

Abstract
Multidrug resistance to current Food and Drug Administration-approved HIV-1 protease (PR) inhibitors drives the need to understand the fundamental mechanisms of how drug pressure-selected mutations, which are oftentimes natural polymorphisms, elicit their effect on enzyme function and resistance. Here, the impacts of the hinge-region natural polymorphism at residue 35, glutamate to aspartate (E35D), alone and in conjunction with residue 57, arginine to lysine (R57K), are characterized with the goal of understanding how altered salt bridge interactions between the hinge and flap regions are associated with changes in structure, motional dynamics, conformational sampling, kinetic parameters, and inhibitor affinity. The combined results reveal that the single E35D substitution leads to diminished salt bridge interactions between residues 35 and 57 and gives rise to the stabilization of open-like conformational states with overall increased backbone dynamics. In HIV-1 PR constructs where sites 35 and 57 are both mutated (e.g. E35D and R57K), x-ray structures reveal an altered network of interactions that replace the salt bridge thus stabilizing the structural integrity between the flap and hinge regions. Despite the altered conformational sampling and dynamics when the salt bridge is disrupted, enzyme kinetic parameters and inhibition constants are similar to those obtained for subtype B PR. Results demonstrate that these hinge-region natural polymorphisms, which may arise as drug pressure secondary mutations, alter protein dynamics and the conformational landscape, which are important thermodynamic parameters to consider for development of inhibitors that target for non-subtype B PR.
Keywords: crystal structure, drug resistance, enzyme inhibitor, enzyme kinetics, HIV-1 protease, nuclear magnetic resonance (NMR), PELDOR, site-directed spin-labeling, backbone dynamics, double electron-electron resonance (DEER)
Introduction
HIV-1 is the causative agent of acquired immunodeficiency syndrome (AIDS), which has been a worldwide epidemic since the 1980s. Although the success of antiretroviral therapy has led to a decrease in the mortality rate from HIV-1 infection, in 2015 there were still over 2.1 million new infections and 1.1 million deaths worldwide (1–3). HIV-1 protease (PR)6 is a major drug target in the battle against HIV-1 infection, where inhibition of HIV-1 PR prevents viral maturation and results in immature noninfectious virions (4–6). Given the effectiveness of PR antiretroviral treatment, studies on the biochemistry and biophysics of this enzyme have been ongoing for several decades (7–11). There is now increasing focus on the impact of genomic differences among subtypes and circular recombinant forms (CRFs) in viral spreading (12–14), where CRFs are the recombinant mosaic of HIV-1 genomes that have been spread to at least three or more persons who are not epidemiologically related (www.hiv.lanl.gov). Because of globalization, different subtypes and CRFs circulate between different geographical regions. The circulation is of great importance because natural polymorphisms carried inside subtypes and CRFs determine the characteristic responses to drug treatment and may accelerate drug resistance development by reducing the time associated with the fitness-restoring processes (15–18).
To date, much knowledge has been gained regarding the effects of drug pressure-selected mutations on protease inhibitor (PI) susceptibility, i.e. gaining an understanding of the effects that primary mutations have on ligand/protease interactions and how secondary mutations both inside and outside the active site region lead to restoring catalytic efficiency with concomitant multidrug resistance (11, 18–26). Natural polymorphisms, which are characteristic of the epidemic map of a given subtype/CRF in a specific geographical region (12–14), are oftentimes similar to these secondary drug pressure-selected mutations. We and others are particularly interested in understanding the roles that natural polymorphisms in non-active site locations have on mechanisms that may lead to drug resistance and resistance-emergent pathways, which include altering HIV-1 PR conformation sampling, modulating PR dynamics, changes in PR stability, and the substrate groove (15, 16, 21, 26, 27–41).
Access of substrates and inhibitors to the catalytic site of HIV-1 PR is regulated by the β-hairpin turns, referred to as the “flaps,” which cover the active site pocket. In the absence and presence of substrate/inhibitors, the flaps and the enzyme adopt different configurations that have been characterized by molecular dynamics (MD) simulation (42, 43) and x-ray crystallography (44, 45). Fig. 1 shows conformations described as “closed,” “semi-open,” and “wide open” ensembles (Fig. 1A) (44, 45). Our laboratory has pioneered the application of site-directed spin labeling with electron paramagnetic resonance (EPR) spectroscopy, particularly double electron-electron resonance (DEER), to characterize the protein's conformational sampling profile of PR in solution (30, 34, 46). Analysis of DEER distance profiles from spin labels incorporated into the flaps leads to a description of the fractional occupancy of the conformational sampling scheme, as shown in Fig. 1A. For subtype B unbound enzyme (without substrate/inhibitor), three characteristic conformations have been observed, which are commonly described as the closed, semi-open, and wide-open states (31, 33, 47). However, our work has shown that drug pressure-selected mutations (30, 31, 33, 34) and natural polymorphisms (30, 33, 47) not only alter the fractional occupancy of the conformational sampling of unbound-subtype B but can also engender an additional distance that we postulate is a “curled” protein conformation (32, 33), where the flaps are proposed to adopt a curling motion that opens up access to the active site (31–33, 47).
FIGURE 1.

HIV-1 PR conformations and sequences. A, conformations of the four states are shown, “closed (PDB code 2BPX),” “semi-open (PDB code 1TW7),” “wide-open (MD coordinates),” and “curled (MD coordinates),” with 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl, MTSL, attached to residue 55 using Multiscale Modeling of Macromolecular systems to generate the anticipated distances. Colored segments with residue span in parentheses in the “closed” conformation highlight distinct regions that are referred to throughout the text: flaps (green, residues 43–59), elbow/hinge (red, residues 34–42), fulcrum (yellow, residues 9–24), and cantilever (blue, residues 60–74). B, amino acid sequences of HIV-1 PR variants B, E35D, PR5, CRF01_AE, and subtype A constructs where the catalytic residue (Asp-25) is highlight in red and natural polymorphisms are underlined in blue. All sequences except for A contain the three stabilizing mutations and CYS substitutions (Q7K, L33I, L63I, C67A, and C95A), where alternative sequences for subtype A are shown in a green background. Note for x-ray studies, EPR, and NMR measurements, the substitution D25N was included to render the enzyme inactive. C, locations and identities of natural polymorphisms of each construct are shown as spheres on HIV-1 PR (PDB code 2PK5 (86)).
The fractional occupancy of this curled conformation was shown to be particularly apparent in CRF01_AE (47) and protease construct PR5 (32), which has five amino acid substitutions relative to subtype B (sequences in Fig. 1B). Both of these constructs include the natural polymorphism at residue 35, glutamate to aspartate (E35D), and both show an increase in the population of the curled state, which is not destabilized upon addition of inhibitors. NMR studies of both of these constructs reveal increased backbone dynamics compared with wild-type subtype B (32). Hence, we suggested that disrupted or altered salt bridge interactions in the hinge regions of these constructs underlie the molecular basis of these altered conformations and dynamics. Hydrogen-deuterium exchange/mass spectrometry (48) also implicated the Glu-35/Arg-57 interaction as important in modulating dynamics in subtype C. This study investigates the impact of the substitution E35D, which is a major natural polymorphism that occurs roughly 30% of the time in subtype B drug-naive patients, increases in patients treated with the inhibitor tipranavir (TPV), and occurs with >90% frequency in subtype A and F drug-naive patients (Stanford University HIV Drug Resistance Database). Additionally, the alterations in protein dynamics and conformational sampling may be exploited in optimizing new inhibitors that more effectively stabilize constructs that harbor these substitutions.
Results and Discussion
E35D Stabilizes Open-like States
The effect of the E35D natural polymorphism on flap conformational sampling was explored by DEER spectroscopy. DEER distance profiles and resultant population analyses for subtype B and the subtype B construct that harbors the single E35D substitution in the absence and presence of the select inhibitors saquinavir (SQV), darunavir (DRV), and a non-hydrolyzable substrate analog Ca-P2 (with sequence H-Arg-Val-Leu-r-Phe-Glu-Ala-Nle-NH2; r = reduced) are given in Fig. 2. These inhibitors were chosen for spectroscopic studies because they were those for which successful crystals were obtained for structural studies. Full data analyses are given in supplemental Figs. S1–S8. Although the conformational sampling of unbound E35D is dominated by the semi-open conformation (52% fractional occupancy, distance near 36 Å), this relative percentage is decreased by 34% relative to subtype B (47), with associated increases in the populations of the other states (Fig. 2C). Specifically, relative to subtype B, re-distribution in the conformational ensemble leads to an increase in the fractional occupancy of the wide-open and closed-like states by 18 and 15%, respectively (Fig. 2C). Compared with the conformational sampling of PR5 and CRF01-AE, E35D possesses less of the curled state with larger fractional occupancy of the wide-open state. Taken together, these comparisons imply that the single amino acid substitution of E35D destabilizes the semi-open conformation with stabilization of the open-like and closed-like states. These results suggest that additional mutations, such as those in PR5 and CRF01_AE, act in concert with E35D to increase fractional occupancy and hence stabilize the curled conformation.
FIGURE 2.

DEER distance profiles show conformational sampling results for unbound, CaP2-bound, DRV-bound, and SQV-bound subtype B (A) and E35D (B). Bar graph comparison of relative percentages (e.g. fractional occupancies) of each conformational state in subtype B, E35D, PR5, and CRF01_AE constructs in unbound form (C) and CaP2-, DRV-, and SQV-bound forms (D). Protein conformational population data of PR5 and CRF01_AE are taken from Refs. 30, 32, 33, 47, 87.
DEER data also show that for E35D, the addition of inhibitors/substrate analogs shifts the predominant conformation to the closed state (distance centered ∼33 Å), an observation also seen for other HIV-1 PR constructs, including subtype B (50). The binding of inhibitors to stabilize the closed state is consistent with results from kinetic and inhibition assays and is comparable with conformational shifts observed for other non-drug-resistant forms of PR (50). For unbound-subtype B, the DEER distance profiles do not contain a detectable population at a distance <30 Å in both the presence and absence of inhibitor, i.e. no evidence for flap curling. In contrast, for E35D, a detectable population at 25 Å appears upon addition of SQV, DRV, and Ca-P2 (Fig. 2, C and D). It is noteworthy to mention that the fractional occupancy of the curled distance in unbound E35D (Fig. 2C) is outside the error of the DEER distance measurements, indicating the result is statistically valid (51). Interestingly, the breadth of the population for this distance is narrowed upon addition of inhibitor or substrate analog (Fig. 2B). This effect was also observed in DEER studies of CRF01_AE and PR5 (32). Additionally, in the presence of both DRV and SQV, E35D contains a lower fractional occupancy of the closed conformation when compared with subtype B (Fig. 2D), potentially indicating that inhibitors may bind to a non-closed or curled conformation as seen for darunavir-resistant PR (52). We propose the persistence of this distance in the presence of inhibitor may suggest that this peak is a signature of inhibitor binding in an altered conformation that contrasts the typical inhibitor-bound closed HIV-1 PR state. Evidence for an asymmetric flap orientation in unbound and inhibitor-bound DRV-resistant PR has been observed crystallographically (52, 53). In these structures, one flap was seen to tuck into the active site cleft where the other sits over the inhibitor, and in one case, DRV was also found in a non-standard orientation sitting perpendicular to the active site cleft (53).
E35D Increases Protein Backbone Dynamics
We have recently shown that PR backbone dynamics vary for constructs that have alterations in conformational sampling profiles, where more rigid dynamics are observed when the protein occupies a predominantly closed-like state, and conversely where increased dynamics are found for constructs that have open-like states stabilized (32, 33). Analysis of NMR-derived heteronuclear order parameter values (S2), reflective of amide bond fluctuations, determined from multifrequency NMR investigations reveal regions of the protein that undergo enhanced or diminished motion relative to subtype B (supplemental Figs. S9 and 10 and supplemental Table S1). Fig. 3A shows color-coded ribbon diagram for subtype B, E35D, and PR5 where the color is indicative of the value of S2. Fig. 3B compares differences in S2 values for these constructs. In general, most sites in E35D exhibit increased backbone dynamics relative to subtype B (indicated by red and orange color along the diagonal in Fig. 3B). E35D has quite similar dynamics to PR5, with some sites (indicated by blue along the diagonal in Fig. 3B) that are more rigid. Strikingly, the tips of the flaps in E35D exhibit slowed dynamics compared with both subtype B and PR5, whereas nearly all other regions of E35D and PR5 are similar to each other and different from subtype B, indicating that the single E35D substitution can account for the majority of the increased backbone dynamics observed.
FIGURE 3.

A, ribbon diagrams of HIV-1 PR (PDB code 1HHP) color-coded according to NMR-derived values of order parameters ranging from 1 (rigid in violet) to 0.4 (flexible in red). Gray indicates the absence of NMR data for those residues. The locations of natural polymorphisms are shown as spheres. B, difference plots of the backbone order parameters comparing E35D to subtype B (E35D-B), PR5 to subtype B (PR5-B), and E35D to PR5 (E35D-PR5). Negative numbers (indicated by red coloring) indicate greater dynamics at that site. The NMR-derived parameter values are given in supplemental Fig. S10.
E35D Minimally Modifies the Overall Backbone Structures
Suitable crystals of HIV-1 PR E35D and PR5 with inhibitors DRV, CaP2, SQV, and amprenavir (APV) were obtained, and x-ray structures were determined and are discussed in reference to four previously reported structures as follows: the DRV-bound subtype B construct (PDB code 3BVB) (54), the subtype A construct (which bears R57K natural polymorphism and 14 other mutations (PDB code 3IXO)) (45), and two DRV-resistant subtype B constructs that each contain Glu-35 (PDB codes 3U7S and 4NPT) (52, 55). The x-ray crystallographic statistics for current and previously reported structures are summarized in Table 1. All crystal structures were refined to reasonable residual factors for both Rfree and Rcryst and Debye-Waller factors (B-factors) of main chains, side chains, ligand, and waters (Table 1).
TABLE 1.
X-ray crystallographic statistics for HIV-1 PR crystals
-- indicates not determined.

Structural imposition of ligand-bound subtype B, E35D, PR5, and unbound subtype A demonstrates a little structural variation, with only minor backbone deviations observed for E35D compared with subtype B and slightly larger deviations occurring upon addition of mutations present in PR5 (Fig. 4A) and subtype A (supplemental Fig. S11). The larger backbone deviation of SQV-bound PR5 and unbound subtype A may partly come from the crystal packing effect in terms of the distinct crystal symmetry (56). For a quantitative assessment of the overall structural variation, root mean square deviation (r.m.s.d.) values were determined in reference to DRV-bound subtype B (PDB code 3BDV). The r.m.s.d. values of inhibitor-bound E35D are 0.15, 0.19, 0.25, and 0.27 Å for DRV, APV, CaP2, and SQV, respectively. These minor deviations indicate very little influence of the amino acid substitution on the overall structure of inhibitor-bound protease. Double difference plots (19) of the backbone α-carbons of HIV-1 PR (Figs. 4B and supplemental Fig. S11) allow for detailed structural comparisons to be made. Regarding DRV-bound subtype B and E35D, Fig. 4B suggests that the E35D natural polymorphism slightly decreases intra-monomer distances between residues 35 and 50, with a minimal decrease of the inter-monomer distances among residues 35 to 16 and 81, and where a similar pattern is observed for APV-bound E35D (supplemental Fig. S11C). However, the impact of E35D when bound with CaP2/SQV leads to slightly increased structural differences where residues from 45 to 55 in the flap region and residues from 80 to 83 near the active pocket of the SQV-bound E35D construct exhibit increased inter-monomer distances. Two common features can be distinguished from DRV-bound and SQV-bound PR5 constructs in supplemental Fig. S11, D and E. The majority of the negative structural alterations (<−1.5 Å) are located inside each monomer around residues from site 15 to site 19 and residues from site 38 to site 42. The majority of the positive structural alterations (>1.5 Å) are around residue 46 between different monomers.
FIGURE 4.

A, side view and top views of overlaid ribbon diagrams for HIV-1 PR for DRV-, CaP2-, SQV-, and APV-bound E35D (yellow), DRV-bound subtype B (black) (left), DRV- and SQV-bound bound PR5 (red), unbound-subtype A (blue), and DRV-bound subtype B (black) (right). B, double difference plot comparisons of the backbone α-carbon inter-distance of DRV-bound subtype B to DRV-bound E35D (left) and PR5 (right), where dimer configuration was considered for HIV-1 PR. Red indicates that backbone distances of the HIV-1 PR constructs between the corresponding residues are shorter than that of the subtype B construct. Sequence numbers run from 1 to 198 to indicate the residues in monomer A (1–99) and monomer B (100–198) such that asymmetry between the two monomer interactions can be readily distinguished.
Additional Mutations in PR5 Slightly Alter the Structure of the Hinge and Fulcrum
The additional mutations in the hinge and flap regions of PR5 and subtype A are seen to lead to moderately larger structural deviations in the flap, hinge, and fulcrum regions when compared with subtype B (Fig. 4B). However, none of these structures show major structural differences compared with the subtype B construct. The additional mutations in PR5 and subtype A lead to increases in r.m.s.d. values. For SQV-bound structures, increased r.m.s.d. values of 0.54 and 0.44 Å are observed for the two dimers of PR5, and an r.m.s.d. value of 0.59 Å is observed for unbound subtype A.
From double difference plots (Figs. 4B and supplemental Fig. S11, C–F), it is apparent that most of the prominent changes of the PR5 construct occur around two natural polymorphisms of I15V and R41K, with only minor changes around sites 35 and 57. More structural alterations (<−1.5 or >1.5 Å) are generated in the subtype A construct (supplemental Fig. S11F) around amino acid residues of 7, 17, 36, 40, 45, 67, and 80 close to these mutation sites in the protein. Nevertheless, the backbone distance between residues 35 and 57 was not significantly changed for E35D, PR5, and subtype A referring to subtype B construct, with a distance variation less than 1 Å.
Salt Bridge Interactions Are Modulated by E35D/R57K Substitutions
The salt bridge interaction in PR between residues Glu-35 and Arg-57 likely serves as an important interaction to modulate the flap and hinge-region interactions that can consequently impact the rigidity and conformational flexibility of the protein backbone. Fig. 5 and supplemental Fig. S11 show the local structure of residues 35 and 57 of the various PR constructs characterized. The results clearly show a systematic disordering of the interactions between residues 35 and 57 as additional mutations are accumulated in the order of subtype B < E35D < PR5 ≈ subtype A. We semi-quantitatively describe the strength of this salt bridge interaction by the N-O distances between the terminal nitrogen and oxygen atoms of side chains. A schematic model of this interaction is depicted in Fig. 6. Results from this analysis are given in Table 2. “Strong” interactions describe N-O distances of <3 Å; “weak” refers to cases when the smallest N-O distance is 3–4 Å; and “absent” reflects situations where all N-O distances are >4 Å. These definitions are consistent with methods reported by others (57).
FIGURE 5.

Local structure of residues 35 and 57 of HIV-1 PR constructs. DRV-bound subtype B is shown in A, and DRV-, CaP2-, SQV-, and APV-bound E35D are shown in B–E, respectively; DRV-bound PR5 is shown in F; two different dimers in the unit cell for SQV-bound PR5 are shown in G and H; unbound subtype A is shown in I; and DRV-bound DRV1 and P51 are shown in J and K. The gray net surface shows the electron density map. The double structures show the arrangement in each monomer and hence asymmetry of the dimer. Coordinates for I are taken from Ref. 45; coordinates for J and K taken from Refs. 55 and 52, respectively.
FIGURE 6.
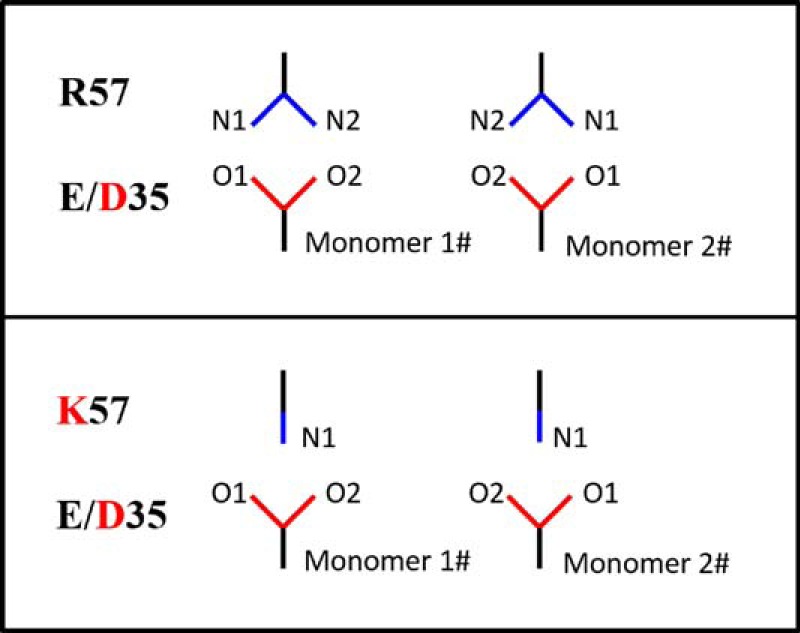
Schematic of side-chain orientations between residues 35 and 57.
TABLE 2.
Nitrogen-oxygen (N-O) atomic distances reported in angstroms between residues 35 and 57
“E/D–R/K” indicate the distance between residue 35 with E/D and residue 57 with R/K. N-O distances less than 3 Å are colored in red; distances between 3 and 4 Å are in blue; the rest of the distances are in black. -- indicates not applicable.
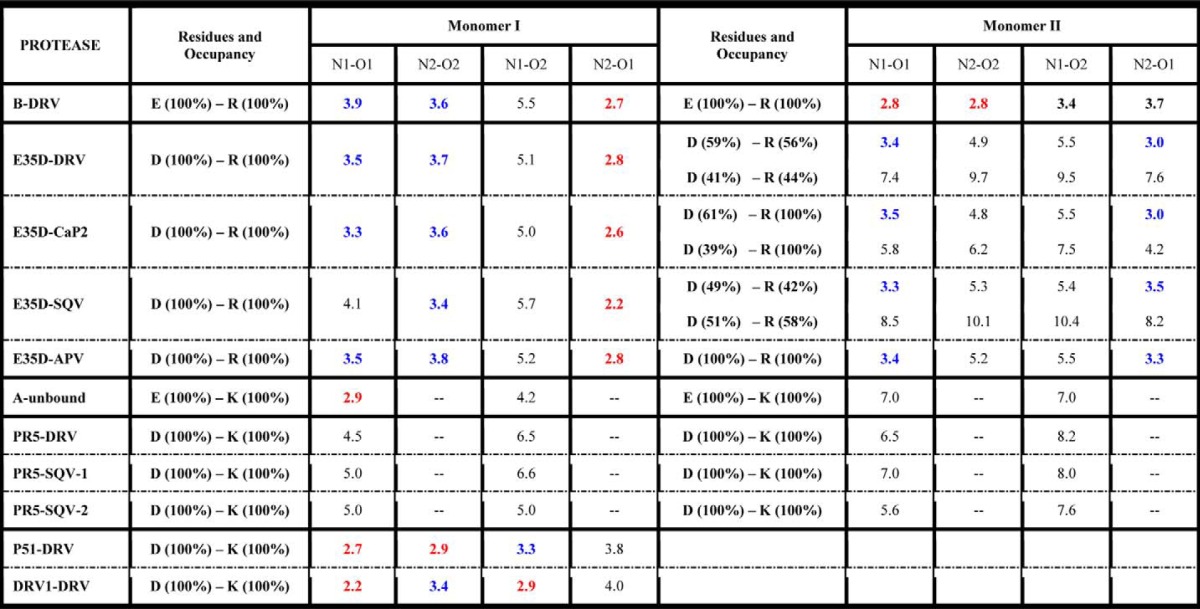
Both “staggered” and “paired” orientations of residues 35 and 57 in the DRV-bound subtype B PR structure (Fig. 5A) can be seen. The staggered configuration contains only one strong N-O interaction with a distance of 2.7 Å, whereas in the paired configuration, two N-O pairs occur with distances of 2.8 Å. The strong salt bridge interactions in the subtype B construct may stabilize the flap with respect to the cantilever region resulting in a more rigid structure with relatively lower backbone dynamics that may be consistent with the dominant semi-open protein conformation and a small fraction of the open-like protein conformation in the unbound subtype B construct characterized by our previous NMR and DEER experiments (30, 47, 50).
Structures of E35D with DRV, CaP2, SQV, and APV determined show how the single E35D natural polymorphism weakens the salt bridge interaction between Asp-35 and Arg-57 (Fig. 5, B–E, respectively) These structures, to varying degrees, reveal multiple conformations in the orientations of both Asp-35 and Arg-57, with higher frequency of alternative conformations seen for Asp-35 in multiple crystals of PR with varied inhibitors. Some evidence exists for the formation of staggered salt bridge interactions, but paired orientations are nearly absent given perpendicular-like orientations of the nitrogen-carbon planes. The weakened interactions give rise to N-O distances of >3 Å. Alternative conformers are generated at both Asp-35 and Arg-57 residues with a population of about 50% in DRV- and SQV-bound E35D. In addition, one alternative conformer is found at the Asp-35 residue in CaP2-bound E35D. These phenomena suggest that weakened salt bridge interactions are generated by a single E35D natural polymorphism, and the impact can be perturbed by different ligands inside the HIV-1 PR.
In inhibitor-bound structures of PR5 (Fig. 5, F–H), which contains the double mutations of E35D and R57K, the salt bridge interactions between residues 35 and 57 are eliminated. Replacing the direct salt bridge interaction between residues 35 and 57 is a series of intermolecular interactions mediated by Pro-79, Gly-78, and Val-77. Specifically, N-O distances between Asp-35, Pro-79, and Gly-78 range within 3 and 4 Å, and the N-O distances between Lys-57 and Val-77 near the active region are <3 Å. These interactions are depicted in Fig. 7 (DRV-bound PR5 and SQV-bound PR5). This network may provide a compensating interaction that retains the stability of the semi-open state but may also contribute to an increase tendency for the flaps to curl (as seen in DEER data) when in solution or interacting with inhibitors.
FIGURE 7.

Local structure of DRV-bound PR5 top view (A) and side view (B) and the local structure of SQV-bound PR5 top view (C) and side view (D) showing network of interactions that may provide a compensating structural and dynamic platform for interactions between Asp-35 and Lys-57 in PR5.
Subtype A carries the natural polymorphism R57K but not E35D. The effect of R57K on weakening the salt bridge interaction is seen Fig. 5I, where a disrupted salt bridge interaction is observed only in one monomer. Given that the average backbone Cα positions are similar to subtype B, it is likely that R57K strongly contributes to the disrupted salt bridge. Hence, the effect of the R57K mutation in subtype A supports our argument that the complete dissociation of the salt bridge interaction seen in crystal structures of PR5 is promoted by double mutation of E35D and R57K.
Results from NMR dynamics indicate that overall the backbone dynamics of HIV-1 PR are increased when E35D is incorporated. The structures reveal that changes in the salt bridge and or molecular interactions within the flap-hinge-cantilever region may provide the molecular basis for this change in dynamics. The alternative interaction network seen in PR5 structures also provides a rationale as to why the dynamics of PR5 and E35D are similar given that the salt bridge interaction is completely abolished in the crystal structure of PR5 and only partially diminished in E35D. We acknowledge that NMR dynamics were determined in unbound enzyme, and structural insights are being taken from crystals of inhibitor-bound states. Nevertheless, we believe the two sets of data are congruent and are useful to paint a structurally based model for understanding how natural polymorphisms are impacting dynamics and conformational sampling.
No Impact on Enzymatic Activity of Resistance Was Generated by E35D/R57K
The impact of natural polymorphisms in E35D and PR5 on catalytic parameters Km, kcat, and kcat/Km was determined to be minimal to modest. Values of dissociation constants, Ki, for inhibitors SQV, APV, ATV, and DRV were also determined and found not to be modulated by these natural polymorphisms (Tables 3 and 4). Overall, the Michaelis-Menten parameters for E35D and PR5 are very similar to subtype B, which are similar to others reported previously (58, 59). Values for kcat and kcat/Km for E35D show a modest decrease of ∼2- and 3-fold, respectively, when compared with subtype B, indicating that disruption of the salt bridge without compensating interactions slightly decreases catalytic turnover. For PR5, where compensating intermolecular interactions between the hinge and cantilever are seen, the values for kcat and kcat/Km show even smaller decreases in values of 1.25- and 2-fold when compared with subtype B. For PR5, Km has increased slightly compared with subtype B. Overall, these are minor changes, but they reveal that changes in dynamics and conformational sampling that originate from alterations in salt bridge interactions in the hinge and cantilever have slightly more of an impact on catalytic turnover than substrate binding.
TABLE 3.
Values for Michaelis-Menten kinetic parameters and inhibition constants (Ki) for HIV-1 PR constructs
Values in parentheses indicate the relative ratio referenced to data for subtype B.
| Sample | Km | kcat | kcat/Km | Ki of SQV | Ki of APV | Ki of ATV | Ki of DRV |
|---|---|---|---|---|---|---|---|
| μm | s−1 | s−1μm−1 | nm | nm | nm | nm | |
| B | 12 ± 3 (1.0) | 7.2 ± 2.8 (1.0) | 0.60 ± 0.12 (1.0) | 3.91 ± 0.05 (1.0) | 0.72 ± 0.05 (1.0) | 0.69 ± 0.09 (1.0) | 0.25 ± 0.03 (1.0) |
| B-E35D | 13.6 ± 1.3 (0.9) | 2.8 ± 0.5 (0.4) | 0.20 ± 0.04 (0.3) | 3.6 ± 1.2 (0.9) | 0.71 ± 0.12 (1.0) | 0.83 ± 0.18 (1.2) | 0.51 ± 0.12 (2.1) |
| PR5 | 18 ± 3 (1.3) | 5.9 ± 0.8 (0.8) | 0.40 ± 0.10 (0.6) | 3.3 ± 0.9 (0.8) | 0.96 ± 0.07 (1.3) | 0.67 ± 0.05 (1.0) | 0.32 ± 0.06 (1.3) |
TABLE 4.
Statistics of the prevalence of E35D natural polymorphisms, where numbers in parentheses indicate the total number of isolates in analysis (one mutation was extracted from one person)
The data analysis is up to March 2015. – indicates no data available in the database.
| E35D mutation in HIV-1 PR subtypes with/without treatment | ||||||||
|---|---|---|---|---|---|---|---|---|
| B | C | D | AG | G | A | AE | F | |
| Naive | 30.2% (29,287) | 25.4% (10,322) | 22.4% (1,976) | 25.1% (4,237) | 49.6% (1665) | 91.8% (4,993) | 83.7% (6,152) | 91% (1,103) |
| TPV | 48.9% (94) | – | 100% (1) | 50% (2) | – | 100% (2) | – | 66.6% (3) |
| APV | 37.8% (193) | 66.6% (3) | – | 0% (1) | 100% (1) | – | – | 100% (5) |
| RTV | 33.4% (1,069) | 28.5% (28) | 46.1% (13) | 14.2% (7) | 66.6% (15) | 87.5% (8) | 88.8% (9) | 90.7% (54) |
| NFV | 32.4% (824) | 37.5% (16) | 50% (6) | 33.3% (3) | 75% (8) | 100% (2) | 100% (1) | 94.1% (51) |
| SQV | 32.6% (925) | 34.7% (23) | 33.3% (9) | 0% (3) | 63.6% (11) | 83.3% (6) | 88.8% (9) | 96.9% (33) |
| IDV | 28.9% (1,852) | 25.9% (54) | 40.0% (25) | 15.2% (46) | 89.2% (84) | 89.1% (37) | 96.4% (28) | 94.2% (105) |
| LPV | 23.1% (320) | 50% (8) | 0% (1) | 0% (2) | 33.3% (9) | 85.7% (14) | 100% (1) | 72.7% (11) |
| DRV | 0% (45) | – | – | – | – | – | – | – |
The Ki values for subtype B with all inhibitors tested were subnanomolar and in agreement with other studies (40, 59). Overall, the E35D and R57K natural polymorphisms, alone or in combination, are found to have no impact on PI resistance, as evidenced by minor to no significantly different changes in Ki values. Kinetic analyses and inhibition assays indicate that E35D and R57K do not modulate interactions of the catalytic pocket. Additionally, drug binding affinity can be assessed by evaluation of the atomic arrangement of inhibitors inside the catalytic pocket of HIV-1 PR (19, 20). As discussed above and shown in Fig. 4, polymorphisms E35D and R57K induce little to no backbone rearrangements around the catalytic pocket (Fig. 8). Nearly identical atomic arrangements of SQV are found in E35D and PR5 constructs. One trivial difference of DRV on the aniline group could be distinguished among conformations when bound to subtype B, E35D, and PR5 constructs. Consequently, the structures are consistent with kinetic analyses indicating little to no impact on inhibitor dissociation. Taking into consideration that patterns of secondary mutations are seen to diverge for various subtypes and drug resistance emerges at different rates than for non-B PR (60–65), our kinetic and inhibition assay results imply that E35D and R57K must act in concert with additional primary mutations and alternative secondary mutations to impact drug resistance.
FIGURE 8.
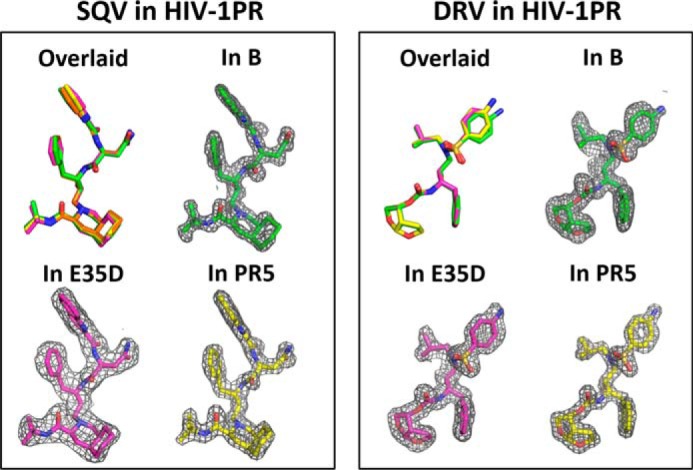
SQV and DRV inhibitor binding within the catalytic pocket of HIV-1 PR. Left, overlaid comparison of overall conformation of SQV, with corresponding electron density maps as found in subtype B, E35D, and PR5. Right, overlaid comparison of overall conformation of DRV, with corresponding electron density maps as found in subtype B, E35D, and PR5. SQV and DRV in subtype B construct are obtained from PDB codes 3TKG (49) and 3BVB (54).
E35D/R57K Are Selected for in Subtype A, F, and G CRF01_AE
The salt bridge interaction between residues 35 and 57 serves as a major interaction between the flap and hinge regions of PR. As reported in the Stanford University HIV Drug Resistance Database, only one natural polymorphism at each of these sites (35 and 57) is found in drug-naive subtypes and CRFs compared with the consensus subtype B sequence, namely E35D and R57K, which on average are found at 41 and 14%, respectively (54). Other amino acids reported for these two residues in drug-naive constructs occur with probabilities of <1%. Analysis of sequences deposited within the HIV Drug Resistance Database shows that the occurrence probability of E35D and R57K differs among subtypes and CRFs. For drug-naive patients, E35D is predominant (>80%) in subtype A (prevalent in Uganda), subtype F (prevalent in Brazil), and CRF01_AE (prevalent in Thailand and Vietnam) (Fig. 9, A and B, and Tables 4 and 5). This occurrence is more than twice the frequency of this mutation seen in subtype B (prevalent in the Americas, west Europe, Australia, and Japan), subtype C (prevalent in India and South Africa), subtype D (prevalent in Uganda), and CRF_AG (prevalent in Cameroon and the Ivory Coast). A similar trend is also observed for R57K, which is nearly absent in subtypes B, C, D and CRF_AG, but occurs at 10–25% prevalence in subtype G and CRF01_AE and with >40% prevalence in subtypes A and F. Based on the structural analysis of the salt bridge interaction between residues 35 and 57, we would suggest that the local protein structure, conformational sampling, and backbone dynamics of subtypes that harbor the E35D/R57K substitutions will be similar to our findings for PR5, CRF01_AE, and E35D.
FIGURE 9.

Bar graphs of the prevalence of E35D (A) and R57K (B) as natural polymorphisms in HIV-1 PR subtypes and CRFs are shown, and data are not shown when the total number of isolates is smaller than 5. C, population percentage change (δP%) of the E35D and R57K natural polymorphisms under PI treatments, where δP% = (P(in PI-treated patients) − P(in drug-naive patients))/(P(in drug-naive patients)) × 100. The insets show the enlarged bar graphs of subtype F. Data were not included for TPV-, APV-, lopinavir-, and DRV-treated subtype F patients due to the limited number of DNA sequences.
TABLE 5.
Statistics of the prevalence of R57K natural polymorphisms, where numbers in parentheses indicate the total number of isolates in analysis (one mutation was extracted from one person)
The data analysis is up to March 2015. – indicates no data available in the database.
| R57K mutation in HIV-1 PR subtypes with/without treatment | ||||||||
|---|---|---|---|---|---|---|---|---|
| B | C | D | AG | G | A | AE | F | |
| Naive | 11.8% (29,309) | 3.5% (10,551) | 11.2% (1,953) | 2.2% (4,302) | 28.1% (1669) | 43.9% (4,934) | 14.3% (6,132) | 85.2% (1,114) |
| TPV | 21.8% (96) | – | 0% (1) | 0% (2) | – | 100% (2) | – | 66.6% (3) |
| APV | 17.0% (194) | 0% (3) | – | 0% (1) | 100% (1) | – | – | 80% (5) |
| RTV | 12.9% (1,075) | 0% (29) | 7.6% (13) | 0% (8) | 18.7% (16) | 62.5% (8) | 11.1% (9) | 88.8% (54) |
| NFV | 13.4% (827) | 0% (17) | 0% (7) | 0% (4) | 33.3% (3) | 100% (2) | 0% (1) | 94.1% (51) |
| SQV | 11.9% (928) | 0% (24) | 0% (9) | 0% (4) | 16.6% (12) | 50% (6) | 11.1% (9) | 93.9% (33) |
| IDV | 11.3% (1,852) | 0% (56) | 8% (25) | 2.1% (47) | 5.8% (86) | 52.6% (38) | 14.2% (28) | 94.3% (106) |
| LPV | 9.8% (315) | 0% (8) | 0% (1) | 0% (2) | 11.1% (9) | 57.1% (14) | 0% (1) | 100% (11) |
| DRV | 6.6% (45) | – | – | – | – | – | – | – |
E35D/R57K Are Selected for in TPV Resistance and Suppressed in DRV Resistance
Further analysis of the data provided by the Stanford database reveals that PI treatment alters the prevalence of E35D and R57K (Tables 4 and 5). Specifically, for subtype B, it appears that E35D/R57K substitutions are selected in response to TPV-induced drug pressure, although these substitutions are suppressed in DRV drug pressure evolution. Fig. 9C shows the percentage change in E35D and R57K prevalence in subtypes B and F as a function of PI exposure. For subtype B, inhibitors SQV, NFV, RTV, APV, and TPV select for these substitutions in increasing prevalence, whereas inhibitors IDV, LPV, and DRV are seen to deselect for the altered salt bridge interaction, again listed in increasing order. In contrast, for subtype F, which contains the E35D and R57K substitutions as natural polymorphisms, drug-pressure is not yet seen to significantly alter the mutational prevalence at these sites. It is noted that insufficient data exist in the Stanford database for a complete analysis of subtype F (Tables 4 and 5). The meaning of these observations is unclear at the moment but may indicate compensating conformational landscape/dynamics/entropic interactions that occur as mutations accumulate in response to different inhibitors. Future work is focused on obtaining structural, biochemical, and biophysical information of TPV bound to E35D, PR5 among other subtypes, and CRFs to gain structural insights into why TPV may select for the altered salt bridge interaction. The E35D/R57K double mutation may serve as an interesting molecular interaction in non-B subtypes to target in future inhibitor design schemes, and it may provide further insights into the balance between enthalpic/entropic protein/inhibitor interactions in drug resistance. It was suggested previously that the current antiretroviral PI drugs, which were designed against HIV-1 subtype B virus, were also actively inhibiting the non-subtype B constructs; however, distinct non-drug-resistant mutation patterns developed (12). Based on the above analysis, it may be beneficial to carry out further studies on possible collaborating effects of E35D and R57K natural polymorphisms in combination with common drug pressure-selected mutations of subtypes and CRFs.
Analysis of Subtype B DRV-resistant Structures
Based on previous studies (11, 54, 55, 66, 67), there are five x-ray crystal structures of DRV-resistant mutants DRV1, DRV2, DRV5, PR20, and P51. Out of these five structures, DRV1 and P51 are the two constructs that not only show DRV resistance but also have similar distances between residues 35 and 57 as that of the subtype B construct (PDB code 3BVB). The local structures around Glu-35 and Arg-57 in DRV1 and P51 are shown in Fig. 5, J and K. A significant salt bridge interaction can be readily observed by the N-O distances (Table 2) and the residue side-chain configurations. Based on the crystal structures, we hypothesize that the DRV-resistant constructs such as P51 and DRV1 should have backbone dynamics more similar to subtype B than PR5 or E35D (future NMR investigations). It is interesting that DRV selects for restabilization of this hinge/flap interaction when eliciting drug resistance, thus potentially implying that the additional mutations may cause destabilization elsewhere in the dimer that needs to be compensated for by regained structural stability between the flap/hinge regions.
Conclusions
The presence of E35D alone is shown by DEER results to destabilize the semi-open conformation. When E35D is combined with other natural polymorphisms, the salt bridge between the flaps and cantilever region is destabilized, likely leading to increased percentages of the curled-open population; however, the possibility does exist that other mutations in PR5 combine to enhance the stability of the curled-open state. NMR analysis suggests that E35D alone and in conjunction with R57K natural polymorphisms are able to enhance the overall protease backbone dynamics especially in the flap and hinge regions, which may confer some selectivity under protease inhibitor pressure. Six HIV-1 protease crystal structures, carrying E35D and/or R57K natural polymorphisms, were analyzed and compared with four other crystals. The increased backbone dynamics in the E35D construct is suggested to be caused by the weakened salt bridge interaction between Asp-35 and Arg-57. The combined effect of the dissociated salt bridge interaction with restabilization via compensating interactions between Asp-35 and Lys-57 via intermediate residues from Val-77 to Pro-79 can explain the similarity in backbone dynamics of PR5 and E35D, although DEER-based conformational ensembles differ. Based on the current statistics from the HIV Drug Resistance Database, occurrence probability of E35D and R57K reveals a distinct characteristic pattern across different HIV-1 subtypes and CRFs indicating possible protein dynamics diversity in HIV-1 PR. Drug-selected mutation evolution patterns for the two latest PIs, including TPV and DRV, demonstrate opposite selection patterns in subtype B of these two natural polymorphisms, possibly suggesting a compensating mechanism for protein/inhibitor flexibility. At this time, there is insufficient data for non-B subtypes to fully analyze the impact of E35D/R57K natural polymorphisms on inhibitor drug resistance development.
Experimental Procedures
Cloning and Site-directed Mutagenesis
Escherichia coli codon-optimized genes of HIV-1 PR subtype B and PR5 constructs were purchased from DNA 2.0 (Menlo Park, CA), which were cloned into pET-23a plasmid between two restriction enzyme digestion sites of NdeI and BamHI. Plasmid carrying the gene for the E35D construct was generated by utilizing the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The D25N inactive constructs were utilized for all DEER, crystallization, and NMR measurements. Active Asp-25 enzymes were utilized for the kinetics measurements. Additionally, three stabling mutations (Q7K, L33I, and L63I) and two other mutations (C67A and C95A) for eliminating native cysteines were designed in all constructs to avoid auto-proteolysis of the protease and to ensure site-specific spin labeling for DEER measurements, respectively. These five substitutions are present in all constructs studied here. Fig. 1A shows amino acid sequences.
Protein Expression, Purification, Spin Labeling, and Sample Preparation for DEER
For DEER measurements the spin label, 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl, methanethiosulfonate (MTSL), was chemically incorporated into PR by mutation at site K55C. MTSL was purchased from Santa Cruz Biotechnology. Protein expression, purification, and spin labeling were carried out by following our previously developed procedures where buffer pH for one step of purification was modified for E35D given its isoelectric point of 9.39 (30, 34). To ensure high spin labeling efficiency, 4-fold molar excess spin label was added into the HIV-1 PR sample solution. The reaction was carried out at 4 °C in the dark for 12 h. Protein precipitates were removed by a high speed centrifuge at 12,000 rpm, and the excess free spin label was removed by buffer exchange (2 mm NaOAc with pH at 5.0) by using a HiPrep 26/10 desalting column (GE Healthcare).
DEER and Data Analysis
HIV-1 PR samples were concentrated and buffer-exchanged to 20 mm NaOAc buffer in D2O at pH 5.0 by using centrifugal membrane concentrators (Millipore, Billerica, MA) to 140 μm. 4-Fold molar excess of inhibitors, proportional to enzyme, were added into HIV-1 PR solutions. The ligand reactions were performed at room temperature for >1 h. Sequentially, 30% v/v deuterated glycerol was added into the protein solution. The final protease concentration was ∼100 μm. The protein sample was then transferred to an EPR tube (3-mm inner diameter and 4-mm outer diameter quartz) (Norell, Marion, NC), flash-frozen in liquid nitrogen, and then inserted into the dielectric ring resonator (ER 4118X-MD-5) in a frozen state. Four-pulse DEER scheme was adopted in the measurements on Bruker EleXsys E580 spectrometer at a temperature of 65 K, as described previously (34, 68). The DEER modulation curve was processed by using DeerAnalysis (69, 70), where the inter-molecular background was subtracted; high frequency noise was filtered by low frequency-pass digital filter, and consequently, the DEER data were converted into distance distribution profiles via Tikhonov regularization. The distance profiles are regenerated by Gaussian-shaped populations that represent conformational states of HIV-1 PR from which DEER modulation curves are reconstructed for error analysis and peak suppression (30, 33, 34, 50). Details regarding DEER data analyses and population validation are provided in supplemental Figs. S1–S8.
Sample Preparation, NMR Spectroscopy, and Data Analysis
Uniformly 15N-labeled E35D was expressed and purified from E. coli grown in modified minimal media as described previously (32, 33). NMR samples contained ∼150 μm 15N-labeled HIV-1 PR in 2 mm deuterated NaOAc buffer at pH 5.0 with 10% D2O and 100 μm 4,4-dimethyl-4-silapentane-1-sulfonic acid as an internal reference. NMR relaxation data were collected on Bruker Avance spectrometers at two frequencies of 600 MHz (AMRIS Facility, University of Florida) and 800 MHz (National High Magnetic Field Laboratory, Florida State University) at 293 K, respectively. Spin-lattice relaxation (R1) and spin-spin (R2) relaxation rates of 15N were measured via HSQC pulse train of hsqct1etf3gpsi and CPMG pulse train of hsqct2etf3gpsi, respectively (33). 1H-15N NOEs were measured in an interleaved manner using the pulse sequence of hsqcnoef3gpsi with a recycle delay of 5 s (33). NMRPipe and Sparky were used to process and analyze the data (71, 72). R1 and R2 relaxation rates were calculated by fitting the peak intensity to a single exponential decay related to delay time by using GUIrelax (73, 74). NOE values were calculated by taking the ratio of resonance intensities with and without 1H pre-saturation. Model-free analysis was performed using GUIrelax (73, 74). Difference plot of NMR order parameters (S2) was calculated by using S2ij = S2ij_HIV-1 PRcompare − S2i_HIV-1 PRreference.
Protein Expression, Purification, and Crystallization
For crystallization trials, the following additional purification steps were added to the NMR/DEER protocols described above. Samples were buffer-exchanged by centrifugal membrane concentrators (Millipore, Billerica, MA) into buffer A (30 mm K2HPO4, 100 mm NaCl, 4 mm EDTA, and 5% glycerol at pH 7.3) for further purification by size-exclusion chromatography with a HiLoad 16/60 Superdex 75 Prep Grade size column (GE Healthcare). Fractions corresponding to dimeric protein were pooled and concentrated by centrifugal membrane concentrators (Millipore, Billerica, MA) to 3–5 mg/ml in buffer B (50 mm sodium acetate and 5% glycerol at pH 5) (19, 40, 75, 76). Inhibitors or non-reducible substrate mimics were added to the protein sample at 3:1 molar excess and allowed to interact at 4 °C for 1 h, after which any precipitant was removed by centrifugation at 14,000 rpm. Hanging drop vapor diffusion was used for crystallization of all samples, using Hampton Crystal Screen Cryo and Crystal Screen (Aliso Viejo, CA) for initial crystallization screening. The gradient reservoir buffer of GRB-1# (ammonium sulfate from 1.0 to 3 m, 20 mm sodium acetate at pH 5) (19, 75) and GRB-2# (sodium chloride from 0.8 to 2.7 m, 30 mm citric acid at pH 5) were chosen as “promising precipitation solution” for further optimization in all crystal trials of E35D and PR5 constructs. SQV-bound E35D crystals were obtained from both GRB-1# and GRB-2#, whereas DRV-, CaP2-, and APV-bound E35D crystals could only be obtained in GRB-2#. SQV- and DRV-bound PR5 crystals were obtained from GRB-1# buffer. Regarding the crystal morphology, rectangular/square sheet-like crystals dominated in E35D crystals, although either non-regularly shaped or rod-like crystals were formed in PR5 crystals.
X-ray Diffraction Data Collection and Analysis
All crystals were pre-soaked in a 30% glycerol cryo-protectant solution before flash cooling to 100 K prior to data collection. Data were collected “in-house” using an RU-H3R rotating copper anode (λ = 1.5418 Å) operating at 50 kV and 22 mA utilizing an R-Axis IV2+ image plate detector (Rigaku Corp.). IMosflm and Scala from CCP4 suite 6.4.0 were used for indexing the diffraction data and merging the diffraction peaks (77). All data were processed and scaled to a maximum resolution of 1.6 Å, with an overall completeness of >92% and Rmerge of <16%, for each data set. Initial phases were obtained using two previously determined HIV-1 PR structures (PDB code 4NJT (78) and PDB code 3K4V (79)) as search models. Phases were generated using PHENIX version 1.9 for E35D and PR5 structures, respectively (80, 81). Structural refinements were also completed using PHENIX.refinement with Rfree calculated with 10% of the unique reflections selected at random and excluded (81–83). Manual refitting of all residues and ligands, including DRV, SQV, CaP2, and APV, into the electron density was complete using Coot 0.7.2.1 (84). PyMOL version 1.7 was used to visualize the crystal structures and to generate figures (85). Double difference analysis of the backbone Cαs was calculated by “CrystalAnalysis” using Dij = Dij_HIV-1 PRcompare − Dij_HIV-1 PRreference, where i and j represent the residue number, and D represents the distance between these two residues. CrystalAnalysis is a Matlab-based program that our laboratory developed and is available upon request or at the Matlab Exchange File on-line website. For the analysis contained within Table 2 regarding HIV-1 P51, we refit the local structure around Glu-35 and Arg-57 using the electron density map in the RCSB Data Bank.
Protein Activity and Inhibition Constants
Protein expression and purification adopted the same protocol as described above except that plasmids encoding active HIV-1 PR were utilized. The Michaelis-Menten constants (Km, kcat, and kcat/Km) for all three constructs as well as the inhibition constant (Ki) for four inhibitors of SQV, APV, ATV, and DRV were determined as described previously (40, 58, 76). The reaction assays were carried on Cary 50 Bio UV-visible spectrophotometer with sodium acetate buffer (50 mm NaOAc, 150 mm NaCl, 2 mm EDTA, and 1 mm DTT at pH 4.7) at a temperature of 37 °C where chromogenic substrate (Lys-Ala-Arg-Val-Leu*Nph-Glu-Ala-NLe-Gly) was used. All experiments were repeated three times, and the averaged value and standard deviation are reported in Table 3.
Author Contributions
G. E. F. conceived the study. G. E. F. and Z. L. designed the experiments and wrote the paper. Z. L., X. H., L. H., and L. P. expressed and purified the HIV-1 PR samples. Z. L. carried out the DEER experiment and analyzed the data. Z. L., X. H., L. H., and W. T. collected and analyzed the NMR data. Z. L. set up crystal trials and optimized the crystal conditions, and N. E. G. trained Z. L. on how to set up the crystal trials. Z. L., R. M., K. M. P., B. P. M., and K. L. collected the x-ray diffraction and solved the protein crystal structures. Z. L., Y. T., and B. M. D. collected the protein kinetics data. B. M. D., B. P. M., and R. M. proofread the manuscript and provided feedback and corrections.
Supplementary Material
Acknowledgments
We thank the Center of Structural Biology for support of the x-ray facility at the University of Florida. We also thank Umar T. Twahir, Justin L. Goodsell, Dr. Alexander Angerhofer, Dr. Joanna R. Long, and Dr. Fengli Zhang for helpful discussions and aid in troubleshooting experimental difficulties.
This work was supported by National Institutes of Health Grants GM105409 (to G. E. F.), S10RR031603 (to G. E. F.), and R37 A128571 (to B. M. D.). A portion of this work was performed at the McKnight Brain Institute at the National High Magnetic Field Laboratory AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and the State of Florida. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1 and Figs. S1–S11.
- PR
- protease
- CRF
- circulating recombinant form
- MD
- molecular dynamics
- r.m.s.d.
- root mean squared deviation
- MTSL
- 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl
- PR5
- protease with five mutations relative to subtype B
- SQV
- saquinavir
- DRV
- darunavir
- APV
- amprenavir
- TPV
- tipranavir
- Ca-P2
- non-hydrolysable substrate mimic
- DEER
- double electron-electron resonance
- PDB
- Protein Data Bank
- PI
- protease inhibitor.
References
- 1. Gallo R. C., Sarin P. S., Gelmann E. P., Robert-Guroff M., Richardson E., Kalyanaraman V. S., Mann D., Sidhu G. D., Stahl R. E., Zolla-Pazner S., Leibowitch J., and Popovic M. (1983) Isolation of human T-cell leukemia-virus in acquired immune-deficiency syndrome (AIDS). Science 220, 865–867 [DOI] [PubMed] [Google Scholar]
- 2. Santos A. F., and Soares M. A. (2010) HIV genetic diversity and drug resistance. Viruses 2, 503–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. UNAIDS (2016) UNAIDS Report on the Global AIDS Epidemic, Joint United Nations Programme on HIV/AIDS (UNAIDS) [Google Scholar]
- 4. Wlodawer A., and Erickson J. W. (1993) Structure-based inhibitors of Hiv-1 protease. Annu. Rev. Biochem. 62, 543–585 [DOI] [PubMed] [Google Scholar]
- 5. Ghosh A. K., Anderson D. D., Weber I. T., and Mitsuya H. (2012) Enhancing protein backbone binding–a fruitful concept for combating drug-resistant HIV. Angew. Chem. Int. Ed. Engl. 51, 1778–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silver N. W., King B. M., Nalam M. N., Cao H., Ali A., Kiran Kumar Reddy G. S., Rana T. M., Schiffer C. A., and Tidor B. (2013) Efficient computation of small-molecule configurational binding entropy and free energy changes by ensemble enumeration. J. Chem. Theory Comput. 9, 5098–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pearl L. H., and Taylor W. R. (1987) A structural model for the retroviral proteases. Nature 329, 351–354 [DOI] [PubMed] [Google Scholar]
- 8. Miller M., Jaskólski M., Rao J. K., Leis J., and Wlodawer A. (1989) Crystal-structure of a retroviral protease proves relationship to aspartic protease family. Nature 337, 576–579 [DOI] [PubMed] [Google Scholar]
- 9. Nicholson L. K., Yamazaki T., Torchia D. A., Grzesiek S., Bax A., Stahl S. J., Kaufman J. D., Wingfield P. T., Lam P. Y., and Jadhav P. K. (1995) Flexibility and function in Hiv-1 protease. Nat. Struct. Biol. 2, 274–280 [DOI] [PubMed] [Google Scholar]
- 10. Dragic T., Litwin V., Allaway G. P., Martin S. R., Huang Y., Nagashima K. A., Cayanan C., Maddon P. J., Koup R. A., Moore J. P., and Paxton W. A. (1996) HIV-1 entry into CD4(+) cells is mediated by the chemokine receptor CC-CKR-5. Nature 381, 667–673 [DOI] [PubMed] [Google Scholar]
- 11. Louis J. M., Aniana A., Weber I. T., and Sayer J. M. (2011) Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 9072–9077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kantor R., Katzenstein D. A., Efron B., Carvalho A. P., Wynhoven B., Cane P., Clarke J., Sirivichayakul S., Soares M. A., Snoeck J., Pillay C., Rudich H., Rodrigues R., Holguin A., Ariyoshi K., et al. (2005) Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: Results of a Global Collaboration. PLoS Med. 2, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lazaro E., Tram L. T., Bellecave P., Guidicelli G. L., Anies G., Thu H. H., Debelleix M. P., Vray M., Recordon-Pinson P., Taupin J. L., Lien T. T., and Fleury H. (2011) Molecular characterization of HIV-1 CRF01_AE in Mekong Delta, Vietnam, and impact of T-cell epitope mutations on HLA recognition (ANRS 12159). PLoS One 6, e26244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. HUGO Pan-Asian SNP Consortium, Abdulla M. A., Ahmed I., Assawamakin A., Bhak J., Brahmachari S. K., Calacal G. C., Chaurasia A., Chen C. H., Chen J., Chen Y. T., Chu J., Cutiongco-de la Paz E. M., De Ungria M. C., Delfin F. C., et al. (2009) Mapping human genetic diversity in Asia. Science 326, 1541–1545 [DOI] [PubMed] [Google Scholar]
- 15. Bandaranayake R. M., Prabu-Jeyabalan M., Kakizawa J., Sugiura W., and Schiffer C. A. (2008) Structural analysis of human immunodeficiency virus type 1 CRF01_AE protease in complex with the substrate p1–p6. J. Virol. 82, 6762–6766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bandaranayake R. M., Kolli M., King N. M., Nalivaika E. A., Heroux A., Kakizawa J., Sugiura W., and Schiffer C. A. (2010) The effect of clade-specific sequence polymorphisms on HIV-1 protease activity and inhibitor resistance pathways. J. Virol. 84, 9995–10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shafer R. W., and Schapiro J. M. (2008) HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 10, 67–84 [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson S. I., Phylip L. H., Mills J. S., Gulnik S. V., Erickson J. W., Dunn B. M., and Kay J. (1997) Escape mutants of HIV-1 proteinase: enzymic efficiency and susceptibility to inhibition. Biochim. Biophys. Acta 1339, 113–125 [DOI] [PubMed] [Google Scholar]
- 19. King N. M., Prabu-Jeyabalan M., Bandaranayake R. M., Nalam M. N., Nalivaika E. A., Özen A., Haliloğlu T., Yilmaz N. K., and Schiffer C. A. (2012) Extreme entropy-enthalpy compensation in a drug-resistant variant of HIV-1 protease. ACS Chem. Biol. 7, 1536–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Q., Anderson C., Zhang H., Li X., Inglis F., Jayagopal A., and Wang S. (2014) Repression of choroidal neovascularization through actin cytoskeleton pathways by MicroRNA-24. Mol. Ther. 22, 378–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ragland D. A., Nalivaika E. A., Nalam M. N., Prachanronarong K. L., Cao H., Bandaranayake R. M., Cai Y., Kurt-Yilmaz N., and Schiffer C. A. (2014) Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc. 136, 11956–11963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kantor R., Fessel W. J., Zolopa A. R., Israelski D., Shulman N., Montoya J. G., Harbour M., Schapiro J. M., and Shafer R. W. (2002) Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob. Agents Chemother. 46, 1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghosh A. K., Osswald H. L., and Prato G. (2016) Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem. 59, 5172–5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurt Yilmaz N., Swanstrom R., and Schiffer C. A. (2016) Improving viral protease inhibitors to counter drug resistance. Trends Microbiol. 24, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber I. T., Kneller D. W., and Wong-Sam A. (2015) Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem. 7, 1023–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laco G. S. (2015) HIV-1 protease substrate-groove: role in substrate recognition and inhibitor resistance. Biochimie 118, 90–103 [DOI] [PubMed] [Google Scholar]
- 27. Batista P. R., Wilter A., Durham E. H., and Pascutti P. G. (2006) Molecular dynamics simulations applied to the study of subtypes of HIV-1 protease common to Brazil, Africa, and Asia. Cell Biochem. Biophys. 44, 395–404 [DOI] [PubMed] [Google Scholar]
- 28. Muzammil S., Ross P., and Freire E. (2003) A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance. Biochemistry 42, 631–638 [DOI] [PubMed] [Google Scholar]
- 29. Naicker P., and Sayed Y. (2014) Non-B HIV-1 subtypes in sub-Saharan Africa: impact of subtype on protease inhibitor efficacy. Biol. Chem. 395, 1151–1161 [DOI] [PubMed] [Google Scholar]
- 30. Kear J. L., Blackburn M. E., Veloro A. M., Dunn B. M., and Fanucci G. E. (2009) Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J. Am. Chem. Soc. 131, 14650–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Vera I. M., Smith A. N., Dancel M. C., Huang X., Dunn B. M., and Fanucci G. E. (2013) Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease. Biochemistry 52, 3278–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang X., Britto M. D., Kear-Scott J. L., Boone C. D., Rocca J. R., Simmerling C., Mckenna R., Bieri M., Gooley P. R., Dunn B. M., and Fanucci G. E. (2014) The role of select subtype polymorphisms on HIV-1 protease conformational sampling and dynamics. J. Biol. Chem. 289, 17203–17214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carter J. D., Gonzales E. G., Huang X., Smith A. N., de Vera I. M., D'Amore P. W., Rocca J. R., Goodenow M. M., Dunn B. M., and Fanucci G. E. (2014) Effects of pre and post therapy drug pressure-selected mutations on HIV-1 protease conformational sampling. FEBS Lett. 588, 3123–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galiano L., Ding F., Veloro A. M., Blackburn M. E., Simmerling C., and Fanucci G. E. (2009) Drug pressure-selected mutations in HIV-1 protease alter flap conformations. J. Am. Chem. Soc. 131, 430–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blackburn M. E., Veloro A. M., and Fanucci G. E. (2009) Monitoring inhibitor-induced conformational population shifts in HIV-1 protease by pulsed EPR spectroscopy. Biochemistry 48, 8765–8767 [DOI] [PubMed] [Google Scholar]
- 36. Cai Y., Myint W., Paulsen J. L., Schiffer C. A., Ishima R., and Kurt Yilmaz N. (2014) Drug resistance mutations alter dynamics of inhibitor-bound HIV-1 protease. J. Chem. Theory Comput. 10, 3438–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen C. H., Chang Y. C., Agniswamy J., Harrison R. W., and Weber I. T. (2015) Conformational variation of an extreme drug resistant mutant of HIV protease. J. Mol. Graph. Model 62, 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piana S., Carloni P., and Rothlisberger U. (2002) Drug resistance in HIV-1 protease: flexibility-assisted mechanism of compensatory mutations. Protein Sci. 11, 2393–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Foulkes-Murzycki J. E., Scott W. R., and Schiffer C. A. (2007) Hydrophobic sliding: a possible mechanism for drug resistance in human immunodeficiency virus type 1 protease. Structure 15, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldfarb N. E., Ohanessian M., Biswas S., McGee T. D. Jr., Mahon B. P., Ostrov D. A., Garcia J., Tang Y., McKenna R., Roitberg A., and Dunn B. M. (2015) Defective hydrophobic sliding mechanism and active site expansion in HIV-1 protease drug resistant variant Gly48Thr/Leu89Met: mechanisms for the loss of saquinavir binding potency. Biochemistry 54, 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chang M. W., and Torbett B. E. (2011) Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 410, 756–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hornak V., Okur A., Rizzo R. C., and Simmerling C. (2006) HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 103, 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ding F., Layten M., and Simmerling C. (2008) Solution structure of HIV-1 protease flaps probed by comparison of molecular dynamics simulation ensembles and EPR experiments. J. Am. Chem. Soc. 130, 7184–7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spinelli S., Liu Q. Z., Alzari P. M., Hirel P. H., and Poljak R. J. (1991) The 3-dimensional structure of the aspartyl protease from the Hiv-1 isolate Bru. Biochimie 73, 1391–1396 [DOI] [PubMed] [Google Scholar]
- 45. Robbins A. H., Coman R. M., Bracho-Sanchez E., Fernandez M. A., Gilliland C. T., Li M., Agbandje-McKenna M., Wlodawer A., Dunn B. M., and McKenna R. (2010) Structure of the unbound form of HIV-1 subtype A protease: comparison with unbound forms of proteases from other HIV subtypes. Acta Crystallogr. D Biol. Crystallogr. 66, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Galiano L., Bonora M., and Fanucci G. E. (2007) Interflap distances in HIV-1 protease determined by pulsed EPR measurements. J. Am. Chem. Soc. 129, 11004–11005 [DOI] [PubMed] [Google Scholar]
- 47. Huang X., de Vera I. M., Veloro A. M., Blackburn M. E., Kear J. L., Carter J. D., Rocca J. R., Simmerling C., Dunn B. M., and Fanucci G. E. (2012) Inhibitor-induced conformational shifts and ligand-exchange dynamics for HIV-1 protease measured by pulsed EPR and NMR spectroscopy. J. Phys. Chem. B 116, 14235–14244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Naicker P., Stoychev S., Dirr H. W., and Sayed Y. (2014) Amide hydrogen exchange in HIV-1 subtype B and C proteases- insights into reduced drug susceptibility and dimer stability. FEBS J. 281, 5395–5410 [DOI] [PubMed] [Google Scholar]
- 49. Agniswamy J., Sayer J. M., Weber I. T., and Louis J. M. (2012) Terminal interface conformations modulate dimer stability prior to amino terminal autoprocessing of HIV-1 protease. Biochemistry 51, 1041–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu Z., Casey T. M., Blackburn M. E., Huang X., Pham L., de Vera I. M., Carter J. D., Kear-Scott J. L., Veloro A. M., Galiano L., and Fanucci G. E. (2016) Pulsed EPR characterization of HIV-1 protease conformational sampling and inhibitor-induced population shifts. Phys. Chem. Chem. Phys. 18, 5819–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Casey T. M., and Fanucci G. E. (2015) Spin labeling and double electron-electron resonance (DEER) to deconstruct conformational ensembles of HIV protease. Methods Enzymol. 564, 153–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y., Chang Y. C., Louis J. M., Wang Y. F., Harrison R. W., and Weber I. T. (2014) Structures of darunavir-resistant HIV-1 protease mutant reveal atypical binding of darunavir to wide open flaps. ACS Chem. Biol. 9, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Agniswamy J., Shen C. H., Aniana A., Sayer J. M., Louis J. M., and Weber I. T. (2012) HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry 51, 2819–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sayer J. M., Liu F., Ishima R., Weber I. T., and Louis J. M. (2008) Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J. Biol. Chem. 283, 13459–13470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sasková K. G., Kozísek M., Rezácová P., Brynda J., Yashina T., Kagan R. M., and Konvalinka J. (2009) Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol. 83, 8810–8818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Venkatakrishnan B., Palii M.-L., Agbandje-McKenna M., and McKenna R. (2012) Mining the protein data bank to differentiate error from structural variation in clustered static structures: an examination of HIV protease. Viruses 4, 348–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barlow D. J., and Thornton J. M. (1983) Ion-pairs in proteins. J. Mol. Biol. 168, 867–885 [DOI] [PubMed] [Google Scholar]
- 58. Clemente J. C., Hemrajani R., Blum L. E., Goodenow M. M., and Dunn B. M. (2003) Secondary mutations M36I and A71V in the human immunodeficiency virus type 1 protease can provide an advantage for the emergence of the primary mutation D30N. Biochemistry 42, 15029–15035 [DOI] [PubMed] [Google Scholar]
- 59. Coman R. M., Robbins A. H., Fernandez M. A., Gilliland C. T., Sochet A. A., Goodenow M. M., McKenna R., and Dunn B. M. (2008) The contribution of naturally occurring polymorphisms in altering the biochemical and structural characteristics of HIV-1 subtype C protease. Biochemistry 47, 731–743 [DOI] [PubMed] [Google Scholar]
- 60. Soares E. A., Santos A. F., Sousa T. M., Sprinz E., Martinez A. M., Silveira J., Tanuri A., and Soares M. A. (2007) Differential drug resistance acquisition in HIV-1 of subtypes B and C. PLoS One 2, e730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lisovsky I., Schader S. M., Martinez-Cajas J. L., Oliveira M., Moisi D., and Wainberg M. A. (2010) HIV-1 protease codon 36 polymorphisms and differential development of resistance to nelfinavir, lopinavir, and atazanavir in different HIV-1 subtypes. Antimicrob. Agents Chemother. 54, 2878–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Langs-Barlow A., and Paintsil E. (2014) Impact of human immunodeficiency virus type-1 sequence diversity on antiretroviral therapy outcomes. Viruses 6, 3855–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Santoro M. M., and Perno C. F. (2013) HIV-1 genetic variability and clinical implications. ISRN Microbiol. 2013, 481314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martinez-Cajas J. L., Pai N. P., Klein M. B., and Wainberg M. A. (2009) Differences in resistance mutations among HIV-1 non-subtype B infections: a systematic review of evidence. (1996–2008). J. Int. AIDS Soc. 12, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ariyoshi K., Matsuda M., Miura H., Tateishi S., Yamada T., and Sugiura W. (2003) Patterns of point mutations associated with antiretroviral drug treatment failure in CRF01_AE (Subtype E) infection differ from subtype B infection. Jaids-J Acq. Imm. Def. 33, 336–342 [DOI] [PubMed] [Google Scholar]
- 66. Agniswamy J., Shen C. H., Wang Y. F., Ghosh A. K., Rao K. V., Xu C. X., Sayer J. M., Louis J. M., and Weber I. T. (2013) Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1′-pyrrolidinone or P2-Tris-tetrahydrofuran. J. Med. Chem. 56, 4017–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kožíšek M., Lepšík M., Grantz Sašková K. G., Brynda J., Konvalinka J., and Rezáčová P. (2014) Thermodynamic and structural analysis of HIV protease resistance to darunavir–analysis of heavily mutated patient-derived HIV-1 proteases. FEBS J. 281, 1834–1847 [DOI] [PubMed] [Google Scholar]
- 68. Tsvetkov Y. D., and Grishin Y. A. (2009) Techniques for EPR spectroscopy of pulsed electron double resonance (PELDOR): A review. Instruments Exp. Techniques. 52, 615–636 [Google Scholar]
- 69. Jeschke G., Panek G., Godt A., Bender A., and Paulsen H. (2004) Data analysis procedures for pulse ELDOR measurements of broad distance distributions. Appl. Magn. Reson. 26, 223–244 [Google Scholar]
- 70. Jeschke G., and Schlick S. (2006) Spatial distribution of stabilizer-derived nitroxide radicals during thermal degradation of poly(acrylonitrile-butadiene-styrene) copolymers: a unified picture from pulsed ELDOR and ESR imaging. Phys. Chem. Chem. Phys. 8, 4095–4103 [DOI] [PubMed] [Google Scholar]
- 71. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) Nmrpipe–a multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 72. Lee W., Tonelli M., and Markley J. L. (2015) MNRFAM-SPARKY: enhanced software for biomolecular NMR. Bioinformatics 31, 1325–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bieri M., d'Auvergne E. J., and Gooley P. R. (2011) relaxGUI: a new software for fast and simple NMR relaxation data analysis and calculation of ps-ns and mus motion of proteins. J. Biomol. NMR 50, 147–155 [DOI] [PubMed] [Google Scholar]
- 74. d'Auvergne E. J., and Gooley P. R. (2008) Optimisation of NMR dynamic models II. A new methodology for the dual optimisation of the model-free parameters and the Brownian rotational diffusion tensor. J. Biomol. NMR 40, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. King N. M., Prabu-Jeyabalan M., Nalivaika E. A., Wigerinck P., de Béthune M. P., and Schiffer C. A. (2004) Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J. Virol. 78, 12012–12021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Clemente J. C., Coman R. M., Thiaville M. M., Janka L. K., Jeung J. A., Nukoolkarn S., Govindasamy L., Agbandje-McKenna M., McKenna R., Leelamanit W., Goodenow M. M., and Dunn B. M. (2006) Analysis of HIV-1CRF_01 A/E protease inhibitor resistance: structural determinants for maintaining sensitivity and developing resistance to atazanavir. Biochemistry 45, 5468–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yedidi R. S., Garimella H., Aoki M., Aoki-Ogata H., Desai D. V., Chang S. B., Davis D. A., Fyvie W. S., Kaufman J. D., Smith D. W., Das D., Wingfield P. T., Maeda K., Ghosh A. K., and Mitsuya H. (2014) A conserved hydrogen-bonding network of P2 bis-tetrahydrofuran-containing HIV-1 protease inhibitors (PIs) with a protease active-site amino acid backbone aids in their activity against PI-resistant HIV. Antimicrob. Agents Chemother. 58, 3679–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Olajuyigbe F. M., Demitri N., Ajele J. O., Maurizio E., Randaccio L., and Geremia S. (2010) Carbamylation of N-terminal proline. ACS Med. Chem. Lett. 1, 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. DeLano W. L. (2002) The PyMOL Molecular Graphics System, version 1.7x DeLano, Scientific LLC, San Carlos, CA [Google Scholar]
- 86. Lafont V., Armstrong A. A., Ohtaka H., Kiso Y., Mario Amzel L., and Freire E. (2007) Compensating enthalpic and entropic changes hinder binding affinity optimization. Chem. Biol. Drug Des. 69, 413–422 [DOI] [PubMed] [Google Scholar]
- 87. Kear J. L. (2010) An Electron Paramagnetic Resonance Study of HIV-1 Protease and the Development of a Soluble Expression System for Prorenin. Ph.D. thesis University of Florida [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.