

Abstract
Background
Idiopathic pulmonary fibrosis is a common interstitial lung disease; it is a chronic, progressive, and fatal lung disease of unknown etiology. Over the last two decades, knowledge about the underlying mechanisms of pulmonary fibrosis has improved markedly and facilitated the identification of potential targets for novel therapies. However, despite the large number of antifibrotic drugs being described in experimental pre-clinical studies, the translation of these findings into clinical practices has not been accomplished yet. NADH:quinone oxidoreductase 1 (NQO1) is a homodimeric enzyme that catalyzes the oxidation of NADH to NAD+ by various quinones and thereby elevates the intracellular NAD+ levels. In this study, we examined the effect of increase in cellular NAD+ levels on bleomycin-induced lung fibrosis in mice.
Methods
C57BL/6 mice were treated with intratracheal instillation of bleomycin. The mice were orally administered with β-lapachone from 3 days before exposure to bleomycin to 1-3 weeks after exposure to bleomycin. Bronchoalveolar lavage fluid (BALF) was collected for analyzing the infiltration of immune cells. In vitro, A549 cells were treated with transforming growth factor β1 (TGF-β1) and β-lapachone to analyze the extracellular matrix (ECM) and epithelial-mesenchymal transition (EMT).
Results
β-Lapachone strongly attenuated bleomycin-induced lung inflammation and fibrosis, characterized by histological staining, infiltrated immune cells in BALF, inflammatory cytokines, fibrotic score, and TGF-β1, α-smooth muscle actin accumulation. In addition, β-lapachone showed a protective role in TGF-β1–induced ECM expression and EMT in A549 cells.
Conclusion
Our results suggest that β-lapachone can protect against bleomycin-induced lung inflammation and fibrosis in mice and TGF-β1–induced EMT in vitro, by elevating the NAD+/NADH ratio through NQO1 activation.
Keywords: Inflammation; Fibrosis; Transforming Growth Factor Beta1; Cell; NAD; Pulmonary Fibrosis; Mice; NQO1 Protein, Human
Introduction
Idiopathic pulmonary fibrosis (IPF) is a common disease of unknown etiology1,2,3,4,5. Thus, there is no established treatment for it; only the degree of its complications can be controlled. In the acute phase of IPF, the mortality rate is very high and has a prevalence of 20 per 10 million in men and 13 per 10 million in women. Moreover, the duration of life after diagnosis is about 2 to 4 years6. IPF is one of the most common interstitial lung diseases (ILDs) and is characterized by the histological pattern of usual interstitial pneumonia7,8; the histological characteristics include remodeling of lung architecture with fibroblastic foci and honeycombing. Treatment options for patients with advanced fibrosis are limited and the clinical management focuses only on the treatment of complications, including heart failure, infection, supportive care, and lung transplantation.
Rodents treated with bleomycin (BLM), an anti-tumor antibiotic, are the most commonly used animal models for IPF research. Intratracheal administration of BLM within tissues induces an inflammatory response and fibrosis, in a short time, by increasing the secretion of inflammatory cytokines and the expression of pro-fibrotic markers, transforming growth factor β1 (TGF-β1), fibronectin, and/or procollagen-1, which appear to be maximized after 14 days9,10. The inflammatory response and fibrotic reaction are triggered about 9 days after the treatment6,11, and wide variations due to the differences in the expression patterns of cytokines or related genes depending on the species of the mice, have been reported6,12.
NAD is a metabolic cofactor that is present in cells in its either oxidized (NAD+) or reduced (NADH) form. NAD+ or NADH functions as a cofactor for a multitude of enzymatic reactions, and therefore critically regulates cellular homeostasis. As NAD+ is critical for a variety of enzymatic reactions, the NAD redox balance, represented as the NAD+/NADH ratio, is tightly regulated13 and its disruption has been associated with multiple clinical disorders and pathologies; for example, pathological conditions, such as diabetes and oxidative stress, are well-correlated with decreased cellular NAD+ levels14,15. It has also been recently reported that the cellular NAD+ level in many tissues declines with age16,17, implying the importance of maintaining optimal intracellular NAD+ levels to prevent age-associated cellular dysfunction. Furthermore, cisplatininduced cochlear and kidney damage are highly associated with decrease in the NAD+/NADH ratio that accompanies inflammation and oxidative stress18,19. Together, these findings suggest that maintenance of the NAD redox balance is very important for general health. NADH:quinone oxidoreductase 1 (NQO1) is a cytosolic antioxidant flavoprotein that catalyzes the reduction of quinones to hydroquinones by utilizing NADH as an electron donor, which consequently increases the intracellular NAD+ levels20,21. Several substrates of the NQO1 enzyme have been identified, of which β-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphthol[1,2-b]pyran-5,6-dione; β-Lap) is the best known22,23. β-Lap was first isolated from the bark of the lapacho tree and reported to inhibit tumor growth24. However, recent reports indicate that the enzymatic action of NQO1 on β-Lap has beneficial effects on several characteristics of metabolic syndromes, for example, prevention of health decline in aged mice, amelioration of obesity or hypertension, prevention of arterial restenosis, and protection against cisplatin-induced renal injury19,25,26,27,28.
Therefore, we investigated the effects of increased NAD+ level via the enzymatic action of NQO1 using β-Lap as a substrate, on BLM-induced lung injury.
Materials and Methods
1. Animals
Wild type C57BL/6 mice were purchased from Hanil Bio Inc. (Koatech Honam Distributor, Jeonbuk, Korea). Experiments were performed using 8-week-old male mice, which were fed a standard commercial diet and housed in a room with an ambient temperature of 20℃–22℃ and a relative humidity of 50±5%, under 12/12-hour light/dark cycle in a specific pathogen-free facility. All animal studies were approved by the Animal Care and Use Committee at Wonkwang University.
2. Development of animal model for lung inflammation and pulmonary fibrosis
To develop the rodent model, mice were anesthetized with ketamine hydrochloride injection (Yuhan Co., Seoul, Korea). For acute lung inflammation, 50 µL of BLM (5 mg/kg body weight of mouse; Nippon Kayaku, Tokyo, Japan) was directly introduced by intratracheal instillation, and β-Lap was administered orally for 10 consecutive days measured from 3 days before BLM administration. On the seventh day after BLM administration, mice were sacrificed and the bronchoalveolar lavage fluid (BALF) and lung tissues were examined. For chronic lung fibrosis (pulmonary fibrosis) model, 50 µL of BLM (2 mg/kg body weight) was introduced in the same way as described above. To examine the effect of β-Lap on BLM-induced lung injury, β-Lap was administered orally to mice for 17 consecutive days measured from 3 days before BLM administration. On the 14th day after BLM administration, mice were sacrificed and lung tissues were further examined. The experimental mice were divided into six groups: control, BLM, BLM+β-Lap (5, 10, and 20 mg/kg body weight), and β-Lap alone (20 mg/kg body weight).
3. Analysis of BALF
BALF was collected by cannulating the trachea and lavaging the lung with 1 mL of sterilized phosphate buffered saline slowly. About 80% of BALF was routinely recovered from each mouse. BALF (100 µL) was centrifuged onto a glass slide at 700 rpm for 10 minutes using a Cytospin II (Shandon Inc., Pittsburgh, PA, USA). Immune cells were fixed and stained with Diff-Quik staining reagent (B4132-1A; IMEB Inc., Deerfield, IL, USA).
4. Histological analysis and hydroxyproline assay
Mouse lung was fixed in 4% formaldehyde and embedded in paraffin wax. The 5-µm-thick sections were deparaffinized in xylene and rehydrated through graded concentrations of ethanol. Hematoxylin and eosin Y staining was performed according to standard protocols. Images were collected and analyzed using a light microscope (OLYMPUS IX71; Olympus, Tokyo, Japan) with DP analyzer software (DP70-BSW; Olympus). Collagen production due to fibrosis was confirmed by picrosirius red staining followed by examination under a light microscope. Hydroxyproline content of lungs was determined as followed by manufacturers instruction (BioVision, Milpitas, CA, USA). In brief, lung tissue homogenates in dH2O were hydrolyzed with concentrated HCl at 120℃ for 3 hours and took supernatants after centrifuge at 10,000 ×g for 3 minutes. After drying 10 µL of supernatant on 96-well plate, samples were assayed for hydroxyproline at 550 nm. The data were expressed as µg of hydroxyproline/mg tissue.
5. Immunohistochemical analysis
To determine the expression of various target molecules, immunohistochemistry (IHC) was performed. In brief, 5-µm-thick lung sections were deparaffinized and rehydrated through graded concentrations of ethanol. Antigen was retrieved by microwaving in Tris-EDTA buffer (10 mmol/L Tris base, 1 mmol/L EDTA solution, 0.05% Tween-20; pH 9.0). Endogenous peroxidase activity was quenched by incubation in 0.3% hydrogen peroxide (H2O2) solution for 5 minutes. Nonspecific antibody binding was blocked with 1.5% goat or rabbit serum for 30 minutes, and then the sections were incubated with antibodies against interleukin 6 (IL-6; Santa Cruz Biotechnology, Santa Cruz, CA, USA), TGF-β1, and α-smooth muscle actin (α-SMA) (Abcam, Cambridge, MA, USA). Sections were washed with phosphate-buffered saline and processed with secondary antibodies conjugated to HRP-labeled polymer (DakoCytomation EnVision+ System-HRP Labeled Polymer; Dako, Carpinteria, CA, USA), according to the manufacturer's instructions. Peroxidase activity was detected using DAB chromogen substrate solution; finally, the sections were counterstained with hematoxylin.
6. Cell culture
The human alveolar basal epithelial cell line A549 was cultured at 37℃ in 5% CO2 atmosphere in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen). A549 cells were synchronized for 24 hours in low-serum medium containing 0.1% FBS and then treated with β-Lap and TGF-β1 for 48 hours. The experimental groups were divided into six categories: control (vehicle only), TGF-β1 (5 ng/mL), TGF-β1+β-Lap (0.5, 1.0, and 2.0 µM), and β-Lap alone (2.0 µM).
7. Western blot analysis
A549 cells were lysed using buffer containing 10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 1 mM ethylenediaminetetraacetic acid, 50 mM β-glycerophosphate, 1 mM dithiothreitol, 1 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail. After centrifugation, the collected lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 10% polyacrylamide gel and then transferred onto a nitrocellulose membrane. The proteins were visualized using chemiluminescence detection kit (West Save Gold; Young In Frontier Co. Ltd., Seoul, Korea), according to the manufacturer's instructions. Specific antibodies against the following antigens were purchased: β-actin (Santa Cruz Inc.), collagen I (Abcam Inc.), Smad2, Smad3, p-Smad2, and p-Smad3 (Cell Signaling Technology, Danvers, MA, USA), and E-cadherin and N-cadherin (BD Transduction Laboratories, Lexington, KY, USA).
8. Assay for NQO1 enzymatic activity and NAD+/NADH ratio
For measuring NQO1 activity in cells, the assay was performed, as described by Gustafson et al.29, using 2,6-dichlorophenolindophenol (DCPIP) as substrate. In brief, cytosolic fraction (CF) was prepared in 0.25 M sucrose buffer, and a reaction mixture containing 25 mM Tris (pH 7.4), 0.07% bovine serum albumin, 200 µM NADH, and 40 µM DCPIP was used for the assay in the presence and absence of 20 µM dicumarol. NQO1 activity was determined by measuring the absorbance at 600 nm with DCPIP as substrate and was expressed as nanomoles of DCPIP reduced per minute. NAD+/NADH was measured using the NAD+/NADH assay kit (BioAssay Systems, Hayward, CA, USA) by following the manufacturer's instructions. Briefly, cells and tissues were homogenized in 100 µL extraction buffer for NAD+ or NADH and neutralized. The concentrations of NAD+ and NADH in the homogenates were measured using a microplate reader.
9. Statistical analysis
All values are represented as mean±standard deviation. one-way analysis of variance (ANOVA) was used to calculate the statistical significance of the results and p-values <0.05 were considered statistically significant.
Results
1. Effect of β-Lap on BLM-induced lung inflammatory responses
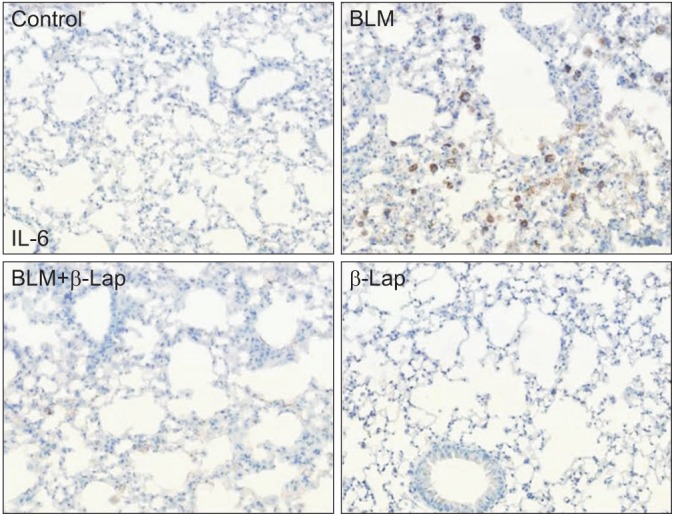
In comparison to the control tissue, BLM-treated lung tissue showed hemorrhagic transformation, which was completely inhibited by co-treatment with β-Lap (Figure 1A). One of the major sources of the inflammatory response is the infiltration of immune cells into lung tissues as a result of chemoattractive signaling; β-Lap abrogated the infiltration of immune cells in the alveoli of BLM-treated mice (Figure 1B). The histological characteristics of the lung tissue treated with β-Lap alone were similar to those of the control tissue. Additionally, to detect the presence of infiltrating immune cells in lung tissue, we performed cellular analysis of the BALF. As shown in Figure 1C and D, β-Lap treatment clearly attenuated the BLM-induced recruitment of macrophages, lymphocytes, and neutrophils into the lung tissues. In addition, the inflammatory responses were assessed by several ways, including measurements of pro-inflammatory cytokines in lung tissue. The increased expression of IL-6, which is one of the pro-inflammatory cytokines, in the BLM-treated tissue, was attenuated by β-Lap (Figure 2). The lung tissues that were treated with β-Lap alone and the controls did not show IL-6 expression. These results suggest that β-Lap has a protective effect on BLM-induced lung inflammation in mice.
Figure 1. Effect of β-Lap on BLM-induced lung inflammation in mice. Gross (A) and histological (B) analyses of lung tissues after bleomycin (5 mg/kg) and/or β-Lap (40 mg/kg) and BALF analysis for infiltration of immune cells (C, D). All procedures were followed, as described in the "Materials and Methods" section. *,**p<0.05 by one-way analysis of variance compared with control and bleomycin groups, respectively. β-Lap: β-lapachone; BALF: bronchoalveolar lavage fluid; BLM: bleomycin.

Figure 2. Effect of β-Lap on bleomycin-induced IL-6 expression in lung tissues. Lung tissues from bleomycin (5 mg/kg) and/or β-Lap (40 mg/kg)-treated mice were immunohistochemically stained with anti-mouse IL-6 antibody, as described in the "Materials and Methods" section. β-Lap: β-lapachone; BLM: bleomycin; IL-6: interleukin 6.
2. Effect of β-Lap on BLM-induced lung fibrosis
We next examined the effect of β-Lap on BLM-induced lung fibrotic response in mice. As shown in Figure 3A, BLM-treated lung tissues showed severe infiltration of immune cells, thickness of alveolar wall, and accumulation of collagen fibers on alveolar tissues. However, β-Lap co-treatment with BLM resulted in a marked improvement in infiltration, wall thickness, and accumulation of collagen, in a dose-dependent manner. Treatment with β-Lap alone did not have any effect on infiltration and extracellular matrix accumulation, similar to the control group. In addition, picrosirius red staining for collagen fibrils showed that BLM-induced collagen deposition was clearly attenuated by β-Lap (Figure 3B); consistent with this result, BLM increased the hydroxyproline content, which is a major component of collagen, in lung tissues, whereas β-Lap attenuated this increase in a dose-dependent manner. Tissues treated with β-Lap alone and the control tissues showed basal levels of collagen fibrils and hydroxyproline. These results suggest that BLM-induced pulmonary fibrosis is alleviated by β-Lap.
Figure 3. Effect of β-Lap on BLM-induced lung fibrosis in mice. Lung tissues from BLM (2 mg/kg) and/or β-Lap (5, 10, and 20 mg/kg)-treated mice were histologically stained with H&E (A) and picrosirius red (B). (C) Lung tissues from bleomycin and/or β-Lap–treated mice were further analyzed for hydroxyproline, as per the manufacture's instruction. *,**p<0.05 by one-way analysis of variance compared with control and BLM groups, respectively. β-Lap: β-lapachone; BLM: bleomycin.

TGF-β isoforms are known to be multifunctional cytokines that play an important role in tissue fibrosis as well as in wound healing and repair. TGF-β stimulates the production of various extracellular matrix proteins and inhibits their degradation. Notably, in many diseases, excessive TGF-β contributes to the pathogenesis of fibrosis. Administration of BLM-induced TGF-β1 expression in lung tissues was significantly reduced by β-Lap co-treatment (Figure 4A). In addition, the expression of α-SMA, a myofibroblast marker, was detected by IHC (Figure 4B); the BLM-induced increase in α-SMA expression was clearly suppressed by co-treatment with β-Lap, suggesting that β-Lap also attenuates BLM-induced pulmonary fibrosis.
Figure 4. Effect of β-Lap on BLM-induced lung fibrosis. Lung tissues from bleomycin (2 mg/kg) and/or β-Lap (20 mg/kg)–treated mice were immunohistochemically stained with anti-mouse TGF-β1 (A) and α-SMA (B) antibodies, as described in the Materials and Methods section. β-Lap: β-lapachone; BLM: bleomycin; TGF-β1: transforming growth factor β1; α-SMA: α-smooth muscle actin.

3. Effect of β-Lap on TGF-β1–mediated epithelial-mesenchymal transition signaling in A549 cells
It is known that repeated cycles of inflammation and repair initiate tissue damage and may progress to fibrotic changes resulting in excessive matrix deposition and scar formation in tissues. In this process, TGF signaling is quite important and epithelial-mesenchymal transition (EMT) is a critical phenomenon. To confirm the effect of β-Lap on TGF-β1–induced EMT, we used the A549 lung epithelial cell line. For synchronization, A549 cells were cultured for 24 hours in low serum media containing 0.1% FBS and then treated with TGF-β1 and β-Lap. TGF-β1 activated the transcription factors, Smad2 and Smad3, by phosphorylation in a time-dependent manner and elevated collagen I protein, one of the well-known extracellular matrix (ECM) proteins, in a dose-dependent manner (data not shown). To address whether β-Lap affected TGF-β1–induced EMT and ECM accumulation in A549 cells, cells were pre-treated with β-Lap (0.5, 1.0, and 2.0 µM) for 30 minutes before treating with TGF-β1 (5 ng/mL) for 48 hours. As indicated in Figure 5A, TGF-β1 treatment resulted in decreased Ecadherin, an epithelial marker, whereas it caused an increase in N-cadherin, a mesenchymal marker. However, β-Lap attenuated the increased N-cadherin expression induced by TGF-β1, and elevated the E-cadherin expression in A549 cells. Moreover, β-Lap treatment not only reduced the TGF-β1–induced collagen I expression but also reversed the TGF-β1–induced change in cellular morphology, from the elongated fibroblast-like appearance back to the classic cobblestone appearance (Figure 5B). These results suggest that β-Lap regulates TGF-β1–induced EMT and ECM accumulation; therefore, it has an antifibrotic activity in vitro.
Figure 5. In vitro study for evaluating the inhibitory effect of β-Lap on TGF-β1–induced ECM expression, EMT, and morphological changes in A549 cells. (A) The effect of β-Lap on TGF-β1–induced collagen I expression and EMT. (B) The effect of β-Lap on TGF-β1–induced cellular morphological changes. β-Lap: β-lapachone; TGF-β1: transforming growth factor β1; ECM: extracellular matrix; EMT: epithelial-mesenchymal transition.

4. Effect of β-Lap on NQO1 enzyme activation and modulation of intracellular NAD+/NADH ratio
The compound β-Lap was developed for use in protective or therapeutic strategies for various diseases by modulating the intracellular NQO1 enzyme activity and thereby regulating metabolites in vivo and in vitro. Therefore, we examined whether β-Lap activates the NQO1 enzyme, and modulates the intracellular NAD+/NADH ratio in vitro and in vivo . As shown in Figure 6A, NQO1 enzyme activity was elevated in the CF of A549 cells, where NQO1 was constitutively expressed, and this activity was further increased by β-Lap treatment; on the other hand, dicumarol, which is a specific inhibitor of NQO1, decreased its enzyme activity (Figure 6B). Because of enzyme activation, the intracellular NAD+/NADH ratio was increased by treatment of A549 cells with β-Lap (Figure 6C). In vivo, BLM induced a reduction of the NAD+/NADH ratio in lung tissue (Figure 6D), whereas β-Lap in combination with BLM, completely restored this ratio to the control level. The NAD+/NADH ratio after treatment with β-Lap alone was similar to that of the control group.
Figure 6. Effect of β-Lap on NQO1 activation and NAD+/NADH ratio, evaluated in vitro and in vivo. (A, B) Expression of NQO1 protein and assay for NQO1 enzymatic activation, and role of β-Lap and dicumarol from cytosolic fraction of A549 cell line. (C) Effect of β-Lap on intracellular NAD+/NADH ratio in A549 cells. (D) Effect of β-Lap on NAD+/NADH ratio in BLM-treated lung tissues of mice. NAD+/NADH ratio was analyzed following manufacture's instruction. *p<0.05 by one-way analysis of variance compared with the control, BLM, and β-Lap. β-Lap: β-lapachone; NQO1: NADH:quinone oxidoreductas; BLM: bleomycin.

Discussion
Lung fibrosis is a common consequence of ILD, and IPF is a common disease with unknown etiology that has few treatment options1,30. Moreover, IPF has been characterized as a chronic and progressive disease caused by the destruction of lung structure and failure of pulmonary function due to the proliferation of fibroblasts and excessive deposition of collagen and other ECM proteins. It has been reported that fibroblasts, the major cells of pulmonary fibrosis, receive signals from a variety of tissue cells and inflammatory cells, to proliferate and differentiate into myofibroblasts, and produce the ECM and TGF-β, thus triggering fibrosis31. The source of lung fibroblasts/myofibroblasts in IPF is not well-defined; however, possible sources include resident mesenchymal cells, marrow-derived circulating fibrocytes, and alveolar epithelial cells that undergo EMT32,33,34. Myofibroblasts, which have a phenotype intermediate between fibroblasts and smooth muscle cells, are defined by the presence of α-SMA, and they secrete excessive amounts of extracellular matrix, mainly collagens, resulting in scarring and destruction of the lung architecture.
Several drugs have been tested for IPF treatment; however, many clinical trials have yielded negative results for pharmacological agents3. Currently, lung transplantation is the best way to improve survival in IPF patients; however, this is not a feasible option for many patients. Therefore, pharmacological management of IPF is accepted as a more realistic approach. The immunosuppressive agents, azathioprine and prednisolone were considered the standard treatment regimen for IPF, given that inflammation is a key event in the pathogenesis of IPF; however, there is limited evidence for their use in IPF treatment. Another therapeutic agent widely used in IPF is N-acetylcysteine (NAC), which increases pulmonary glutathione levels and may restore the oxidant-antioxidant imbalance. Based on these findings, triple therapy with NAC and the two immunosuppressive agents was conducted in IPF patients; unfortunately, recent results suggest that triple therapy offers no benefit in treating IPF and is potentially harmful35. On the other hand, two antifibrotic agents (nintedanib and pirfenidone) were found to reduce the decline in lung function36,37. Pirfenidone was the first drug to be licensed for the treatment of IPF. It has antifibrotic, anti-inflammatory, and antioxidant effects, via inhibitory roles in multiple pathways related to the pathogenesis of IPF38. Nintedanib is a small molecular inhibitor that was initially found as an anticancer drug. It inhibits multiple tyrosine kinases associated with fibrogenesis, including the receptors for vascular endothelial growth factor, fibroblast growth factor, and platelet-derived growth factor36. Further research is required to assess the efficacy of these drugs in more advanced stages of the disease.
However, a recent study clearly demonstrated that the regulation of metabolism and subsequent control of intracellular metabolites is an emerging tool that might be more efficient than conventional strategies related to disease signaling cascades39. In light of this finding, our compound was designed to activate a flavoprotein NQO1 and elevate the NAD+ pool. NQO1 is an enzyme that detoxifies various quinonic compounds and their derivatives by catalyzing two-electron reductions, resulting in the prevention of redox cycling. In addition, NQO1 has several kinds of biological activities, including scavenging superoxide anions and stabilizing the tumorsuppressor p53. However, several studies have reported that intracellular NAD+ is important for cell survival in various diseases, such as axonal degeneration, cerebral ischemia, and cardiac hypertrophy. There are three pathways for NAD+ biosynthesis: the de novo pathway (from tryptophan), salvage pathway (from nicotinamide and nicotinic acid), and via NADH oxidation by NQO1. The latter pathway is very slow; therefore, NQO1 does not play a major role in the regulation of cellular NAD+ levels, in normal condition. However, many recent studies have reported that the increase of cellular NAD+ level by NQO1 enzymatic action using a strong substrate has a critical role in aging, arterial restenosis, hypertension, obesity, and renal injury25,26,27,28; thus, raising the possibility of its therapeutic use in treating various diseases via enzymatic activation of NAD+-dependent enzymes and downstream regulation of target proteins, including sirtuins (SIRTs). Kim et al.18 and Oh et al.19 reported that cisplatin-induced side effects, including ototoxicity and nephrotoxicity, are attenuated by the elevations of SIRT1 and 3 enzyme activities by the intracellular NAD+ increase. As an anticancer drug, the major limitation of cisplatin is its adverse side-effects due to oxidative stress and DNA damage. These studies showed that the enzymatic action of NQO1 by β-Lap elevates the intracellular NAD+/NADH ratio and subsequently activates SIRT1 and SIRT3 enzymes in the nucleus and mitochondria. SIRT1 and SIRT3, which are deacetylases, target acetylated nuclear factor κB (NF-κB) p65 in nuclei and acetylated p53 in mitochondria. Both acetylated p65 and p53 are major downstream molecules in cisplatinmediated auditory hearing loss and acute renal injury. In this study, we focused on the role of β-Lap in BLM-induced lung inflammation and fibrosis in the mouse model. β-Lap treatment attenuates BLM-induced lung injury, as assessed by gross of lung tissues, histology, and the number of infiltrating cells in the BALF (Figure 1). In addition, β-Lap co-administration suppressed the expression of IL-6, one of the important pro-inflammatory cytokines, in lung tissues (Figure 2). We also demonstrate that β-Lap attenuates BLM-induced lung fibrosis, evaluated by histology, picrosirius red staining, and hydroxyproline assay (Figure 3). Additionally, β-Lap reduces the expression of pro-inflammatory cytokine, tumor necrosis factor α, and myofibroblast differentiation marker, α-SMA, in lung tissues (Figure 4). These results suggest that β-Lap may prevent BLM-induced acute lung injury and chronic fibrosis by inhibiting inflammatory signaling and fibrosis. In addition to developing an in vivo mouse model, we showed that β-Lap reverses the TGF-β1–induced expression of collagen I, which is one of the well-known ECM proteins, and inhibits EMT and the morphological changes in A549 cells (Figure 5). Taken together, these results strongly suggest that β-Lap protects the lungs from BLM-induced inflammation and fibrosis.
In addition, we investigated whether β-Lap may activate NQO1 and elevate NAD+ in vitro and in vivo . β-Lap increased NQO1 enzyme activity in the CF of A549 cells, in a dosedependent manner; dicumarol, which is an NQO1 specific inhibitor, markedly inhibited its enzymatic activity (Figure 6A, B). Because of NQO1 activation, β-Lap elevated the intracellular NAD+/NADH ratio (Figure 6C); in addition, β-Lap restored the BLM-induced reduction of NAD+/NADH ratio in lung tissue (Figure 6D). In future studies, we will investigate the detailed mechanism of the effects of β-Lap in BLM-induced lung fibrosis.
In conclusion, we demonstrate that β-Lap activates NQO1 enzyme and subsequently protects against BLM-induced acute inflammatory responses and lung fibrosis by regulating potential mediators. BLM causes lung injury by acute inflammatory responses triggered by pro-inflammatory cytokines and a vicious cycle of NF-κB activation, and oxidative stress induces myofibroblast differentiation, ECM production, and fibrosis. Our results suggest that β-Lap elevates cellular NAD+ levels via NQO1 and may activate downstream target enzymes, such as SIRTs. Taken together, our results strongly suggest a potential therapeutic strategy for preventing BLM-induced lung inflammation and fibrosis by pharmacological activation of NQO1.
Acknowledgements
This study was supported by a 2013 Grant from The Korean Academy of Tuberculosis and Respiratory Diseases and the National Research Foundation of Korea (NRF) and funded by the Korean Government (Ministry of Science, ICT and Future Planning) [2011-0030130], and [2012R1A1A2043961]).
Footnotes
Conflicts of Interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Spagnolo P, Sverzellati N, Rossi G, Cavazza A, Tzouvelekis A, Crestani B, et al. Idiopathic pulmonary fibrosis: an update. Ann Med. 2015;47:15–27. doi: 10.3109/07853890.2014.982165. [DOI] [PubMed] [Google Scholar]
- 2.Prasad R, Gupta N, Singh A, Gupta P. Diagnosis of idiopathic pulmonary fibrosis: current issues. Intractable Rare Dis Res. 2015;4:65–69. doi: 10.5582/irdr.2015.01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Re. 2014;6:16. doi: 10.12703/P6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. 2012;5:11. doi: 10.1186/1755-1536-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenkins G, Goodwin A. Novel approaches to pulmonary fibrosis. Clin Med (Lond) 2014;14(Suppl 6):S45–S49. doi: 10.7861/clinmedicine.14-6-s45. [DOI] [PubMed] [Google Scholar]
- 6.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 8.Troy L, Corte T. Interstitial lung disease in 2015: where are we now? Aust Fam Physician. 2015;44:546–552. [PubMed] [Google Scholar]
- 9.Williamson JD, Sadofsky LR, Hart SP. The pathogenesis of bleomycin-induced lung injury in animals and its applicability to human idiopathic pulmonary fibrosis. Exp Lung Res. 2015;41:57–73. doi: 10.3109/01902148.2014.979516. [DOI] [PubMed] [Google Scholar]
- 10.Impellizzeri D, Talero E, Siracusa R, Alcaide A, Cordaro M, Maria Zubelia J, et al. Protective effect of polyphenols in an inflammatory process associated with experimental pulmonary fibrosis in mice. Br J Nutr. 2015;114:853–865. doi: 10.1017/S0007114515002597. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhary NI, Schnapp A, Park JE. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med. 2006;173:769–776. doi: 10.1164/rccm.200505-717OC. [DOI] [PubMed] [Google Scholar]
- 12.Phan SH, Kunkel SL. Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp Lung Res. 1992;18:29–43. doi: 10.3109/01902149209020649. [DOI] [PubMed] [Google Scholar]
- 13.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ido Y. Pyridine nucleotide redox abnormalities in diabetes. Antioxid Redox Signal. 2007;9:931–942. doi: 10.1089/ars.2007.1630. [DOI] [PubMed] [Google Scholar]
- 15.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 16.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Oh GS, Shen A, Lee SB, Choe SK, Kwon KB, et al. Augmentation of NAD(+) by NQO1 attenuates cisplatin-mediated hearing impairment. Cell Death Dis. 2014;5:e1292. doi: 10.1038/cddis.2014.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh GS, Kim HJ, Choi JH, Shen A, Choe SK, Karna A, et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014;85:547–560. doi: 10.1038/ki.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 21.Gaikwad A, Long DJ, 2nd, Stringer JL, Jaiswal AK. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem. 2001;276:22559–22564. doi: 10.1074/jbc.M101053200. [DOI] [PubMed] [Google Scholar]
- 22.Pardee AB, Li YZ, Li CJ. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets. 2002;2:227–242. doi: 10.2174/1568009023333854. [DOI] [PubMed] [Google Scholar]
- 23.Li LS, Bey EA, Dong Y, Meng J, Patra B, Yan J, et al. Modulating endogenous NQO1 levels identifies key regulatory mechanisms of action of beta-lapachone for pancreatic cancer therapy. Clin Cancer Res. 2011;17:275–285. doi: 10.1158/1078-0432.CCR-10-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Planchon SM, Wuerzberger S, Frydman B, Witiak DT, Hutson P, Church DR, et al. Beta-lapachone-mediated apoptosis in human promyelocytic leukemia (HL-60) and human prostate cancer cells: a p53-independent response. Cancer Res. 1995;55:3706–3711. [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang JH, Kim DW, Jo EJ, Kim YK, Jo YS, Park JH, et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes. 2009;58:965–974. doi: 10.2337/db08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SY, Jeoung NH, Oh CJ, Choi YK, Lee HJ, Kim HJ, et al. Activation of NAD(P)H:quinone oxidoreductase 1 prevents arterial restenosis by suppressing vascular smooth muscle cell proliferation. Circ Res. 2009;104:842–850. doi: 10.1161/CIRCRESAHA.108.189837. [DOI] [PubMed] [Google Scholar]
- 27.Kim YH, Hwang JH, Noh JR, Gang GT, Kim DH, Son HY, et al. Activation of NAD(P)H:quinone oxidoreductase ameliorates spontaneous hypertension in an animal model via modulation of eNOS activity. Cardiovasc Res. 2011;91:519–527. doi: 10.1093/cvr/cvr110. [DOI] [PubMed] [Google Scholar]
- 28.Lee JS, Park AH, Lee SH, Kim JH, Yang SJ, Yeom YI, et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS One. 2012;7:e47122. doi: 10.1371/journal.pone.0047122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustafson DL, Siegel D, Rastatter JC, Merz AL, Parpal JC, Kepa JK, et al. Kinetics of NAD(P)H:quinone oxidoreductase I (NQO1) inhibition by mitomycin C in vitro and in vivo. J Pharmacol Exp Ther. 2003;305:1079–1086. doi: 10.1124/jpet.103.050070. [DOI] [PubMed] [Google Scholar]
- 30.Green FH. Overview of pulmonary fibrosis. Chest. 2002;122(6 Suppl):334S–339S. doi: 10.1378/chest.122.6_suppl.334s. [DOI] [PubMed] [Google Scholar]
- 31.Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coward WR, Saini G, Jenkins G. The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2010;4:367–388. doi: 10.1177/1753465810379801. [DOI] [PubMed] [Google Scholar]
- 33.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517–523. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 35.Idiopathic Pulmonary Fibrosis Clinical Research Network. Raghu G, Anstrom KJ, King TE, Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 37.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:821–829. doi: 10.1183/09031936.00005209. [DOI] [PubMed] [Google Scholar]
- 39.Zhu H, Li Y. NAD(P)H: quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc Toxicol. 2012;12:39–45. doi: 10.1007/s12012-011-9136-9. [DOI] [PubMed] [Google Scholar]