

Abstract
Metformin is a first line oral anti-diabetic agent that has been used clinically to treat patients with type 2 diabetes for over 60 years. Due to its efficacy in therapy and affordable price, metformin is taken by over 150 million people each year. Metformin improves hyperglycemia mainly through the suppression of hepatic gluconeogenesis along with the improvement of insulin signaling. However, its mechanism of action remains partially understood and controversial, especially in regards to the role of AMPK in metformin’s action and the mechanism of AMPK activation. In this review, we will discuss recent advances in the understanding of metformin’s suppression of hepatic glucose production and the mechanism related to the improvement of insulin signaling.
1. Metformin, a time-proven effective agent for the treatment of patients with type 2 diabetes (T2D)
In the medieval era, the Gallega officinalis plant was used in Europe as a folk medicine to treat diabetes. This plant was found to be rich in guanidine in the late 1800s, and subsequent animal study in 1918 showed that guanidine has a blood glucose-lowering effect (Watanabe 1918). However, guanidine proved to be too toxic for clinical use. In 1929, the dimethylbiguanide, including phenformin and metformin, was synthesized. These compounds preserve the glucose-lowering effect of their parent compound with reduced toxicity. Phenformin is more potent than metformin in improving hyperglycemia, but with the high occurrence of the serious side effect- lactic acidosis led to the withdrawal of phenformin from the market in the 1970s.
After over 60 years of clinical use for the treatment of T2D, metformin has proven to be safe along with affordable, making it now the most commonly prescribed oral anti-diabetic agent worldwide, taken by over 150 million people each year. The guidelines for the treatment of T2D, published by the American Diabetes Association and the European Association for the Study of Diabetes in 2012, recommended metformin as the initial drug for T2D treatment (Inzucchi et al. 2012). Moreover, many studies have shown a reduction in cancer incidence in patients with type 2 diabetes treated with metformin (Evans et al. 2005, Landman et al. 2010).
2. Metformin alleviates hyperglycemia in T2D mainly through the direct suppression of hepatic glucose production
T2D accounts for more than 90% of diabetes cases (Successes and Opportunities for Population-Based Prevention and Control At A Glance 2011), and increased hepatic glucose production is the major cause of fasting hyperglycemia in these patients (Magnusson et al. 1992, Saltiel & and Kahn 2001, Wajngot et al. 2001). It is now clear that metformin improves hyperglycemia mainly through the suppression of gluconeogenesis in the liver. In a human study, treatment of patients with type 2 diabetes with metformin led to a decrease in hepatic glucose output via the suppression of gluconeogenesis by 37% (Stumvoll et al. 1995). In another human study, patients with diabetes exhibited a two-fold increase in the rate of gluconeogenesis, but metformin administration led to a decrease in gluconeogenesis by 33% (Hundal et al. 2000). In animal studies, metformin reduced hepatic glucose production by over 60% in a euglycemic-clamp study (Takashima et al. 2010) and led to 50% suppression of endogenous glucose production in high-fat-diet-fed rats (Song et al. 2001). Furthermore, we have found that metformin suppressed cAMP-stimulated glucose production by 35% in primary hepatocytes (Cao et al. 2014). These in vivo and in vitro data suggest that metformin acts mainly through the suppression of hepatic gluconeogenesis.
3. Metformin suppresses hepatic glucose production through AMPK activation
3.1 AMPK—a master regulator of cellular energy homeostasis
AMP-activated protein kinase, AMPK, was first named and reported in 1989 for its AMP-activation property and its role in regulating three key enzymes in lipid metabolism (Brown et al. 1975, Hardie et al. 1989). Numerous studies over the past decades have confirmed the function of AMPK as a principal energy sensor of the cell and its role in regulating cellular and whole-body energy balance by sensing and responding to changes in the AMP/ADP concentration relative to ATP (Hardie et al. 2012). AMPK is activated when cellular energy levels are low, prompting a switch from ATP-consuming anabolic pathways to ATP-producing catabolic pathways by stimulating glucose uptake and utilization and fatty acid oxidation together with suppression of hepatic glucose production.
Phylogenetically, AMPK is a highly conserved serine/threonine kinase and presents in virtually all eukaryotes. Functional AMPK is a heterotrimeric complex comprised of an α catalytic subunit and regulatory βγ non-catalytic subunits, with each subunit having several isoforms (α1, α2, β1, β2, γ1, γ2, γ3, encoded by separated genes) (Hardie et al. 2012). The catalytic α-subunit consists of an N-terminal protein kinase domain (conventional Ser/Thr kinase domain), followed by an auto-inhibitory domain (AID), a linker domain, and a C-terminal regulatory domain (α-CTD) (Chen et al. 2009, Hardie et al. 2012). The β-subunits contain a carbohydrate-binding module (CBM) and a C-terminal domain (β-CTD). The CBM domain allows AMPK to act as a glycogen sensor, while the β-CTD works as a scaffold, interacting with α-CTD and the γ-subunit, thus forming the core of the heterotrimeric complex. The γ-subunit has a tandem of four cystathionine-β-synthase domains, functioning as the adenine nucleotide-binding region (Hardie et al. 2012). When the cellular energy status changes, an increase in the AMP/ATP or ADP/ATP ratio leads to AMP or ADP binding to the γ-subunit. This results in an allosteric change in AMPK and augments α-subunit phosphorylation at T172 by an upstream kinase or prevents dephosphorylation by a protein phosphatase (Corton et al. 1994, Xiao et al. 2011). Phosphorylation of the α-subunit at T172 leads to an increase in AMPK enzymatic activity of over several-hundred-fold; in comparison, AMP only causes a 2- to 5-fold allosteric activation of AMPK, which also depends on the nature of the isoforms present in the AMPK complex (Xiao et al. 2011).
In mammals, the upstream kinases for AMPK α-subunit phosphorylation at T172 are the LKB1–STRAD–MO25 complex (Lizcano et al. 2004), the Ca2+/calmodulin-activated protein kinases, especially CaMKKβ (also termed as CaMKK2) (Hawley et al. 2005), or the transforming growth factor-β activated protein kinase-1 (TAK1) (Momcilovic et al. 2006). The LKB1/AMPK pathway plays a major role in metabolic regulation.
3.2 The role of AMPK in mediating metformin action
After oral administration, metformin is absorbed into enterocytes and then delivered directly to the liver through the portal vein; plasma concentrations in the portal vein are between 40–70 μM in animals after a therapeutic dose (Wilcock & Bailey 1994, He & Wondisford 2015). Over a decade ago, Zhou et al. reported that metformin at concentrations found in the portal vein (≤ 80 μM) activates AMP-activated kinase (AMPK) by increasing α-subunit phosphorylation at T172 (Zhou et al. 2001). A subsequent study reported that hepatic knockout of LKB1, an upstream kinase for AMPK α-subunit phosphorylation at T172, abrogated the effect of metformin on the alleviation of hyperglycemia in mice fed a high-fat diet (Shaw et al. 2005). In agreement with this result, our study showed that depletion of AMPK catalytic α-subunits abolished the suppression of cAMP- or glucagon-stimulated glucose production by metformin at concentrations typically found in the portal vein (≤ 80 μM) (Cao et al. 2014). Furthermore, metformin-mediated activation of AMPK increases CBP phosphorylation at S436 via atypical protein kinase ι/λ, resulting in the disassembly of the CREB co-activator complex, inhibition of gluconeogenic gene expression, and a reduction in glucose production (He et al. 2009). A growing body of evidence supports the notion that metformin suppresses mRNA or protein levels of rate-limited gluconeogenic gene expression in the liver of mice and in primary hepatocytes (Cheng et al. 2006, Heishi et al. 2008, He et al. 2009, Martin-Montalvo et al. 2013). Corroborating our findings, a P300G422S knock-in mouse model, which harbors a reconstituted metformin phosphorylation site found in CBP, exhibits exaggerated hypoglycemia in a metformin tolerance test (He et al. 2014). The above evidence supports a mechanism for metformin suppression of hepatic glucose production via activation of the LKB-AMPK pathway.
3.3 Activation of AMPK by low metformin concentrations (≤ 80 μM) via the promotion of the formation of the AMPK αβγ heterotrimeric complex
Metformin has no direct effect on AMPKα enzymatic activity (Meng et al. 2015). But, metformin is able to increase AMPK enzymatic activity by stimulating the phosphorylation of the α subunit at T172 (Zhou et al. 2001). Considering that the upstream kinase LKB1 is a constitutively active kinase (Lizcano et al. 2004), we asked whether metformin could affect the formation of functional AMPK heterotrimeric complex, consisting of an α catalytic subunit and βγ non-catalytic subunits, to increase the phosphorylation of the α subunit at T172 by upstream kinase (Davies et al. 1994, Mitchelhill et al. 1994, Stapleton et al. 1996). However, previous studies showed that AMPK α1β1γ1 or α2β1γ1 subunits exist in a 1:1:1 molar ratio (Davies et al. 1994, Stapleton et al. 1996), and it is generally thought that the heterotrimeric complex assembles immediately following translation in order to keep the subunits stable; free subunits may not be stable and will therefore degrade rapidly (Neumann et al. 2003). Interestingly, we were able to express AMPK-α1,-β1,-γ1 individually in both primary hepatocytes and Hepa1-6 cells (Cao et al. 2014, Meng et al. 2015). Purified AMPK subunits did not associate with other subunits in a coomassie stained gel, indicating that free AMPK subunits can exist and are stable. Furthermore, we examined the protein levels of the AMPK subunits in different mouse tissues and found that the protein levels of different AMPK subunits vary from tissue to tissue (Meng et al 2015), suggesting that cellular protein levels of AMPK subunits are unlikely to exist in a 1:1:1 molar ratio (Davies et al. 1994, Stapleton et al. 1996).
To assess the cellular ratio of endogenous AMPK subunits, we generated and purified FLAG-tagged -α1,-β1,-γ1 since they account for over 90% of AMPK activity in hepatocytes (Davies et al. 1994, Stapleton et al. 1996). We first determined the amounts of purified FLAG-tagged -α1,-β1, and -γ1 subunits that were equivalent to the corresponding protein levels in the same amount of cellular lysates. Subsequently, these amounts of purified FLAG-tagged -α1,-β1, and -γ1 subunits were used and immunoblotted with anti-FLAG antibody. Our data reveal that the -α1,-β1, and -γ1 subunits exist approximately in a 1:5:0.6 molar ratio in Hepa1-6 cells and in a 1:31:0.5 molar ratio in mouse liver (Meng et al 2015). Our data suggest that the β1 subunit is much more abundant than α1and γ1 subunits in both Hepa1-6 cells and liver.
The unequal expression of endogenous α1, β1, and γ1 protein levels suggest that not all of the α1, β1, and γ1 are in the heterotrimeric complex. Therefore, we reasoned that metformin might be able to affect the assembly of the AMPK heterotrimeric complex. In immunoprecipitation studies, we found that both α and γ subunits were co-immunoprecipitated when using β1/2 antibodies in metformin-treated Hepa1-6 cells. In contrast, in untreated Hepa1-6 cells, the γ1 but not the α subunit co-immunoprecipitated with β1 subunit. These data suggest that β1 and γ1 subunits are preassembled, and metformin promotes the association of the α1 subunit with β1 and γ1 complex to form the functional heterotrimeric complex (Meng et al 2015). In agreement with these results, we found that mice fed a high-fat diet were given metformin in the drinking water exhibited an increase in α subunit phosphorylation at T172 as well as the association of the α subunit with the β subunit in the liver (Meng et al 2015). Furthermore, low concentrations of metformin also increased the formation of AMPKαβγ heterotrimeric complex in in vitro assembly assay. Interestingly, neither AMP/ADP, nor phenformin affected the heterotrimeric assembly, suggesting that they activate AMPK through different mechanisms.
As we found that metformin was able to promote the formation of the AMPKαβγ heterotrimeric complex, we further tested whether the formation of the AMPKαβγ heterotrimeric complex had any effect on the phosphorylation of the α1 subunit at T172 by an upstream kinase. In in vitro phosphorylation assays, the phosphorylation of the α1 subunit at T172 by LKB1 is 10-fold greater in the presence of β1and γ1 subunits than α1 subunit alone. The presence of the γ1 subunit is essential for metformin’s effect on the phosphorylation of the α1 subunit at T172 by LKB1, as metformin’s effect was lost when the γ1 subunit was absent in the reaction. A previous study showed that cells expressing a mutant AMPKγ subunit (R531G) were resistant to metformin activation of AMPK (Hawley et al. 2010), suggesting that R531 in the γ subunit is critical for metformin binding. Furthermore, low metformin concentrations (≤100 μM) prevented the dephosphorylation of the catalytic α1 subunit by PP2C in the presence of β1and γ1 subunits, and this effect was lost in the absence of β1and γ1 subunits. Taken together, our data support that metformin binds directly to AMPK subunits, resulting in increased assembly of the AMPK αβγ heterotrimeric complex as well as allosteric changes, making it more accessible for upstream kinases, such as LKB1, to phosphorylate the α1 subunit at T172 (Figure 1). The formation of the AMPK αβγ heterotrimeric complex also hinders the dephosphorylation of the α1 subunit at T172 by protein phosphatase (Meng et al 2015). Overall, the collective effects of metformin action lead to the activation of AMPK.
Figure 1. Metformin Suppression of Hepatic Glucose Production by Promoting the Formation of the AMPKαβγ Heterotrimeric Complex.
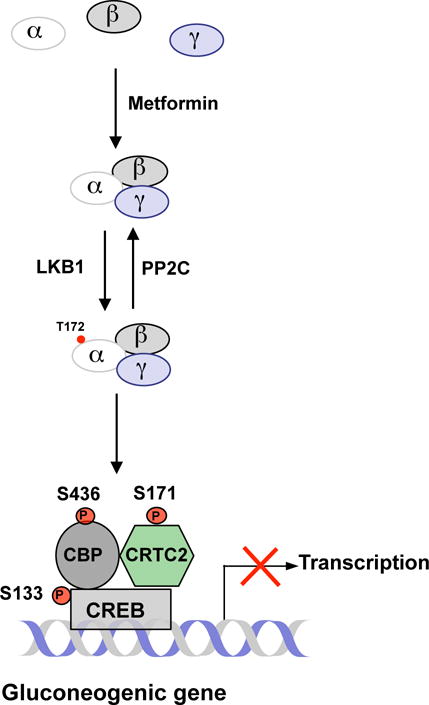
Metformin increases the phosphorylation of AMPK by promoting the formation of the AMPK heterotrimeric complex. Activated AMPK leads to the phosphorylation of CREB co-activators and the inhibition of gluconeogenic gene expression.
4. Metformin suppresses hepatic glucose production through the inhibition of mitochondrial respiratory chain complex 1 and AMP deaminase
Mitochondria respiratory-chain complex 1, also termed NADH:ubiquinone oxidoreductase, is the first membrane-embedded supramolecular complex in the electron transport chain. Complex 1 catalyzes the transfer of a hydride ion from NADH and a proton from the matrix to ubiquinone. Ubiquinol (QH2), a lipid-soluble carrier ubiquinone, can diffuse quickly in the inner mitochondrial membrane from Complex 1 to Complex 3. Complex 1 also transfers four protons from the matrix to the intermembrane space to generate a high electron gradient, which is the energy source in ATP synthesis. In 1955, it was documented that guanidine can inhibit the oxygen uptake of mitochondria; this was the first study that showed that guanidine may have an effect on the mitochondrial respiratory chain (Hollunger 1955). In 2000, two studies showed that metformin’s action is through the disruption of mitochondrial complex I (Owen et al. 2000, El-Mir et al. 2000), leading to a decrease in ATP production. More recently, Foretz et al. suggested that the change in the AMP/ATP or ADP/ATP ratio after metformin’s inhibition of the mitochondrial complex is primarily responsible for metformin’s effect and that this occurs through an AMPK independent pathway (Foretz et al. 2010). It was also shown that metformin increases AMP levels via the inhibition of AMP deaminase (Figure 2) (Ouyang et al. 2011). In addition, elevated AMP levels after metformin treatment led to the inhibition of adenylate cyclase and a decrease in cAMP levels, resulting in the suppression of the cAMP-PKA pathway, and the inhibition of gluconeogenesis was proposed (Miller et al. 2013). We also found that metformin is able to decrease cellular ATP levels and increase the ratios of AMP/ATP or ADP/ATP (Cao et al. 2014). Since gluconeogenesis is an energy-demanding process in which synthesis of one molecule of glucose from lactate or pyruvate requires 4 molecules of ATP and 2 molecules of GTP, reduction of cellular ATP levels by metformin will lead to the suppression of hepatic gluconeogenesis (Figure 2).
Figure 2. Direct Suppression of Hepatic Glucose Production by Metformin through the Inhibition of Mitochondrial Respiratory Chain Complex 1, AMP Deaminase, and Mitochondrial Glycerol 3-Phosphate Dehydrogenase.

Inhibition of mitochondrial complex 1 also leads to an increase in AMP levels or the AMP/ATP ratio, as does the inhibition of AMP deaminase, resulting in the activation of AMPK. However, high metformin concentrations are needed to inhibit the mitochondrial complex and AMP deaminase. Metformin inhibition of mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH) will increase NADH levels in the cytoplasm and suppress the conversion of lactate from pyruvate. This mechanism of metformin action is important for patients with high levels of serum lactate.
However, metformin’s inhibition of mitochondrial complex I or AMP deaminase activity occurred at concentrations ≥ 5 mM in hepatocytes (Owen et al. 2000, El-Mir et al. 2000, Ouyang et al. 2011). The concentrations of metformin required to affect the ratios of AMP/ATP or ADP/ATP are ≥ 250 μM (Foretz et al. 2010, Cao et al. 2014). These metformin concentrations are unreachable in the portal vein after a therapeutic dose (Wilcock & Bailey 1994, He & Wondisford 2015). Interestingly, using typical pharmacologic dosing in mice, metformin administration did not decrease, but significantly increased mitochondrial complex I activity in the liver (Martin-Montalvo et al. 2013). We also found that a low metformin concentration (80 μM) significantly increased ATP levels in primary hepatocytes treated with cAMP. This may be due to the inhibition of gluconeogenesis and activation of fatty acid oxidation (Cao et al. 2014), but metformin still significantly activated AMPK (Zhou et al. 2001, Cao et al. 2014) and the suppression of glucose production relative to cAMP treatment alone in primary hepatocytes (Cao et al. 2014). These data indicate that inhibition of mitochondrial complex I activity is unlikely at pharmacologic metformin concentrations found in the portal vein, and that the inhibition of mitochondrial complex I is unnecessary for metformin’s suppression of gluconeogenesis. Since very high metformin concentrations are required to inhibit mitochondrial complex I and perhaps, the AMP deaminase activity in hepatocytes as well (El-Mir et al. 2000, Ouyang et al. 2011), we suggest the effects of metformin seen at high concentrations might be either an off-target phenomenona or signals of cell toxicity, and is therefore clinically irrelevant.
5. Metformin suppresses hepatic glucose production through the inhibition of mitochondrial glycerol 3-phosphate dehydrogenase and through affecting gut-brain communication
It has been reported that metformin suppresses hepatic glucose production by inhibiting the enzymatic activity of mitochondrial glycerol 3-phosphate dehydrogenase (mG3PDH), which blocks the transport of NADH from the cytoplasm into mitochondria (Madiraju et al. 2014). This mechanism of metformin action may be important for the suppression of hepatic glucose production in patients with diabetes who have high levels of serum lactate, as the conversion of lactate to pyruvate leads to the generation of NADH. The inhibition of mG3PDH will result in an increase in NADH levels and a decrease in NAD+ levels, resulting in no NAD+ available for converting lactate to pyruvate. Subsequently, this action will halt glucose production from lactate (Figure 2, right panel). Interestingly, AMPK negatively regulates the activity of yeast glycerol 3-phosphate dehydrogenase 2 (an analog of mammalian mG3PDH) by phosphorylating S72 (Lee et al. 2012). However, it remains to be determined whether AMPK can affect mG3PDH enzymatic activity by phosphorylation.
Moreover, intestinal nutrients can regulate glucose homeostasis in a manner involving the gut-brain-liver axis (Wang et al. 2008). Affecting gut-brain communication will lead to a change in hepatic glucose production. Duca et al. tested whether metformin has any effect on the gut-brain-liver axis and found that metformin suppression of hepatic glucose production occurs in part through the gut-brain-liver axis (Duca et al. 2015). Gut-brain communication is critical for acute metformin action, as metformin suppression of hepatic glucose production was negated in rats with blockade of the gut-brain-liver axis. The acute metformin effect is AMPK-dependent, involving the activation of PKA by GLP-1 in duodenal enterocytes (Duca et al. 2015). However, the importance of the gut-brain-liver axis in chronic metformin action remains to be determined.
6. Metformin alleviates hyperglycemia in T2D through the improvement of insulin signaling—an indirect pathway of metformin action
Even though metformin improves hyperglycemia mainly through the suppression of gluconeogenesis in the liver, it has also been found to be able to increase insulin sensitivity. This effect would improve insulin-mediated suppression of hepatic glucose production and enhance insulin-stimulated glucose disposal in skeletal muscle (Stumvoll et al. 1995, Inzucchi et al. 1998, Hundal et al. 2000). The potential mechanism of a metformin-mediated increase in insulin sensitivity will be discussed.
6.1 Metformin alters gut microbiota and maintains the integrity of the intestinal barrier
Obese individuals have a different composition of gut microbiota, and in a rodent model, obesity is associated with altered gut microbiota (Ley et al. 2005, 2006, Ridaura et al. 2013). High-fat diet feeding leads to a similar change in gut microbiota composition and to a 2–3 fold increase in blood LPS (endotoxin) levels in mice (Cani et al. 2007, 2008). In addition, a single oral administration of lipids in human or mice acutely increased serum LPS concentration (Cani et al. 2007, Laugerette et al. 2011). Germ-free mice resisted high-fat-diet induced insulin resistance (Backhed et al. 2007). A low dose LPS infusion mimicked high-fat-diet feeding and induced fasting hyperglycemia and hepatic insulin resistance in a mouse model (Ley et al. 2005). This line of evidence suggests that LPS from the gut is a critical etiologic factor for the development of insulin resistance. Increased LPS leakage from the gut has emerged as one of the most appealing mechanisms to explain the connections between changes in the intestinal microbiota and insulin resistance (Cani et al. 2007).
Recently, metformin has been shown to modulate the composition of gut microbiota in high-fat-diet fed and diabetic mice and decreases serum LPS levels (Shin et al. 2014). In addition, metformin can activate mucosal AMPK in the intestine (Duca et al. 2015). An earlier study showed that high-fat-diet feeding increases the intestinal permeability of LPS (Cani et al. 2007, 2008, Creely et al. 2007, Oliveira et al. 2011), and AMPK plays a critical role in maintaining intestinal barrier integrity (Peng et al. 2009, Elamin et al 2013). Therefore, metformin-mediated activation of AMPK should decrease LPS leakage from the gut (Figure 3).
Figure 3. Metformin Improves Insulin Signaling in the Liver.
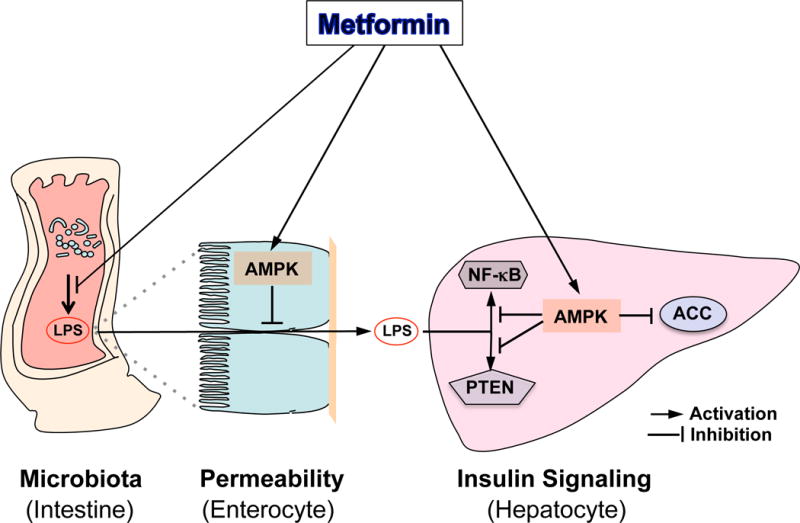
Metformin can alter the microbiota in the intestine, resulting a reduction in LPS production and translocation across the intestinal barrier. Activation of AMPK by metformin also blocks LPS-mediated activation of the NF-κB signaling pathway and PTEN induction.
6.2 Metformin blocks the activation of the NF-κB pathway and affects the expression of PTEN
The liver is the major organ responsible for LPS clearance. After being delivered to the liver, LPS binds to the multireceptor complex constituted of CD14, TLR4 and MD2 within the lipid rafts of hepatocytes and triggers the signal activation of IRAK, and subsequently, its downstream target, the NF-κB pathway (Scott & Billiar 2008). NF-κB was firstly drew attention in the development of insulin resistance and type 2 diabetes after the landmark finding that the anti-inflammatory agent aspirin inhibits NF-κB. Activation of NF-κB leads to insulin resistance (Arkan et al. 2005), and inactivation of this pathway protects against the development of insulin resistance, as evident in mice with NF-κB (p50) knockout (Gao et al. 2009). This mouse model exhibited increased insulin sensitivity in the liver and produced significantly less glucose in a hyperinsulinemic-euglycemic clamp. Furthermore, inhibition of the NF-κB pathway improved insulin resistance in db/db mice (Kim et al. 2013). Of particular interest is the finding that the activation of AMPK by AICAR inhibited the NF-κB pathway (Cacicedo et al. 2004); aligning with this, metformin-mediated AMPK activation attenuates the activation of the NF-κB pathway (Huang et al. 2009, Hattori et al. 2006). Therefore, the inhibition of the NF-κB pathway by metformin-mediated AMPK activation would lead to an improvement in hepatic insulin signaling (Figure 3).
Phosphatase and tensin homolog (PTEN), a tumor suppressor, can reverse PI3K (Phosphatidylinositol-4, 5-bisphosphate 3-kinase) function by dephosphorylating the PI(3,4,5)P3 to PI(4,5)P2, therefore, suppressing the PI3K-PKB/AKT pathway (Myers et al. 1998, Stiles et al. 2004). Intriguingly, LPS can induce the expression of PTEN (Okamura et al. 2007), and metformin can suppress PTEN expression in pre-adipocyte 3T3 cells (Okamura et al. 2007, Lee et al. 2011). This metformin action is AMPK dependent, as the metformin effect is lost in cells treated with Compound C (an AMPK inhibitor) or with AMPK depletion by shRNA. This report showed that PTEN is a downstream regulator of AMPK and that the AMPK-PTEN pathway plays a critical role in regulating the inflammatory response (Figure 3). However, further studies will be needed to demonstrate conclusively that how metformin’s effect on PTEN occurs in the liver as well as muscle and determine how activated AMPK suppresses PTEN expression.
7. Perspective
Since the maximum metformin dose prescribed to patients with diabetes is ~2.5 g/day, this high therapeutic dose might affect multiple targets. As an oral agent, metformin can change the composition of gut microbiota (Shin et al. 2014) and activate mucosal AMPK (Duca et al. 2015) that will maintain intestinal barrier integrity (Peng et al. 2009, Elamin et al 2013). Together, these metformin effects will decrease LPS levels in the circulation as well as hepatic LPS levels, since LPS is delivered directly to the liver through the portal vein. In addition, the activation of duodenal AMPK by acute metformin treatment will stimulate the gut-brain-liver axis to regulate hepatic glucose production (Duca et al. 2015). After being delivered to the liver from the intestines, metformin can inhibit gluconeogenesis through different mechanisms. First, it can activate hepatic AMPK, which will then inhibit gluconeogenesis by phosphorylating the critical co-activators CBP and CRTC2 (He et al. 2009) (Figure 1). Second, high metformin concentrations can suppress hepatic gluconeogenesis by inhibiting mitochondrial respiratory chain complex 1 to reduce cellular ATP levels and increase the AMP/ATP ratio (Owen et al. 2000, El-Mir et al. 2000, Foretz et al. 2010) due to the high energy demand of gluconeogenesis. We found that depletion of AMPKα1/2 decreased the suppression of glucose production in primary hepatocytes at high metformin concentrations (Cao et al. 2014). Thus, the increased ratio of AMP/ATP or ADP/ATP should activate AMPK (Xiao et al. 2011, Oakhill et al. 2011, Gowans et al. 2013) to augment the inhibition of gluconeogenesis. Third, inhibition of mG3PDH enzymatic activity by metformin will affect transport of NADH from the cytoplasm into mitochondria (Madiraju et al. 2014), suppressing gluconeogenesis process from lactate.
Reducing LPS leakage from the gut and blocking the activation of the NF-κB pathway, in concert with down-regulating the expression of PTEN, will result in an increase in insulin sensitivity. Moreover, metformin administration will activate AMPK to increase fatty acid oxidation by phosphorylating acetyl-CoA carboxylase and consequently improve insulin sensitivity in the liver (Fullerton et al. 2013). Collectively, suppression of gluconeogenesis by metformin’s direct effects and the indirect improvement of insulin signaling in the liver will lead to the amelioration of hyperglycemia in patients with type 2 diabetes. Even though our studies demonstrate that suppression of gluconeogenesis by low metformin concentrations is through the activation of AMPK by promoting the formation of the AMPKαβγ heterotrimeric complex in both in vivo and in vitro assays (Cao et al. 2014, Meng et al. 2015), and the γ subunit plays a critical role in the phosphorylation of the α subunit at T172 by upstream kinase LKB1. However, the exact metformin binding site(s) on AMPK subunits and the detailed mechanism leading to the improvement of insulin signaling by metformin still remain unknown and need to be unraveled in future studies.
Acknowledgments
Funding
This work was supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases R00DK085142 and R01DK107641.
Footnotes
Authors have no conflict of interest to declare.
References
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Brunschede GY, Goldstein JL. Inactivation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in vitro. An adenine nucleotide-dependent reaction catalyzed by a factor in human fibroblasts. J Biol Chem. 1975;250:2502–2509. [PubMed] [Google Scholar]
- Cacicedo JM, Yagihashi N, Keaney JF, Rudermann NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappa B transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–1209. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Cao J, Meng SM, Chang E, Beckwith-Fickas K, Xiong LS, Cole RN, Radovick S, Wondisford FE, He L. Low Concentrations of Metformin Suppress Glucose Production in Hepatocytes through AMP-activated Protein Kinase (AMPK) Journal of Biological Chemistry. 2014;289:20435–20446. doi: 10.1074/jbc.M114.567271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Jiao ZH, Zheng LS, Zhang YY, Xie ST, Wang ZX, Wu JW. Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature. 2009;459:1146–U1139. doi: 10.1038/nature08075. [DOI] [PubMed] [Google Scholar]
- Cheng JT, Huang CC, Liu IM, Tzeng TF, Chang CJ. Novel mechanism for plasma glucose-lowering action of metformin in streptozotocin-induced diabetic rats. Diabetes. 2006;55:819–825. doi: 10.2337/diabetes.55.03.06.db05-0934. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hardie DG. Role of the Amp-Activated Protein-Kinase in the Cellular Stress-Response. Current Biology. 1994;4:315–324. doi: 10.1016/s0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- Creely SJ, McTernan PG, Kusminski CM, Fisher fM, Da Silva NF, Khanolkar M, Evans M, Harte AL, Kumar S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292:E740–747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- Davies SP, Hawley SA, Woods A, Carling D, Haystead TAJ, Hardie DG. Purification of the Amp-Activated Protein-Kinase on Atp-Gamma-Sepharose and Analysis of Its Subunit Structure. European Journal of Biochemistry. 1994;223:351–357. doi: 10.1111/j.1432-1033.1994.tb19001.x. [DOI] [PubMed] [Google Scholar]
- Duca FA, Cote CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, Lam TK. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nature Medicine. 2015;21:506–511. doi: 10.1038/nm.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elamin EE, Masclee AA, Dekker J, Pieters HJ, Jonkers DM. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J Nutr. 2013;143:1872–1881. doi: 10.3945/jn.113.179549. [DOI] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. Journal of Biological Chemistry. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Br Med J. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. Journal of Clinical Investigation. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature Medicine. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZG, Yin J, Zhang J, He Q, McGuinness OP, Ye JP. Inactivation of NF-kappa B p50 Leads to Insulin Sensitization in Liver through Post-translational Inhibition of p70S6K. Journal of Biological Chemistry. 2009;284:18368–18376. doi: 10.1074/jbc.M109.007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP Is a True Physiological Regulator of AMP-Activated Protein Kinase by Both Allosteric Activation and Enhancing Net Phosphorylation. Cell Metab. 2013;18:556–566. doi: 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Sim ATR. The Amp-Activated Protein-Kinase – a Multisubstrate Regulator of Lipid-Metabolism. Trends in Biochemical Sciences. 1989;14:20–23. [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappa B activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, et al. Use of Cells Expressing gamma Subunit Variants to Identify Diverse Mechanisms of AMPK Activation. Cell Metab. 2010;11:554–565. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Meng SM, Germain-Lee EL, Radovick S, Wondisford FE. Potential biomarker of metformin action. Journal of Endocrinology. 2014;221:363–369. doi: 10.1530/JOE-14-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Sabet A, Djedjos S, Miller R, Sun XJ, Hussain MA, Radovick S, Wondisford FE. Metformin and Insulin Suppress Hepatic Gluconeogenesis through Phosphorylation of CREB Binding Protein. Cell. 2009;137:635–646. doi: 10.1016/j.cell.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wondisford FE. Metformin Action: Concentrations Matter. Cell Metab. 2015;21:159–162. doi: 10.1016/j.cmet.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Heishi M, Hayashi K, Ichihara J, Ishikawa H, Kawamura T, Kanaoka M, Taiji M, Kimura T. Comparison of gene expression changes induced by biguanides in db/db mice liver. Journal of Toxicological Sciences. 2008;33:339–347. doi: 10.2131/jts.33.339. [DOI] [PubMed] [Google Scholar]
- Hollunger G. Guanidines and oxidative phosphorylations. Acta Pharmacol Toxicol (Copenh) 1955;11:1–84. doi: 10.1111/j.1600-0773.1955.tb02972.x. [DOI] [PubMed] [Google Scholar]
- Huang NL, Chiang SH, Hsueh CH, Liang YJ, Chen YJ, Lai LP. Metformin inhibits TNF-alpha-induced I kappa B kinase phosphorylation, I kappa B-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol. 2009;134:169–175. doi: 10.1016/j.ijcard.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, Peters AL, Tsapas A, Wender R, Matthews DR. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetologia. 2012;55:1577–1596. doi: 10.1007/s00125-012-2534-0. [DOI] [PubMed] [Google Scholar]
- Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. New England Journal of Medicine. 1998;338:867–872. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- Kim JE, Lee MH, Nam DH, Song HK, Kang YS, Lee JE, Kim HW, Cha JJ, Hyun YY, Han SY, et al. Celastrol, an NF-kappa B Inhibitor, Improves Insulin Resistance and Attenuates Renal Injury in db/db Mice. Plos One. 2013;8 doi: 10.1371/journal.pone.0062068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landman GWD, Kleefstra N, van Hateren KJJ, Groenier KH, Gans ROB, Bilo HJG. Metformin Associated With Lower Cancer Mortality in Type 2 Diabetes – ZODIAC-16. Diabetes Care. 2010;33:322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugerette F, Vors C, Geloen A, Chauvin MA, Soulage C, Lambert-Porcheron S, Peretti N, Alligier M, Burcelin R, Laville M, et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem. 2011;22:53–59. doi: 10.1016/j.jnutbio.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Lee SK, Lee JO, Kim JH, Kim SJ, You GY, Moon JW, Jung JH, Park SH, Uhm KO, Park JM, et al. Metformin Sensitizes Insulin Signaling Through AMPK-Mediated PTEN Down-Regulation in Preadipocyte 3T3-L1 Cells. Journal of Cellular Biochemistry. 2011;112:1259–1267. doi: 10.1002/jcb.23000. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeschke GR, Roelants FM, Thorner J, Turk BE. Reciprocal Phosphorylation of Yeast Glycerol-3-Phosphate Dehydrogenases in Adaptation to Distinct Types of Stress. Molecular and Cellular Biology. 2012;32:4705–4717. doi: 10.1128/MCB.00897-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology – Human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo Journal. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–+. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4 doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng S, Cao J, He Q, Xiong L, Chang E, Radovick S, Wondisford FE, He L. Metformin activates AMP-activated protein kinase by promoting formation of the alphabetagamma heterotrimeric complex. J Biol Chem. 2015;290:3793–3802. doi: 10.1074/jbc.M114.604421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu QW, Xie JX, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelhill KI, Stapleton D, Gao G, House C, Michell B, Katsis F, Witters LA, Kemp BE. Mammalian Amp-Activated Protein-Kinase Shares Structural and Functional Homology with the Catalytic Domain of Yeast Snf1 Protein-Kinase. Journal of Biological Chemistry. 1994;269:2361–2364. [PubMed] [Google Scholar]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. Journal of Biological Chemistry. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann D, Woods A, Carling D, Wallimann T, Schlattner U. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr Purif. 2003;30:230–237. doi: 10.1016/s1046-5928(03)00126-8. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. AMPK Is a Direct Adenylate Charge-Regulated Protein Kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- Okamura H, Yoshida K, Sasaki E, Qiu LH, Amorim BR, Morimoto H, Haneji T. Expression of PTEN and Akt phosphorylation in lipopolysaccharide-treated NIH3T3 cells. Cell Biology International. 2007;31:119–125. doi: 10.1016/j.cellbi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Oliveira AG, Carvalho BM, Tobar N, Ropelle ER, Pauli JR, Bagarolli RA, Guadagnini D, Carvalheira JB, Saad MJ. Physical exercise reduces circulating lipopolysaccharide and TLR4 activation and improves insulin signaling in tissues of DIO rats. Diabetes. 2011;60:784–796. doi: 10.2337/db09-1907. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ouyang JY, Parakhia RA, Ochs RS. Metformin Activates AMP Kinase through Inhibition of AMP Deaminase. Journal of Biological Chemistry. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochemical Journal. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. 2009;139:1619–1625. doi: 10.3945/jn.109.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng JY, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science. 2013;341:1079–U1049. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Scott MJ, Billiar TR. beta 2-Integrin-induced p38 MAPK Activation Is a Key Mediator in the CD14/TLR4/MD2-dependent Uptake of Lipopolysaccharide by Hepatocytes. Journal of Biological Chemistry. 2008;283:29433–29446. doi: 10.1074/jbc.M803905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DePinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63:727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- Song SM, Andrikopoulos S, Filippis C, Thorburn AW, Khan D, Proietto J. Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. American Journal of Physiology-Endocrinology and Metabolism. 2001;281:E275–E282. doi: 10.1152/ajpendo.2001.281.2.E275. [DOI] [PubMed] [Google Scholar]
- Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, et al. Mammalian AMP-activated protein kinase subfamily. Journal of Biological Chemistry. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, Sherwin R, Devaskar S, Lesche R, Magnuson MA, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected] Proc Natl Acad Sci U S A. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic Effects of Metformin in Non-Insulin-Dependent Diabetes-Mellitus. New England Journal of Medicine. 1995;333:550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- Successes and Opportunities for Population-Based Prevention and Control At A Glance. 2011 http://www.cdc.gov/chronicdisease/resources/publications/aag/ddt.htm.
- Takashima M, Ogawa W, Hayashi K, Inoue H, Kinoshita S, Okamoto Y, Sakaue H, Wataoka Y, Emi A, Senga Y, et al. Role of KLF15 in Regulation of Hepatic Gluconeogenesis and Metformin Action. Diabetes. 2010;59:1608–1615. doi: 10.2337/db09-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajngot A, Chandramouli V, Schumann WC, Ekberg K, Jones PK, Efendic S, Landau BR. Quantitative contributions of gluconeogenesis to glucose production during fasting in type 2 diabetes mellitus. Metabolism-Clinical and Experimental. 2001;50:47–52. doi: 10.1053/meta.2001.19422. [DOI] [PubMed] [Google Scholar]
- Wang PYT, Caspi L, Lam CKL, Chari M, Li XS, Light PE, Gutierrez-Juarez R, Ang M, Schwartz GJ, Lam TKT. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012–1016. doi: 10.1038/nature06852. [DOI] [PubMed] [Google Scholar]
- Watanabe CK. Studies in the metabolic changes induced by administration of guanidine bases. I. Influence of injected guanidine hydrochloride upon blood sugar content. Journal of Biological Chemistry. 1918;33:253–265. [Google Scholar]
- Wilcock C, Bailey CJ. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica. 1994;24:49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou GC, Myers R, Li Y, Chen YL, Shen XL, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]