

Abstract
Cellular signaling leading to the initiation of apoptosis typically results in the activation of caspases, which in turn leads to the proteolytic generation of protein fragments with new or altered cellular functions. Increasing numbers of reports are demonstrating that the activity of many of these proteolytically activated protein fragments can be attenuated by their selective degradation by the N-end rule pathway. Here we report the first evidence that selective degradation of a caspase product by the N-end rule pathway can be modulated by phosphorylation. We demonstrate that the pro-apoptotic fragment of the bone marrow kinase on chromosome X (BMX) generated by caspase cleavage in the prostate cancer-derived PC3 cell line is metabolically unstable in cells because its N-terminal tryptophan targets it for proteasomal degradation via the N-end rule pathway. In addition, we have demonstrated that phosphorylation of tyrosine 566 relatively inhibits degradation of the C-terminal BMX catalytic fragment, and this phosphorylation is crucial for its pro-apoptotic function. Overall, our results demonstrate that cleaved BMX is a novel N-end rule substrate, and its degradation exhibits a novel interplay between substrate phosphorylation and N-end rule degradation, revealing an increasing complex regulatory network of apoptotic proteolytic signaling cascades.
Keywords: apoptosis, phosphorylation, protein degradation, tyrosine-protein kinase (tyrosine kinase), ubiquitin ligase, BMX, Etk, N-end Rule
Introduction
Bone marrow kinase on chromosome X (BMX) is a member of the Tec non-receptor tyrosine kinase family that also includes BTK, ITK, TEC, and TXK (1–4). Members of this family share a modular structure, including an N-terminal pleckstrin homology domain, a Tec homology domain, Src homology 2 and 3 (SH2 and SH3)3 domains, and a C-terminal catalytic kinase domain (5). Although there is tissue-specific expression of several of the Tec family kinases, BMX is expressed in a variety of tissues and cell types, in contrast to what is implied by its name. BMX expression has been observed in epithelial cells, endothelial cells, cells of hematopoietic origins, and various prostate cancer cell lines (5–7).
Investigations have revealed that BMX plays pivotal roles in signaling for diverse cellular processes, such as cell differentiation, stem cell renewal, proliferation, and transformation (1, 5, 8, 9). BMX is involved in IL-6-induced neuroendocrine differentiation and neuropeptide-induced androgen-independent growth of prostate cancer cells (7). Additionally, BMX has been linked to Src-induced transformation of epithelial cells and fibroblasts via activation of STAT3 and to the transformation of human mammary epithelial cancer cells through activation of Pak1 (5, 10).
BMX activity has been reported to have what appear to be contradictory roles in cell survival and apoptosis. The BMX kinase has been shown to play a protective role against radiation-induced apoptosis in prostate cancer, nasopharyngeal carcinoma, and breast cancer cells (7, 11–13). However, it has also been reported that the BMX kinase can mediate a pro-apoptotic function as well (14, 15). How BMX kinase plays such a paradoxical role in cell survival has been attributed, at least in part, to the proteolytic activation of BMX kinase during apoptosis. For example, during apoptosis in PC3 prostate cancer cells, BMX kinase is cleaved by caspases after Asp-242, generating an active truncated BMXΔN kinase containing the complete SH2 and tyrosine kinase domains but lacking the intact pleckstrin homology and SH3 domains (15). The truncated BMXΔN has been shown to exhibit increased kinase activity and sensitizes prostate cancer cells toward apoptosis in response to various apoptosis-inducing stimuli (15). Although the function of the cleaved BMXΔN has been investigated, the stability of this protein fragment and the role of the neo-N termini generated by proteolysis have never been investigated.
Here we report the first study on the interplay and role of N-end rule-mediated degradation and phosphorylation in BMXΔN metabolic stability and drug-induced apoptosis in prostate cancer cells. The N-end rule pathway is one branch of N terminus-dependent protein degradation (16–18). We have determined that the pro-apoptotic BMXΔN fragment is unstable in cells because its N-terminal tryptophan targets it for proteasomal degradation via the N-end rule pathway. Additionally, we have demonstrated that the metabolic stabilization of BMXΔN, either through the mutation of the destabilizing N-terminal amino acid to a stabilizing residue or via the inhibition of the N-end rule E3 ubiquitin ligases UBR1 and UBR2, augments the apoptosis-inducing effect of docetaxel in prostate cancer cells. Interestingly, we also determined that phosphorylation at tyrosine 566 of BMXΔN inhibits its degradation by the N-end rule pathway, and this phosphorylation is also crucial for its pro-apoptotic function.
Results and Discussion
BMX Is Proteolytically Cleaved in Docetaxel-treated PC3 Cells to Generate the Pro-apoptotic BMXΔN Fragment
Proteolytic generation of BMXΔN by caspase cleavage was reported over a decade ago (15), where cleavage releases the C-terminal BMXΔN fragment (amino acids 243–675) containing part of the SH3 domain, the SH2 domain, and the kinase domain of the protein. To investigate this BMXΔN fragment, we constructed plasmids to express either the full-length BMX kinase with a C-terminal 3×FLAG tag or a ubiquitin (Ub)-BMXΔN 3×FLAG tag fusion protein as schematically shown in Fig. 1A. The Ub-BMXΔN fusion mirrors the caspase-cleaved form of the BMX kinase as endogenous ubiquitin hydrolases proteolytically remove the N-terminal ubiquitin to expose the N-terminal tryptophan. Both constructs were expressed in the androgen-independent PC3 prostate cancer cell line via electroporation. The cells expressing the full-length BMX were also treated with 10 nm docetaxel (or DMSO control), a chemotherapeutic that induces cell death in PC3 cancer cells by inducing mitotic catastrophe and caspase-dependent apoptosis (19–21). As predicted, expression of the Ub-BMXΔN fusion construct results in a protein fragment with an electrophoretic mobility, of an approximate molecular mass of 50 kDa, that matches that of the docetaxel-induced caspase-cleaved form of the full-length BMX kinase (Fig. 1B).
FIGURE 1.
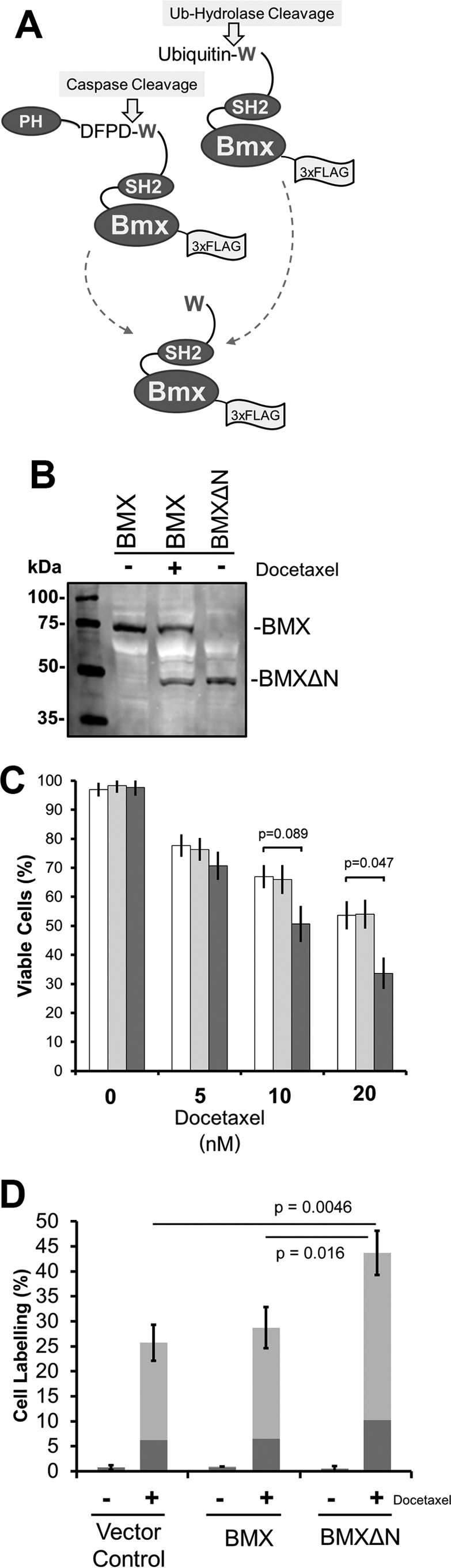
Protease-dependent generation of BMXΔN. A, schematic depiction of the generation of BMXΔN by either caspase cleavage after Asp-242 or as a ubiquitin fusion to generate BMXΔN with an N-terminal tryptophan. PH, pleckstrin homology domain. B, transient transfection with BMX-FLAG or Ub-BMXΔN-FLAG in PC3 cells. After transfection, the cells were treated with 10 nm docetaxel or left untreated before lysis and analysis by SDS-PAGE and Western blotting (WB) analysis with an anti-FLAG antibody. WB analysis reveals that docetaxel treatment results in cleavage of full-length BMX, which results in a cleaved product with electrophoretic mobility identical to that of BMXΔN. C, PC3 cells were transfected with plasmids to express full-length BMX (light gray bars), BMXΔN (dark gray bars), or a vector control (white bars). Cells were then treated with the indicated concentrations of docetaxel for 48 h and then assayed for viability using trypan blue staining. The data represent the average and S.D. (error bars) from three independent experiments, and p values were derived from paired two-tailed t tests. D, FACS analysis of docetaxel-treated PC3 cells. PC3 cells were transfected with the indicated expression plasmids and then left untreated or were treated with 10 nm docetaxel for 48 h. The percentage of cells that were only stained with annexin V (dark gray) or with both annexin V and propidium iodide (light gray) are shown. The data represent the average and S.D. from three independent experiments, and p values were derived from paired two-tailed t tests.
Increased Apoptosis Sensitivity in BMXΔN-expressing Cells
Previous investigations have reported that the proteolytic removal of the N-terminal domain of the BMX kinase produces a catalytically active protein fragment, BMXΔN, which sensitizes PC3 cells toward Fas ligand-induced apoptosis (15). To verify whether this enhanced sensitivity also applies to docetaxel treatment, we investigated the effects of expressing full-length BMX and BMXΔN on docetaxel-induced cell death in PC3 cells. PC3 cells transfected to express either full-length BMX, BMXΔN, or a vector control were treated with 0–20 nm docetaxel for 48 h, upon which the cells were evaluated for viability by a trypan blue exclusion staining assay and counted on an automated counter. As shown in Fig. 1B, there was no difference in cell viability when expressing either BMX construct when no docetaxel was present. In agreement with the previous investigation, reduced cell viability was observed in the BMXΔN-expressing cells in the presence of 10 or 20 nm docetaxel (Fig. 1C). An analogous experiment was also performed in the presence or absence of 10 nm docetaxel and was analyzed by FACS after annexin V and propidium iodide (PI) staining. The data in Fig. 1D concur with the data from trypan blue staining, where the cells expressing BMXΔN exhibited increased annexin V-positive cells (dark gray) and double-positive cells stained for both PI and annexin V (light gray) when treated with docetaxel. Again the data reveal no observable cell death, versus vector control, when BMXΔN is expressed in the absence of docetaxel. Together, our data indicate that the expression of BMXΔN enhances drug-induced apoptosis in PC3 cells but is not toxic in untreated or unstressed cells, as was previously observed for cells treated with Fas ligand (15).
BMXΔN Is Unstable and Is Degraded by the Proteasome
The proteolytic generation of BMXΔN gives rise to an N-terminal tryptophan, the reported specificity of the N-end rule pathway (22) predicts that this protein fragment will be unstable in cells, but the stability of BMXΔN has never previously been investigated.
The stability of both full-length BMX and BMXΔN was investigated by expressing the proteins in PC3 cells and then adding cycloheximide (CHX) to the cells to block protein synthesis. Cells were then lysed at 0, 1, and 4 h after the addition of CHX, and the amounts of protein present were relatively quantified by Western blotting analysis after resolving the samples by SDS-PAGE. The data in Fig. 2A demonstrate that the full-length BMX protein was stable, whereas BMXΔN was degraded over the 4-h time course. Western blotting analysis for actin was performed to verify equal protein loading on the gel. This is the first demonstration that BMXΔN is unstable in cells.
FIGURE 2.

BMXΔN is degraded by the N-end rule pathway. A, the stability of full-length BMX and BMXΔN was determined in transfected PC3 cells by treating the cells with 100 μg/ml CHX, to block protein synthesis, and then the cell cell lysates were analyzed by WB analysis at the indicated times. An anti-FLAG antibody was used to detect BMX, and an anti-actin antibody was used as a loading control. B, stability of BMXΔN was investigated in PC3 cells in the presence and absence of MG132 (10 μm) and analyzed as in A. C, wild type BMXΔN with N-terminal tryptophan (type II destabilizing N termini) and N-terminal mutants of arginine (type I destabilizing N termini) and valine (stabilizing N termini) were transfected into the cell lines indicated, and BMXΔN stability was determined as described in A. D, verification of shRNA knockdown of UBR1 in F by WB analysis with an anti-UBR1 antibody. E, verification of shRNA knockdown of UBR2 in F by WB analysis with an anti-UBR2 antibody. F, stability of BMXΔN was visualized in PC3 cells that also expressed shRNAs targeting UBR1 and UBR2, control shRNAs, or a pcDNA 3.1 vector control. G, stability of wild type BMXΔN and the listed N-terminal mutants in the presence and absence of 200 μm Phe-NH3 (preincubated for 4 h before the addition of CHX), an inhibitor reported to block the degradation of type II N termini while not affecting the degradation of proteins with type I N termini.
To verify that BMXΔN degradation is via the proteasome, we investigated whether the addition of a proteasome inhibitor, MG132, inhibits degradation. The stability of BMXΔN in PC3 cells was investigated in the presence or absence 5 μm MG132. The data in Fig. 2B reveal that the addition of MG132 to the cells stabilized BMXΔN in contrast to the DMSO control. Together, the data demonstrate that the BMXΔN protein fragment is unstable and is degraded by the proteasome.
The N-end Rule Targets BMXΔN to the Proteasome for Degradation
Because the proteolytic generation of BMXΔN exposes an N-terminal tryptophan residue, we hypothesized that it is targeted to the proteasome for degradation by the components of the N-end rule pathway (16). The N-end rule pathway is a protein degradation pathway that recognizes proteins with a specific destabilizing N-terminal residues and then ubiquitinates these proteins for proteasome-dependent degradation (16). In eukaryotes, the primary N-terminal destabilizing residues are classified into positively charged amino acids (type I), such as Arg and Lys, or bulky hydrophobic residues (type II), such as Trp and Tyr. Because BMXΔN has an N-terminal tryptophan (a type II destabilizing residue), we investigated whether the N-end rule pathway targets BMXΔN for degradation.
To investigate the potential for N-end rule degradation, we created a couple of BMXΔN N-terminal mutants where the Trp was mutated to either Val or Arg. If BMXΔN is degraded via the N-end rule pathway, an N-terminal Val would be predicted to stabilize BMXΔN because this is a stabilizing N terminus (16). Mutating Trp to Arg, a type I destabilizing residue, is predicted to continue to render BMXΔN unstable (16). The wild type BMXΔN and the two N-terminal mutants were expressed in PC3 cells, and the stability of the proteins was investigated after the addition of CHX to the cells. As predicted, the N-terminal Val mutant was stabilized (Fig. 2C), and the N-terminal Arg mutant remained unstable. This finding is in agreement with the hypothesis that BMXΔN is degraded by the N-end rule pathway.
To rule out the possibility that the BMXΔN degradation is specific to the androgen-independent prostate cancer cell line PC3, we also investigated the degradation of wild type BMXΔN, Val-BMXΔN, and Arg-BMXΔN in the androgen-dependent prostate cancer cell line, LNCaP, and the unrelated HEK293T cell line. Despite the presence of some cell-specific differences in the actual rates of degradation, the overall trends of N terminus-dependent degradation were identical (Fig. 2C).
Selective degradation via the N-end rule pathway involves the recognition of the N-terminal destabilizing residue on the substrate protein by the UBR protein domain containing E3 ubiquitin ligases. The mammalian UBR E3 ubiquitin ligases have two key recognition domains, the UBR box domain and the N-domain, which function independently as recognition domains for either type I or type II N-terminal destabilizing residues, respectively (22). To verify that BMXΔN is a bona fide N-end rule substrate, we verified that its degradation was dependent on these E3 ubiquitin ligases. We investigated the two functionally redundant E3 ubiquitin ligases UBR1 and UBR2, because they play an important role in recognition of type II N-terminal destabilizing residues in mammals (22). BMXΔN was expressed in PC3 cells stably expressing control shRNAs, a mixture of shRNAs to target both UBR1 and UBR2, or vector control cells. The simultaneous knockdown of both UBR1 and UBR2 by shRNA expression was verified by Western blotting analysis for endogenous UBR1 (Fig. 2D) and UBR2 (Fig. 2E). The stability of BMXΔN was then investigated in these cells after treatment with CHX. Fig. 2F reveals that the simultaneous knockdown of UBR1 and UBR2 inhibits the degradation of BMXΔN, whereas the protein is still rapidly degraded in either the shRNA control-expressing cells or the vector control cells. This result parallels what we have observed for the caspase-cleaved form of the Lyn kinase, which is targeted for degradation by the N-end rule pathway (23). This finding along with the stability of the N-terminal mutants above is strong evidence that BMXΔN is an N-end rule substrate.
Inhibition of Degradation of BMXΔN by the UBR Inhibitor
The N-domains of UBR1 and UBR2, which are structurally and functionally homologous to the bacterial ClpS protein (24), recognize N termini with hydrophobic destabilizing residues, such as tryptophan. A recent investigation has reported that some phenylalanine derivatives can inhibit the recognition and degradation of an N-end rule model substrate, nsP4, with an N-terminal Tyr in rabbit reticulocyte lysates (25). We investigated whether one of the compounds identified in this study, l-phenylalaninamide (Phe-NH3), can inhibit the degradation of BMXΔN in living cells. The stability of BMXΔN and four different N-terminal mutants, which included Tyr (type I), Lys (type II), Arg (type II), and Val (stabilizing), was investigated in PC3 cells in the presence and absence of Phe-NH3. The data in Fig. 2G reveal that when cells are pretreated with Phe-NH3, only the degradation of BMXΔN with type II destabilizing N termini (Trp and Tyr) are inhibited. Degradation of BMXΔN mutants with type I N termini (Arg and Lys) is unaffected by Phe-NH3, and the N-terminal Val mutant remains stable. Although not a potent inhibitor, these are the first data that verify the use of Phe-NH3 to selectively inhibit the degradation of proteins with type II N termini in cultured cells. The results for the different methods to inhibit the N-end rule (N termini mutations, shRNA knockdown of UBR1 and UBR2, or pharmacological inhibition with Phe-NH3) are all in agreement with a model where BMXΔN is targeted for N-rule degradation by the UBR1 and UBR2 E3 ligases.
The Role of the N-end Rule in BMXΔN-induced Sensitivity to Apoptosis
Given the degradation of BMXΔN by the N-end rule, we investigated the impact of degradation on its pro-apoptotic function. PC3 cells stably expressing shRNAs targeting UBR1 and UBR2 or shRNA controls were transfected with either a vector control or plasmids to express either wild type BMXΔN or the stable N-terminal Val-BMXΔN mutant. After the transfection, the cells were treated with 10 nm docetaxel for 48 h and then investigated by trypan blue staining to quantify cell viability. The data in Fig. 3A reveal that stabilizing BMXΔN by the N-terminal valine mutation increased the sensitivity of the cells to docetaxel. When BMXΔN and Val-BMXΔN were expressed in UBR1/2 knockdown cells, the sensitivity to docetaxel was essentially the same. Together, the data suggest that BMXΔN results in lower docetaxel sensitivity than the Val-BMXΔN mutant as a result of its degradation.
FIGURE 3.
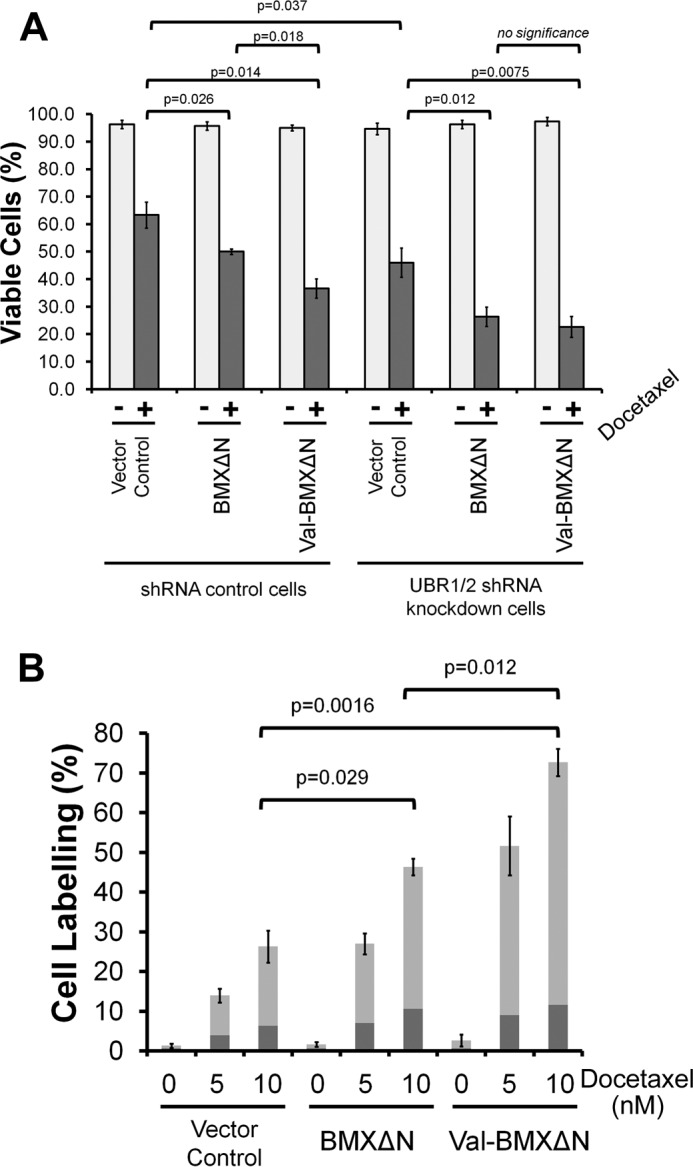
BMXΔN stability and the N-end rule influencing apoptosis. A, PC3 cells expressing control or UBR1/2 targeting shRNAs were transfected to express either wild type BMXΔN, the valine N-terminal mutant, or a vector control. 24 h after transfection, cells were treated with 10 nm docetaxel for 48 h. The cells were then analyzed by trypan blue staining. The data represent the average and S.D. (error bars) from three independent experiments, and p values were determined from paired two-tailed t tests. B, FACS analysis of docetaxel treated PC3 cells. PC3 cells were transfected to express either wild type BMXΔN, the valine N-terminal mutant, or a vector control. Cells were then treated with the indicated concentrations of docetaxel for 48 h. Cells were then analyzed by FACS for annexin V and propidium iodine staining. The percentage of cells that were stained with either annexin V (dark gray) or doubly labeled with both annexin V and propidium iodide (gray) are indicated. The data represent the average and S.D. from three independent experiments and p values were derived from paired two-tailed t tests.
A more detailed investigation of the effect of stabilizing BMXΔN on apoptosis was done by cell staining and FACS analysis. PC3 cells were transfected with wild type BMXΔN, the stable N-terminal Val-BMXΔN mutant, or a vector control. The cells were then treated with 5 or 10 nm docetaxel for 48 h and then stained with PI and annexin V before FACS analysis. The data in Fig. 3B reveal that stabilizing BMXΔN with the valine N-terminal mutation results in increased numbers of annexin V (dark gray) and PI-annexin V doubly labeled (light gray) cells at both docetaxel concentrations when compared with wild type BMXΔN. The presence of the increased amounts of both early and late apoptotic cells upon docetaxel treatment when BMXΔN is stabilized indicates that the N-end rule is attenuating the pro-apoptotic activity of BMXΔN. As already mentioned above, despite its instability, wild type BMXΔN expression also leads to increased docetaxel sensitivity in Fig. 3B when compared with the vector control. The difference between the wild type BMXΔN and vector control indicates that even in the presence of active N-end rule degradation of BMXΔN, it can still function as a pro-apoptotic molecule.
The N-end Rule Impacts Apoptosis
Our data on cell viability also reveal that UBR1 and UBR2 exhibit a BMXΔN-independent influence on apoptosis. The data in Fig. 3A reveal a difference in docetaxel sensitivity between the UBR1/2 knockdown cells versus the shRNA control cells, even when BMXΔN is not expressed. Although we have not further investigated this observation, this finding is in agreement with previous reports. Specifically, UBR1/UBR2 knock-out mouse embryonic fibroblasts were shown to be hypersensitive to apoptosis-inducing agents, such as UV or staurosporine, a kinase inhibitor known to induce apoptotic cell death (26). Second, mouse spermatocytes that lack UBR2 ligase undergo apoptosis during meiosis, which eventually leads to male infertility (27).
N-end Rule Degradation of Caspase-generated BMXΔN
In addition to studying the degradation of BMXΔN using the recombinant ubiquitin fusion constructs, we investigated whether caspase-mediated generation of BMXΔN from full-length BMX kinase in PC3 cells is accompanied by degradation by the N-end rule pathway. PC3 cells were transfected to express full-length BMX or a W243V mutant. Caspase cleavage of the W243V mutant will generate the stable Val-BMXΔN variant. After 24 h, the cells were treated with 10 nm doctaxel for 48 h, upon which cleavage of BMX is observed (Fig. 4A). When Z-VAD-fmk, a pan-caspase inhibitor (28), was added for 2 h to halt the ongoing production of BMXΔN by caspase cleavage of full-length BMX, a decrease in BMXΔN was observed (Fig. 4A). This Z-VAD-fmk-dependent reduction in BMXΔN is hypothesized to be a result of N-end rule degradation and, as predicted, is inhibited by the addition of MG132 or UBR1/2 shRNA knockdown. The Z-VAD-fmk-dependent drop in BMXΔN is also not observed with the W243V mutant, which is in agreement with N-end rule degradation because the resulting BMXΔN with a valine N-terminal residue is predicted to be stable.
FIGURE 4.
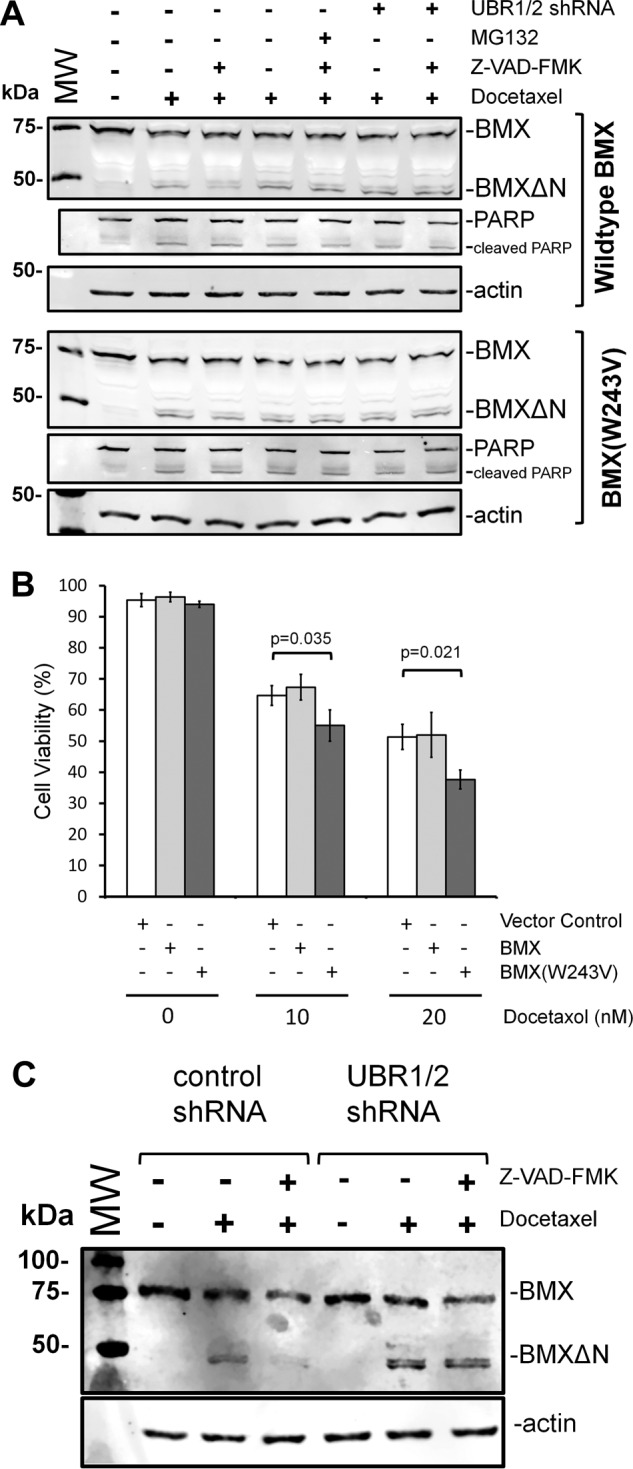
Cleavage of full-length recombinant or endogenous BMX and C-terminal fragment degradation. A, PC3 cells that stably express either shRNAs targeting UBR1 and UBR2 or shRNA controls were transfected to express full-length BMX (top) or the W243V mutant BMX. 24 h after transfection, the cells were either untreated or treated with 10 nm docetaxel for 48 h. The indicated samples were also treated with MG132 or Z-VAD-fmk. The pan-caspase inhibitor Z-VAD-fmk was added, for 2 h, to the indicated samples to prevent ongoing formation of BMXΔN by continued caspase activity. Western blotting analysis of cell lysates from the resulting experiments was performed to detect BMX and BMXΔN amounts, PARP, or an actin loading control. When Z-VAD-fmk was added to prevent the ongoing formation of BMXΔN, the disappearance of the C-terminal fragment is inhibited by knockdown of UBR1/2 with shRNAs or the W243V mutation, which results in a BMXΔN fragment with a stabilizing valine N terminus. B, PC3 cells expressing either recombinant full-length BMX or the W243V mutant were treated with the indicated amounts of docetaxel for 48 h and then analyzed by trypan blue staining. The data represent the average and S.D. (error bars) from three independent experiments, and p values were determined from paired two-tailed t tests. C, PC3 cells that stably express either shRNAs targeting UBR1 and UBR2 or shRNA controls were either untreated or treated with 10 nm docetaxel for 48 h to induce apoptosis. The pan-caspase inhibitor Z-VAD-fmk was added, for 3 h, to the indicated samples after docetaxel to prevent ongoing formation of BMXΔN by continued caspase activity. Western blotting analysis of cell lysates from the resulting experiments was performed to detect endogenous BMX and BMXΔN resulting from caspase cleavage.
To investigate whether N-end rule degradation of caspase-generated BMXΔN impacts cell viability, PC3 cells expressing either full-length wild type or W243V BMX were treated with docetaxel for 48 h and assayed for cell viability by trypan blue staining (Fig. 4B). As shown, the expression of full-length wild type BMX had no measurable impact on cell viability, similar to what was also demonstrated above by FACS analysis (Fig. 1D). In contrast, when the W243V BMX mutant was expressed, a statistically significant decrease in cell viability was observed upon docetaxel treatment (Fig. 4B). A parsimonious interpretation for this data is that whereas BMXΔN generated by caspase cleavage has the potential to be pro-apoptotic, the active degradation of this fragment by the N-end rule pathway effectively counters this function.
N-end Rule Degradation of Endogenous BMXΔN Generated by Caspase Cleavage
To investigate whether BMXΔN generated from caspase cleavage of endogenous BMX was similarly targeted for degradation by the N-end rule, the following experiment was performed. PC3 cells expressing the UBR1/2 shRNAs (or control shRNAs) were treated with 10 nm docetaxel for 48 h to induce apoptosis and BMX cleavage (Fig. 4C). As described above, Z-VAD-fmk was utilized to prevent the ongoing formation of BMXΔN after docetaxel treatment. 3 h after the addition of Z-VAD-fmk, the cells were then also harvested for Western blotting analysis (Fig. 4C). The data in Fig. 4C reveal that treatment of the cells with docetaxel results in the formation of BMXΔN from endogenous BMX in both the control and UBR1/2 knockdown cells. When the cells were then treated with Z-VAD-fmk, the BMXΔN fragment disappeared in the control shRNA-expressing cells, as was observed for recombinant full-length BMX (Fig. 4A). In contrast, when the UBR1/2 knockdown cells are similarly treated, BMXΔN remains present after the addition of Z-VAD-fmk. This is in agreement with our model that BMXΔN is stabilized in the absence of UBR1 and UBR2. In sum, the data support a model where the caspase-mediated cleavage of endogenous BMX kinase generates a cleaved fragment that is a bona fide N-end rule substrate in cells.
Catalytic Activity of BMXΔN Is Dispensable for Degradation
With the pro-apoptotic role for BMXΔN, we next investigated roles for its catalytic activity in both protein degradation and docetaxel sensitivity. An inactivating mutation (K455R), as described previously (15), was introduced into the wild type ubiquitin-BMXΔN construct. The stability of the wild type and K455R mutant BMXΔN was then investigated. The data in Figs. 5A and 6E do not reveal an observable difference in the stabilities of the proteins, leading to the conclusion that BMXΔN activity was not required for its degradation. Concomitantly, further analysis reveals that the abrogation of the catalytic activity of BMXΔN does not impact the stability of long lived mutant Val-BMXΔN (Fig. 5A).
FIGURE 5.

BMXΔN kinase activity is required for its pro-apoptotic activity. A, wild type (top) and the N-terminal valine mutant (bottom) BMXΔN along with their corresponding kinase inactive mutants (K455R) were expressed in PC3 cells. The stability of the proteins was investigated by treating the cells with 100 μg/ml CHX and collecting lysates at the indicated times. The lysates were resolved by SDS-PAGE, and the amount of protein remaining was determined by WB analysis with either an anti-FLAG or anti-actin antibody. B, PC3 cells were transfected to express the indicated BMX variants. Cells were then treated with 10 nm docetaxel for 48 h and were evaluated for viability by staining with trypan blue. The data represent the average and S.D. (error bars) from three independent experiments, and p values were derived from paired two-tailed t tests.
FIGURE 6.

BMXΔN phosphorylation regulates its degradation and pro-apoptotic function. A, cell lysates from wild type BMXΔN-expressing cells treated with λ-phosphatase (or untreated control) were subjected to SDS-PAGE and WB analysis with an anti-FLAG antibody to detect BMXΔN. Phosphatase treatment resulted in the loss of the band doublet. B, PC3 cells expressing BMXΔN cells were treated with the kinase inhibitor staurosporine (1 μm) for 30 min before lysis and WB analysis with an anti-FLAG antibody. C, the stability of wild type BMXΔN was investigated in PC3 cells after a 60-min pretreatment with the phosphatase inhibitor sodium orthovanadate (or untreated control). The stability of the proteins was investigated by treating the cells with 100 μg/ml CHX and collecting lysates at the indicated times. The lysates were resolved by SDS-PAGE, and the amount of protein remaining was determined by WB analysis with either an anti-FLAG or anti-actin antibody. D, wild type BMXΔN, Y566F, and the valine N-terminal Y566F double mutant BMXΔN were expressed in PC3 cells. The stability of the proteins was determined as described for C. E, quantification of the degradation of wild type, K455R, Y566F, and valine N-terminal Y566F double mutant BMXΔN from a minimum of three individual experiments. The data were plotted to fit an apparent first order reaction to determine apparent rates and half-life. The amounts remaining of each of the different mutants (wild type, K455R, Y566F, and valine N-terminal Y566F double mutant BMXΔN) are quantified relative to an actin loading control, where the rates of disappearance are depicted as follows: the Y566F BMXΔN mutant (▵), the valine N-terminal Y566F double mutant BMXΔN (×), the wild type BMXΔN (○) mutant, and the K455R mutant (□). F, stability of wild type BMXΔN and the Y566F mutant was determined in the presence and absence of 10 μm MG132 (left) or in UBR1 and UBR2 shRNA-expressing cells (right). In all cases for the Y566F mutant, the doublet band is no longer observed. G, PC3 cells were transfected to express either BMXΔN, the Y566F mutant, the valine N-terminal Y566F double mutant, or a vector control. 24 h after transfection, the cells were treated with docetaxel for 48 h. The cells were then stained with trypan blue to quantify cell viability. The data represent the average and S.D. (error bars) from three independent experiments, and p values were derived from paired two-tailed t tests.
In contrast to protein degradation, the catalytic activity of BMXΔN is essential for its pro-apoptotic function. This is demonstrated in Fig. 5B, where expression of the kinase-dead BMXΔN (K455R) mutant in PC3 cells results in the same cell viability after docetaxel treatment as the vector control. In contrast to this is the decreased viability upon docetaxel treatment, when cells are expressing either the wild type or the stable Val-BMXΔN mutant. It is noteworthy that the expression of double mutant Val-BMXΔN (K455R) in PC3 cells also does not result in a significant difference in cell death after docetaxel treatment with respect to cells expressing vector control (Fig. 5B). This finding has 2-fold consequences. First it precludes the possibility that the lack of pro-apoptotic activity by the K455R mutant is a simple result of its instability, because the K455R mutant does not exhibit pro-apoptotic activity when it is stabilized with an N-terminal valine. Second, it further confirms the pivotal role of catalytic activity of BMXΔN in mediating its pro-apoptotic function in PC3 cells upon docetaxel treatment.
After the induction of apoptotic cell death in mammalian cells, proteases, such as caspases and calpains, cleave a large number of proteins (29). A number of cleaved fragments have pro-apoptotic activity (i.e. they can amplify apoptosis signaling, presumably via participating in positive feedback loops). For instance, cleaved RIPK1 enhances caspase activation (30, 31), and cleaved BAX augments the mitochondrial outer membrane permeability (32). Our findings with BMXΔN reveal that whereas the catalytic activity of the kinase is required for its function in a positive feedback loop for apoptotic signaling, this activity is being attenuated by the N-end rule, as has been observed for proteolytic products of RIPK1 and LIMK1 (26).
Phosphorylation of BMXΔN
Given the observation that the BMXΔN fragment migrates as a 50 kDa doublet of bands in our Western blotting analysis of both recombinant and endogenous BMXΔN (Fig. 4, A and C), we hypothesized that this doublet is a result of a posttranslational modification, such as phosphorylation. The observation of a doublet also suggests that the modification is incomplete and that BMXΔN exists in both modified and unmodified populations.
To address this possibility of phosphorylation, λ-phosphatase treatment was performed with cell lysate from PC3 cells where the wild type BMXΔN was expressed. As opposed to the untreated control, the data in Fig. 6A reveal a single band upon phosphatase treatment. To further investigate BMXΔN phosphorylation, wild type BMXΔN was expressed in PC3 cells treated with 1 μm staurosporine, a nonspecific kinase inhibitor (33), for 30 min. As seen in Fig. 6B, staurosporine treatment culminated in a relative accumulation of the lower band, presumably the non-phosphorylated band, of the doublet as well as a reduction in the net level of BMXΔN, which may be presumably attributed to its phosphorylation-inhibitory effect and the consequences for BMXΔN metabolic stability. Crucially, to further demonstrate the role of phosphorylation in the regulation of BMXΔN doublet band turnover, BMXΔN was again expressed in PC3 cells that were either untreated or treated with 1 μm sodium orthovanadate, a tyrosine phosphatase inhibitor (34), for 1 h. After the sodium orthovanadate preincubation, CHX was added to the cells to monitor BMXΔN stability. Fig. 6C reveals that pretreatment with the phosphatase inhibitor, when compared with the untreated control, leads to the accumulation of the upper band of the doublet, presumably the phosphorylated form of BMXΔN. Additionally, the data in Fig. 6C reveal that sodium orthovanadate treatment stabilizes BMXΔN, where the upper, presumably phosphorylated, band persists even at the 3 h time point after CHX treatment. From this finding, it appears that the phosphorylated form of BMXΔN is degraded more slowly, but at this point, we cannot rule out the possibility of secondary indirect cellular effects of the phosphatase inhibitor. Nonetheless, the data with λ-phosphatase, staurosporine, and sodium orthovanadate strongly suggest that the BMXΔN doublet observed by Western blotting is a result of phosphorylation. The presence of the doublet with the kinase-dead mutant (Fig. 5A) indicates that this phosphorylation is not a result of autophosphorylation by the proteolytically activated BMXΔN kinase.
BMXΔN Phosphorylation at Tyrosine 566
Previous reports identified that BMX phosphorylation at Tyr-566, within the activation loop, is essential for full-length BMX kinase activity (1, 5, 35, 36). This highly conserved tyrosine residue has been shown to be activated by Src family kinases and is required for subsequent full-length BMX autophosphorylation (5, 37). To investigate whether potential Tyr-566 phosphorylation plays a role in BMXΔN degradation and docetaxel sensitization, a non-phosphorylatable mutation (Y566F) was introduced into the ubiquitin-BMXΔN fusion constructs. The wild type and Y566F BMXΔN constructs were transfected into PC3 cells investigated for protein stability. The data in Fig. 5D reveal two essential points. First, the Y566F mutant is no longer observed as a complete doublet band, and second, the Y566F mutant is more rapidly degraded than wild type BMXΔN. The quantified data from at least three replicate experiments are shown in Fig. 6E. Our interpretation of these data is that phosphorylation at Tyr-566 is crucial for the formation of a complete doublet band observed by Western blotting analysis. There may be additional phosphorylations that occur after Tyr-566 phosphorylation, but our current data cannot address this possibility. Autophosphorylation events are known to occur with the Tec family of kinases after Src kinase phosphorylation of Tyr-566 (37), but in BMX, both of these reported sites, Tyr-216 and Tyr-224, are located in the N-terminal fragment that is removed by proteolysis at Asp-242, so the presence of additional phosphorylation sites in BMXΔN remains undetermined.
Taken together, our data demonstrating increased degradation of Y566F BMXΔN (Fig. 6, D and E) along with the reduced degradation of wild type BMXΔN in the presence of sodium orthovanadate (Fig. 6C) are consistent with a model that phosphorylation of Tyr-566 inhibits BMXΔN degradation. The stability of a valine N-terminal mutant of the Y566F construct was also investigated to ensure that the degradation of the Y566F mutant was still a result of N-end rule degradation and not by an alternative mechanism (Fig. 6, D and E).
To additionally verify that the inhibition of protein degradation by phosphorylation is up- or downstream on N-end rule recognition, we evaluated the stability of wild type and the Y566F BMXΔN mutant in the presence of MG132 or upon UBR1/2 shRNA knockdown. Fig. 6F reveals that both MG132 and shRNA knockdown of UBR1/2 inhibit degradation of both wild type and the Y566F mutant BMXΔN. Inhibition of degradation upon UBR1/2 knockdown suggests that Y566F phosphorylation prevents degradation via the N-end rule. Inhibition may be at the point of substrate recognition or ubiquitination by UBR1 and UBR2. This is the first demonstrated example of phosphorylation regulating the recognition of a caspase proteolytic product by the N-end rule pathway.
Given that the data for BMXΔN generated by caspase cleavage of endogenous BMX also reveal the presence of the doublet band (Fig. 4C), we believe this phosphorylation is not an experimental artifact as a result of expressing BMXΔN as a ubiquitin fusion. What we cannot determine from our data is whether the phosphorylation was already present in the full-length protein or whether it occurs subsequent to cleavage.
With the Y566F being more rapidly degraded than wild type, with half-lives of 0.55 and 1.5 h, respectively (Fig. 6E), we then investigated the effect of its expression on docetaxel sensitivity. Wild type, the Y566F mutant BMXΔN, and a vector control were transfected into PC3 cells, which were then evaluated for viability by trypan blue staining after a 48-h treatment with 10 nm docetaxel. Fig. 6G reveals that the expression of the Y566F mutant did not alter docetaxel sensitivity, in comparison with a vector control. Wild type BMXΔN expression again resulted in increased docetaxel sensitivity. Moreover the data in Fig. 6G rule out the possibility that the lack of pro-apoptotic activity by the Y566F mutant was a simple result of its instability, because the Y566F mutant did not exhibit pro-apoptotic activity when it was stabilized with an N-terminal valine (Fig. 6G). Thus, like full-length BMX, it appears that BMXΔN requires Tyr-566 phosphorylation on the activation loop for activity. The specific mechanism of how phosphorylation of Tyr-566 inhibits N-end rule degradation remains to be determined. In addition, the more rapid turnover of the Y566F mutant and the apparent similar turnover of the BMXΔN band doublets in all of our experiments suggest that the cellular equilibrium of BMXΔN phosphorylation and dephosphorylation is probably rapid in comparison with the time scale of BMXΔN degradation.
Conclusions
Cross-talk between phosphorylation and ubiquitin-dependent protein degradation plays a prominent role in cellular signaling and may take many forms (38). Notably, phosphorylation can regulate protein ubiquitination and degradation machinery via regulating the activity of E3-ubiquitin ligases, as reported previously (39). Alternatively, the coordinated targeting of a protein substrate by phosphorylation and ubiquitin-dependent degradation provides another avenue of cross-regulation (40). Although there are a number of examples of how phosphorylation impacts, either positively or negatively, target protein substrate degradation by the ubiquitin-proteasome system (40, 41), herein we describe, to the best of our knowledge, the first clear example of phosphorylation regulating recognition of a caspase proteolytic product by the N-end rule degradation pathway, which was discovered about 3 decades ago (42). Although the impact on degradation is clear, the mechanism of how phosphorylation at this internal residue inhibits recognition and proteasome targeting by UBR1 and UBR2 has yet to be determined. It remains unclear whether degradation of the phosphorylated form of BMXΔN is slower or whether it must first be dephosphorylated before degradation (Fig. 7). Tellingly, it also remains to be determined whether there are other phosphorylation sites that can play a role in the regulation of stability of BMXΔN. Future investigations may identify potential mechanisms, such as the inhibition of recognition of an essential lysine by the N-domain preventing ubiquitination.
FIGURE 7.
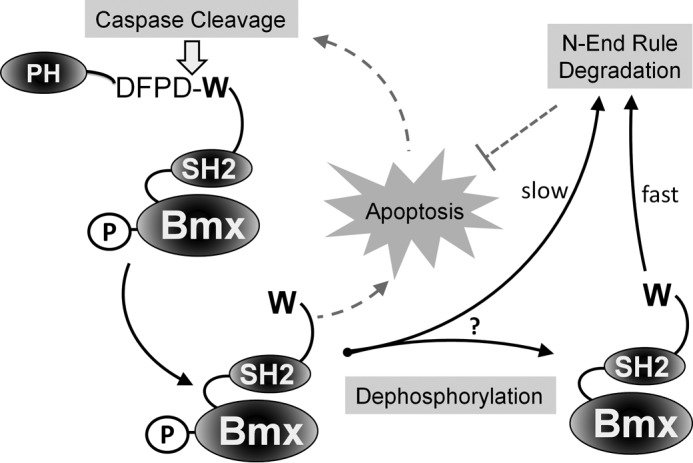
Model for BMXΔN degradation. Our proposed model is that the caspase-generated BMXΔN is recognized by the degenerate UBR1 and UBR2 E3 ubiquitin ligases. UBR1/2 recognize the N-terminal tryptophan of BMXΔN, which then ubiquitinates the protein, targeting it to the proteasome for degradation and thus attenuating its pro-apoptotic function. Phosphorylation of BMXΔN at tyrosine 566 is required its pro-apoptotic function and relatively inhibits its degradation by the N-end rule pathway. PH, pleckstrin homology domain.
Overall, our findings have revealed an unforeseen interplay between phosphorylation and N-end rule degradation of a caspase product. Our observations are suggestive of increased complexity of signaling networks. Although proteomics techniques have identified examples of how phosphorylation can regulate caspase cleavage (43), our data now demonstrate that phosphorylation can also regulate the outcome of some caspase products.
The degradation of the caspase-generated BMXΔN is also a novel example of a protease product generated upon the activation of the apoptotic program, which is actively degraded by the N-end rule pathway. This emerging role for the N-end rule pathway in attenuating the apoptotic program by degrading proteolytically activated proteins (26, 44, 45) is revealing this pathway to be a potential target to sensitize cells to cell death-promoting therapies.
Experimental Procedures
Generation of Ubiquitin Fusion Cleaved BMX Expression Vector
To express the cleaved BMX fragment, we cloned the cleaved BMX as a fusion between an N-terminal ubiquitin and C-terminal triple FLAG tag (3×FLAG) to generate a Ub-BMX-FLAG pcDNA 3.1 vector, as we have described previously (45). A cDNA clone (cloneID MHS1010-BC016652BE894841, Open Biosystems) was used to clone the BMX sequence corresponding to the caspase-3 cleavage site to the C-terminal end of the protein, amino acids 243–675. The final Ub-BMX-FLAG pcDNA 3.1 vector was verified by DNA sequencing. The full-length BMX was similarly cloned with a C-terminal 3×FLAG in a pcDNA 3.1 plasmid.
Site-directed Mutagenesis
Mutagenesis of the codon for the tryptophan corresponding to the N termini of the cleaved BMX protein was performed by site-directed mutagenesis to change the codon to Arg (CGG), Val (GTG), Tyr (TAC), or Lys (AAA). Similarly, the kinase-dead form of cleaved BMX was obtained through mutating lysine 455 in the putative ATP-binding site to arginine (CGG) by site-directed mutagenesis. To prevent phosphorylation at tyrosine 566, this residue was mutated to Phe (TTT). In full-length BMX-FLAG, tryptophan 243 was mutated to valine (GTG) to change the neo-N termini after caspase cleavage.
Cell Culture
HEK293T, PC3, and LNCaP cells were obtained from the ATCC. The HEK293T cells were cultured in DMEM supplemented with 10% FBS. PC3 cells were cultured in DMEM/F-12 with 10% FBS, and LNCaP cells were cultured in RPMI 1640 supplemented with 10% FBS.
Cell Transfection
HEK293T were transfected using the calcium phosphate-based method. PC3 and LNCaP cells were transfected using electroporation (Neon transfection system) according to the manufacturer's procedures. The transfection efficiencies for the PC3 cells ranged from 55 to 75% between experiments, as determined by transfecting with a GFP-expressing vector and FACS analysis. To ensure identical transfection of samples in each well of a culture plate for protein stability measurements and/or cell death measurements, a single batch of cells was transfected with a single Neon tip and then seeded into individual wells (equal number of cells/well) in a 12- or 6-well plate.
Antibodies and Inhibitors
PARP antibody (catalog no. 9542) was purchased from Cell Signaling Technologies. Mouse anti-FLAG M2 antibody (F1804) was purchased from Sigma. Rabbit anti-β-actin (I-19, sc-1616-R), anti-UBR1 (sc-100626), anti-UBR2 (sc-135594), and anti-BMX (sc-8874) were purchased from Santa Cruz Biotechnology, Inc. Secondary antibodies for Western blotting analysis (goat anti-mouse and goat anti-rabbit) coupled to IRDyes® were purchased from LI-COR.
Protein Stability Assays and Western Blotting Analysis
24 h after transfection, 5 × 105 cells were treated with 100 μg/ml CHX for the indicated amounts of time. Cells were harvested and then lysed in 150 μl of lysis buffer (50 mm Tris, pH 6.8, 8% glycerol (v/v), 0.016% SDS (w/v), 0.125% β-mercaptoethanol (v/v), 0.125% bromphenol blue (w/v), 1 mm PMSF, and 1 μg/ml leupeptin). The samples were sonicated and then resolved by SDS-PAGE on 10% gels along with Precision Plus All Blue protein prestained standards (Bio-Rad).
After SDS-PAGE, proteins were transferred onto nitrocellulose membranes (LI-COR Biosciences). The membranes were blocked with 2.5% fish skin gelatin (Truin Science) in 1× PBS with 0.1% Triton X-100), probed with primary and secondary antibodies, and imaged with an Odyssey® infrared imaging system using the manufacturer's recommended procedures (LI-COR).
UBR1 and UBR2 shRNAs
Four UBR1 unique 29-mer shRNA constructs in GFP-V-RS vectors (catalog no. TG300681) were purchased from Origene. Four unique UBR2 29-mer shRNA constructs in RFP-C-RS vectors (catalog no. TF300680) were also purchased from Origene. Because individual shRNA resulted in only moderate knockdown of either UBR1 or UBR2, mixtures of the four shRNAs for each target were empirically screened to obtain the maximal knockdown of each protein. Specifically, the combinations of A, B, and C (GFP-V-RS) and A, B, and D (RFP-C-RS) gave the most effective knockdowns. These six shRNA-expressing vectors were then transfected into PC3 cells and cultured in 1 μg/ml puromycin for 4 weeks to select for cells stably incorporating these plasmid vectors. FACS was then used to isolate the cells expressing both GFP and RFP (the reporters for each shRNA vector, respectively). The PC3 cells expressing the shRNAs for UBR1 and UBR2 exhibited reduced proliferation rates when compared with either the wild type PC3 cells or PC3 cells similarly selected for using vector control plasmids. The doubling time for wild type PC cells was determined to be 27 ± 3 h, and that for the vector control cells was determined to be 29 ± 2 h. In contrast, the doubling time for the UBR1 and UBR2 shRNA-expressing cells was determined to be 55 ± 9 h. This impaired proliferation is not entirely surprising, considering the previous well documented role of the N-end rule in cell division and chromosome stability (46, 47).
Flow Cytometry
An annexin V-FITC apoptosis detection kit (eBioscience) was used for apoptosis analysis by flow cytometry using the manufacturer's recommended procedures and analyzed on an LSR-Fortessa instrument. 10,000 events were acquired for statistical analysis.
Cell Viability Assay
Cell counting and the trypan blue exclusion test were performed with a TC20TM automated cell counter (Bio-Rad).
Protein Dephosphorylation
24 h after transfection, 5 × 105 cells were harvested and then lysed in 150 μl of lysis buffer as described previously (15) and then incubated at 4 °C for 30 min. The sample was then incubated with 400 units of λ-phosphatase (New England BioLabs) in λ-phosphatase reaction buffer at 30 °C for 60 min.
Chemicals
The l-phenylalaninamide inhibitor, cycloheximide, staurosporine, and sodium orthovanadate were purchased from Sigma.
Author Contributions
M. A. E. and R. P. F. designed the experiments. M. A. E. performed the experiments. M. A. E and R. P. F. wrote the manuscript.
This work was supported by Natural Sciences and Engineering Research Council (NSERC) of Canada Discovery Grant 341453-12 (to R. P. F.). The authors declare that they have no conflicts of interest with the contents of this article.
- SH2 and SH3
- Src homology 2 and 3, respectively
- Ub
- ubiquitin
- PI
- propidium iodide
- CHX
- cycloheximide
- Phe-NH3
- l-phenylalaninamide
- Z-VAD-fmk
- N-Benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone
- WB
- Western blotting.
References
- 1. Chen K. Y., Huang L. M., Kung H. J., Ann D. K., and Shih H. M. (2004) The role of tyrosine kinase Etk/Bmx in EGF-induced apoptosis of MDA-MB-468 breast cancer cells. Oncogene 23, 1854–1862 [DOI] [PubMed] [Google Scholar]
- 2. Qiu Y., and Kung H. J. (2000) Signaling network of the Btk family kinases. Oncogene 19, 5651–5661 [DOI] [PubMed] [Google Scholar]
- 3. Lewis C. M., Broussard C., Czar M. J., and Schwartzberg P. L. (2001) Tec kinases: modulators of lymphocyte signaling and development. Curr. Opin. Immunol. 13, 317–325 [DOI] [PubMed] [Google Scholar]
- 4. Smith C. I., Islam T. C., Mattsson P. T., Mohamed A. J., Nore B. F., and Vihinen M. (2001) The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 23, 436–446 [DOI] [PubMed] [Google Scholar]
- 5. Tsai Y. T., Su Y. H., Fang S. S., Huang T. N., Qiu Y., Jou Y. S., Shih H. M., Kung H. J., and Chen R. H. (2000) Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol. Cell. Biol. 20, 2043–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robinson D., He F., Pretlow T., and Kung H. J. (1996) A tyrosine kinase profile of prostate carcinoma. Proc. Natl. Acad. Sci. U.S.A. 93, 5958–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qiu Y., Robinson D., Pretlow T. G., and Kung H. J. (1998) Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95, 3644–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guryanova O. A., Wu Q., Cheng L., Lathia J. D., Huang Z., Yang J., MacSwords J., Eyler C. E., McLendon R. E., Heddleston J. M., Shou W., Hambardzumyan D., Lee J., Hjelmeland A. B., Sloan A. E., et al. (2011) Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 19, 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Potter D. S., Kelly P., Denneny O., Juvin V., Stephens L. R., Dive C., and Morrow C. J. (2014) BMX acts downstream of PI3K to promote colorectal cancer cell survival and pathway inhibition sensitizes to the BH3 mimetic ABT-737. Neoplasia 16, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bagheri-Yarmand R., Mandal M., Taludker A. H., Wang R. A., Vadlamudi R. K., Kung H. J., and Kumar R. (2001) Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity of breast cancer cells. J. Biol. Chem. 276, 29403–29409 [DOI] [PubMed] [Google Scholar]
- 11. Xue L. Y., Qiu Y., He J., Kung H. J., and Oleinick N. L. (1999) Etk/Bmx, a PH-domain containing tyrosine kinase, protects prostate cancer cells from apoptosis induced by photodynamic therapy or thapsigargin. Oncogene 18, 3391–3398 [DOI] [PubMed] [Google Scholar]
- 12. Zhang Z., Zhu W., Zhang J., and Guo L. (2011) Tyrosine kinase Etk/BMX protects nasopharyngeal carcinoma cells from apoptosis induced by radiation. Cancer Biol. Ther. 11, 690–698 [DOI] [PubMed] [Google Scholar]
- 13. Fox J. L., and Storey A. (2015) BMX negatively regulates BAK function thereby increasing apoptotic resistance to chemotherapeutic drugs. Cancer Res. 75, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ekman N., Arighi E., Rajantie I., Saharinen P., Ristimäki A., Silvennoinen O., and Alitalo K. (2000) The Bmx tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 19, 4151–4158 [DOI] [PubMed] [Google Scholar]
- 15. Wu Y. M., Huang C. L., Kung H. J., and Huang C. Y. (2001) Proteolytic activation of ETK/Bmx tyrosine kinase by caspases. J. Biol. Chem. 276, 17672–17678 [DOI] [PubMed] [Google Scholar]
- 16. Varshavsky A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eldeeb M., and Fahlman R. (2016) The N-end rule: the beginning determines the end. Protein Pept. Lett. 23, 343–348 [DOI] [PubMed] [Google Scholar]
- 18. Gibbs D. J., Bacardit J., Bachmair A., and Holdsworth M. J. (2014) The eukaryotic N-end rule pathway: conserved mechanisms and diverse functions. Trends Cell Biol. 24, 603–611 [DOI] [PubMed] [Google Scholar]
- 19. Gan L., Wang J., Xu H., and Yang X. (2011) Resistance to docetaxel-induced apoptosis in prostate cancer cells by p38/p53/p21 signaling. Prostate 71, 1158–1166 [DOI] [PubMed] [Google Scholar]
- 20. Mediavilla-Varela M., Pacheco F. J., Almaguel F., Perez J., Sahakian E., Daniels T. R., Leoh L. S., Padilla A., Wall N. R., Lilly M. B., De Leon M., and Casiano C. A. (2009) Docetaxel-induced prostate cancer cell death involves concomitant activation of caspase and lysosomal pathways and is attenuated by LEDGF/p75. Mol. Cancer 8, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fabbri F., Carloni S., Brigliadori G., Zoli W., Lapalombella R., and Marini M. (2006) Sequential events of apoptosis involving docetaxel, a microtubule-interfering agent: a cytometric study. BMC Cell Biol. 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tasaki T., Sriram S. M., Park K. S., and Kwon Y. T. (2012) The N-end rule pathway. Annu. Rev. Biochem. 81, 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eldeeb M. A., and Fahlman R. P. (2014) The anti-apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget 5, 2714–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Román-Hernández G., Grant R. A., Sauer R. T., and Baker T. A. (2009) Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc. Natl. Acad. Sci. U.S.A. 106, 8888–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sriram S., Lee J. H., Mai B. K., Jiang Y., Kim Y., Yoo Y. D., Banerjee R., Lee S. H., and Lee M. J. (2013) Development and characterization of monomeric N-end rule inhibitors through in vitro model substrates. J. Med. Chem. 56, 2540–2546 [DOI] [PubMed] [Google Scholar]
- 26. Piatkov K. I., Brower C. S., and Varshavsky A. (2012) The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. U.S.A. 109, E1839–E1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwon Y. T., Xia Z., An J. Y., Tasaki T., Davydov I. V., Seo J. W., Sheng J., Xie Y., and Varshavsky A. (2003) Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol. 23, 8255–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polverino A. J., and Patterson S. D. (1997) Selective activation of caspases during apoptotic induction in HL-60 cells. Effects Of a tetrapeptide inhibitor. J. Biol. Chem. 272, 7013–7021 [DOI] [PubMed] [Google Scholar]
- 29. Crawford E. D., and Wells J. A. (2011) Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 80, 1055–1087 [DOI] [PubMed] [Google Scholar]
- 30. Lin Y., Devin A., Rodriguez Y., and Liu Z. G. (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim J. W., Choi E. J., and Joe C. O. (2000) Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene 19, 4491–4499 [DOI] [PubMed] [Google Scholar]
- 32. Gao G., and Dou Q. P. (2000) N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J. Cell Biochem. 80, 53–72 [DOI] [PubMed] [Google Scholar]
- 33. Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., et al. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 34. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 35. Gottar-Guillier M., Dodeller F., Huesken D., Iourgenko V., Mickanin C., Labow M., Gaveriaux S., Kinzel B., Mueller M., Alitalo K., Littlewood-Evans A., and Cenni B. (2011) The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J. Immunol. 186, 6014–6023 [DOI] [PubMed] [Google Scholar]
- 36. Chen R., Kim O., Li M., Xiong X., Guan J. L., Kung H. J., Chen H., Shimizu Y., and Qiu Y. (2001) Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 3, 439–444 [DOI] [PubMed] [Google Scholar]
- 37. Nore B. F., Mattsson P. T., Antonsson P., Bäckesjö C. M., Westlund A., Lennartsson J., Hansson H., Löw P., Rönnstrand L., and Smith C. I. (2003) Identification of phosphorylation sites within the SH3 domains of Tec family tyrosine kinases. Biochim. Biophys. Acta 1645, 123–132 [DOI] [PubMed] [Google Scholar]
- 38. Hunter T. (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 39. Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., and Isobe T. (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 [DOI] [PubMed] [Google Scholar]
- 40. Jiang S., Zhao L., Lu Y., Wang M., Chen Y., Tao D., Liu Y., Sun H., Zhang S., and Ma Y. (2014) Piwil2 inhibits keratin 8 degradation through promoting p38-induced phosphorylation to resist Fas-mediated apoptosis. Mol. Cell. Biol. 34, 3928–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sheng J., Kumagai A., Dunphy W. G., and Varshavsky A. (2002) Dissection of c-MOS degron. EMBO J. 21, 6061–6071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bachmair A., Finley D., and Varshavsky A. (1986) In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 [DOI] [PubMed] [Google Scholar]
- 43. Dix M. M., Simon G. M., Wang C., Okerberg E., Patricelli M. P., and Cravatt B. F. (2012) Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell 150, 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piatkov K. I., Oh J. H., Liu Y., and Varshavsky A. (2014) Calpain-generated natural protein fragments as short-lived substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 111, E817–E826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu Z., Payoe R., and Fahlman R. P. (2012) The C-terminal proteolytic fragment of the breast cancer susceptibility type 1 protein (BRCA1) is degraded by the N-end rule pathway. J. Biol. Chem. 287, 7495–7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y. J., Liu C., Chang Z., Wadas B., Brower C. S., Song Z. H., Xu Z. L., Shang Y. L., Liu W. X., Wang L. N., Dong W., Varshavsky A., Hu R. G., and Li W. (2016) Degradation of the separase-cleaved Rec8, a meiotic cohesin subunit, by the N-end rule pathway. J. Biol. Chem. 291, 7426–7438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. An J. Y., Kim E., Zakrzewska A., Yoo Y. D., Jang J. M., Han D. H., Lee M. J., Seo J. W., Lee Y. J., Kim T. Y., de Rooij D. G., Kim B. Y., and Kwon Y. T. (2012) UBR2 of the N-end rule pathway is required for chromosome stability via histone ubiquitylation in spermatocytes and somatic cells. PLoS One 7, e37414. [DOI] [PMC free article] [PubMed] [Google Scholar]