

Abstract
Soluble guanylyl cyclase (sGC) plays a crucial role in cyclic nucleotide signaling that regulates numerous important physiological processes. To identify new sGC inhibitors that may prevent the formation of the active catalytic domain conformation, we carried out an in silico docking screen targeting a ‘backside pocket’ of the inactive sGC catalytic domain structure. Compounds 1 and 2 were discovered to inhibit sGC even at high/saturating nitric oxide concentrations. Both compounds also inhibit the BAY 58-2667-activated sGC as well as BAY 41-2272-stimulated sGC activity. Additional biochemical analyses showed that compound 2 also inhibits the isolated catalytic domain, thus demonstrating functional binding to this domain. Both compounds have micromolar affinity for sGC and are potential leads to develop more potent sGC inhibitors.
INTRODUCTION
sGC is the main mammalian receptor for the gaseous signaling molecule nitric oxide (NO) [1, 2]. Binding of NO to sGC augments the production of cGMP by several hundred fold [3–5]. cGMP is an important second messenger molecule in cells and binds to protein kinase G, ion channels, and phosphodiesterases [6]. This regulation by cGMP leads to various physiological responses including vasodilation, photosensitivity, and cell growth and differentiation [7].
sGC is a heterodimeric enzyme comprised of 4 domains: the HNOX, PAS, coiled-coil, and catalytic guanylyl cyclase domains. Structures of individual domains from sGC or from bacterial homologs have been solved [8–15]. The precise signal transduction events required for activation are not known although some insight has been gained from low resolution structural studies [16, 17].
In contrast to pharmaceutical activation of sGC [18], targeting inhibition of sGC is less well explored. In diseases such as sepsis and cancer, inhibition of sGC could potentially be beneficial [19–21]. For example, time-dependent inhibition of sGC improves bactericidal activity, restores vasoconstriction in sepsis, and reduces mortality in both a rat sepsis model and mouse model [19, 20] although the opposite strategy, i.e. activating sGC, was beneficial in a different study [22]. Inhibition of the cGMP/sGC pathway decreases tumor cell migration and invasion [21, 23] whereas stimulating of this pathway does the opposite for invasion of melanoma cells [24]. Furthermore, inhibition of sGC decreases angiogenesis [25, 26] which could also aid in cancer treatment [23, 24, 26]. In addition, inhibition of sGC attenuates dysfunctions in basal ganglia in Parkinson’s disease, by reversing many molecular dysfunctions in the murine nervous system [27].
The most commonly used sGC inhibitors, ODQ and NS2028, are effective both in vitro and in vivo. These inhibitors are similar in structure and decrease sGC activity by oxidizing the heme cofactor in the regulatory H-NOX domain of the β1 subunit of the enzyme (potentially resulting in heme loss). Due to this mechanism of inhibition, these compounds are unlikely to affect basal sGC activity, and indeed ODQ was found to be effective in inhibiting only NO activated sGC activity [28] and ODQ inhibition can be overcome by increasing the concentration of NO [29]. ODQ may not be sGC specific as it can also act on other heme containing molecules like hemoglobin [30]. These observations suggest the need for new inhibitors of sGC, especially compounds that can attenuate enzymatic function independent of allosteric activation.
Candidate target sites most useful for identifying activation-independent inhibitors of sGC are most likely to reside in the catalytic cyclase domain. Crystal structures of both a wild type and a mutated inactive catalytic domain of human sGC have been solved both in an inactive conformation [14, 15]. These structures suggest that upon sGC activation, the catalytic domain undergoes a conformational change via a 26 rotation of the α1 subunit of the catalytic domain [15]. Although not in an active conformation, we reasoned that the sGC catalytic domain crystal structure could be used to target the dimer interface for binding of small molecules to act as allosteric inhibitors and prevent the inter-domain reorientation needed for activation. The crystal structure of the catalytic domain reveals a partially occluded active site, as well as a pocket on the other side of the heterodimer which we term a “backside pocket” (Fig 1). This backside pocket at the dimer interface could be used to develop allosteric inhibitors, since a small molecule bound there is likely to prevent reorienting of the two interfaces thereby locking the catalytic domains in an inactive conformation.
Fig 1. In silico screening targeting the “backside pocket” of sGC catalytic domain.

Molecular surface of the sGC catalytic domain showing the active site and backside pocket: A, View of the active site (outlined by white dotted line). The α1 catalytic domain is shown in blue, the β1 catalytic domain is in orange. B, Opposite face of the sGC catalytic domain showing the backside pocket (outlined by red dotted line). View in panel B is a 180° vertical rotation compared to A. C, Slabbed view of the catalytic domain dimer showing both the active site and the backside pocket. The view is obtained by a roughly 90° vertical rotation with respect to B. The active site and backside pocket are separated by a short segment of amino acids that includes residue α1 T527 (labeled as 1). D, Chemical structure of compounds 1–4. E, Predicted interactions of compound 1(left) and compound 2 (right) docking in the catalytic domain: hydrogen bonds are depicted as dashed lines. The terminal primary amine group of α K524 was not present in the structure of the catalytic domain (3UVJ), but was modelled in this figure as it can potentially form an interaction with chloride atom of compound 2. Similarly, the side chain of E473 was not included in the crystal structure but added here for illustrative purposes (this side chain is not anticipated to interact with either of the compounds). All the backside pocket residues are conserved in bovine and human sGC.
Here, we used in silico docking screening with the University of Cincinnati compound library to identify compounds that could potentially bind to this backside pocket site, and characterized the matches biochemically. Our efforts serve dual purposes: the first is to probe the hypothesized rotational catalytic domain activation mechanism, as successfully inhibiting sGC in this manner would provide evidence for such a mechanism; the second is to discover new lead compounds that could be developed into a potent new class of sGC inhibitors that affect both the NO stimulated and basal sGC activities. Such compounds could serve as molecular tools to probe sGC function and also have therapeutic potential.
MATERIALS AND METHODS
In silico compound screening
The GLIDE/SCHRODINGER software package [31] was used for docking the compounds of the University of Cincinnati compound database (~350,000 compounds). We used an inactive mutant human catalytic domain structure (PDB ID 3UVJ) as the target for our initial computational screens, as that was the only structure of the catalytic domain available at the time. We choose a 14Å×14Å×14Å outer box in the catalytic domain region near β1 residues S541 and T474 (both side chain hydroxyl moieties were kept fixed) for the docking, and used default settings for the HTVS, SP, and XP (extra-precision) docking searches. The compounds identified were obtained from the University of Cincinnati (compounds 1–4 with IDs #AF-407/12044005, #5192950, #384033, and #5141573, respectively). Additional quantities of compounds 1, 2 and 4 were subsequently purchased from Specs and Chembridge.
Expression and purification of sGC catalytic domain
The vectors for wt catalytic domain expression were obtained from the Structural Genomics Consortium (SGC). The individual subunits were expressed and purified as previously described [15]. In short, the heterodimeric protein was prepared by mixing together cell pellets containing either the expressed β1 or α1 catalytic domain, and resuspension in 50mM sodium phosphate pH 7.5, 500mM NaCl, and 30mM imidazole. Cells were lysed using a microfluidizer followed by centrifugation of the lysate at 20,000g for 1 hour. The supernatant was applied to Ni-NTA beads (Qiagen) pre-equilibrated with the cell lysis buffer. The beads were washed with 50mM sodium phosphate pH 7.5, 500mM NaCl, and 60mM imidazole and the protein eluted with a similar buffer containing 300mM imidazole. The His tag was subsequently cleaved overnight with TEV protease in 20mM Tris-HCl pH 7.5, 150mM NaCl, 10% ethylene glycol, and 1mM TCEP. After cleavage, the protein was loaded onto a Superdex 200 column (GE Life Sciences) pre-equilibrated with 50mM HEPES pH 7 and 150mM NaCl. The fractions containing the protein were pooled and stored at −80°C. Aliquots were thawed for activity assays and other experiments.
Activity measurements
We measured NO stimulated sGC activitiy using bovine lung sGC (Enzo Life Sciences). sGC was diluted 17-fold in 50mM Tris pH 7.5, 5mM MgCl2, 2mM β-mercaptoethanol (BME), 20% sucrose, and 0.5mg/ml bovine serum albumin (BSA). We pre-incubated sGC (1nM final enzyme concentration) with varying concentrations of each inhibitor in 50mM Tris pH 7.5, 2mM BME, and 5mM MgCl2 for 10 min at 37°C. The uninhibited sGC activity was measured in the presence of 1–2.5% dimethyl sulfoxide (DMSO) as a control to match the DMSO concentrations present in the inhibitor experiments due to the DMSO in the inhibitor stock solutions. The NO stimulated sGC reaction was initiated by the addition of varying concentrations of GTP in the presence of 100μM of the NO donor S-nitroso-N-acetylpenicillamine (SNAP), and continued for 10min at 37°C before boiling the sample for 10 min to stop the reaction. The activity assays with heme independent activator BAY 58-2667 were done using varying concentrations of BAY 58-2667 with 100μM inhibitor. 1nM bovine lung sGC was incubated with both BAY 58-2667 and inhibitors for 10 min at 37°C in the presence of 5mM MgCl2. The reaction was started by addition of 600μM GTP. The activity assays with the sGC stimulator BAY 41-2272 were done using varying concentrations of BAY 41-2272 (both in the presence and absence of the NO donor SNAP) with 100μM of compound 1 or 2 in the presence of 5mM MgCl2. 1nM bovine lung sGC was preincubated with different compounds and BAY 41-2272 for 10 min at 37°C. The reaction was started by the addition of 500μM GTP and after 10 min were stopped by boiling.
The catalytic domain activity assays utilized 1μM final concentration of purified catalytic domain. The inhibitors were pre-incubated with the catalytic domain for 10 min at 37°C in the presence of Mg2+. The reaction was started by the addition of GTP, and stopped after 10 minutes by boiling. All above cyclase reactions were repeated in 3 independent experiments. The cGMP generated was measured using an ELISA (EIA) kit (Cayman).
Analysis of sGC inhibition
The inhibition data from NO activated full-length sGC activity assays were fit using GraphPad Prism 6. The models were fit individually in a non-competitive inhibition mode, competitive inhibition mode, and mixed inhibition mode. The goodness of fit was examined by the R-squared value in each mode. The data were interpreted using a mixed model of inhibition and the value of α. The Ki was determined by fitting the data to the mode of inhibition suggested by the value of α.
RESULTS
In silico screening
An initial docking screen yielded ~ 30 compounds predicted to bind to several sites along the dimer interface including the backside pocket. The active site and the backside pocket are on opposite sides of the dimer, yet they are proximal as both the active site and backside pocket are quite deep and approach each other near the middle of the heterodimeric catalytic domain (Figs 1A, B, C). Nonetheless, the two pockets are separated by a short section of residues including residue T527. We chose molecules that yielded promising docking scores for their relatively small (fragment-like) size to use as lead compounds. The structures of the initial lead compounds 1 and 2 and their computationally docked binding mode to the catalytic domain are shown in Figs 1D and 1E. From our inspection of the molecular docking of inhibitors and the catalytic domain, compound 1 forms hydrogen bonds with the side chain and/or main chain atoms of α1 T527, α1 G529, α1 K524 and β1 L542, β1 E473. In addition, the phenyl moiety of compound 1 makes van der Waals interactions with α1 Y510 and Y532, and β1 I533 and F543 (Fig 1E). Compound 2 is docked in the same site and hydrogen bonds to side chain and/or main chain atoms of residues β1 S541, β1 L542, β1 T474, and α1 G529, and forms van der Waals interactions with α1 Y510, α1 I528, α1 Y532, β1 I533, and β1 F543. For both inhibitors, most of the ligand-protein interactions are predicted to be with the peptide backbone.
We obtained the initial compounds from the University of Cincinnati as 10mM stock solutions in DMSO. These compounds were tested for inhibition in vitro using reactions containing 1nM bovine lung sGC, 25μM GTP, 100μM SNAP, and 10μM inhibitor. After this initial screen, compounds 1 and 2 yielded substantial inhibition of NO stimulated sGC activity (Fig 2). We subsequently searched the University of Cincinnati compound database for analogs, resulting in the identification of compounds 3 and 4 (Fig 1). Compounds 3 and 4 (Fig 1D) were obtained to test whether variation in the structures of compounds 1 and 2 might improve or decrease inhibition of sGC in a manner consistent with chemical specificity in their mode of action. Compound 3 has an additional methyl group at the C6 position compared to compound 2. The compound 1 analog, compound 4, has the same 6-azacytosine moiety but with a carboxyphenol ribose moiety attached to a different ring-nitrogen. Upon testing, both compounds 3 and 4 demonstrated inhibition of sGC, although the observed inhibition was significantly weaker compared to compounds 1 and 2 (Fig 2).
Fig 2. Reduction of SNAP activated sGC activity by novel small molecule inhibitors.

Inhibition of SNAP (100μM) activated sGC by compounds with 150μM GTP (compounds 1 and 2) and 100μM GTP (compounds 3 and 4). The activity assays were performed as independent experiments in triplicate, error-bars represent standard error of the mean (SEM).
Testing for inhibition by non-specific inhibition/aggregation
We tested compounds 1 and 2 for undesired, promiscuous inhibition, a behavior often observed for hits from compound libraries [32–34]. We first tested the unrelated enzyme lactate dehydrogenase (LDH) and found that none of the compounds inhibited LDH (Fig 3). In addition, we probed the unrelated enzymes SHV-1 β-lactamase and β-galactosidase (Fig S1). Compound 2 showed no inhibition of β-lactamase or β-galactosidase (Fig S1). Compound 1 also did not inhibit SHV-1 β-lactamase (Fig S1); we were however unable to test compound 1 with β-galactosidase because its intrinsic absorption interfered with the spectroscopic assay. Next, we tested whether compounds 1 and 2 could form aggregates using a dynamic light scattering device (DYNAPRO) as this provides a means of identifying promiscuous aggregators which can cause loss of protein function [35, 36]. The results show that the inhibitors do not form detectably large aggregates (Fig S2). We also used size exclusion chromatography with full length sGC and 200μM concentrations of compounds 1 and 2 to confirm that the inhibitors did not cause sGC aggregation (data not shown). As a final quality control method, we confirmed the purity of all purchased compounds using LC-MS to check their chemical integrity (Fig S3). The molecular weights matched the calculated molecular weights and combined with the Chembridge and Specs supplied NMR spectra, indicate the correct chemical composition of the compounds. The above experiments confirm that compounds 1 and 2 bind with chemical specificity to inhibit sGC activity and are not promiscuous inhibitors.
Fig 3. Analysis of promiscuous inhibition of lactate dehydrogenase.

The lactate dehydrogenase (LDH) enzyme (1nM), unrelated to sGC, was used to probe for non-specific enzyme inhibition by the compounds, possibly due to promiscuous aggregation. A, LDH activity in the presence of 100μM compound 1 measured by absorbance at 500nm. B, LDH activity in the presence of 200μM compound 2. C, LDH activity in the presence of 200μM compound 3. D, LDH activity in the presence of 200μM compound 4. The inhibitor oxamate was used as a positive control as it is a known inhibitor of LDH. The DMSO bars represent uninhibited LDH with the DMSO concentration matching the DMSO concentration in the compound containing experiment. The error bars represent SEM; the experiment was done in triplicate.
Inhibition of sGC is independent of NO activation
To gain additional insight into the mechanism of sGC inhibition we tested the ability of compounds 1 and 2 to inhibit sGC that was activated by the NO-independent activator BAY 58-2667. The data in Fig 4 show that 100μM of compound 1 or 2 inhibited BAY 58-2668 activated sGC by 96% and 99%, respectively. These results indicate that the inhibitors act by a different mechanism than inhibitors such as ODQ, which do not inhibit sGC that is activated by BAY 58-2667. Note that their analogs, compounds 3 and 4, showed only modest inhibition at only the lowest BAY 58-2667 concentration (Fig 4). To probe whether compounds 1 and 2 could inhibit NO-dependent sGC stimulation by BAY 41-2272, we tested a range of BAY 41-2272 concentrations in the presence and absence of a low (1μM) concentration of NO (Fig 4). Similar to inhibiting BAY 58-2667 activated sGC, compounds 1 and 2 also inhibited BAY 41-2272 stimulated sGC.
Fig 4. Inhibition of BAY 58-2667 stimulated sGC.

A, The activity of 100μM inhibitors (labeled 1–4 for compounds 1–4), or the control with 0.5% DMSO (labeled D) incubated with sGC and with varying concentrations of BAY 58-2667. B, The inhibition activity of 100μM compounds 1, 2, or 0.5% DMSO (labeled D) incubated with sGC and indicated concentrations of BAY 41-2272 in the absence of NO donor. C, The inhibitory activity of 100μM compounds 1, 2, or 0.5% DMSO (labeled D) incubated with sGC and varying concentrations of BAY 41-2272 in the presence of 1μM SNAP. The error bars represent SEM; the rate measurements were performed in triplicate.
Analysis of inhibition mechanism using steady state kinetics
Steady state kinetics were used to probe the inhibitory mechanism of compounds 1 and 2. Reactions contained 1nM full length bovine lung sGC in the presence of 100μM SNAP with varying substrate and inhibitor concentrations (Fig 5). The matrix of steady state kinetic data was globally fit to different models of steady state inhibition to identify the mechanism of inhibition and estimate the apparent Ki for these compounds. The goodness of fit, measured with the non-linear R-squared value for the global fit for each curve was >80% for each inhibitor (Fig S4). We observed that the Ki for each inhibitor determined on different days was subject to contributions to experimental error from variation in Vmax likely due to differences in enzyme specific activity. Accordingly, relative measurements were analyzed using parallel control reactions in the absence of inhibitor. Additionally, to further reduce experimental error it was necessary to optimize the activity assays for presence of BME and BSA in the sGC dilution buffer and the activity buffer (Fig S5). Reactions at the highest concentrations of inhibitor were limited by low signal to noise and enzyme stability due to the long incubation times required to observe product formation. Data at >100 μM compound 1 and 2 were collected but excluded from the global fitting since they contained a proportionally higher level of experimental error due to these limitations. Although only a relatively narrow range of rate constants are accessible using standard ELISA assays for sGC activity, the data nonetheless show that compounds 1 and 2 inhibit SNAP activated sGC activity at micromolar concentrations.
Fig 5. Steady state inhibition kinetic analyses of SNAP-activated sGC.

The substrate dependence of the steady state velocity of SNAP activated sGC in the presence of increasing concentrations of inhibitor. A, Compound 1. B, Compound 2. The error bars represent SEM; each point represents a rate measurement performed in triplicate. The lines represent a global fit of the data sets to a model for mixed inhibition as described in the text.
In order to test for different mechanisms of inhibition the reaction velocities observed over a range of inhibitor and substrate concentrations were globally fit to mixed-model of inhibition represented by the following equilibrium reaction scheme:
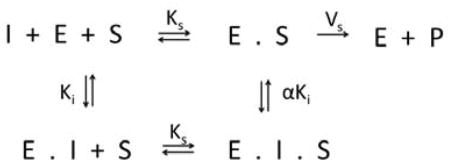
In this general mechanism the inhibitor can bind both to the free enzyme as well as the enzyme-substrate complex with varying affinities and behaves according to the following rate equation:
In this expression the affinity of the inhibitor for the free enzyme is defined as Ki. The magnitude of the term α allows the inhibition mechanism to be evaluated. When α approaches 1, the inhibitor is defined as non-competitive, whereas when α approaches infinity the mechanism becomes competitive. When the steady state inhibition data were globally fit to the above equation, the α-value for compound 1 was 3.6. In contrast, the α-value for compound 2 was very high (3.0×1024). The magnitude of these values suggest a mostly non-competitive inhibition mechanism for compound 1 with a moderately higher affinity for the E.S complex (α>1). However, a competitive inhibition mechanism is indicated for compound 2 (α≫1).
Both compound 1 and 2 provided inhibition of SNAP activated sGC activity at micromolar concentrations (Figs 5A, B). The observed Ki values for compounds 1 and 2, assuming non-competitive inhibition for compound 1 and competitive inhibition for compound 2, were 28±11μM and 19±5μM, respectively. The calculated Ki for compounds 3 and 4 indicated that they were weaker inhibitors (284±85μM and 193±56μM respectively) (Figs S4, S6). Overall, the steady state kinetics indicate that compounds 1 and 2 function as the best inhibitors and they provide inhibition of SNAP-activated sGC in the micromolar range, albeit by different mechanisms.
Probing inhibition with the catalytic domain
We next tested whether compounds 1 and 2 could inhibit the purified catalytic domain since this domain contains the targeted site used for in silico screening. We note that the activity of the purified catalytic domain by itself was much weaker than the full length sGC as was previously observed by others [14]. We tested the inhibition of 1μM catalytic domain at 3 different GTP substrate concentrations and varied the inhibitor concentrations from 0–600μM. Only compound 2 showed inhibition at the higher inhibitor concentrations with >50% inhibition of the purified catalytic domain at 400μM compound 2 (Fig 6). In contrast to compound 2, we did not observe significant inhibition with compound 1, even up to 600μM concentration of compound 1 (Fig 6). Compounds 3 and 4 did not significantly inhibit the catalytic domain (Fig S7) although compound 4’s inhibition profile was similar to that of compound 2.
Fig 6. Inhibition of the sGC catalytic domain.

1μM purified catalytic domain was used to determine the activity of the cGMP activity of the catalytic domain with each of the inhibitors. A, Compound 1. B, Compound 2. The error bars represent SEM; each point represents a rate measurement performed in triplicate.
DISCUSSION
We set out to develop allosteric inhibitors directed against the catalytic domain of sGC with the goal of probing the rotational catalytic domain conformational activation mechanism as well as developing a novel inhibitor tool that can be used to probe sGC basal and stimulated activity functions in vitro. The data shown here identify two molecules described that inhibit SNAP-activated sGC at micromolar concentrations (27±11μM and 19±5μM for compounds 1 and 2, respectively).
Our hypothesis is that these inhibitors bind to the backside pocket of the sGC catalytic domain, as targeted and predicted by in silico docking. In this model the compounds can bind to free E or the E.S complex, ultimately preventing the formation of the active conformation of the catalytic domain making this pocket an allosteric site for the development of inhibitors. This perspective is supported by the following observations. When the inhibition data were fitted to a mixed-inhibition model, to probe for either competitive or non-competitive inhibition, the α-value for compound 1 was relatively close to 1, while that of compound 2 was very high. This indicates that compound 2 likely inhibits sGC in a mostly competitive manner, whereas compound 1 is likely to be predominantly a non-competitive inhibitor and preferentially binds the sGC-GTP complex. It is also possible that compound 1 captures sGC between a completely activated state and a completely inactivated state, where the binding of GTP to the active site is reduced. Furthermore, both inhibitors decrease BAY 58-5667 stimulated sGC activity by >90%, suggesting that both compounds do not merely inhibit sGC by inactivating NO/SNAP or interfering with the heme redox state. In addition, both inhibitors also inhibit BAY 41-2272 stimulated sGC activity. BAY 41-2272 stimulation of sGC occurs via a different mechanism, compared to BAY 58-2276, that is not yet fully understood yet does require low concentrations of NO either added or already present in the environment [37]. BAY 41-2272, or its analog YC-1, is postulated to bind to either the β1 chain near or in the H-NOX domain [38] or to the catalytic domain [37, 39]. That compounds 1 and 2 inhibit both BAY 58-2276 activated sGC and BAY 41-2272 stimulated sGC suggest that compounds 1 and 2 act on a common part of these different activation/stimulation mechanisms; this is consistent with compounds 1 and 2 acting on the catalytic domain.
Additional evidence for the compounds targeting the catalytic domain, at least for compound 2, comes from assays with the purified catalytic domain which showed that, unlike compound 1, compound 2 does inhibit catalytic domain activity (Fig 6). However, this compound 2 inhibition occurs at higher concentrations than required for its inhibition of SNAP activated full length sGC. We do not have an explanation for this observation except to note that the catalytic domain sGC heterodimer is likely not a uniform oligomeric mixture as previously observed [14]. Note also that we used a 1μM catalytic domain concentration for our assay, which is below the dimerization constant of heterodimers [14] and suggests a predominance of inactive monomers in solution. However, the addition of GTP and Mg2+ has been shown to shift the catalytic dimerization constant to 0.45μM [40] indicating that at the concentrations we used to measure activity, the subunits may mostly be in a dimer state.
We next carried out a similarity search to find molecules that were similar to compounds 1 and 2 to gain additional insight into which chemical moieties are important for activity. We found one analog for each that did exhibit inhibition of SNAP stimulated sGC, albeit with a weaker affinity. The compound 2 analog compound 3 yielded Ki values of 284±85μM, whereas the compound 1 analog compound 4 had a Ki of 193±56μM. We speculate that the weaker affinity of compound 3, compared to compound 2, might be due to unfavorably burying a hydrogen bond donor (i.e. carbonyl oxygen of α1 T527) upon addition of the methyl group at the C6 position of the tetrahydropyran ring. The analog of compound 1, compound 4, has an additional ribose ring connected to the azacytosine ring. The ribose group contains an ester of benzoic acid at the 5′ carbon. Compound 4 is considerably larger than compound 1, and we speculate that the steric hindrance could decrease binding to the catalytic domain. Furthermore, the ribose sugar moiety branches off from the azacytosine-nitrogen that is involved in hydrogen bonding with β1 E473 in compound 1. This may considerably re-orient the inhibitor compared to compound 1 and could also explain the lower affinity.
Unlike the original compounds, compounds 3 and 4 do not cause inhibition of BAY 58-2667 activated sGC, at high BAY 58-2667 concentrations. However, at the lowest concentration of BAY 58-2667 (0.1nM), these analogs did show a modest inhibition in sGC activity: a 38% decrease in activity for compound 3, and 24% for compound 4 (Fig 4). Furthermore, we observed that our analogs did not inhibit purified catalytic domain, even up to 1mM inhibitor concentration. Overall, these results highlight chemical specificity of the compounds 1 and 2 for sGC inhibition.
Compound 2 is the most promising lead because of its highest affinity and its ability to inhibit full length sGC activated by either SNAP, BAY 58-2667, or BAY 41-2272 as well as purified sGC catalytic domain. Compound 2 behaves mostly as a competitive inhibitor, as judged by its α value. This suggests that compound 2 does not bind to sGC when bound to GTP, and thus that binding of compound 2 to the backside pocket prevents binding of GTP. However, since we do not have direct evidence that compound 2 binds in the backside pocket of the catalytic domain, it is also possible that compound 2 can bind to the active site and compete with GTP for its binding site. We have also ruled out other non-specific modes of inhibition, for example via promiscuous inhibition, by testing the effect of all compounds on several unrelated enzymes. Despite our promising in vitro results, none of the compounds provided inhibition of sGC activity in transiently transfected COS7 cells when tested up to 100μM concentrations (data not shown). Therefore, these compounds need to be improved in terms of affinity and possibly also cell permeability and/or stability. Nevertheless, these molecules could serve as lead molecules for iterative drug design to develop new sGC inhibitors. While preparing this manuscript, a similar approach to target the catalytic domain was also published [41].
Supplementary Material
Acknowledgments
We thank the Structural Genomics Consortium (SGC) for the plasmids and the University of Cincinnati Drug Discovery Center for the compounds. We thank the Tochtrop lab for help with the LC/MS machine and Vivien Yee for critical reading of the manuscript. This research was supported by the NIH Grants HL075329 to FVDA and GM096000 to MEH. JV was supported by NIGMS training grant T32 GM008056.
References
- 1.Katsuki S, Arnold W, Mittal C, Murad F. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J Cyclic Nucleotide Res. 1977;3:23–35. [PubMed] [Google Scholar]
- 2.Denninger JW, Marletta MA. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim Biophys Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 3.Humbert P, Niroomand F, Fischer G, Mayer B, Koesling D, Hinsch KD, Gausepohl H, Frank R, Schultz G, Bohme E. Purification of soluble guanylyl cyclase from bovine lung by a new immunoaffinity chromatographic method. Eur J Biochem. 1990;190:273–278. doi: 10.1111/j.1432-1033.1990.tb15572.x. [DOI] [PubMed] [Google Scholar]
- 4.Stone JR, Marletta MA. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry. 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 5.Lee YC, Martin E, Murad F. Human recombinant soluble guanylyl cyclase: expression, purification, and regulation. Proc Natl Acad Sci U S A. 2000;97:10763–10768. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feil R, Kemp-Harper B. cGMP signalling: from bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006;7:149–153. doi: 10.1038/sj.embor.7400627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boerrigter G, Costello-Boerrigter LC, Cataliotti A, Lapp H, Stasch JP, Burnett JC., Jr Targeting heme-oxidized soluble guanylate cyclase in experimental heart failure. Hypertension. 2007;49:1128–1133. doi: 10.1161/HYPERTENSIONAHA.106.083832. [DOI] [PubMed] [Google Scholar]
- 8.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci U S A. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Sayed N, Baskaran P, Beuve A, van den Akker F. PAS-mediated dimerization of soluble guanylyl cyclase revealed by signal transduction histidine kinase domain crystal structure. J Biol Chem. 2008;283:1167–1178. doi: 10.1074/jbc.M706218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purohit R, Weichsel A, Montfort WR. Crystal structure of the Alpha subunit PAS domain from soluble guanylyl cyclase. Protein Sci. 2013;22:1439–1444. doi: 10.1002/pro.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma X, Beuve A, van den Akker F. Crystal structure of the signaling helix coiled-coil domain of the beta1 subunit of the soluble guanylyl cyclase. BMC Struct Biol. 2010;10:2. doi: 10.1186/1472-6807-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seeger F, Quintyn R, Tanimoto A, Williams GJ, Tainer JA, Wysocki VH, Garcin ED. Interfacial residues promote an optimal alignment of the catalytic center in human soluble guanylate cyclase: heterodimerization is required but not sufficient for activity. Biochemistry. 2014;53:2153–2165. doi: 10.1021/bi500129k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allerston CK, von Delft F, Gileadi O. Crystal structures of the catalytic domain of human soluble guanylate cyclase. PLoS One. 2013;8:e57644. doi: 10.1371/journal.pone.0057644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell MG, Underbakke ES, Potter CS, Carragher B, Marletta MA. Single-particle EM reveals the higher-order domain architecture of soluble guanylate cyclase. Proc Natl Acad Sci U S A. 2014;111:2960–2965. doi: 10.1073/pnas.1400711111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fritz BG, Roberts SA, Ahmed A, Breci L, Li W, Weichsel A, Brailey JL, Wysocki VH, Tama F, Montfort WR. Molecular model of a soluble guanylyl cyclase fragment determined by small-angle X-ray scattering and chemical cross-linking. Biochemistry. 2013;52:1568–1582. doi: 10.1021/bi301570m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263–2273. doi: 10.1161/CIRCULATIONAHA.110.981738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandes D, Sordi R, Pacheco LK, Nardi GM, Heckert BT, Villela CG, Lobo AR, Barja-Fidalgo C, Assreuy J. Late, but not early, inhibition of soluble guanylate cyclase decreases mortality in a rat sepsis model. J Pharmacol Exp Ther. 2009;328:991–999. doi: 10.1124/jpet.108.142034. [DOI] [PubMed] [Google Scholar]
- 20.Paula-Neto HA, Alves-Filho JC, Souto FO, Spiller F, Amendola RS, Freitas A, Cunha FQ, Barja-Fidalgo C. Inhibition of guanylyl cyclase restores neutrophil migration and maintains bactericidal activity increasing survival in sepsis. Shock. 2011;35:17–27. doi: 10.1097/SHK.0b013e3181e37ea8. [DOI] [PubMed] [Google Scholar]
- 21.Babykutty S, Suboj P, Srinivas P, Nair AS, Chandramohan K, Gopala S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin Exp Metastasis. 2012;29:471–492. doi: 10.1007/s10585-012-9464-6. [DOI] [PubMed] [Google Scholar]
- 22.Vandendriessche B, Rogge E, Goossens V, Vandenabeele P, Stasch JP, Brouckaert P, Cauwels A. The soluble guanylate cyclase activator BAY 58–2667 protects against morbidity and mortality in endotoxic shock by recoupling organ systems. PLoS One. 2013;8:e72155. doi: 10.1371/journal.pone.0072155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwappacher R, Rangaswami H, Su-Yuo J, Hassad A, Spitler R, Casteel DE. cGMP-dependent protein kinase Ibeta regulates breast cancer cell migration and invasion via interaction with the actin/myosin-associated protein caldesmon. J Cell Sci. 2013;126:1626–1636. doi: 10.1242/jcs.118190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arozarena I, Sanchez-Laorden B, Packer L, Hidalgo-Carcedo C, Hayward R, Viros A, Sahai E, Marais R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45–57. doi: 10.1016/j.ccr.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Morbidelli L, Pyriochou A, Filippi S, Vasileiadis I, Roussos C, Zhou Z, Loutrari H, Waltenberger J, Stossel A, Giannis A, Ziche M, Papapetropoulos A. The soluble guanylyl cyclase inhibitor NS-2028 reduces vascular endothelial growth factor-induced angiogenesis and permeability. Am J Physiol Regul Integr Comp Physiol. 2010;298:R824–32. doi: 10.1152/ajpregu.00222.2009. [DOI] [PubMed] [Google Scholar]
- 26.Pyriochou A, Beis D, Koika V, Potytarchou C, Papadimitriou E, Zhou Z, Papapetropoulos A. Soluble guanylyl cyclase activation promotes angiogenesis. J Pharmacol Exp Ther. 2006;319:663–671. doi: 10.1124/jpet.106.108878. [DOI] [PubMed] [Google Scholar]
- 27.Tseng KY, Caballero A, Dec A, Cass DK, Simak N, Sunu E, Park MJ, Blume SR, Sammut S, Park DJ, West AR. Inhibition of striatal soluble guanylyl cyclase-cGMP signaling reverses basal ganglia dysfunction and akinesia in experimental parkinsonism. PLoS One. 2011;6:e27187. doi: 10.1371/journal.pone.0027187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakane M, Hsieh G, Miller LN, Chang R, Terranova MA, Moreland RB, Kolasa T, Brioni JD. Activation of soluble guanylate cyclase causes relaxation of corpus cavernosum tissue: synergism of nitric oxide and YC-1. Int J Impot Res. 2002;14:121–127. doi: 10.1038/sj.ijir.3900843. [DOI] [PubMed] [Google Scholar]
- 29.Lies B, Groneberg D, Gambaryan S, Friebe A. Lack of effect of ODQ does not exclude cGMP signalling via NO-sensitive guanylyl cyclase. Br J Pharmacol. 2013;170:317–327. doi: 10.1111/bph.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Brandish PE, DiValentin M, Schelvis JP, Babcock GT, Marletta MA. Inhibition of soluble guanylate cyclase by ODQ. Biochemistry. 2000;39:10848–10854. doi: 10.1021/bi9929296. [DOI] [PubMed] [Google Scholar]
- 31.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 32.Coan KE, Shoichet BK. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol Biosyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 33.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nat Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 34.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 35.Shoichet BK. Screening in a spirit haunted world. Drug Discov Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sassano MF, Doak AK, Roth BL, Shoichet BK. Colloidal aggregation causes inhibition of G protein-coupled receptors. J Med Chem. 2013;56:2406–2414. doi: 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy B, Halvey EJ, Garthwaite J. An enzyme-linked receptor mechanism for nitric oxide-activated guanylyl cyclase. J Biol Chem. 2008;283:18841–18851. doi: 10.1074/jbc.M801712200. [DOI] [PubMed] [Google Scholar]
- 38.Purohit R, Fritz BG, The J, Issaian A, Weichsel A, David CL, Campbell E, Hausrath AC, Rassouli-Taylor L, Garcin ED, Gage MJ, Montfort WR. YC-1 binding to the beta subunit of soluble guanylyl cyclase overcomes allosteric inhibition by the alpha subunit. Biochemistry. 2014;53:101–14. doi: 10.1021/bi4015133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamothe M, Chang FJ, Balashova N, Shirokov R, Beuve A. Functional characterization of nitric oxide and YC-1 activation of soluble guanylyl cyclase: structural implication for the YC-1 binding site? Biochemistry. 2004;43:3039–3048. doi: 10.1021/bi0360051. [DOI] [PubMed] [Google Scholar]
- 40.Winger JA, Marletta MA. Expression and characterization of the catalytic domains of soluble guanylate cyclase: interaction with the heme domain. Biochemistry. 2005;44:4083–4090. doi: 10.1021/bi047601d. [DOI] [PubMed] [Google Scholar]
- 41.Mota F, Gane P, Hampden-Smith K, Allerston CK, Garthwaite J, Selwood DL. A new small molecule inhibitor of soluble guanylate cyclase. Bioorg Med Chem. 2015;23:5303–5310. doi: 10.1016/j.bmc.2015.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.