

Abstract
Class III myosins (MYO3A and MYO3B) are proposed to function as transporters as well as length and ultrastructure regulators within stable actin-based protrusions such as stereocilia and calycal processes. MYO3A differs from MYO3B in that it contains an extended tail domain with an additional actin-binding motif. We examined how the properties of the motor and tail domains of human class III myosins impact their ability to enhance the formation and elongation of actin protrusions. Direct examination of the motor and enzymatic properties of human MYO3A and MYO3B revealed that MYO3A is a 2-fold faster motor with enhanced ATPase activity and actin affinity. A chimera in which the MYO3A tail was fused to the MYO3B motor demonstrated that motor activity correlates with formation and elongation of actin protrusions. We demonstrate that removal of individual exons (30–34) in the MYO3A tail does not prevent filopodia tip localization but abolishes the ability to enhance actin protrusion formation and elongation in COS7 cells. Interestingly, our results demonstrate that MYO3A slows filopodia dynamics and enhances filopodia lifetime in COS7 cells. We also demonstrate that MYO3A is more efficient than MYO3B at increasing formation and elongation of stable microvilli on the surface of cultured epithelial cells. We propose that the unique features of MYO3A, enhanced motor activity, and an extended tail with tail actin-binding motif, allow it to play an important role in stable actin protrusion length and ultrastructure maintenance.
Keywords: actin, cytoskeleton, hair cell, molecular motor, myosin
Introduction
Several classes of vertebrate sensory cells detect chemical or mechanical stimuli through actin bundle-based protrusions (1, 2). The vertebrate inner ear sensory hair cells consist of parallel actin bundle-based mechanoreceptive structures on their apical surfaces called stereocilia. These structures are critical for allowing hair cells to respond to the sound-evoked mechanical stimuli (3–6). Myosin Ic, myosin XVa, myosin VIIa, myosin VI, and myosin III are some of the myosin superfamily members that are involved in various aspects of stereocilia structure-function maintenance and when mutated cause hearing loss (7–15). The role that these myosins play in formation and elongation of parallel actin bundle-based protrusions is still unclear.
Class III myosins (MYO3) were discovered in Drosophila and named NINAC (Neither inactivation nor after potential C), because of the defect in the electroretinogram of the null mutant flies (16–19). There are two vertebrate MYO3 isoforms, MYO3A and MYO3B, that are encoded by two separate genes (20). In addition to the inner ear hair cells, MYO3 isoforms are also expressed in vertebrate photoreceptor calycal processes, brain, testis, and intestine (21–23). Calycal processes are parallel actin-based microvillus-like protrusions in vertebrate photoreceptors. Mutations in human MYO3A are associated with non-syndromic hearing loss (DFNB30) (24, 25).
MYO3 isoforms contain an N-terminal kinase domain, a conserved motor domain, a neck region, and a class specific C-terminal tail domain (Fig. 1, A and B) (20). Our earlier reports have shown the differences in the motor ATPase activity of human MYO3A and mouse MYO3B (26); however, no studies have directly compared the ATPase properties of human MYO3A and MYO3B or compared their in vitro motility properties. The MYO3A tail domain is encoded by six different exons (482 amino acids (aa)6) and contains two conserved domains, tail homology I (THD1) and tail homology II (THD2) (21). MYO3B contains a much shorter tail, which includes THD1, but lacks THD2 as well as a large portion of the MYO3A tail with unknown function. Previous reports have hypothesized that MYO3A and MYO3B may have overlapping functions, but MYO3B can only partially compensate for MYO3A (26, 27). Thus it is critically important to reveal functional differences between the human isoforms to determine how the loss of MYO3A leads to deafness.
FIGURE 1.

Diagrammatic representation of the domain structure of class III myosins and the COS7 cell expression of various constructs. A, schematic map including kinase domain, motor domain, IQ motifs, THD1, and THD2 of various MYO3A and MYO3B constructs used in this study. The amino acid residue number is shown at the start and end of each domain. The vertical black lines represent IQ motifs. MYO3A tail magnified diagram shows that the exon 30-amino acid sequence length is greater than that of combined sequences of exons 31–34. To represent the diagram to scale and to highlight all the features, complete exon 30 is not shown. Overlapping residues between exons 31–32, 33–34, and 34–35 in the MYO3A tail sequence are underlined. Diagrams are drawn to scale. B, Western blot of COS7 lysates expressing various MYO3 constructs probed with anti MYO3A tail tip antibody. Predicted molecular masses (in Da) are as follows: MYO3A, 182,230; MYO3A,Δ30, 148,080; MYO3A,Δ31, 176,370; MYO3A,Δ32, 177,910; MYO3A,Δ33, 180,350; MYO3A,Δ34, 176,480; and MYO3B.3A Tail, 176,290.
MYO3A motor activity is reduced by concentration-dependent autophosphorylation (26, 28–31). To avoid the autophosphorylation-induced reduction in motor activity, many studies have focused on kinase-deleted or kinase-dead MYO3 constructs (6, 26, 30).
MYO3 is proposed to bind ESPN1 and ESPNL (ectoplasmic specialization protein) and to transport it along actin bundles from the base to the tips (6, 32). Once the ESPN1 is transported to the barbed-ends, it is thought that the ESPN1 WH2 domain stimulates elongation of actin bundles (6, 26). Interestingly, recent papers identifying novel MYO3-binding partners highlight the complex nature of combined MYO3 isoform specificity, and its cargos in stereocilia formation and length regulation (26, 32).
There is a strong rationale for understanding the role of MYO3 in actin bundle-based protrusions in the absence of ESPN isoforms. During mouse embryonic development, the emergence and maturation of the outer and inner ear hair cells overlap well with the MYO3A expression and its proposed functions. MYO3A expression begins at postnatal day 0 and reaches its peak during postnatal days 6–10 as it progresses into adult stages (7). The Jerker mouse lacking all ESPN isoforms had short and thin stereocilia from P0 onward; however, the first phase of stereocilia elongation appeared to be normal (33).
Examining the properties of the tail domain of MYO3 has improved our understanding of MYO3 motor-based transport and its interactions with cargo. We demonstrated that the conserved THD1 of MYO3 binds to both ESPN1 and ESPNL (6, 26, 32) (34). We also found that the membrane occupation recognition nexus (MORN4) protein interacts with a region of the MYO3A tail upstream of THD1 and may allow tethering of MYO3A to the membrane (35). The THD2 is known to bind F-actin, and it is required for MYO3A to tip localize in actin protrusions independent of ESPN isoforms (22, 26, 36). MYO3B lacks THD2, and hence it cannot tip localize in the absence of ESPN1 or ESPNL (26, 37). In addition, fusing THD2 to the C terminus of MYO3B enhances its tip localization (26). Interestingly, there has been no detailed examination of the role of THD2 and other regions of the MYO3A tail in actin protrusion formation and elongation.
In this study, we compared the properties of the motor and tail domains of human MYO3A and MYO3B to determine their role in actin protrusion formation and elongation. We hypothesized that the unique extended tail of MYO3A containing THD2 is critical to promote the formation, elongation, and stabilization of actin protrusions. We also predicted that the MYO3 motor activity would correlate with the ability to enhance the formation and elongation of actin protrusions. To determine the impact of differing motor properties, we examined the motile and ATPase properties of human MYO3A and MYO3B. To characterize MYO3-based functions in actin protrusions, we examined tip localization and actin protrusion formation and elongation activity of a range of fluorescently tagged MYO3A and MYO3B deletion, mutation, and chimeric constructs. We also examined the filopodia dynamics in MYO3A- and MYO10-expressing cells. MYO10 is a known filopodia inducer, and it is implicated in Ena/VASP transport as well as actin filament cross-linking functions within filopodia protrusions (38–40). In addition to the frequently used (filopodia-forming) COS7 cell model, we also used microvilli forming W4 cells, providing a model more similar to stereocilia. Based on our biochemical, biophysical, and cell biological results, we suggest that the motor activity and the extended tail of MYO3A are precisely engineered to regulate stable actin protrusion formation and elongation.
Results
MYO3A Is a Faster Motor with Enhanced Actin Affinity Compared with MYO3B
We performed biochemical analysis of MYO3A 2IQ and MYO3B 2IQ (kinase domain deleted, truncated after second IQ domain, and containing a C-terminal GFP). The maximal ATPase activity (kcat) of MYO3A 2IQ was ∼3–4-fold higher than MYO3B 2IQ (Fig. 2A and Table 1). MYO3A 2IQ also contains a 20-fold higher actin affinity compared with MYO3B 2IQ as assessed by the KATPase (Table 1). However, the determined ATPase parameters for MYO3B 2IQ are subject to large uncertainties because the ATPase activity did not saturate at higher actin concentrations. The kcat values for human MYO3A 2IQ without the C-terminal GFP tag and in KMg50 buffer were previously reported to be slightly lower (29) than the current results (1.5 and 1.8 s−1, respectively). The sliding velocity of MYO3A 2IQ was ∼2-fold higher compared with MYO3B 2IQ (Fig. 2B and Table 1).
FIGURE 2.
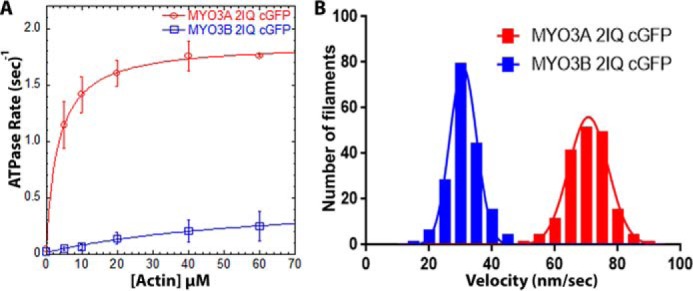
Actin-activated ATPase and in vitro motility properties of MYO3A and MYO3B. A, steady state actin-activated ATPase rate was measured and plotted as a function of actin concentration and fit to the Michaelis-Menten equation to determine kcat and KATPase. Error bars represent standard deviation. B, in vitro motility assay was performed with the same constructs, and the actin filament sliding velocity was determined (n = 177 filaments). ATPase and in vitro motility data were reported from at least two protein preparations and 2–3 independent experiments. The velocities for each construct were fit to a Gaussian distribution, and the mean velocity was determined. See Table 1 for summary of ATPase and motility values.
TABLE 1.
Summary of actin-activated ATPase activity and in vitro motility results
ATPase (represented as mean ± S.D.) and in vitro motility (represented as mean ± S.E.; n = 177 filaments) were reported from at least two protein preparations and 2–3 independent experiments.
| Construct | V0 | kcat | KATPase | Velocity |
|---|---|---|---|---|
| s−1 | s−1 | μm | nm/s | |
| MYO3A 2IQ cGFP | 0.04 ± 0.05 | 1.83 ± 0.02 | 3.3 ± 0.2 | 70.62 ± 0.48 |
| MYO3B 2IQ cGFP | 0.02 ± 0.01 | 0.50 ± 0.13 | 66.8 ± 31.0 | 31.20 ± 0.38 |
MYO3A Requires Enhanced Motor Activity and THD2 to Induce and Elongate Actin Protrusions
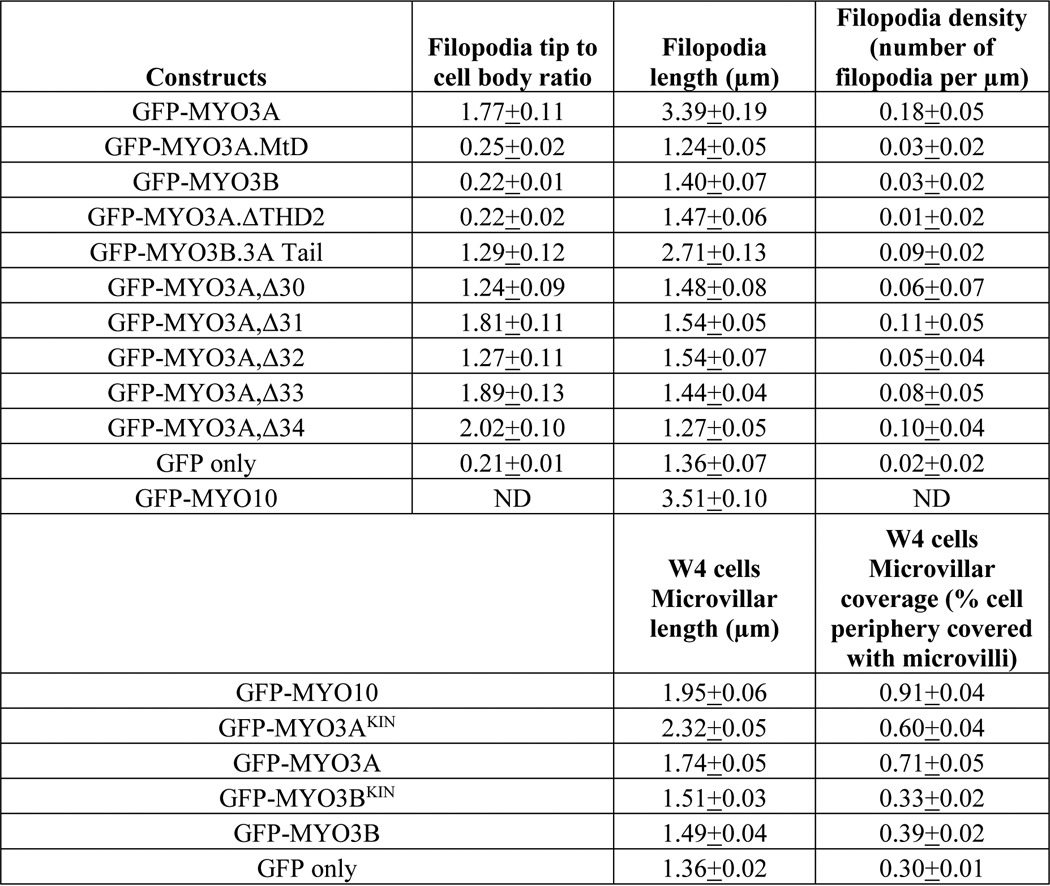
MYO3A (kinase domain deleted) demonstrated the highest tip localization, as well as filopodia formation (density) and elongation (length) activity compared with the rest of the constructs (Fig. 3, A–L). It has been shown previously that the motor activity is required for MYO3A tip localization; however, its impact on actin protrusion formation and elongation has never been examined before. Our results demonstrate that MYO3A motor dead (MYO3A.MtD) lost actin protrusion formation (Fig. 3L) and elongation activity (Fig. 3K) in addition to the loss of tip localization activity (Fig. 3J). Our MYO3A.MtD construct carried a point mutation (G720A) in the motor domain which was expected to result in a loss of motor activity as was found in the corresponding mutation in Dictyostelium MYO2 (aa 457) (41) and Chicken MYO5A (aa 440) (42, 43). We found that MYO3A.MtD 2IQ resulted in loss of ATPase activity (∼0.05 s−1) in the presence and absence of 20 μm actin, and it was able to bind actin in an ATP-dependent manner based on co-sedimentation assays (data not shown). It is unclear why the MYO3A.MtD did not co-localize with actin in COS7 cells, but similar results were obtained with other MYO3A motor-dead mutant constructs (7, 36).
FIGURE 3.

Role of MYO3A motor domain and THD2 in actin protrusion formation and elongation. Representative confocal images of paraformaldehyde-fixed and phalloidin-stained COS7 cells transfected with GFP-MYO3A (A–C), GFP-MYO3A.MtD (D–F), and GFP-MYO3B.3A Tail (G–I) (left panel, merged GFP-MYO3 (red) and 568-phalloidin actin (blue); middle panel, phalloidin-568 actin; right panel, GFP-MYO3). Scale bar, 5 μm. J, MYO3A exhibited significantly higher tip localization (*, p < 0.01) compared with all the other constructs. MYO3B.3ATail demonstrated tip localization greater than MYO3A.MtD, MYO3A.ΔTHD2, MYO3B, and GFP only (∧, p < 0.0001). A similar trend was observed in filopodia length (K) and filopodia density (L) measurements. MYO3A-expressing cells demonstrated the highest filopodia length (K, **, p < 0.0001) and density (L, ***, p < 0.0001). MYO3B.3ATail-expressing cells exhibited enhanced filopodia length (K, ∧∧, p < 0.001) and density (L, ∧∧∧, p < 0.0001) compared with MYO3A.MtD, MYO3A.ΔTHD2, MYO3B, and GFP only. J, filopodia tip to cell body ratio plot (J) and filopodia length plot (K) error bars indicate mean ± S.E. Error bars in the filopodia density plot (L) indicate mean ± S.D. For all the parameters, data were collected from ≥60 filopodia from ≥10 cells for each condition in three independent experiments. Numerical data are shown in Table 2.
Removal of THD2 from MYO3A (MYO3A.ΔTHD2) abolished its tip localization (Fig. 3J) as shown previously (26). Interestingly, we observed that its filopodia formation and elongation activity were also abolished compared with MYO3A-expressing cells (Fig. 3, K and L). MYO3B (kinase domain deleted) did not tip localize as it lacks THD2 (26), whereas the MYO3B.3ATail (MYO3B motor with entire MYO3A Tail) chimeric construct demonstrated enhanced filopodia tip localization, formation, and elongation activity compared with MYO3B (Fig. 3, J–L). However, the tip localization, filopodia formation, and elongation activity of the MYO3B.3ATail construct was significantly reduced compared with MYO3A (Fig. 3, A–C and G–L). MYO3B.3ATail is a unique chimeric construct that allowed us to compare the motor activity of MYO3A and MYO3B while keeping the tail domains identical. Overall, our results support our hypothesis that the more robust motor activity of MYO3A correlates with its enhanced ability to function in actin protrusion formation and elongation.
MYO3A Extended Tail Domain Is Crucial for Formation and Elongation of Stable Actin Protrusions
Apart from higher motor activity, the MYO3A extended tail domain is a structurally distinct feature compared with MYO3B. MYO3A tail exons 32–34 encode for THD1 (6, 34), and exon 35 encodes for THD2 (36). It is known that MYO3A requires THD2 to tip localize (36); however, the functional role of other regions of the extended tail in actin protrusion formation or elongation is not known. Walsh et al. (24) have reported one naturally occurring alternative splice variant of mouse MYO3A that lacks complete tail exon 32, providing further rationale for examining the function of individual MYO3A tail exons.
Deletion of MYO3A tail exons demonstrated minimal impact on tip localization (Table 2). However, all the tail exon deletion constructs lost the ability to induce and elongate actin protrusions (Table 2). We confirmed by Western blotting that the expressed MYO3A tail exon deletion constructs were the correct size (Fig. 1B), and fluorescence intensity levels demonstrated similar expression levels. We have successfully used these constructs in our previous study (35) to determine the binding site of a novel MYO3A cargo-MORN4. Finally, the ability of these constructs to tip localize suggests that the structure of the tail region in each of the constructs was unperturbed except for the removal of specific regions. Interestingly, our live cell imaging data revealed that the actin protrusions formed by MYO3A-expressing cells were longer and more stable compared with the protrusions formed by MYO3A,Δ(tail exon) constructs expressing cells (supplemental video 1–3). Our results indicate that the integrity of the MYO3A tail may be critical for its ability to induce, elongate, and stabilize the protrusions.
TABLE 2.
Summary of parameters used to quantify MYO3-associated actin protrusions
Filopodia tip to cell body ratio and filopodia length values are presented as mean ± S.E. Filopodia density values are presented as mean ± S.D. Filopodia data were collected from >60 filopodia from >10 cells for each condition in three independent experiments. Statistical comparisons are shown in respective data figures. ND, not determined.
MYO3A Enhances Actin Protrusion Lifetime
We examined the filopodia dynamics of MYO3A-, MYO10-, and GFP only-expressing cells (supplemental Videos 1 and 4). MYO10-expressing cells and GFP only-expressing cells (control cells) demonstrated ∼6- and ∼3-fold greater filopodia extension velocities, respectively, compared with MYO3A (Fig. 4A). MYO10 and control cells also demonstrated greater retraction velocity compared with MYO3A (Fig. 4B). Interestingly, we did not observe any protrusions in MYO3A-expressing cells with a lifetime less than 5 min. However, MYO10-expressing cells displayed 44% protrusions with a lifetime of <3 min, 36% protrusions with a 3–5-min lifetime, and only 20% protrusions with a lifetime of >5 min. Control cells showed 22% protrusions with a lifetime of <3 min, 31% protrusions with 3–5-min lifetime, and 47% protrusions with a lifetime of >5 min (Fig. 4C). These data also reinforce our proposal that MYO3A can impact actin-protrusion dynamics in the absence of ESPN1.
FIGURE 4.
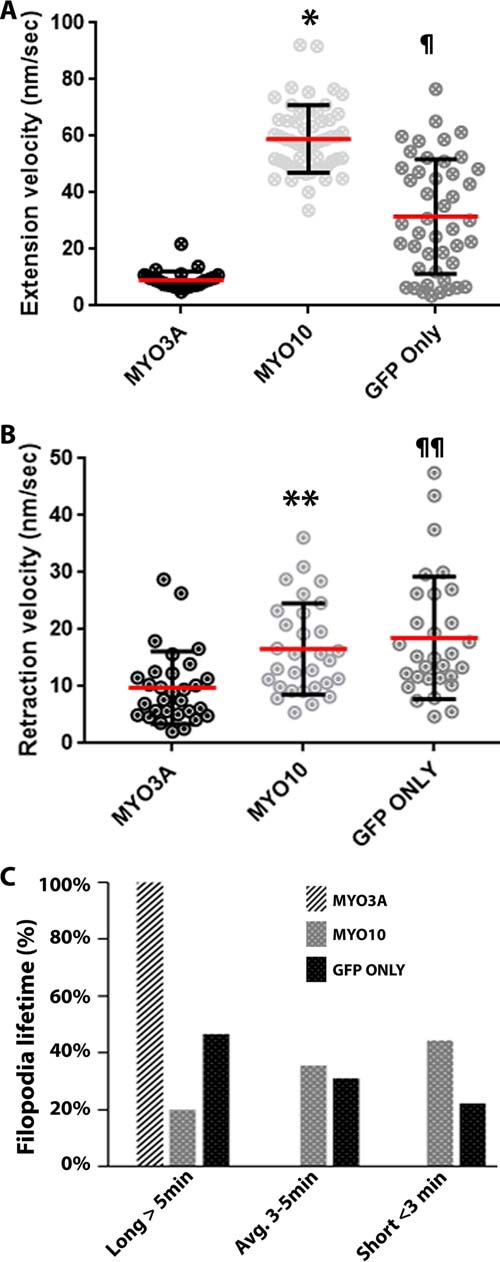
Impact of MYO3A on actin protrusion dynamics. A, scattered dot plots showing the filopodia extension velocities of MYO3A-, MYO10-, and GFP only-expressing COS7 cells. MYO10-expressing cells exhibited the highest filopodia extension velocity (*, p < 0.0001). GFP only-expressing cells had a filopodia extension velocity greater than MYO3A (¶, p < 0.0001) (extension velocity (nm/s): MYO3A, 8.78 ± 3.39; MYO10, 58.97 ± 11.91; GFP only, 21.46 ± 15.10. B, scattered dot plot showing the filopodia retraction velocity of MYO3A-, MYO10-, and GFP only-expressing COS7 cells. MYO10 and GFP only exhibited significantly greater retraction velocity compared with MYO3A (**, p < 0.01, and ¶¶, p < 0.001, respectively) (retraction velocity (nm/s): MYO3A, 9.76 ± 6.38; MYO10, 16.59 ± 8.00; GFP only, 18.51 ± 10.76). Extension and retraction velocity data were collected from ≥31 filopodia from ≥10 cells for each condition in three independent experiments. Scattered dot plots are shown as mean ± S.D., and the mean is represented by a red line. C, quantification of filopodia lifetimes by live-cell imaging of COS7 cells expressing the indicated GFP-tagged constructs. MYO3A-expressing cells exhibited protrusions with a longer lifetime (>5 min) compared with MYO10- and GFP only-expressing cells. Data were collected from 45 filopodia from ≥7 cells for each condition in three independent experiments.
Role of MYO3A in Inducing and Elongating Stable Actin Bundle-based Microvilli in Cultured W4 Cells
To demonstrate whether MYO3A can function as an inducer and elongator of self-supporting actin-based protrusions, we turned to W4 epithelial cells, which build a microvillus-rich brush border during differentiation (44). W4 cells expressing GFP-tagged MYO3AKIN (full-length, kinase active), MYO3A, MYO3BKIN (full-length, kinase active), MYO3B, MYO10, and GFP only were imaged using structured illumination microscopy (Fig. 5, A–F). MYO10 expression in W4 cells led to a profuse microvilli generation on the cell surface (Fig. 5A). We hypothesized that MYO3A transfection in W4 cells would also lead to the generation of microvilli along the cell surface if it is capable of enhancing parallel actin bundle-based protrusions. Average microvillar length was the highest in MYO3AKIN-expressing cells and the lowest in GFP only-expressing cells (Fig. 5S). MYO3A-expressing cells formed shorter microvilli compared with MYO10-expressing cells (Fig. 5S) and longer microvilli compared with MYO3BKIN-, MYO3B-, and GFP only-expressing cells, respectively (Fig. 5S). MYO3B-expressing cells demonstrated longer microvilli compared with GFP only (Fig. 5S). Average microvillar coverage was the highest in MYO10-expressing cells (Fig. 5T) and lowest in GFP only-expressing cells. MYO3AKIN and MYO3A induced greater microvillar formation compared with MYO3B-, MYO3BKIN-, and GFP only-expressing cells (Fig. 5T). However, MYO3B induced greater microvilli formation compared with GFP only (Fig. 5T). Overall, MYO3A-expressing cells demonstrated significantly longer and higher numbers of microvilli compared with MYO3B- and GFP only-expressing W4 cells. Villin is a major actin cross-linking protein present in the actin core of the brush border microvilli (45). The presence of villin in the actin protrusions of MYO10- and MYO3A-expressing W4 cells (Fig. 6) confirmed that the protrusions are indeed microvilli-like and not filopodia.
FIGURE 5.

Impact of MYO3A on microvilli formation and elongation in W4 cells. A–R, range of GFP-tagged MYO3 and MYO10 constructs were transfected in induced W4 cells and imaged using super-resolution structured illumination microscopy to examine their microvilli formation and elongation activity. The zoomed in image of the surface features are shown at the bottom of each panel. In the merged image, actin is shown in magenta and MYO3/MYO10 shown in green. Scale bar, 3 μm. S, MYO3AKIN exhibited the highest microvilli length compared with the other constructs (*, p < 0.001). MYO10 demonstrated longer microvilli compared with MYO3A, MYO3B, and GFP only constructs (∧, p < 0.01). MYO3A demonstrated longer microvilli length compared with MYO3B constructs and GFP only (#, p < 0.001), whereas MYO3B constructs showed longer microvilli length compared with GFP only (**, p < 0.02). T, MYO10 expression resulted in the highest microvillar coverage (*, p < 0.01). Expression of MYO3A constructs resulted in microvillar coverage greater than MYO3B constructs and GFP only, whereas MYO3B constructs induced microvillar coverage greater than GFP only (∧, p < 0.05). Data were collected from ≥54 microvilli from ≥9 cells in two independent sets of experiments. Error bars in both the plots indicate mean ± S.E. Numerical data are shown in Table 2.
FIGURE 6.
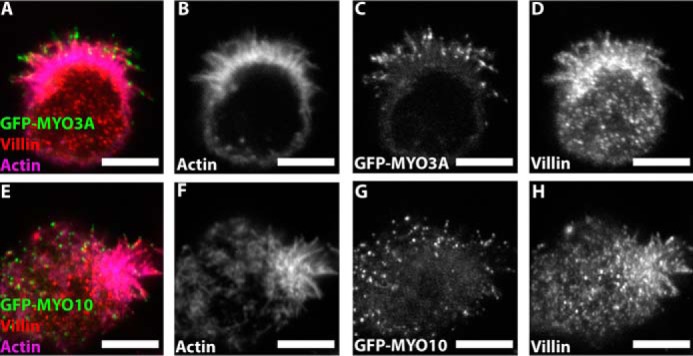
Villin immunostaining in MYO3A- and MYO10-expressing W4 cells. Confocal images of paraformaldehyde-fixed and phalloidin-stained GFP-MYO3A- (A–D) and GFP-MYO10 (E–H)-expressing W4 cells immunostained for villin, a microvilli-specific actin cross-linking protein. (merged image: GFP-MYO3A or GFP-MYO10 (green), actin (magenta), and villin (red)). Scale bar, 5 μm.
Discussion
The findings from recent MYO3 studies (25, 27, 32, 34) and an overall growing interest in the function of myosins in actin-based protrusions (8, 39, 46) demanded a systematic study of the functional differences of MYO3 motor and tail domains. Our results highlight the enhanced motor activity of human MYO3A compared with MYO3B, which correlates with its enhanced ability to localize to actin protrusion tips as well as induce the formation and elongation of actin protrusions. In addition, the extended tail domain of MYO3A, which contains an actin-binding motif, is required for the formation, elongation, and stabilization of actin protrusions. Thus, we propose that MYO3A is uniquely engineered to function as a motorized actin cross-linker that can control the dynamics of actin protrusions even in the absence of its binding partners ESPN1 and ESPNL. We demonstrate that MYO3A can enhance the formation and elongation of more stable actin protrusions such as the microvilli of W4 cells. Our results shed light on the crucial role that MYO3 plays in controlling the lengths and dynamics of the stereocilia of inner ear hair cells.
Role of MYO3 Motor Domain in Regulating Actin Protrusion Dynamics
Our results demonstrate that MYO3A is a faster motor compared with MYO3B (Fig. 2 and Table 1), and an active motor domain is absolutely required for allowing MYO3A to induce and elongate actin protrusions (Fig. 3, K and L). To examine the role of the motor domain in class III myosins, we compared MYO3A and MYO3B.3ATail constructs, which were designed to have an identical tail domain but differing motor domains. The results (Fig. 3, J–L) support our hypothesis that motor activity correlates with protrusion formation and elongation activity.
The correlation between MYO3 motor activity and actin protrusion formation and elongation was consistently observed in microvilli forming W4 cells. Greater microvilli formation and elongation activity of MYO3A constructs compared with MYO3B constructs (Fig. 5) suggested that MYO3A can induce and elongate stable actin protrusions as well. Based on our observation of MYO3B distribution along the microvilli in W4 cells, it is likely that some form of ESPN is expressed in these cells (47), because MYO3B is known to localize to actin bundles only in the presence of ESPN1 or ESPNL (26). MYO3AKIN exhibited greater microvilli elongation activity compared with MYO3A (Fig. 5S), suggesting that the presence of the kinase domain may further enhance elongation activity. Interestingly, no such difference was observed between the elongation activity of MYO3BKIN and MYO3B. MYO10 demonstrated higher elongation activity compared with MYO3A in W4 cells (Fig. 5S), which is consistent with the proposed higher motor activity of MYO10 (39, 48) compared with MYO3A.
Role of the MYO3A Tail Domain in Regulating Actin Protrusion Dynamics
Our results demonstrate that THD2 is not only critical for tip localization, as demonstrated previously in fish and mouse MYO3A (22, 36), but also for the actin protrusion formation and elongation activity of MYO3A (Fig. 3, G–L). In addition, analysis of the exon deletion constructs demonstrated that the extended tail is also required for actin protrusion formation and elongation. We propose that MYO3A may be involved in actin protrusion initiation via a mechanism similar to the convergent elongation model (49, 50) in which actin protrusion formation occurs by a gradual organization of branched actin filaments in the cell cortex. Although microvilli do not form via a convergent elongation mechanism (51, 52), MYO3A likely stabilizes actin-based structures important for the early stages of microvilli formation. MYO3A may be able to stabilize actin-based structures utilizing its motor and tail domain to cross-link actin filaments. MYO3 are not anticipated to function as dimers because they lack a predicted coiled-coil domain, and thus MYO3A is the first monomeric myosin determined to be capable of enhancing the formation of protrusions. The deafness-associated G488E MYO3A mutant, which does not tip localize in filopodia (COS7 cells), is not rescued in the presence of WT MYO3A and ESPN1 (25), supporting the idea that MYO3A is monomeric.
Our investigation of the actin protrusion dynamics in control cells compared with MYO10- and MYO3A-expressing cells (Fig. 4) revealed interesting differences that suggest potential mechanisms for how these two motors influence actin protrusion dynamics. The changes in actin protrusion length are thought to be dependent upon the balance between cytoskeletal assembly at the tips and the rate of actin retrograde flow, with assembly being a dominant influence (53–58). Our results demonstrate that MYO3A dramatically increases protrusion lifetime while slowing protrusion dynamics (extension and retraction velocity) (Fig. 4). MYO10 can enhance the filopodia extension velocity by increasing the actin polymerization rate through mechanisms that have been previously proposed, such as transporting the anti-capping protein Ena/VASP to the tips (48). Interestingly, MYO3A may be able to stabilize actin bundles by cross-linking actin filaments at the tips of protrusions. It is also possible that the MYO3A-mediated slow extension velocity may provide a greater opportunity for incorporation of actin-bundling proteins in the growing actin bundle, thus further stabilizing the protrusions. The tip localized MYO3A may slow elongation if it interferes with the addition of monomers at the barbed ends. Interestingly, all the tail exon deletion constructs expressing cells displayed shorter (Table 2) and less stable (supplemental video 1–3) protrusions compared with MYO3A. These results suggest that disrupting the tail domain structure abolishes its ability to stabilize the actin protrusions. It was previously reported that the rearward intrafilopodial movement of MYO3A is similar to MYO10 and VASP (30, 31, 38, 48), suggesting that the retrograde flow rate is unaffected by MYO3A. Thus, we propose that MYO3A is precisely engineered to maintain stable actin protrusions that demonstrate slow elongation rates.
A recent study demonstrated severe defects in stereocilia shape and size in MYO3A−/−MYO3B−/− double knock-out mice (27). On embryonic stage 16.5 of the double knock-out mice, the cochlear hair bundles showed abnormal shape and exaggerated elongation. Interestingly, a mouse model of DFNB30, which lacks functional MYO3A, demonstrated changes in stereocilia ultrastructure and inner ear hair cell degeneration as the animals aged (59). Because our work suggests MYO3A can enhance protrusion elongation by stabilizing the tips, the double knock-out phenotype may seem to contradict our results. However, a possible interpretation is that, in the absence of MYO3A, the stereocilia may elongate at a faster rate although there may be a reduction in its stability. Thus, in the absence of MYO3A, the stereocilia elongate abnormally in early development, and because the ultrastructure is perturbed, they are more likely to degenerate as an adult. It should be noted that length regulation in stereocilia is a highly complex process that involves many different players, including multiple unconventional myosins and their binding partners (7, 8, 27). Indeed, a recent study has shown that MYO3 isoforms are capable of elongating or restricting actin protrusion length depending upon the presence of its specific cargo (ESPN1 or ESPNL) (32).
Conclusions
Based on our results we envision MYO3A as a motorized monomeric actin cross-linker that specifically localizes to the barbed ends of actin filaments and stabilizes growing actin filaments to facilitate length maintenance. We also suggest that MYO3A can bring actin filaments together near the membrane to initiate the formation of actin protrusions, a process that is critical during stereocilia formation. We suggest that in addition to the differences in the MYO3A and MYO3B motor activity, the difference in their tail domain may also be critical for function. These key differences may prevent MYO3B from compensating for MYO3A in DFNB30 patients, which lack functional MYO3A and experience age-dependent stereocilia degeneration. Our results may be crucial for establishing a functional mechanism of MYO3 in the generation and maintenance of various actin-based protrusions in native vertebrate sensory and neuronal cell types. It remains to be seen whether other tip localizing myosins such as MYO7 and MYO15 have the ability to regulate actin protrusion stability and dynamics.
Experimental Procedures
Expression Plasmids
GFP-tagged human MYO3A (aa 340–1616 of NM_017433, kinase-deleted) and MYO3AKIN (full-length, kinase-active) were constructed as described previously (6, 30). The following naturally occurring variants exist in the MYO3A sequence (I348V, V369I, S956N, and R1313S) (60).
All the GFP-MYO3A tail exon deletion constructs were designed to avoid insertion of a premature stop codon as described previously (35), with the following amino acids deleted: 1) MYO3A,Δ30, deleted aa 1134–1431; 2) MYO3A,Δ31, deleted aa 1432–1479; 3) MYO3A,Δ32, deleted aa 1481–1515; 4) MYO3A,Δ33, deleted aa 1515–1528); and 5) MYO3A,Δ34, deleted aa 1529–1576.
GFP-MYO3A was used as template to generate the GFP-MYO3A.MtD (motor-dead) construct by introducing a point mutation (G720A) (Table 3). GFP-MYO3A lacking THD2 (GFP-MYO3A.ΔTHD2) was generated by deleting THD2 (deleted aa 1595–1616) (Table 3). GFP-MYO3BKIN (full-length, kinase-active) and GFP-MYO3B (kinase-deleted) (NM_138995) constructs were developed from the template DNA provided by the Kachar laboratory. Briefly, MYO3BKIN was inserted into the pEGFP vector, and this construct was further modified to remove the N-terminal kinase domain (aa 345–1314) (Table 3). A GFP-MYO3B.3A Tail chimeric construct encoding MYO3B motor-neck (aa 346–1148 of NM_138995) and C-terminal MYO3A tail (aa 1146–1616 of NM_017433.4) was also generated (Table 3).
TABLE 3.
List of primers used to generate novel constructs for this study
| Gene/species (accession no.) | Plasmid (template vector) | Primers used (5′ to 3′), forward (F) and reverse (R) |
|---|---|---|
| MYO3A/human (NM_017433) | MYO3A.MtD (pEGFP-C1) | F, GCATTGGCATTCTTGATATATTTGCCTTTGAAAATTTCAAAAAAAATT |
| R, AATTTTTTTTGAAATTTTCAAAGGCAAATATATCAAGAATGCCAATGC | ||
| MYO3A.ΔTHD2 (pEGFP-C1) | F, AGAGAGCCAGCAGCCGGCGGCCGCAACCCCTACGACTTC | |
| R, GAAGTCGTAGGGGTTGCGGCCGCCGGCTGCTGGCTCTCT | ||
| MYO3A 2IQ (pFastBac) | F, TCGGGCGCGGATCCCATGGTAGATGATTTAGC | |
| R, GCTAAATCATCTACCATGGGATCCGCGCCCGA | ||
| MYO3B.3A Tail (pEGFP-C1) | F, TTTCGTGAAGAAACAAGCAGAAAATGGATCCTCTGCTAATGAAAGATTCATTTCAG | |
| R, CTGAAATGAATCTTTCATTAGCAGAGGATCCATTTTCTGCTTGTTTCTTCACGAAA | ||
| MYO3B/human (NM_138995) | MYO3BKIN (pEGFP-C1) | F, TAGGATCCAAACATCTGTATGGA |
| R, TAGCGGCCGCTTAATGTTGAGCAAAAGAGTC | ||
| MYO3B (pEGFP-C1) | F, TAGGATCCAAACATCTGTATGGA | |
| R, TAGGATCCGATGATTTGGTCAACCTAGA | ||
| MYO3B 2IQ (pFastBac) | F, TAGGATCCATGGATGATTTGGTCAACCTAGA | |
| R, TAGCGGCCGCGTCCCCTGCTTGATT | ||
| MYO3B.3A Tail (pEGFP-C1) | F, AGTGCCGAGGTTCAAGGATCCAGCGAGCCTGGTGAC | |
| R, GTCACCAGGCTCGCTGGATCCTTGAACCTCGGCACT |
Both pFBMYO3A 2IQ and pFBMYO3B 2IQ constructs (for biochemical examination) were without the kinase domain, truncated after the second IQ domain (29), and contain a C-terminal GFP tag (Table 3). The GFP-MYO10 (bovine) construct used in this study was gifted by Richard Cheney (38).
Protein Expression and Purification
The FastBac system (Invitrogen) was used to generate the recombinant baculoviruses (26, 30, 31). Recombinant MYO3A 2IQ and MYO3B 2IQ c-GFP with C-terminal FLAG tag were expressed in the baculovirus SF9 insect cell system and purified with anti-FLAG affinity chromatography as described previously (26, 30). The affinity-purified MYO3A and MYO3B 2IQ c-GFP constructs were also purified by actin co-sedimentation and released with ATP to ensure 100% active myosin heads.
COS7 Cell Culture, Transfection, Imaging, and Analysis
COS7 cells were cultured as described previously (30). Briefly, cells were cultured in DMEM (Corning) supplemented with 10% fetal bovine serum, 100 units of penicillin/streptomycin, 4 mm l-glutamine, 4.5 g/liter d-glucose, and 1 mm sodium pyruvate. For transfection, ∼30,000 COS7 cells were plated onto acid-washed 22-mm #1.5 glass coverslips placed in each well of a 6-well plate. FuGENE HD (Promega) transfection reagent was used for transient transfections according to the manufacturer's protocol, and the cells were imaged ∼20–30 h after transfection. For live cell imaging, coverslips with transfected cells were placed in rose chambers (61) filled with Opti-MEM media supplemented with 100 units of penicillin/streptomycin and 5% fetal bovine serum (Gemini). Single and time-lapse images were acquired at room temperature using a Nikon TE2000-PFS fluorescence microscope with a ×60 1.4 N.A. phase objective and equipped with CoolSnap HQ2 cooled charge-coupled device digital camera (Photometrics).
For fixed cell imaging, transfected cells were fixed for 20 min in 4% formaldehyde in 1× PBS, permeabilized for 30 min in 0.5% Triton X-100 in 1× PBS, counterstained with Alexa Fluor 568 phalloidin (Life Technologies, Inc.), and mounted using ProLong Gold Antifade reagent (Invitrogen). Fixed and live cell confocal imaging was accomplished using a TiE inverted fluorescence microscope (Nikon Instruments) equipped with either 1) a swept-field confocal scan head (Prairie Technologies), DU-897 EMCCD (Andor), and ×100 Plan Apo 1.45NA objective or 2) a spinning disk confocal head (PerkinElmer Life Sciences), DU-888 (Andor), and Apo TIRF 1.49 NA objective. Image acquisition was managed through NIS-Elements software (Nikon Instruments), and ImageJ (with ND2 reader plugin) was used to analyze the images and to quantify tip to cell body ratio, and filopodia length, and density (number of filopodia/μm), as described previously (30).
ImageJ (with MTrackJ plugin) was used to measure the filopodia extension and retraction velocities. Retracting and stationary filopodia were excluded from extension velocity measurements. Extending and stationary filopodia were excluded from retraction velocity measurements. Five minute live cell time-lapse videos were used to analyze filopodia lifetime. An extending protrusion reaching 1-μm length was categorized as an initiation event, which marked the beginning of the lifetime. The same extending protrusion was then followed over time until it retracted back to 1-μm length, which is then categorized as end of lifetime. Stationary protrusions were included for filopodia lifetime measurements. Protrusions <1-μm were excluded from all the measurements. The data were compared by using one-way analysis of variance with the subsequent Tukey post hoc analysis tool of GraphPad Prism 6 software.
COS7 Fluorescence Intensity Measurement
Uniformity in expression levels of our experimental constructs was confirmed by examining the mean fluorescence intensity (brightness per cell area) from cell body (excluding protrusions) of >10 cells per condition from which the data were reported. No significant difference was observed in the fluorescence intensity among different conditions, except MYO3B and MYO3A,Δ32, which demonstrated ∼1.2-fold higher intensity compared with the mean of the rest of the constructs. However, neither of the constructs showed higher tip localization or filopodia formation and elongation activity compared with the wild type constructs.
COS7 Lysate Preparation and Western Blotting
COS7 cell lysates were prepared as described previously (6). Briefly, 24–30 h after transfection of COS7 cells with various constructs, cell were treated with 300 μl of ice-cold lysis buffer (50 mm Tris, pH 7.4, 5 mm DTT, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, 1 mm PMSF, aprotinin, and leupeptin). To prepare the lysate, cells were scraped off the surface using a cell scraper, and the lysates were homogenized by pipetting. Lysates were examined using NuPAGE BisTris 4–12% gels (Invitrogen) and transferred to a nitrocellulose membrane for analysis by Western blotting. Anti-MYO3A tail tip antibody (21) was used to detect various MYO3 fusion proteins. HRP-linked goat anti-rabbit secondary antibodies (Cell Signaling) and Lumi GLO chemiluminescent substrate (Cell Signaling) were used to visualize the blots.
W4 Cell Transfection
Ls174T-W4 cells (generously provided by Dr. Hans Clevers) were cultured in DMEM with high glucose and 2 mm l-glutamine supplemented with 10% tetracycline-free FBS, G418 (1 mg/ml), blasticidin (10 μg/ml), and phleomycin (20 μg/ml). Cells were grown at 37 °C and 5% CO2. One day before transfection, cells were plated in a T-25 such that on the next day, cells were at ∼80% confluency. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The following day, cells were plated onto coverslips in the presence of 1 μg/ml doxycycline and allowed to adhere and induce for 8 h. Doxycycline activates STRAD (STE20-related kinase adaptor), which in turn activates LKB1 (liver kinase B1). Upon activation of LKB1, the W4 cell actin cytoskeleton rapidly remodels to form an apical brush border (microvilli), thus completely polarizing the cells (44).
Fixing, Staining, and Imaging of W4 Cells
Cells were first washed once with warm PBS and then fixed with 4% paraformaldehyde in PBS for 15 min at 37 °C. Following fixation, cells were washed three times with PBS and permeabilized with 0.1% Triton X-100/PBS for 15 min at room temperature. Cells were then washed three times with PBS and blocked overnight at 4 °C in 5% BSA/PBS. Cells were washed once with PBS and incubated with anti-GFP (1:200, Aves, GFP-1020) or anti-villin (1:50, Santa Cruz Biotechnology, BDID2C3) in PBS for 2 h at room temperature, followed by four 5-min washes with PBS. Alexa Fluor 488 goat anti-chicken (1:200) and Alexa Fluor 568 phalloidin (1:200) or Alexa Fluor 568 donkey anti-mouse (1:200) and Alexa Fluor 647 phalloidin (1:100) (Life Technologies, Inc.) were diluted in PBS and incubated with the cells at 37 °C for 1 h, followed by four 5-min washes with PBS. Coverslips were mounted using ProLong Gold Antifade Mountant (Life Technologies, Inc.). Structured illumination microscopy was performed using an Applied Precision DeltaVision OMX (GE Healthcare) located in the Vanderbilt Cell Imaging Shared Resource equipped with a ×60 Plan-Apochromat N/1.42 NA objective and processed using softWoRx software (GE Healthcare). Confocal imaging was performed using a Nikon A1R laser-scanning confocal microscope with a ×100 Apo TIRF 1.49 NA objective. Percent microvillar coverage was calculated by dividing the percentage of total W4 cell margin by the percentage of the margin covered by microvilli. Microvillar actin bundles were traced from the projected structured illumination microscopy images (z-stacks), and lengths were measured using ImageJ (length tool) (62). Edges of microvilli actin bundles were visualized and defined with phalloidin staining. Individual microvilli actin bundles (base to tips) were first identified from maximum intensity projection images (example Fig. 5, A–R). The base and tips of the microvilli were further traced and confirmed from the projected F-actin z-stacks (visualized with phalloidin staining). Microvilli, whose base and tips could not be traced clearly from both maximum intensity and z-stack projections, were excluded from the analysis.
Myosin ATPase Assay
The steady state NADH-linked ATPase assay was used to examine MYO3A 2IQ and MYO3B 2IQ c-GFP actin-activated ATPase activity in modified KMg50 buffer (10 mm imidazole, pH 7.0, 50 mm KCl, 1 mm EGTA, 1 mm MgCl2, 1 mm DTT) with additional KCl (final 72 mm) and ATP (1.85 mm) at 25 °C (28, 30, 31). Briefly, the motor ATPase was examined in the presence of a range of actin concentrations in an Applied Photophysics stopped-flow. The Michaelis-Menten equation was used to determine the kcat (maximal actin-activated ATPase rate) and KATPase (actin concentration at which the ATPase activity is half-maximal) using a hyperbolic fit of the ATPase rates as a function of actin concentration.
In Vitro Motility Assay
The in vitro motility assay (63) was performed as described previously (64, 65). Briefly, C-terminal GFP-tagged MYO3A 2IQ or MYO3B 2IQ was attached to the nitrocellulose-coated glass coverslip surface using an anti-GFP antibody (Invitrogen), and the surface was then blocked with 1 mg ml−1 BSA solution in KMg50 buffer. The activation buffer consisted of KMg50 supplemented with 0.35% methylcellulose, 10 μm calmodulin, 1 mg ml−1 BSA, 2 mm ATP, and 20 units ml−1 pyruvate kinase and 2.5 mm phosphoenolpyruvate. To reduce photobleaching, 1 mg ml−1 glucose, 0.1 mg ml−1 glucose oxidase, and catalase were also added. Finally, after the addition of activation buffer, the motility of the rhodamine-phalloidin-labeled F-actin filaments was observed using a Nikon TE2000 microscope (65). The time-lapse images were acquired at 5-s intervals for a period of 10 min. The velocity of moving actin filaments was measured using ImageJ (MTrackJ plugin) (66).
Author Contributions
M. H. R., O. A. Q., B. K., M. J. T., and C. M. Y. designed the research; M. H. R., O. A. Q., M. L. W., W. C. U., J. W. G., R. C., B. K., and C. M. Y. performed the research; M. H. R., O. A. Q., and C. M. Y. analyzed the data; M. H. R. wrote the paper with critical input from O. A. Q., B. K., M. L. W., M. J. T., and C. M. Y. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Lina Jamis and Anja M. Swenson for helpful discussions.
This work was supported in part by National Institutes of Health Grant EYE018141, Pennsylvania Lions Hearing Research Foundation (to C. M. Y.), and National Institutes of Health Grants DK075555 and R01-DK095811 (to M. J. T.). This work was authored, in whole or in part, by National Institutes of Health staff. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Videos S1–S4.
- aa
- amino acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Bartles J. R. (2000) Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 12, 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sekerková G., Zheng L., Loomis P. A., Mugnaini E., and Bartles J. R. (2006) Espins and the actin cytoskeleton of hair cell stereocilia and sensory cell microvilli. Cell. Mol. Life Sci. 63, 2329–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin H. W., Schneider M. E., and Kachar B. (2005) When size matters: the dynamic regulation of stereocilia lengths. Curr. Opin. Cell Biol. 17, 55–61 [DOI] [PubMed] [Google Scholar]
- 4. Rzadzinska A. K., Schneider M. E., Davies C., Riordan G. P., and Kachar B. (2004) An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J. Cell Biol. 164, 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneider M. E., Belyantseva I. A., Azevedo R. B., and Kachar B. (2002) Rapid renewal of auditory hair bundles. Nature 418, 837–838 [DOI] [PubMed] [Google Scholar]
- 6. Salles F. T., Merritt R. C. Jr., Manor U., Dougherty G. W., Sousa A. D., Moore J. E., Yengo C. M., Dosé A. C., and Kachar B. (2009) Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat. Cell Biol. 11, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneider M. E., Dosé A. C., Salles F. T., Chang W., Erickson F. L., Burnside B., and Kachar B. (2006) A new compartment at stereocilia tips defined by spatial and temporal patterns of myosin IIIa expression. J. Neurosci. 26, 10243–10252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Belyantseva I. A., Boger E. T., Naz S., Frolenkov G. I., Sellers J. R., Ahmed Z. M., Griffith A. J., and Friedman T. B. (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 7, 148–156 [DOI] [PubMed] [Google Scholar]
- 9. Nal N., Ahmed Z. M., Erkal E., Alper O. M., Lüleci G., Dinç O., Waryah A. M., Ain Q., Tasneem S., Husnain T., Chattaraj P., Riazuddin S., Boger E., Ghosh M., Kabra M., Riazuddin S., Morell R. J., and Friedman T. B. (2007) Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum. Mutat. 28, 1014–1019 [DOI] [PubMed] [Google Scholar]
- 10. Grati M., and Kachar B. (2011) Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. U.S.A. 108, 11476–11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boëda B., El-Amraoui A., Bahloul A., Goodyear R., Daviet L., Blanchard S., Perfettini I., Fath K. R., Shorte S., Reiners J., Houdusse A., Legrain P., Wolfrum U., Richardson G., and Petit C. (2002) Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 21, 6689–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seiler C., Ben-David O., Sidi S., Hendrich O., Rusch A., Burnside B., Avraham K. B., and Nicolson T. (2004) Myosin VI is required for structural integrity of the apical surface of sensory hair cells in zebrafish. Dev. Biol. 272, 328–338 [DOI] [PubMed] [Google Scholar]
- 13. Sakaguchi H., Tokita J., Naoz M., Bowen-Pope D., Gov N. S., and Kachar B. (2008) Dynamic compartmentalization of protein tyrosine phosphatase receptor Q at the proximal end of stereocilia: implication of myosin VI-based transport. Cell Motil. Cytoskeleton 65, 528–538 [DOI] [PubMed] [Google Scholar]
- 14. Batters C., Arthur C. P., Lin A., Porter J., Geeves M. A., Milligan R. A., Molloy J. E., and Coluccio L. M. (2004) Myo1c is designed for the adaptation response in the inner ear. EMBO J. 23, 1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gillespie P. G., and Cyr J. L. (2004) Myosin-1c, the hair cell's adaptation motor. Annu. Rev. Physiol. 66, 521–545 [DOI] [PubMed] [Google Scholar]
- 16. Lee S. J., and Montell C. (2004) Light-dependent translocation of visual arrestin regulated by the NINAC myosin III. Neuron 43, 95–103 [DOI] [PubMed] [Google Scholar]
- 17. Porter J. A., Hicks J. L., Williams D. S., and Montell C. (1992) Differential localizations of and requirements for the two Drosophila ninaC kinase/myosins in photoreceptor cells. J. Cell Biol. 116, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Porter J. A., and Montell C. (1993) Distinct roles of the Drosophila ninaC kinase and myosin domains revealed by systematic mutagenesis. J. Cell Biol. 122, 601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wes P. D., Xu X. Z., Li H. S., Chien F., Doberstein S. K., and Montell C. (1999) Termination of phototransduction requires binding of the NINAC myosin III and the PDZ protein INAD. Nat. Neurosci. 2, 447–453 [DOI] [PubMed] [Google Scholar]
- 20. Dosé A. C., and Burnside B. (2000) Cloning and chromosomal localization of a human class III myosin. Genomics 67, 333–342 [DOI] [PubMed] [Google Scholar]
- 21. Dosé A. C., Hillman D. W., Wong C., Sohlberg L., Lin-Jones J., and Burnside B. (2003) Myo3A, one of two class III myosin genes expressed in vertebrate retina, is localized to the calycal processes of rod and cone photoreceptors and is expressed in the sacculus. Mol. Biol. Cell 14, 1058–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin-Jones J., Parker E., Wu M., Dosé A., and Burnside B. (2004) Myosin 3A transgene expression produces abnormal actin filament bundles in transgenic Xenopus laevis rod photoreceptors. J. Cell Sci. 117, 5825–5834 [DOI] [PubMed] [Google Scholar]
- 23. Sahly I., Dufour E., Schietroma C., Michel V., Bahloul A., Perfettini I., Pepermans E., Estivalet A., Carette D., Aghaie A., Ebermann I., Lelli A., Iribarne M., Hardelin J. P., Weil D., et al. (2012) Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 199, 381–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walsh T., Walsh V., Vreugde S., Hertzano R., Shahin H., Haika S., Lee M. K., Kanaan M., King M. C., and Avraham K. B. (2002) From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. U.S.A. 99, 7518–7523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grati M., Yan D., Raval M. H., Walsh T., Ma Q., Chakchouk I., Kannan-Sundhari A., Mittal R., Masmoudi S., Blanton S. H., Tekin M., King M. C., Yengo C. M., and Liu X. Z. (2016) Myo3a causes human dominant deafness and interacts with protocadherin 15-Cd2 isoform. Hum. Mutat. 37, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Merritt R. C., Manor U., Salles F. T., Grati M., Dose A. C., Unrath W. C., Quintero O. A., Yengo C. M., and Kachar B. (2012) Myosin IIIB uses an actin-binding motif in its espin-1 cargo to reach the tips of actin protrusions. Curr. Biol. 22, 320–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lelli A., Michel V., Boutet de Monvel J., Cortese M., Bosch-Grau M., Aghaie A., Perfettini I., Dupont T., Avan P., El-Amraoui A., and Petit C. (2016) Class III myosins shape the auditory hair bundles by limiting microvilli and stereocilia growth. J. Cell Biol. 212, 231–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dosé A. C., Ananthanarayanan S., Moore J. E., Burnside B., and Yengo C. M. (2007) Kinetic mechanism of human myosin IIIA. J. Biol. Chem. 282, 216–231 [DOI] [PubMed] [Google Scholar]
- 29. Dosé A. C., Ananthanarayanan S., Moore J. E., Corsa A. C., Burnside B., and Yengo C. M. (2008) The kinase domain alters the kinetic properties of the myosin IIIA motor. Biochemistry 47, 2485–2496 [DOI] [PubMed] [Google Scholar]
- 30. Quintero O. A., Moore J. E., Unrath W. C., Manor U., Salles F. T., Grati M., Kachar B., and Yengo C. M. (2010) Intermolecular autophosphorylation regulates myosin IIIa activity and localization in parallel actin bundles. J. Biol. Chem. 285, 35770–35782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quintero O. A., Unrath W. C., Stevens S. M. Jr, Manor U., Kachar B., and Yengo C. M. (2013) Myosin 3A kinase activity is regulated by phosphorylation of the kinase domain activation loop. J. Biol. Chem. 288, 37126–37137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ebrahim S., Avenarius M. R., Grati M., Krey J. F., Windsor A. M., Sousa A. D., Ballesteros A., Cui R., Millis B. A., Salles F. T., Baird M. A., Davidson M. W., Jones S. M., Choi D., Dong L., et al. (2016) Stereocilia-staircase spacing is influenced by myosin III motors and their cargos espin-1 and espin-like. Nat. Commun. 7, 10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sekerková G., Richter C. P., and Bartles J. R. (2011) Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet. 7, e1002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu H., Li J., Raval M. H., Yao N., Deng X., Lu Q., Nie S., Feng W., Wan J., Yengo C. M., Liu W., and Zhang M. (2016) Myosin III-mediated cross-linking and stimulation of actin bundling activity of Espin. eLife 5, e12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mecklenburg K. L., Freed S. A., Raval M., Quintero O. A., Yengo C. M., and O'Tousa J. E. (2015) Invertebrate and vertebrate class III myosins interact with MORN repeat-containing adaptor proteins. PLoS ONE 10, e0122502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Les Erickson F., Corsa A. C., Dose A. C., and Burnside B. (2003) Localization of a class III myosin to filopodia tips in transfected HeLa cells requires an actin-binding site in its tail domain. Mol. Biol. Cell 14, 4173–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Manor U., Grati M., Yengo C. M., Kachar B., and Gov N. S. (2012) Competition and compensation: dissecting the biophysical and functional differences between the class 3 myosin paralogs, myosins 3a and 3b. Bioarchitecture 2, 171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berg J. S., and Cheney R. E. (2002) Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 4, 246–250 [DOI] [PubMed] [Google Scholar]
- 39. Tokuo H., Mabuchi K., and Ikebe M. (2007) The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J. Cell Biol. 179, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bohil A. B., Robertson B. W., and Cheney R. E. (2006) Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. U.S.A. 103, 12411–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sasaki N., Shimada T., and Sutoh K. (1998) Mutational analysis of the switch II loop of Dictyostelium myosin II. J. Biol. Chem. 273, 20334–20340 [DOI] [PubMed] [Google Scholar]
- 42. Trivedi D. V., David C., Jacobs D. J., and Yengo C. M. (2012) Switch II mutants reveal coupling between the nucleotide- and actin-binding regions in myosin V. Biophys. J. 102, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yengo C. M., De la Cruz E. M., Safer D., Ostap E. M., and Sweeney H. L. (2002) Kinetic characterization of the weak binding states of myosin V. Biochemistry 41, 8508–8517 [DOI] [PubMed] [Google Scholar]
- 44. Baas A. F., Kuipers J., van der Wel N. N., Batlle E., Koerten H. K., Peters P. J., and Clevers H. C. (2004) Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 116, 457–466 [DOI] [PubMed] [Google Scholar]
- 45. Bretscher A., and Weber K. (1980) Villin is a major protein of the microvillus cytoskeleton which binds both G and F actin in a calcium-dependent manner. Cell 20, 839–847 [DOI] [PubMed] [Google Scholar]
- 46. Courson D. S., and Cheney R. E. (2015) Myosin-X and disease. Exp. Cell Res. 334, 10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Revenu C., Ubelmann F., Hurbain I., El-Marjou F., Dingli F., Loew D., Delacour D., Gilet J., Brot-Laroche E., Rivero F., Louvard D., and Robine S. (2012) A new role for the architecture of microvillar actin bundles in apical retention of membrane proteins. Mol. Biol. Cell 23, 324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tokuo H., and Ikebe M. (2004) Myosin X transports Mena/VASP to the tip of filopodia. Biochem. Biophys. Res. Commun. 319, 214–220 [DOI] [PubMed] [Google Scholar]
- 49. Svitkina T. M., Bulanova E. A., Chaga O. Y., Vignjevic D. M., Kojima S., Vasiliev J. M., and Borisy G. G. (2003) Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 160, 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gupton S. L., and Gertler F. B. (2007) Filopodia: the fingers that do the walking. Sci STKE 2007, re5. [DOI] [PubMed] [Google Scholar]
- 51. Tilney L. G., Connelly P. S., and Guild G. M. (2004) Microvilli appear to represent the first step in actin bundle formation in Drosophila bristles. J. Cell Sci. 117, 3531–3538 [DOI] [PubMed] [Google Scholar]
- 52. Shifrin D. A. Jr., Crawley S. W., Grega-Larson N. E., and Tyska M. J. (2014) Dynamics of brush border remodeling induced by enteropathogenic E. coli. Gut Microbes 5, 504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mallavarapu A., and Mitchison T. (1999) Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J. Cell Biol. 146, 1097–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Faix J., and Rottner K. (2006) The making of filopodia. Curr. Opin. Cell Biol. 18, 18–25 [DOI] [PubMed] [Google Scholar]
- 55. Waterman-Storer C. M., Desai A., Bulinski J. C., and Salmon E. D. (1998) Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr. Biol. 8, 1227–1230 [DOI] [PubMed] [Google Scholar]
- 56. Pollard T. D., and Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 57. Mogilner A., and Rubinstein B. (2005) The physics of filopodial protrusion. Biophys. J. 89, 782–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vignjevic D., Yarar D., Welch M. D., Peloquin J., Svitkina T., and Borisy G. G. (2003) Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 160, 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walsh V. L., Raviv D., Dror A. A., Shahin H., Walsh T., Kanaan M. N., Avraham K. B., and King M. C. (2011) A mouse model for human hearing loss DFNB30 due to loss of function of myosin IIIA. Mamm. Genome 22, 170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Greenman C., Stephens P., Smith R., Dalgliesh G. L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E. E., Avis T., et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rose G. G., Pomerat C. M., Shindler T. O., and Trunnell J. B. (1958) A cellophane-strip technique for culturing tissue in multipurpose culture chambers. J. Biophys. Biochem. Cytol. 4, 761–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grega-Larson N. E., Crawley S. W., Erwin A. L., and Tyska M. J. (2015) Cordon bleu promotes the assembly of brush border microvilli. Mol. Biol. Cell 26, 3803–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kron S. J., Toyoshima Y. Y., Uyeda T. Q., and Spudich J. A. (1991) Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 196, 399–416 [DOI] [PubMed] [Google Scholar]
- 64. Yengo C. M., Takagi Y., and Sellers J. R. (2012) Temperature dependent measurements reveal similarities between muscle and non-muscle myosin motility. J. Muscle Res. Cell Motil. 33, 385–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Trivedi D. V., Muretta J. M., Swenson A. M., Thomas D. D., and Yengo C. M. (2013) Magnesium impacts myosin V motor activity by altering key conformational changes in the mechanochemical cycle. Biochemistry 52, 4710–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meijering E., Dzyubachyk O., and Smal I. (2012) Methods for cell and particle tracking. Methods Enzymol. 504, 183–200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.