

Abstract
Docetaxel has been the standard first-line therapy in metastatic castration resistant prostate cancer. The survival benefit is, however, limited by either primary or acquired resistance. In this study, Du145 prostate cancer cells were converted to docetaxel-resistant cells Du145-R and Du145-RB by in vitro culturing. Next generation RNAseq was employed to analyze these cell lines. Forty-two genes were identified to have acquired mutations after the resistance development, of which thirty-four were found to have mutations in published sequencing studies using prostate cancer samples from patients. Fourteen novel and 2 previously known fusion genes were inferred from the RNA-seq data, and 13 of these were validated by RT-PCR and/or re-sequencing. Four in-frame fusion transcripts could be transcribed into fusion proteins in stably transfected HEK293 cells, including MYH9-EIF3D and LDLR-RPL31P11, which were specific identified or up-regulated in the docetaxel resistant DU145 cells. A panel of 615 gene transcripts was identified to have significantly changed expression profile in the docetaxel resistant cells. These transcriptional changes have potential for further study as predictive biomarkers and as targets of docetaxel treatment.
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-016-3543-0) contains supplementary material, which is available to authorized users.
Keywords: Docetaxel resistance, Prostate cancer, RNAseq, Gene fusion, Mutation, Altered expression
Background
Most metastatic prostate cancers respond to androgen deprivation therapy (ADT) but eventually develop castration resistance and become metastatic castration resistant prostate cancers (mCRPC) about 24–36 months after the treatment start (Harris et al. 2009; Attar et al. 2009; Watson et al. 2010). mCRPC is the major cause of cancer death in prostate cancer patients. Median survival time of patients with mCRPCs is 16–18 months from the start of progression (Amaral et al. 2012). Docetaxel chemotherapy can further prolong the median overall survival by 3–5 months (Galsky et al. 2012). However, docetaxel resistance is a critical problem because half of patients will not respond to docetaxel treatment (intrinsic resistance), while the other half, which responds initially, become resistant ultimately (acquired resistance) (Tannock et al. 2004). Failure of docetaxel treatment has been thought to be caused by either intrinsic or acquired resistance.
Docetaxel is a member of taxane family and widely been used to treat mCRPC patients. Docetaxel induces cancer cell death by binding β-tubulin, stabilizing microtubule assembly, suppressing dynamics of individual micro-tubules in G2-M phase tumor cells and preventing disassembly (Yvon et al. 1999; Eisenhauer and Vermorken 1998). Despite a decade of clinical use, the mechanism of resistance to docetaxel has not been fully investigated and there are no clinically reliable biomarkers to predict the drug resistance. Limited data suggests that the resistance may be caused by the following mechanisms: (1) decreased drug concentration due to high expression of drug export pump proteins ABCB1, ABCB4, ABCC1 (Gottesman et al. 2002); (2) mutations in the drug targets (Berrieman et al. 2004); (3) inhibition of apoptotic pathways (Bhalla 2003); (4) altered expression profile of tubulins or microtubule-associated proteins (MAPs) (Seve and Dumontet 2008; Verrills et al. 2006). So far, only a few drugs have been developed with modest survival benefit in docetaxel resistant mCRPC.
This study applied next generation RNA sequencing (RNAseq) technology in combination with specific software (Ozsolak and Milos 2011) to determine gene expression changes, mutations and fusions in docetaxel sensitive cell lines versus docetaxel resistant cell lines. The comparison between these cell lines identified a panel of genes potentially involved in the development of docetaxel resistance. The clinical importance was further addressed by comparing with published RNA sequencing results in prostate cancer samples from patients.
Results
Mutations acquired in docetaxel resistant cell lines
We generated docetaxel resistant variants of Du145 prostate cancer cells as described in M&M. We used triplicates of each cell line (Du145, Du145-R and Du145-RB) for whole transcriptome RNA-sequencing and found 4864 mutations totally (Additional file 1). We compared TaxR (docetaxel resistant) and TaxS (docetaxel sensitive) cell lines to find mutations acquired after docetaxel treatment. Only mutations, which were absent in TaxS (Du145) but present in all Du145-R triplicates and Du145-RB triplicates, were chosen as “stably acquired mutations”. Forty-two such mutations were identified (Table 1) and 4 randomly selected mutations were validated by PCR followed by SANGER sequencing (Fig. 1).
Table 1.
Mutations acquired after docetaxel treatment
| Gene | Full name | Mutation | AA | Type | Non-synonymous | In prostate tumor samples? | |
|---|---|---|---|---|---|---|---|
| GALE | UDP-galactose-4-epimerase | TTT G[G/A]C AAT | Single AA change | G > D | Damaging | Yes | yes |
| KIAA1522 | KIAA1522 | Deletion | Frameshift | NA | Yes | yes | |
| ATP5F1 | ATP synthase, H + transporting, mitochondrial Fo complex subunit B1 | GCC AA[G/T] TGC | Single AA change | K > N | Damaging | Yes | |
| ZNF669 | Zinc finger protein 669 | GAA [C/T]AG TGT | Nonsense | Q > * | NA | Yes | yes |
| MDK | Midkine (neurite growth-promoting factor 2) | Deletion | Frameshift | NA | – | ||
| DDX23 | DEAD-box helicase 23 | GCT G[A/T]C AAA | Single AA change | D > V | Damaging | Yes | yes |
| SFSWAP | Splicing factor, suppressor of white-apricot homolog | GAG [A/G]GG AGT | Single AA change | R > G | Damaging | Yes | yes |
| TJP1 | Tight junction protein 1 | CCA C[G/A]T TTT | Single AA change | R > H | Damaging | Yes | yes |
| CASC4 | Cancer susceptibility candidate 4 | AAT AT[G/A] CCT | Single AA change | M > I | Damaging | Yes | |
| MRPL28 | Mitochondrial ribosomal protein L28 | CAG G[A/G]C CCC | Single AA change | D > G | Damaging | Yes | yes |
| STUB1 | STIP1 homology and U-box containing protein 1 | ATC G[C/T]G AAG | Single AA change | A > V | Damaging | Yes | yes |
| UQCRC2 | Ubiquinol-cytochrome C reductase core protein II | ACA A[A/C]A GGA | Single AA change | K > T | Damaging | Yes | yes |
| CHTF8 | Chromosome transmission fidelity factor 8 | CCC A[G/T]G TCA | Single AA change | R > M | Damaging | – | yes |
| KLHDC4 | Kelch domain containing 4 | GAC G[T/C]G TAT | Single AA change | V > A | Damaging | Yes | yes |
| SPATA20 | Spermatogenesis associated 20 | GTC [C/T]CT CAC | Single AA change | P > S | Damaging | Yes | yes |
| SMAD4 | SMAD Family Member 4 | Deletion | Frameshift | NA | Yes | yes | |
| LSM14A | LSM14A MRNA processing body assembly factor | CAG T[C/T]C ATG | Single AA change | S > F | Damaging | Yes | yes |
| CALM3 | Calmodulin 3 (phosphorylase kinase, delta) | GGG [G/A]AG AAG | Single AA change | E > K | NA | Yes | |
| MYADM | Myeloid-associated differentiation marker | TCC C[C/T]T CGG | Single AA change | P > L | Damaging | Yes | yes |
| ODC1 | Ornithine decarboxylase 1 | CAT G[T/C]G GGT | Single AA change | V > A | Damaging | Yes | yes |
| FOSL2 | FOS like antigen 2 | GAC [C/A]TG CAG | Single AA change | L > M | Damaging | Yes | yes |
| CYBRD1 | Cytochrome B reductase 1 | TTC [G/A]GG GCC | Single AA change | G > R | Damaging | Yes | |
| BOK | BCL2-related ovarian killer | GAC [T/C]GT GTG | Single AA change | C > R | Damaging | Yes | |
| ITCH | Itchy E3 ubiquitin protein ligase | AAT G[G/A]T GAA | Single AA change | G > D | Damaging | Yes | yes |
| DIP2A | Disco interacting protein 2 homolog A | AAC [G/A]TC TTC | Single AA change | V > I | Damaging | Yes | yes |
| BID | BH3 interacting domain death agonist | ACC [G/A]TA GCA | Single AA change | V > I | Damaging | Yes | yes |
| NUP210 | Nucleoporin 210 kDa | ATA [G/T]CC TAC | Single AA change | A > S | Damaging | Yes | yes |
| HYAL2 | Hyaluronoglucosaminidase 2 | CTG [C/T]GA CCT | Nonsense | R > * | NA | Yes | |
| RBM15B | RNA binding motif protein 15B | ACC CA[G/T] CTG | Single AA change | Q > H | Damaging | Yes | yes |
| CTBP1 | C-terminal binding protein 1 | TCC A[C/T]G CAG | Single AA change | T > M | Damaging | Yes | yes |
| TACC3 | Transforming acidic coiled-coil containing protein 3 | AGC T[C/T]T TCC | Single AA change | S > F | Damaging | Yes | yes |
| AFAP1 | Actin filament associated protein 1 | TCA [G/C]AG GCC | Single AA change | E > Q | Damaging | Yes | yes |
| MCTP1 | Multiple C2 and transmembrane domain containing 1 | ATG G[G/T]C TCA | Single AA change | G > V | Damaging | Yes | yes |
| MAN2A1 | Mannosidase alpha class 2A member 1 | CTT A[T/A]C CAG | single aa change | I > N | Damaging | Yes | yes |
| C5orf15 | Chromosome 5 open reading frame 15 | Deletion | Frameshift | NA | Yes | ||
| UIMC1 | Ubiquitin interaction motif containing 1 | GAA G[C/A]T AGG | Single AA change | A > D | Damaging | Yes | yes |
| MAML1 | Mastermind like transcriptional coactivator 1 | Deletion | Frameshift | NA | Yes | yes | |
| C6orf141 | Chromosome 6 open reading frame 141 | CGG [G/T]GG CCT | Single AA change | G > W | Damaging | Yes | yes |
| CROT | Carnitine O-octanoyltransferase | TTT [T/C]CC AAA | Single AA change | S > P | Damaging | Yes | yes |
| CAPZA2 | Capping actin protein of muscle Z-line alpha subunit 2 | AGG A[A/C]G GAG | Single AA change | K > T | Damaging | Yes | yes |
| ASB6 | Ankyrin Repeat and SOCS box containing 6 | AAC [C/T]GC TTC | Single AA change | R > C | Damaging | Yes | yes |
| ABCA2 | ATP binding cassette subfamily a member 2 | GGC C[G/A]C TTC | Single AA change | R > H | Damaging | Yes | yes |
Mutation and amino acid changing were showed in column 3 and 4. Mutation Type was analyzed by program SIFT and gained by setting cutoff as 0.05. ‘Damaging’ means that the substitution is predicted to affect protein function. NA not analyzed. The last column shows the connection between cell lines and tumor samples. It is labeled “yes” if mutation can be found in both cell line sequence and tumor sample sequence
Fig. 1.
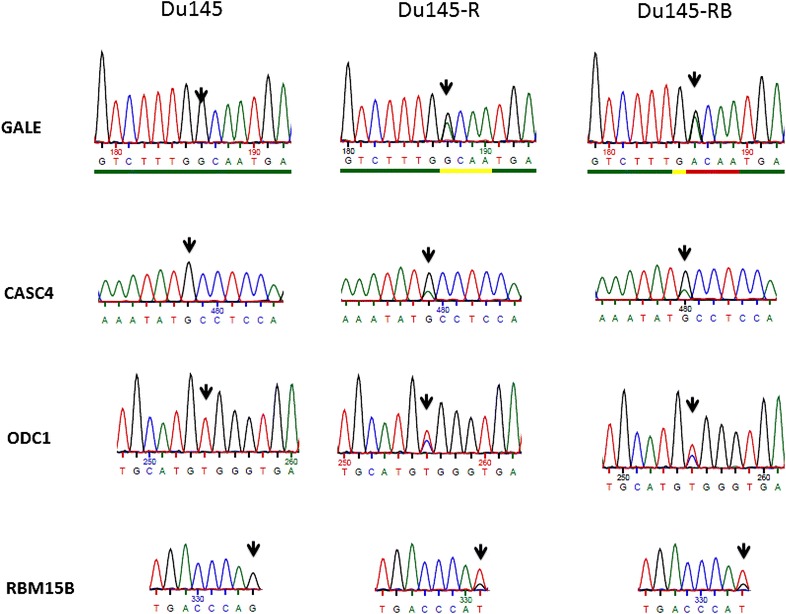
Mutation list validated by PCR followed by Sanger sequencing. Black arrow shows the mutation positions
By matching with previously published whole transcriptome analyses, we could identify that 34 of these genes had mutations in prostate cancer samples from patients (Table 1) (Robinson et al. 2015). For many genes, e.g. ABCB2, there are published data to support their importance in the development of drug resistance (Aberuyi et al. 2014; Rahgozar et al. 2014).
Fusion transcript detection and validation
ChimeraScan software was employed to find fusions from RNAseq data. Selecting gene–gene pairs supported by two or more unique alignment reads provided an initial list of 48, 75 and 66 fusion candidates in DU145, Du145-R and Du145-RB cell lines respectively (Additional file 2). We validated all fusion candidates that had a ChimeraScan score above 5 in at least 1 out of 3 cell lines. Of 16 fusion candidates selected (Table 2), 13 (81.25 %) were verified by Reverse Transcription PCR (RT-PCR) with primers covering the fusion break points (Additional file 3), and 5 of validated genes were further verified by Sanger sequencing in the Du145, Du145-R and Du145-RB cell lines (Fig. 2). Two gene fusions had been found by previous studies: UBE2L3-KRAS (Wang et al. 2011) expressed in all three cell lines and TAF15-AP2B1 (http://54.84.12.177/PanCanFusV2/Fusions!fusion) specific expressed in Du145 (Additional file 3). The other fourteen fusions were novel discovered.
Table 2.
Fusion transcripts identified by NGS and validated by PCR
| 5′ gene (full name) | 5′ chr | 3′ gene (full name) | 3′ chr | Type | Verified | Express in TaxS | Express in TaxR |
|---|---|---|---|---|---|---|---|
| TAF15 (TATA-box binding protein associated factor 15) | 17 | AP2B1 (adaptor related protein complex 2 beta 1 subunit) | 17 | Read through | Yes | Yes | No |
| VCL (vinculin) | 10 | ADK (adenosine kinase) | 10 | Read through | Yes | Yes | No |
| MYH9 (myosin, heavy chain 9, non-muscle) | 22 | EIF3D (eukaryotic translation initiation factor 3 subunit D) | 22 | Read through | Yes | No | Yes |
| C14orf166 (chromosome 14 open reading frame 166) | 14 | SLC25A21 (solute carrier family 25 member 21) | 14 | Intra chromosomal | Yes | Yes | Yes |
| UBE2L3 (ubiquitin conjugating enzyme E2 L3) | 22 | KRAS (kirsten rat sarcoma viral oncogene homolog) | 12 | Inter chromosomal | Yes | Yes | Yes |
| LDLR (low density lipoprotein receptor) | 19 | RPL31P11 (ribosomal protein l31 pseudogene 11) | 15 | Read through | Yes | Yes | Yes (up regulated) |
| IGSF9B (immunoglobulin superfamily member 9B) | 11 | FAM177A1 (family with sequence similarity 177 member A1) | 14 | Inter chromosomal | No | ||
| CTSD (cathepsin D) | 11 | IFITM10 (interferon induced transmembrane protein 10) | 11 | Read through | Yes | Yes | Yes (up regulated) |
| FLJ39739 | 1 | BC065231 | 1 | Intra chromosomal | Yes | Yes | Yes |
| LOC100286793 | 1 | BC065231 | 1 | Intra chromosomal | Yes | Yes | Yes |
| UBE2H (ubiquitin conjugating enzyme E2 H) | 7 | WIZ (widely interspaced zinc finger motifs) | 19 | Inter chromosomal | No | ||
| SFPQ (splicing factor proline/glutamine-rich) | 1 | AL831889 (LOC100996496) | 1 | Read through | Yes | Yes | Yes |
| CADM4 (cell adhesion molecule 4) | 19 | ZNF428 (zinc finger protein 428) | 19 | Read through | Yes | Yes | Yes |
| GOLT1A (golgi transport 1A) | 1 | KISS1 (KiSS-1 metastasis-suppressor) | 1 | Read through | Yes | Yes | Yes |
| SRGAP2P2 (SLIT-ROBO Rho GTPase activating protein 2B) | 1 | SRGAP2 (SLIT-ROBO Rho GTPase activating protein 2) | 1 | Intra chromosomal | Yes | Yes | Yes (up regulated) |
| BTNL8 (butyrophilin like 8) | 5 | HMGA1 (high mobility group AT-Hook 1) | 6 | Inter chromosomal | No |
5′ end genes and their information were listed in column 1 and 2, and 3′ end genes in column 3 and 4. Read Through, new fusion gene can be read through when translated. Intrachromosomal, fusion partners come from same chromosomes. Interchromosomal, fusion formed between different chromosomes. Last column was marked ‘Yes’ if fusions can be verified in Du145, Du145-R or Du145-RB
Fig. 2.
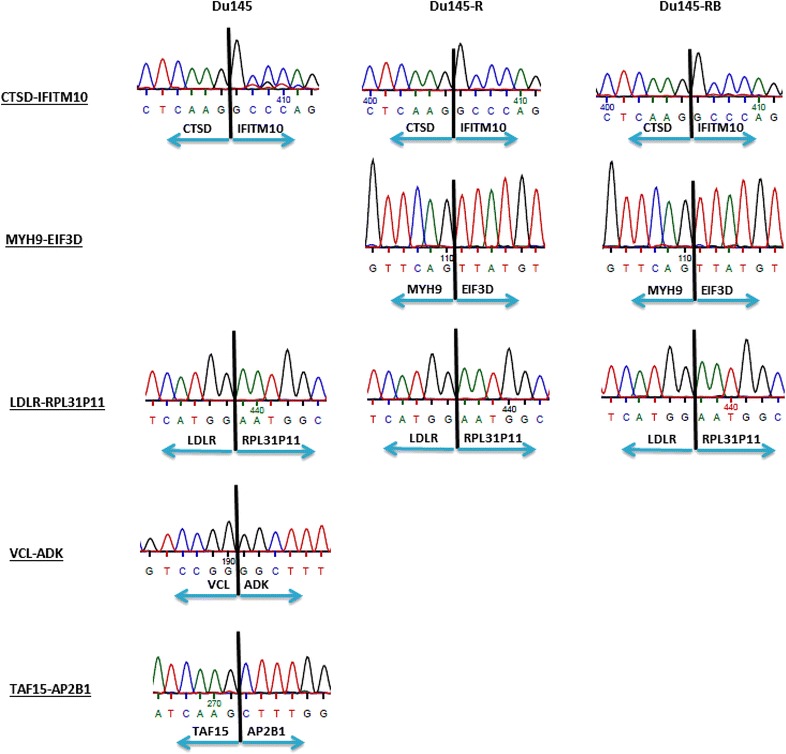
Sanger-sequencing validation of 5 fusion candidates discovered by NGS. Black lines indicates the fusion points between 2 genes
Figure 3a showed that all chromosomes were involved in gene fusion except chr 21 and chr Y. The two largest fusion groups were distributed in chr1 (27, 14.3 %) and chr6 (44, 23.3 %), and most of the fusions were intra-chromosomal (26 out of 27 in chr1; 39 out of 44 in chr6). Of the 16 chosen fusion candidates (3 of them could not be validated by PCR), 10 of them were commonly expressed in all 3 cell lines, one expressed only in TaxR cell line (MYH9-EIF3D) and 2 specifically in TaxS cell line (TAF15-AP2B1, VCL-ADK) (Table 2; Fig. 3b). Among 10 commonly expressed fusions, two were up-regulated in TaxR cell lines compared to TaxS cell line (LDLR-RPL31P11, SRGAP2P2-SRGAP2). Eight out of 16 were predicted to be in-frame suggesting their potential to produce functional fusion proteins (Table 2).
Fig. 3.
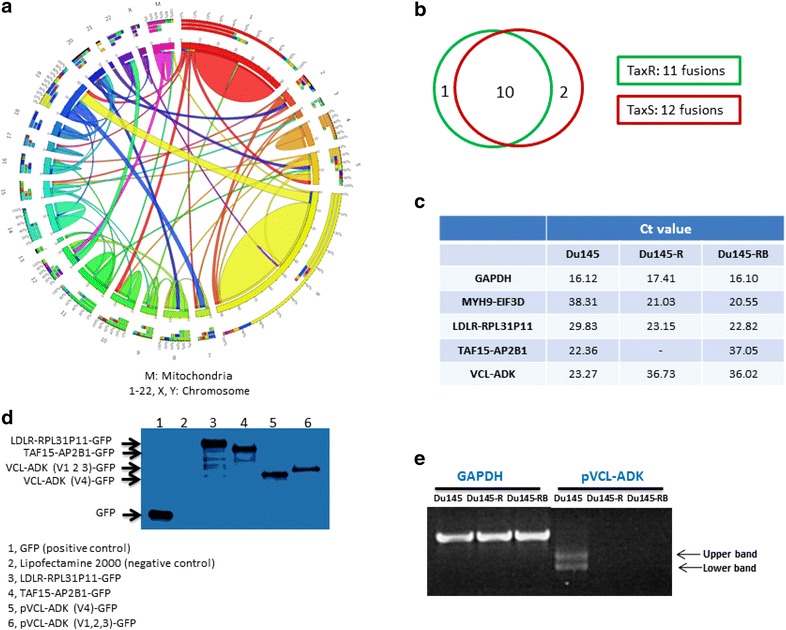
Landscape and validation of fusion candidates. a Circos plot of the genomic landscape of gene fusions discovered by RNAseq. b Venn diagram analysis of 13 fusions which could be validated by PCR. c qPCR validation of fusions in Du145, Du145-R and Du145-RB. d Fusion proteins detected in transfected HEK293 cells by Western blot. e Two different fusion transcript types, that is between VCL-ADK variants 1, 2, 3 and VCL-ADK variant 4
Four fusion candidates were validated by qPCR in Du145, Du145-R and Du145-RB cells, as well as at the protein level by western blot in plasmid transfected HEK293 cells (Fig. 3c, d), but their translation into protein could not be validated by western blot in Du145, Du145-R and Du145-RB cell lines.
Interestingly, when we validated the VCL-ADK fusion candidate by PCR, we found that there were two bands in the same PCR lane (Fig. 3e). Sanger sequencing results showed that both of the two bands were VCL-ADK fusions. The upper band was a fusion between VCL and ADK variant 1, 2 and 3, while the lower band was another fusion with ADK variant 4. Western blot showed that both fusions (VCL-ADK variant 1, 2, 3 and VCL-ADK variant 4) could be detected as protein in plasmid transfected HEK293 cell lines (Fig. 3d).
Identification of stably up- or down-regulated genes in the TaxR cell lines
Using gene expression of parental Du145 (TaxS) cells as a baseline, we identified 453 up-regulated and 473 down-regulated genes in the Du145-R cells, and 483 up- and 365 down-regulated genes in the Du145-RB cells (Additional file 4). In addition, we found 216 genes with significantly different expression levels between DU145-RB and DU145-R. These 216 genes were presumably not related to the development of docetaxel resistance. By matching the three gene lists we further identified 615 (329 up-regulated and 286 down-regulated) genes that were shared by both DU145-R and DU145-RB as compared with DU145 (Additional file 4). These genes were thought to have stable expression changes after acquiring resistance to docetaxel. Of the 615 genes, the 40 most up- and down-regulated in the TaxR cell lines were chosen for verification by RT-PCR and 37/40 (92.5 %) were confirmed (Additional file 5).
Information about the most-differentially-expressed genes is shown in Additional file 6. The second most up-regulated gene was ABCB1, which encodes an ATP-dependent drug efflux pump that mediates the development of resistance to anticancer drugs (Gottesman et al. 2002). The average fold changes (log) in TaxR cell lines were up to 8.9 and 10.2 in up-regulated genes and down-regulated genes, respectively. The largest functional group was transcription factors (Additional file 7). Twenty-one oncogenes and 16 translocated cancer genes were also among the enriched functional groups in the set of 615 stably differentially-expressed genes.
The 615 most significantly deregulated genes were put into the Panther Online tool (www.pantherdb.org), which yielded 528 functional hits distributed on 11 GO-terms, where the two largest groups were Binding (GO: 0005488) and Catalytic Activity (GO: 0003824) (Fig. 4b). Thomson Reuters was employed to analyze enriched networks of expression changing genes and showed that the NF-κb, EGR1 (Early Growth Response 1) and ETS (ETS family of transcription factors) were the three most enriched networks in the docetaxel resistant cells (Fig. 4c and Additional file 8). PLAU and PLAUR (Plasminogen Activator, Urokinase Receptor), a ligand—membrane receptor pair, are the only ‘Convergence hubs’ and MDR1 (ABCB1) was connected to all three pathways.
Fig. 4.
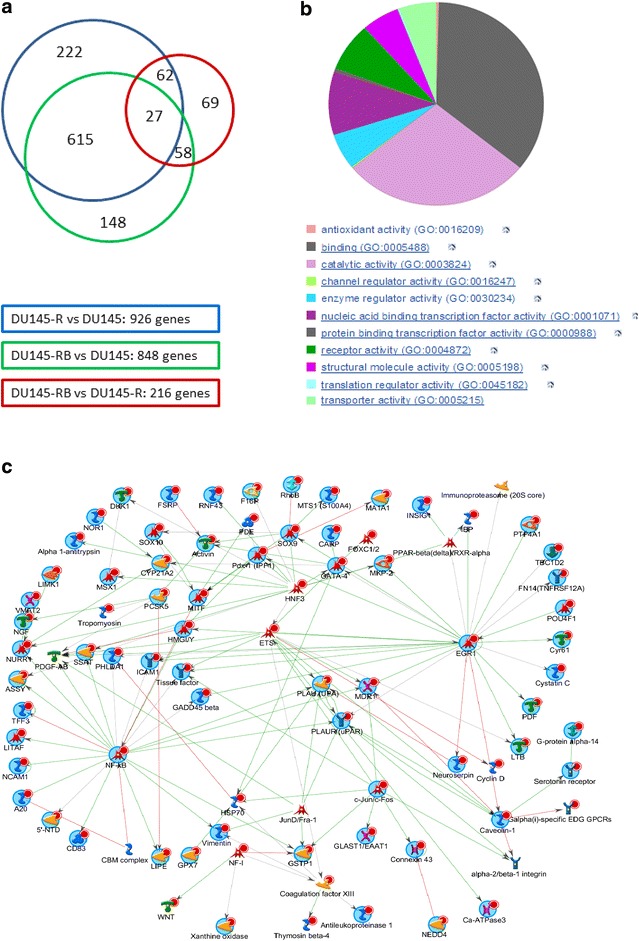
NGS analysis in TaxS and TaxR prostate cancer cell lines identifies gene expression changes and pathway networks involved in docetaxel resistance. a Venn diagram of overlap of significantly differently regulated genes by matching 3 gene lists. b Pie chart of 615 genes with stable gene expression changes in TaxR cells lines by Panther (www.pantherdb.org). c GeneGo (Thomsom Reuters) network analysis of the most deregulated genes in TaxR cell lines
Next generation sequencing data of PC3 and LNCaP, two docetaxel-sensitive cell lines similar to Du145, were added into further analysis. Multivariate modeling with SIMCA resulted in a model, which separated all TaxS and TaxR cell lines into two classes and extracted those genes that contributed most to the model (Table 3).
Table 3.
Genes contributed most to separate prostate cancer cell lines into TaxR and TaxS classes
| Gene symbol | Full name | M1.VIPpred |
|---|---|---|
| ABCB1 | ATP-binding cassette, sub-family B (MDR/TAP), member 1 | 1.48587 |
| GPSM2 | G-protein signaling modulator 2 | 1.48037 |
| IL31RA | Interleukin 31 receptor A | 1.47976 |
| LIMK1 | LIM domain kinase 1 | 1.47921 |
| GRK5 | G protein-coupled receptor kinase 5 | 1.47789 |
| SKAP1 | Src kinase associated phosphoprotein 1 | 1.47782 |
| ST6GALNAC5 | Sialyltransferases | 1.4765 |
| C9orf125 | Transmembrane protein 246 | 1.47636 |
| ICAM1 | Intercellular adhesion molecule 1 | 1.47543 |
M1.VIPpred, score shows the contribution to the SIMCA model. All 9 genes in this table were up-regulated in the TaxR cells
When comparing the list of 615 stably up- or down-regulated gene lists with the fusion gene list, we found 6 genes in common (Table 4), all of which were up-regulated.
Table 4.
Fusion genes whose expression levels were also up-regulated in TaxR cell lines
| Gene | Full name | Fold up-regulation | Fusion partner | Full names of fusion partners |
|---|---|---|---|---|
| HIVEP2 | Human immunodeficiency virus type I enhancer binding protein 2 | 1.4 | JA040725 | JA040725 |
| HMGA1 | High mobility group AT-hook 1 | 1.8 | BTNL8 | Butyrophilin like 8 |
| PTRF | Polymerase I and transcript release factor | 1.7 | ABCA9 | ATP binding cassette subfamily A member 9 |
| RPL31P11 | Ribosomal protein L31 pseudogene 11 | 3.9 | LDLR | Low density lipoprotein receptor |
| VCL | Vinculin | 2.3 | ADK | Adenosine kinase |
| VIM | Vimentin | 4.9 | SYCP1 | Synaptonemal complex protein 1 |
Fold up-regulation, log2 fold expression change of genes in column 1 comparing TaxS cell line and TaxR cell lines
Methods
Prostate cancer cell lines
Table 5 summarizes the prostate cancer cell lines used in this study. LNCaP, PC3 and DU145 cell lines were originally ordered from the ATCC (American Type Culture Collection). Du145 was cultured in medium containing docetaxel (from low concentration to high concentration, increased gradually) for one year, until Du145 acquired docetaxel resistance (Du145-R). We also cultured Du145-R in normal medium without docetaxel for one month (Du145-RB) to see if it would revert to docetaxel sensitive again (Kharaziha et al. 2015). DU145-RB was frozen after one month cultured without docetaxel. Every time when we needed to use DU145-RB, we would thaw and culture it in normal medium (without docetaxel) and the culture time would not extend 4 weeks. While, DU145-R cells were always cultured in medium with 1000 ng/ml docetaxel. DU145, PC3 and LNCaP cells were cultured in medium without docetaxel.
Table 5.
Prostate cancer cell lines which were analyzed by whole transcriptome sequencing
| Cell line | Androgen-dependent | Docetaxel-sensitive | Triplicates |
|---|---|---|---|
| LNCaP | Yes | Yes | Yes |
| PC3 | No | Yes | Yes |
| DU145 | No | Yes | Yes |
| DU145-R | No | No | Yes |
| Du145-RB | No | No | Yes |
Androgen-dependent, cell line is sensitive to hormone treatment (Yes) or not (No). Docetaxel-sensitive, cell line is sensitive to docetaxel (Yes) or not (No). Triplicates, all cell lines were triplicates when sent to be sequenced
RNAseq
Total RNA from prostate cancer cell lines was isolated by TRIzol (Invitrogen, Catalog #15596018) and extracted by subsequent phenol/chloroform. RNase-free DNase set (Qiagen, Catalog #79254) was used to remove DNA by DNase digestion. RNA quality was controlled by RNA Integrity Number (RIN) analysis by Agilent 2100 Bioanalyzer System. Total RNA samples were sent to SciLifeLab, Stockholm, Sweden and polyA selection was done at SciLifeLab. Samples were clustered on cBot and sequenced on HiSeq 2000 according to manufacturer’s instructions. Between 16.0 and 76.3 million reads were obtained per sample sequenced on HiSeq 2000.
Variant calling method
Removal of PCR duplicates was performed with Picard (picard.sf.net). After that the reads were extracted from bam file, imported into CLC Genomics workbench (CLC, Aarhus, Denmark) and aligned to the human reference genome (build 37p5) using Large Gap Read Mapping. Variant calling was conducted using Probabilistic Variant Detection tool within CLC Genomics workbench. The following criteria were applied for variant calling: (1) ignore non-specific matches, (2) Minimum coverage, and (3) Variant probability 90. The variations were filtered out if detected in any of 190 control exomes from non-cancer patients, or were dbSNP v137 reported SNPs, with a population frequency higher than 1 % in dbSNP v137. The variants were annotated according to their overlap with genes and transcripts (UCSC, refSeq at http://genome.ucsc.edu/, and Sanger cancer census gene at http://cancer.sanger.ac.uk/cancergenome/projects/census/), conservation scores (UCSC), segmental duplications (UCSC), exon number, splice sites, amino acid change, cosmic database v63, ClinVar (a database of mutations and their clinical relevance at ftp://ncbi.nlm.nih.gov/pub/clinvar/), dbSNP v137 and predictions from Provean (http://provean.jcvi.org), Sift (http://sift.jcvi.org) and Polyphen (http://genetics.bwh.harvard.edu/pph2/bgi.shtml).
Analysis of differentially expressed genes
We analyzed RNAseq data according to a published TopHat and Cufflinks protocol (Trapnell et al. 2012). In summary, we used TopHat to align reads to the reference genome, Cufflinks to assemble and obtain expression values for all transcripts, Cuffdiff for testing differential expression of genes and transcripts and finally the CummeRbund R package for downstream analysis and visualization.
Fusion detection method
We used ChimeraScan, which aligns paired-end reads to a reference genome-transcriptome with Bowtie in an iterative process where read pairs that could not be aligned were trimmed into smaller fragments and realigned (Iyer et al. 2011). ChimeraScan uses a filter to avoid false-positive chimeras.
Statistical analysis
The online services Panther (http://www.pantherdb.org) and Thomson Reuters were applied for functional enrichment analysis (Mi et al. 2013; Huber-Keener et al. 2012). Fusion transcripts from Du145, Du145-R and Du145-RB were visualized by Circos online (http://mkweb.bcgsc.ca/tableviewer) (Krzywinski et al. 2009). OPLS-DA model was established by SIMCA software, and 2 classes (TaxS and TaxR) were set in the model to obtain VIP scores by whichvariables (genes) are sorted based on importance (contribution to the model) of genes (Bylesjo et al. 2006).
PCR and qPCR validation
Total RNA was isolated from cell lines by TRIzol (Invitrogen, Catalog #15596018) according to the manufacturer’s instructions. Cloned AMV First-Strand Synthesis Kit (Life Technologies, Catalog #12328) was used to transcribe mRNA to cDNA.
PCR primers for fusion validation were designed according to the sequence of fusion transcripts. Forward primer was located on the 5′ gene of the fusion gene and reverse primer on the 3′ gene of the fusion gene. Primers for validation of mutations covered mutation points. PCR was conducted using Platinum Taq DNA polymerase (Life Technologies, Catalog #10966018) and was followed by Sanger sequencing (conducted by Eurofins Genomics). To differentiate gene expression levels of selected genes, 20–32 amplifying cycles were used based on gene expression level.
qPCR primers for fusion validation were purchased from Applied Biosystems (Custom plus TaqMan RNA Assays). LightCycler 480 Probes Master was used combined with TaqMan primer on LightCycler 480 instrument from Roche according to manufacturer’s instructions.
Plasmid construction and western blot validation
PCR product was ligated into multiple cloning sites of pCMV-AC-GFP after digestion of restriction enzymes, Sgf I and Mlu I. pCMV-AC-GFP was purchased from ORIGENE (Catalog #PS100010), Sgf I enzyme from NEB (Catalog #R0630S), Mlu I enzyme from NEB (Catalog #R0198S), and T4 ligase from Promega (Catalog #M180A). We transfected HEK293 cells with constructed plasmid 48 h before collecting cells in lysis buffer. Western blot experiments were conducted using these cell lysates. Anti-TurboGFP antibody was purchased from Evrogen (Catalog #AB513).
Discussion
We have identified 42 genes with specific and stable mutations in TaxR cells. The functions of these genes may support their importance in the development of docetaxel resistance. Among these genes, SMAD4 is a co-activator and mediator of signal transduction by TGF-beta and acts as a tumor suppressor. Experiments have shown that SMAD4 inactivation promotes drug resistance in cancer (Zhang et al. 2014; Raz et al. 2014). ABCA2 is a member of ATP-binding cassette (ABC) transporters that transports many kinds of small molecules through membranes and is involved in drug resistance in leukemia cell lines (Dharmapuri et al. 2015).
Approximately 50 % of prostate cancer has primary resistance to docetaxel treatment. The other half is sensitive to docetaxel but eventually develops secondary (acquired) resistance (Marin-Aguilera et al. 2012). In this study, 34 out of the 42 mutations discovered in the resistant cell lines can be found in tumor samples from patients (Table 1), implicating that primary and acquired resistance may share the same molecular mechanism(s). In the case of primary resistance, most cancer cells carry the resistant genomic changes before the treatment, whereas for acquired resistance, just a few cancer cells carry these resistant genomic changes before treatment. By treatment selection or new mutational events, most cancer cells become carriers of resistant genomic changes. This hypothesis can be further tested in studies using tumor samples from patient cohorts with data of docetaxel treatment.
The four fusion transcripts (listed in Fig. 3c: MYH9-EIF3D, LDLR-RPL31P11, TAF15-AP2B1, VCL-ADK) could be detected by PCR and qPCR in the cell lines, but their translation into protein could not be validated by western blot in Du145, Du145-R and Du145-RB, probably due to the low expression of the fusion proteins. Fusion transcripts could be translated into protein in stably transfected HEK293 cells analyzed by western blot. Moreover, several genes involved in the fusion events have shown important functions in cancer development. TAF15, a member of the FET family, has been found rearranged with various transcription factors with cancer promoting functions in sarcomas as well as in rare hematopoietic and epithelial cancers (Kovar 2011). MYH9 is a member of the myosin superfamily and its function is related to migration, invasion and metastasis of cancer cells. EIF3D is associated with cell cycle regulation and motility of prostate cancer cells (Gao et al. 2015). MYH9 fusion proteins have been found in anaplastic large cell lymphoma and one example is the MYH9-ALK fusion protein that has tyrosine kinase activity in vivo (Lamant et al. 2003). The MYH9-USP6 detected by a previous study and MYH9-EIF3D found in the present study have the same fusion point in MYH9. MYH9, which is located in the 5′ part of the fusion product, functions as a regulator to manipulate gene expression and function of USP6, as well as EIF3D (Erickson-Johnson et al. 2011). These functional implications may encourage further verification by using tumor samples from the patients.
When we compared the expression of DU145-RB and DU145-R, we found 216 genes that were differently expressed. We tested that DU145-RB was still docetaxel resistant, indicating these genes were not involved in maintaining docetaxel resistance of the two resistant cell lines. As expected, ABCB1 (MDR1) was confirmed as one of the top 10 differentially expressed genes that could separate TaxR from TaxS cells. Its functional importance was further supported by its connection with the NF-κb, EGR1 and ETS pathways (Fig. 4). ABCB1, which shows overexpression in some cancers, is involved in a common resistance mechanism. However, limited studies showed significant connection between ABCB1 and clinical outcomes, such as survival (Shaffer et al. 2012), indicating the importance of other molecular and biological changes. Researchers and pharmaceutical companies are trying to circumvent this strategy and find new potential genes or pathways to overcome resistance in cancer.
TGPSM2 and GRK5 are members of G-protein signaling pathway important in cancer progression. SKAP1 encodes a src kinase associated phosphoprotein 1 and is a member of the Ras signaling pathway and B cell receptor signaling pathway. LIMK1 is a serine/threonine kinase associated with the cytoskeletal structure in many cellular processes, and may have importance in the sensitivity of lung cancer and osteosarcoma cells to chemotherapy treatment (Chen et al. 2013; Zhang et al. 2011). The analysis further showed that PLAU and PLAUR (Plasminogen Activator, Urokinase Receptor), a pair of ligand and membrane receptor, constituted the only ‘Convergence hub’ by statistical analysis using the Thomson Reuters software. This novel finding may suggest that they may play a unique role in docetaxel resistance. It would be interesting to further study if they alone or, together with other important genomic findings in this study, can be further verified as important biomarkers to predict primary docetaxel resistance. Most importantly, they can even become attractive targets for the development of new drugs to overcome both primary and acquired docetaxel resistance.
Conclusion
The present study found both previous and novel mutations, genes with altered expression levels, and fusion proteins in docetaxel resistant prostate cancer cell lines, and provide some understanding of acquired docetaxel resistance at the gene transcription level. If some of these changes can be further verified with importance in primary resistance, they can be considered as predictive biomarkers for docetaxel treatment as well as targets for the development of new treatments to overcome the docetaxel resistance.
Authors’ contributions
Yuanjun Ma, Chunde Li and Sten Nilsson designed the study plan. Sten Nilsson, Lena Lennartsson, Zhuochun Peng and Chunde Li provided materials. Yuanjun Ma, Yali Miao and Zhuochun Peng performed all experiments. Yuanjun Ma, Johanna Sandgren, Teresita Díaz De Ståhl, Mikael Huss, Yanling Liu and Chunde Li analyzed data together. Yuanjun Ma and Chunde Li wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank SciLifeLab, Stockholm for their RNAseq technical support. The study was supported by research grants from the Swedish Cancer Society (Cancerfonden), the The Cancer Research Foundations of Radiumhemmet and the Stockholm’s County Council.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1. Mutation in all TaxS and TaxR cell lines.
Additional file 2. Fusions in all TaxS and TaxR cell lines.
Additional file 3. PCR validation of fusion candidates.
Additional file 4. Expression changing genes in TaxR cell lines.
Additional file 5. PCR validation of expression changing genes.
Additional file 6. Most differentially-expressed genes in TaxR cell lines.
Additional file 7. Distribution of 615 genes with stable expression level changes in TaxR cell lines.
Additional file 8. Hubs in the network analysis.
Footnotes
An erratum to this article is available at http://dx.doi.org/10.1186/s40064-016-3759-z.
Contributor Information
Yuanjun Ma, Email: yuanjun.ma@ki.se.
Yali Miao, Email: miaoyali2006@163.com.
Zhuochun Peng, Email: zhuo-chun.peng@ki.se.
Johanna Sandgren, Email: johanna.sandgren@ki.se.
Teresita Díaz De Ståhl, Email: teresita.diaz.de.stahl@ki.se.
Mikael Huss, Email: mikael.huss@scilifelab.se.
Lena Lennartsson, Email: Lena.Lennartsson@ki.se.
Yanling Liu, Email: yleen.liu@outlook.com.
Monica Nistér, Email: Monica.Nister@ki.se.
Sten Nilsson, Email: Sten.Nilsson@ki.se.
Chunde Li, Email: chunde.li@ki.se.
References
- Aberuyi N, Rahgozar S, Moafi A. The role of ATP-binding cassette transporter A2 in childhood acute lymphoblastic leukemia multidrug resistance. Iran J Pediatr Hematol Oncol. 2014;4:118–126. [PMC free article] [PubMed] [Google Scholar]
- Amaral TM, Macedo D, Fernandes I, Costa L. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer. 2012;2012:327253. doi: 10.1155/2012/327253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res. 2009;15:3251–3255. doi: 10.1158/1078-0432.CCR-08-1171. [DOI] [PubMed] [Google Scholar]
- Berrieman HK, Lind MJ, Cawkwell L. Do beta-tubulin mutations have a role in resistance to chemotherapy? Lancet Oncol. 2004;5:158–164. doi: 10.1016/S1470-2045(04)01411-1. [DOI] [PubMed] [Google Scholar]
- Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- Bylesjo M, Rantalainen M, Cloarec O, Nicholson JK, Holmes E, Trygg J. Opls discriminant analysis: combining the strengths of PLS-DA and simca classification. J Chemom. 2006;20:341–351. doi: 10.1002/cem.1006. [DOI] [Google Scholar]
- Chen Q, Jiao D, Hu H, Song J, Yan J, Wu L, et al. Downregulation of LIMK1 level inhibits migration of lung cancer cells and enhances sensitivity to chemotherapy drugs. Oncol Res. 2013;20:491–498. doi: 10.3727/096504013X13657689382699. [DOI] [PubMed] [Google Scholar]
- Dharmapuri G, Doneti R, Philip GH, Kalle AM. Celecoxib sensitizes imatinib-resistant K562 cells to imatinib by inhibiting MRP1-5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk Res. 2015;39:696–701. doi: 10.1016/j.leukres.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Vermorken JB. The taxoids. Comparative clinical pharmacology and therapeutic potential. Drugs. 1998;55:5–30. doi: 10.2165/00003495-199855010-00002. [DOI] [PubMed] [Google Scholar]
- Erickson-Johnson MR, Chou MM, Evers BR, Roth CW, Seys AR, Jin L, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9–USP6 gene fusion. Lab Invest. 2011;91:1427–1433. doi: 10.1038/labinvest.2011.118. [DOI] [PubMed] [Google Scholar]
- Galsky MD, Small AC, Tsao CK, Oh WK. Clinical development of novel therapeutics for castration-resistant prostate cancer: historic challenges and recent successes. CA Cancer J Clin. 2012;62:299–308. doi: 10.3322/caac.21141. [DOI] [PubMed] [Google Scholar]
- Gao Y, Teng JF, Hong Y, Qu FJ, Ren JZ, Li L, et al. The oncogenic role of EIF3D is associated with increased cell cycle progression and motility in prostate cancer. Med Oncol. 2015;32:518. doi: 10.1007/s12032-015-0518-x. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP—dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber-Keener KJ, Liu X, Wang Z, Wang Y, Freeman W, Wu S, et al. Differential gene expression in tamoxifen-resistant breast cancer cells revealed by a new analytical model of RNA-seq data. PLoS ONE. 2012;7:e41333. doi: 10.1371/journal.pone.0041333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Chinnaiyan AM, Maher CA. Chimerascan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharaziha P, Chioureas D, Rutishauser D, Baltatzis G, Lennartsson L, Fonseca P, et al. Molecular profiling of prostate cancer derived exosomes may reveal a predictive signature for response to docetaxel. Oncotarget. 2015;6:21740–21754. doi: 10.18632/oncotarget.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar H. Dr. Jekyll and Mr. Hyde: the two faces of the fus/ews/taf15 protein family. Sarcoma. 2011;2011:837474. doi: 10.1155/2011/837474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamant L, Gascoyne RD, Duplantier MM, Armstrong F, Raghab A, Chhanabhai M, et al. Non-muscle myosin heavy chain (MYH9): a new partner fused to ALK in anaplastic large cell lymphoma. Genes Chromosomes Cancer. 2003;37:427–432. doi: 10.1002/gcc.10232. [DOI] [PubMed] [Google Scholar]
- Marin-Aguilera M, Codony-Servat J, Kalko SG, Fernandez PL, Bermudo R, Buxo E, et al. Identification of docetaxel resistance genes in castration-resistant prostate cancer. Mol Cancer Ther. 2012;11:329–339. doi: 10.1158/1535-7163.MCT-11-0289. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. Panther in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahgozar S, Moafi A, Abedi M, Entezar EGM, Moshtaghian J, Ghaedi K, et al. Mrna expression profile of multidrug-resistant genes in acute lymphoblastic leukemia of children, a prognostic value for ABCA3 and ABCA2. Cancer Biol Ther. 2014;15:35–41. doi: 10.4161/cbt.26603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz S, Stark M, Assaraf YG. Binding of a Smad4/Ets-1 complex to a novel intragenic regulatory element in exon12 of FPGS underlies decreased gene expression and antifolate resistance in leukemia. Oncotarget. 2014;5:9183–9198. doi: 10.18632/oncotarget.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seve P, Dumontet C. Is class III beta-tubulin a predictive factor in patients receiving tubulin-binding agents? Lancet Oncol. 2008;9:168–175. doi: 10.1016/S1470-2045(08)70029-9. [DOI] [PubMed] [Google Scholar]
- Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updates. 2012;15:62–69. doi: 10.1016/j.drup.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrills NM, Po’uha ST, Liu MLM, Liaw TYE, Larsen MR, Ivery MT, et al. Alterations in γ-actin and tubulin-targeted drug resistance in childhood leukemia. J Natl Cancer I. 2006;98:1363–1374. doi: 10.1093/jnci/djj372. [DOI] [PubMed] [Google Scholar]
- Wang XS, Shankar S, Dhanasekaran SM, Ateeq B, Sasaki AT, Jing XJ, et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov. 2011;1:35–43. doi: 10.1158/2159-8274.CD-10-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Chen YNF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon AM, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang Y, Xing F, Wang J, Wang Y, Wang H, et al. Overexpression of LIMK1 promotes migration ability of multidrug-resistant osteosarcoma cells. Oncol Res. 2011;19:501–509. doi: 10.3727/096504012X13286534482511. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zhang B, Chen X, Bae S, Singh K, Washington MK, et al. Loss of Smad4 in colorectal cancer induces resistance to 5-fluorouracil through activating Akt pathway. Br J Cancer. 2014;110:946–957. doi: 10.1038/bjc.2013.789. [DOI] [PMC free article] [PubMed] [Google Scholar]