulose column (P11, Whatman), and tau was eluted with 0.3 m sodium chloride. Tau was fractioned from the effluent by ammonium sulfate precipitation and loaded onto a reverse-phase HPLC column (COSMOSIL Protein-R Waters, Nacalai Tesque Inc.) following gel filtration chromatography to exchange buffers (NAP-10 column, GE Healthcare). After freeze-drying, tau was dissolved in water and stored at −80 °C. Tau protein concentration was determined using a BCA protein assay kit (Pierce).
Mass Spectrometric Analysis of the S-guanylated Peptide of the Tau Microtubule Binding Domain
A partial tau region R3′ peptide ((0.4 mm) SKVTSKCGSLGN; MW, 1180.4) was mixed with immobilized tris(2-carboxyethyl)phosphine disulfide reducing gel (Thermo) for 1 h at room temperature to reduce spontaneously formed disulfide bonds. After centrifugation, the peptide was incubated with 0.4 or 2.0 mm 8-nitro-cGMP in sodium phosphate buffer (pH 7.4) for 2 h at room temperature. The reaction mixture was investigated by LC/MS analysis. LC/MS was performed in positive scan mode (m/z, 500–1600) with triple quadrupole mass spectrometer (LCMS-8060, Shimadzu) after reverse-phase HPLC on a reverse-phase column (YMC-Triart C18 column, 50 × 2.0 mm inner diameter, YMC Co., Ltd.) under the following elution conditions: mobile phases A (0.1% formic acid) and B (0.1% formic acid in methanol) with a linear gradient from 5% to 90% mobile phase B in 15 min at a flow rate of 0.2 ml/min at 40 °C.
S-guanylation of Recombinant Tau Isoforms
Recombinant tau isomers were treated in 1 mm dithiothreitol in 50 mm sodium phosphate buffer (pH 7.4) at 37 °C for 4 h to reduce any spontaneously formed cysteine disulfide and then desalted by gel filtration chromatography (PD Spintrap G-25, GE Healthcare). After determining tau concentration using a BCA protein assay kit, wild-type and mutant tau isomers (each at 10 μm) were incubated with 100 μm 8-nitro-cGMP in 100 mm sodium phosphate buffer (pH 7.4) at 37 °C for 16 h. Gel filtration chromatography (PD Spintrap G-25 column) was then performed to remove excess 8-nitro-cGMP and to exchange for an optimal buffer. To confirm S-guanylation, tau was loaded into a SuperSep Ace 5–20% gel (WAKO Pure Chemical Industries), subjected to PAGE under reducing conditions, and visualized through immunoblot analysis using RS-cGMP and HT-7 antibody.
Thioflavin T Assay
The ThT assay was performed by a modified method described previously (9). Unmodified or S-guanylated 2N3R (5 μm) and 2N4R (10 μm) were mixed into 10 mm HEPES buffer (pH 7.4) containing 100 mm sodium chloride and 10 μm ThT. The assay was immediately started after addition of 0.06 mg/ml heparin sodium, a trigger for tau aggregation. The reaction mixture was incubated at 37 °C throughout. The fluorescence intensity was measured (excitation at 440 nm, emission at 486 nm) at various time points by a Wallac 1420 ARVO MX multilabel counter (PerkinElmer Life Sciences).
To observe tau oligomer formation, unmodified and S-guanylated tau (10 μm) was mixed into 10 mm HEPES buffer (pH 7.4) containing 100 mm sodium chloride and incubated at 37 °C immediately after addition of 0.06 mg/ml heparin sodium. SDS-PAGE loading buffer lacking the reducing agent was added at the earlier time points indicated (up to 4 h) to stop the reaction. These samples were loaded into Novex 3–8% Tris acetate gel (Invitrogen) and subjected to PAGE under non-reducing conditions. Immunoblot analysis was then carried out using HT-7 (2N3R), POX-Tau5 (2N4R), and RS-cGMP antibody.
Observation of Reaction Mixtures from the ThT Assay by AFM
To observe tau granules and fibrils after performing the ThT assay, the reaction mixtures were observed by AFM as described previously (9). A scanning probe microscope (SPM-970, Shimadzu Corp.) equipped with a dynamic mode was used for AFM analyses. A microcantilever (OMCL-TR800PSA, Olympus) was used. Reaction mixtures from three different ThT assays were dropped (20 μl) onto freshly cleaved mica and incubated at room temperature for at least 30 min before being visualized under AFM.
Measurement of the Microtubule-stabilizing Activity of Tau
The effect of S-guanylated tau on microtubule formation was investigated using a tubulin polymerization assay kit (BK011P, Cytoskeleton) according to the protocol of the manufacturer. Briefly, 1 mg/ml tubulin, 1 mg/ml tau, and 1 mm GTP were mixed in tubulin polymerization buffer (80 mm PIPES (pH 6.9), 0.5 mm EGTA, 2.0 mm magnesium chloride, and 10 μm fluorescent reporter), and tubulin polymerization was measured at an emission wavelength of 460 nm (the excitation wavelength was 355 nm) after incubation for 60 min at 37 °C by a Wallac 1420 ARVO MX multilabel counter.
Detection of Intracellular Tau S-guanylation in 8-Nitro-cGMP-treated HEK293T Cells
HEK293T cells were cultured at 37 °C in humidified 5% CO2 in DMEM (Nacalai Tesque Inc.) supplemented with 10% FBS and 1% penicillin-streptomycin (Invitrogen). 24 h before the experiment, HEK293T cells were plated at a density of 4–5 × 105 cells/well in 12-well plates. Tau isomers (2N3R and 2N4R) and a histidine tag-conjugated microtubule binding domain (four-repeat) fragment (His-4rp, residues 252–376 of the longest tau) were cloned into a pcDNA3 mammalian cell expression vector. This vector was then transfected into HEK293T cells using FuGENE-HD transfection reagent (Promega Corp.) according to the recommendation of the manufacturer and cultured for 36 h at 37 °C. Cells were washed twice with DMEM without supplements before being cultured for 6 h with 200 μm 8-nitro-cGMP dissolved in DMEM without supplements. After 8-nitro-cGMP treatment, cells were washed with ice-cold PBS and collected.
2N3R- or 2N4R-expressing cells were dissolved in 50 mm Tris-HCl (pH 7.4) supplemented with 500 mm sodium chloride and a protease inhibitor (25955, Nacalai Tesque Inc.) then sonicated and centrifuged (20,000 × g at 4 °C for 30 min). The supernatant was heated in boiling water for 15 min, and any denatured protein was removed by centrifugation. RS-cGMP antibody was added to the supernatant and incubated overnight at 4 °C. Any immune complexes present were trapped by protein G-Sepharose gel (GE Healthcare) and washed by a batch method twice with 100 mm Tris-HCl (pH 7.4) supplemented with 150 mm sodium chloride (TBS) after washing three times with RIPA buffer (100 mm Tris-HCl (pH 7.4), 150 mm sodium chloride, 1 mm EDTA, 1% Nonidet P-40, and 0.25% deoxycholate). After centrifugation, SDS-PAGE loading buffer containing 2-mercaptoethanol was mixed into protein G-Sepharose gel. The supernatant was loaded into SuperSep Ace 5–20% gel and subject to PAGE under reducing conditions. Immunoblot analysis using HT-7 antibody was carried out.
His-4rp-expressing cells were suspended in 10 mm sodium phosphate buffer (pH 7.4) supplemented with 500 mm sodium chloride, 20 mm imidazole, and a protease inhibitor (Nacalai Tesque Inc.) and then sonicated and centrifuged (20,000 × g on 4 °C for 30 min) to remove cell debris. The supernatant was heated in boiling water for 15 min, and any denatured protein was removed by centrifugation. His-4rp was trapped by Ni-NTA-agarose (Qiagen) (2.5-h incubation at 4 °C), washed by a batch method three times with 10 mm sodium phosphate buffer (pH 7.4) supplemented with 500 mm sodium chloride, 20 mm imidazole, and 0.05% Tween 20, and then washed once more with PBS. The Ni-NTA-agarose collected was mixed with SDS-PAGE loading buffer containing 2-mercaptoethanol. The supernatant was loaded into SuperSep Ace 5–20% gel and subject to PAGE under reducing conditions. Immunoblot analysis using RS-cGMP antibody or 2B11 antibody was carried out.
Detection of Sarcosyl-insoluble Tau in P301L-Neuro2A Cells
Neuroblastoma-derived Neuro2A cell stably expressing a proline 301-to-leucine substituted mutant tau (P301L-Neuro2A cell) was established by a method reported previously (17). P301L-Neuro2A cells were cultured at 37 °C in humidified 5% CO2 in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. 24 h before the experiment, cells were plated at a density of 1 × 106 cells/well in 6-well plates. After washing twice with DMEM without supplements, P301L-Neuro2A cells were cultured in DMEM supplemented with 1% FBS in the presence or absence of 150 μm 8-nitro-cGMP. After 48 h of culturing, cells were scraped, collected, and lysed in RIPA buffer supplemented with a protease inhibitor after washing twice with PBS and then centrifuged (20,000 × g on 4 °C for 30 min). The supernatant was then layered on 10 mm HEPES (pH 7.4) supplemented with 150 mm sodium chloride and 320 mm sucrose and centrifuged (20,000 × g at 4 °C for 10 min). The cell lysate on the upper layer was collected, and the protein concentration was measured using the BCA assay.
The cell lysate (500 μg of protein) was ultracentrifuged at 200,000 × g for 20 min at 4 °C, and the supernatant was collected as a RIPA-soluble fraction. Part of the RIPA-soluble fraction was mixed into SDS-PAGE loading buffer containing 2-mercaptoethanol for immunoblotting. The remainder was used for immunoprecipitation according to the method described above to detect S-guanylated P301L-Tau. Briefly, RS-cGMP antibody was added to the RIPA-soluble fraction. Immune complexes were trapped by protein G-Sepharose and mixed with SDS-PAGE loading buffer containing 2-mercaptoethanol after washing with RIPA buffer and TBS.
The precipitate after ultracentrifugation of the cell lysate was dissolved in 20 mm Tris-HCl (pH 7.4) supplemented with 500 mm sodium chloride, 1 mm EGTA, and 1% lauroylsarcosine sodium salt (sarcosyl buffer). This was sonicated and then ultracentrifuged (200,000 × g for 20 min at 4 °C). The supernatant was removed, and the precipitate was dissolved again in sarcosyl buffer and sonicated. After ultracentrifugation, the pellet was dissolved in 70% formic acid and then sonicated and centrifuged (20,000 × g for 20 min at 4 °C). The supernatant was dried by centrifugal concentration (CC-105, TOMY) and dissolved in SDS-PAGE loading buffer containing 2-mercaptoethanol.
PAGE under reducing conditions was performed using SuperSep Ace 5–20% gel for immunoblot analyses with TauC and biotin-TauC. Densitometry of the immunoblots was determined using ImageJ software (Wayne Rasband, National Institutes of Health), and the intensity of 8-nitro-cGMP-treated cells was normalized to that of the vehicle. Statistical analysis was performed using Prism 6 version 6 (GraphPad Software, Inc.).
Immunoblot Analyses
Samples were separated by SDS-PAGE using SuperSep Ace 5–20% gel (detection of S-guanylated tau in vitro and in vivo) or Novex 3–8% Tris-Acetate Gel (observation of tau oligomer formation) and then transferred to an Immobilon-P membrane (IPVH00010, Millipore). The membrane was blocked with TBS supplemented with 0.05% Tween 20 containing 2% skim milk (blocking buffer) and incubated overnight at 4 °C with blocking buffer containing the primary antibody. Subsequently, the membrane was washed three times with the blocking buffer and incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody or horseradish peroxidase-conjugated avidin. After washing three times with TBS supplemented with 0.05% Tween 20, immunoreactive bands were detected using a chemiluminescence reagent (ImmunoStar® Zeta, Wako Pure Chemical Industries) and a luminescent image analyzer (LAS3000, Fujifilm Corp.).
Author Contributions
A. T. and J. Y. designed the study and wrote the manuscript. T. I. performed the mass spectrometric analysis. A. T., Y. S., A. S., M. Y., and K. M. provided technical assistance and critical suggestions during the experiments. T. A. provided 8-nitro-cGMP and RS-cGMP antibody. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Maiko Imai and Tamiko Saji for helping to prepare this work.
This work was supported by Mext grant-in-aid projects Scientific Research on Innovation Area (Brain Protein Aging and Dementia Control (to A. T.) and Brain Environment (to A. T.)), Mext Strategic Research Program for Brain Science (Integrated Research on Neuropsychiatric Disorders) (to A.T.), and an intramural grant of the National Center for Geriatrics and Gerontology (to A. T.).
- NFT
- neurofibrillary tangle
- AD
- Alzheimer's disease
- 8-nitro-cGMP
- 8-nitroguanosine 3′,5′-cyclic monophosphate
- MW
- molecular weight
- Ni-NTA
- nickel-nitrilotriacetic acid
- AFM
- atomic force microscopy
- RIPA
- radioimmune precipitation assay
- ThT
- thioflavin T.
References
- 1. Perl D. P. (2010) Neuropathology of Alzheimer's disease. Mt. Sinai J. Med. 77, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Braak H., and Braak E. (1996) Evolution of the neuropathology of Alzheimer's disease. Acta Neurol. Scand. Suppl. 165, 3–12 [DOI] [PubMed] [Google Scholar]
- 3. Samuel W., Masliah E., Hill L. R., Butters N., and Terry R. (1994) Hippocampal connectivity and Alzheimer's dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 44, 2081–2088 [DOI] [PubMed] [Google Scholar]
- 4. Siuda J., Fujioka S., and Wszolek Z. K. (2014) Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat. Disord. 20, 957–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Götz J., Deters N., Doldissen A., Bokhari L., Ke Y., Wiesner A., Schonrock N., and Ittner L. M. (2007) A decade of Tau transgenic animal models and beyond. Brain Pathol. 17, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kimura T., Fukuda T., Sahara N., Yamashita S., Murayama M., Mizoroki T., Yoshiike Y., Lee B., Sotiropoulos I., Maeda S., and Takashima A. (2010) Aggregation of detergent-insoluble Tau is involved in neuronal loss but not in synaptic loss. J. Biol. Chem. 285, 38692–38699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., and Feany M. B. (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714 [DOI] [PubMed] [Google Scholar]
- 8. Sahara N., Maeda S., Murayama M., Suzuki T., Dohmae N., Yen S. H., and Takashima A. (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 25, 3020–3029 [DOI] [PubMed] [Google Scholar]
- 9. Maeda S., Sahara N., Saito Y., Murayama M., Yoshiike Y., Kim H., Miyasaka T., Murayama S., Ikai A., and Takashima A. (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861 [DOI] [PubMed] [Google Scholar]
- 10. Takashima A. (2013) Tauopathies and tau oligomers. J. Alzheimers Dis. 37, 565–568 [DOI] [PubMed] [Google Scholar]
- 11. Ignarro L. J., Buga G. M., Wood K. S., Byrns R. E., and Chaudhuri G. (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 84, 9265–9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hardingham N., Dachtler J., and Fox K. (October 31, 2013) The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell Neurosci. 10.3389/fncel.2013.00190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alam M. S., Akaike T., Okamoto S., Kubota T., Yoshitake J., Sawa T., Miyamoto Y., Tamura F., and Maeda H. (2002) Role of nitric oxide in host defense in murine salmonellosis as a function of its antibacterial and antiapoptotic activities. Infect. Immun. 70, 3130–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sawa T., Zaki M. H., Okamoto T., Akuta T., Tokutomi Y., Kim-Mitsuyama S., Ihara H., Kobayashi A., Yamamoto M., Fujii S., Arimoto H., and Akaike T. (2007) Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol. 3, 727–735 [DOI] [PubMed] [Google Scholar]
- 15. Fujii S., Sawa T., Ihara H., Tong K. I., Ida T., Okamoto T., Ahtesham A. K., Ishima Y., Motohashi H., Yamamoto M., and Akaike T. (2010) The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J. Biol. Chem. 285, 23970–23984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito C., Saito Y., Nozawa T., Fujii S., Sawa T., Inoue H., Matsunaga T., Khan S., Akashi S., Hashimoto R., Aikawa C., Takahashi E., Sagara H., Komatsu M., Tanaka K., et al. (2013) Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Mol. Cell 52, 794–804 [DOI] [PubMed] [Google Scholar]
- 17. Hatakeyama S., Matsumoto M., Kamura T., Murayama M., Chui D. H., Planel E., Takahashi R., Nakayama K. I., and Takashima A. (2004) U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J. Neurochem. 91, 299–307 [DOI] [PubMed] [Google Scholar]
- 18. Ueno H., Murayama O., Maeda S., Sahara N., Park J. M., Murayama M., Sanda A., Iwahashi K., Matsuda M., and Takashima A. (2007) Novel conformation-sensitive antibodies specific to three- and four-repeat tau. Biochem. Biophys. Res. Commun. 358, 602–607 [DOI] [PubMed] [Google Scholar]
- 19. Schweers O., Mandelkow E. M., Biernat J., and Mandelkow E. (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein τ controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walker S., Ullman O., and Stultz C. M. (2012) Using intramolecular disulfide bonds in tau protein to deduce structural features of aggregation-resistant conformations. J. Biol. Chem. 287, 9591–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barghorn S., and Mandelkow E. (2002) Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 22. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., and Mandelkow E. (2000) Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. von Bergen M., Barghorn S., Li L., Marx A., Biernat J., Mandelkow E. M., and Mandelkow E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J. Biol. Chem. 276, 48165–48174 [DOI] [PubMed] [Google Scholar]
- 24. Soeda Y., Yoshikawa M., Almeida O. F., Sumioka A., Maeda S., Osada H., Kondoh Y., Saito A., Miyasaka T., Kimura T., Suzuki M., Koyoma H., Yoshiike Y., Sugimoto H., Ihara Y., and Takashima A. (December 16, 2015) Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 10.1038/ncomms10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bredt D. S., Hwang P. M., and Snyder S. H. (1990) Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347, 768–770 [DOI] [PubMed] [Google Scholar]
- 26. Zhao Y., and Zhao B. (July 25, 2013) Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid. Med. Cell Longev. 10.1155/2013/316523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crouch P. J., Blake R., Duce J. A., Ciccotosto G. D., Li Q. X., Barnham K. J., Curtain C. C., Cherny R. A., Cappai R., Dyrks T., Masters C. L., and Trounce I. A. (2005) Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β1–42. J. Neurosci. 25, 672–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Manczak M., Anekonda T. S., Henson E., Park B. S., Quinn J., and Reddy P. H. (2006) Mitochondria are a direct site of Aβ accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449 [DOI] [PubMed] [Google Scholar]
- 29. Nakashima H., Ishihara T., Yokota O., Terada S., Trojanowski J. Q., Lee V. M., and Kuroda S. (2004) Effects of α-tocopherol on an animal model of tauopathies. Free Radic. Biol. Med. 37, 176–186 [DOI] [PubMed] [Google Scholar]
- 30. Colton C. A., Vitek M. P., Wink D. A., Xu Q., Cantillana V., Previti M. L., Van Nostrand W. E., Weinberg J. B., Weinberg B., and Dawson H. (2006) NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 12867–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balez R., and Ooi L. (2015) Getting to NO Alzheimer's disease: neuroprotection versus neurotoxicity mediated by nitric oxide. Oxid. Med. Cell Longev. 10.1155/2016/3806157 [DOI] [PMC free article] [PubMed] [Google Scholar]