

Abstract
A microfluidic system consisting of generic single use cartridges which interface with a workstation allows the automatic performance of all necessary sample preparation, PCR analysis and interpretation of multiplex PCR assays. The cartridges contain a DNA array with 20 different 16mer DNA “universal” probes immobilized at defined locations. PCR amplicons can be detected via hybridization of user-defined “reporter” probes that are complementary at their 3′ termini to one or more of the universal probes and complementary to the target amplicons at their 5′ termini. The system was able to detect single-plex and multiplex PCR amplicons from various infectious agents as well as wild type and mutant alleles of single nucleotide polymorphisms. The system's ease of use was further demonstrated by converting a published PCR assay for the detection of Mycobacterium genitalium in a fully automated manner. Excellent correlation between traditional manual methods and the automated analysis performed by the workstation suggests that the system can provide a means to easily design and implement a variety of customized PCR-based assays. The system will be useful to researchers or clinical investigators seeking to develop their own user defined assays. As the U.S. FDA continues to pursue regulatory oversight of LDTs, the system would also allow labs to continue to develop compliant assays.
Keywords: Bioengineering, Biotechnology
1. Introduction
The ability to amplify and detect specific nucleic acids by polymerase chain reaction (PCR) or other gene amplification methods has greatly expanded the scope and use of nucleic acid assays into a variety of diverse fields. Molecular diagnostics has become a valuable tool in healthcare by providing a means to identify infectious agents [1], characterize malignant tumors [2], predict sensitivity to pharmacologic agents [3], and stratify oncology patients into responders and non-responders [4], thus allowing physicians to make well-informed decisions about medical treatment. Bioinformatics information gleaned from the human genome project can also provide valuable information about possible health risk factors [5]. In addition to clinical applications, nucleic acid assays have also expanded to fields such as food and beverage testing [6, 7], forensics [8, 9], and quality control in personal care products production [10]. In general, each of these applications relies upon gene amplification technology, often PCR, to first increase the total number of targets to detectable levels and then either simultaneously detect those molecular targets via “real time” fluorescence methods or at the completion of the amplification process by “end point” analysis of the resulting amplicons using DNA arrays. Both approaches have distinct advantages. Real time methods provide an effective means to quantitate the concentration of the target sequences. One example of the clinical utility of real time methods is the monitoring of HIV viral loads, thus providing a means to monitor the efficacy of therapeutic interventions in the treatment of HIV infection [11]. In the food and beverage industry, real time methods also provide a means to quantitate the level of pathogen contamination [12] in materials intended for human consumption. Despite the power of such real time methods, however, one drawback is the limited number of targets that can be simultaneously detected. Since quantitation relies upon the ability to detect and distinguish the emission of fluorescent dyes bound to probe molecules, it is necessary to use fluorescent dyes that have non-overlapping emission spectra in order to achieve multiplex detection of multiple targets in a single reaction. The total number of targets detected in a fluorescence-based multiplex PCR is often in the range of 4–6 [13]. A variety of novel methods have been developed, however, that can increase the number of detectable targets in real time assays. The use of melting profiles has been used to distinguish multiple amplicons generated in a single PCR using the same detection dye [14]. Other methods, including Target Enriched Multiplex (TEM) PCR can be used to further enrich the target population and allow detection of up to 20 individual targets within the same sample [15, 16]. Finally, Multiple Detection Temperature (MuDT) technology has also been used to detect multiple targets in a single reaction using a single dye [17]. In contrast to real time methods, detection of multiple targets in “end point” detection methods provides a simple means to detect a large number of individual targets. Since detection does not rely upon fluorescence, there is no need to utilize expensive optical equipment or strive to deconvolute overlapping emission spectra. But, in order to achieve effective end point detection, specific probes complementary to the target sequences need to be immobilized on either a fixed substrate such as a DNA array [18, 19] or onto magnetic beads [20, 21]. Regardless of the specific method used, it is necessary for the user to design and immobilize the probes to a suitable substrate before the assay is performed. From a commercial standpoint, this represents a disadvantage since a manufacturer seeking to provide a broad spectrum of assay kits or products would require knowledge of the user's specific targets of interest in order for the correct probes to be provided on the arrays. Furthermore, manufacturers of fully integrated systems that can automatically process a raw specimen through lysis, purification, amplification and detection are at a further disadvantage since the power of such fully integrated systems is diminished because a multitude of different probe sets would be required for the systems to be suitable for widespread commercial use. While xMAP technology provides a means for investigators to functionalize microspheres for detection of multiple targets [22], our goal was to provide a simplified means to not only functionalize the DNA array, but to also include a seamless, fully integrated analysis of the user's specimens. Such a system becomes especially useful in research laboratories that need to develop assays “on the fly” for their particular use. Moreover, in clinical settings where laboratories, regulated by the Clinical Laboratory Improvement Amendment (CLIA) develop their own laboratory developed tests (LDTs), such labs would have to rely on such commercial entities to provide specific probes within their systems in order to detect the user's targets of interest.
The U.S. Food and Drug Administration (FDA) has recently released draft guidance documents indicating that enforcement discretion of user defined assays is likely to end, at least for Class II and Class III medical devices, and reiterated its position that a “traditional LDT” is defined as a test that was developed and used within a single laboratory without assistance from outside parties [23, 24]. Although there has been considerable opposition to the new draft guidance documents by CLIA laboratories and multiple laboratory associations, it is apparent that changes to oversight of LDTs, either by the FDA, the Centers of Medicare and Medicaid Services (CMS) or both is likely to occur in the future. In order to overcome the limitations of current commercial systems and to provide universal “plug and play” functionality, we set out to expand the capabilities of our current fully automated system [25, 26, 27] to allow end users to automatically functionalize DNA arrays contained within generic microfluidic cartridges interfaced with a programmable workstation. Another goal of this effort was to allow the system to achieve the required functionalization of the universal array while the rest of the assay was underway, thus eliminating any delay in having the DNA arrays functionalized and available for hybridization. Such a system will provide considerable versatility to users in multiple fields, allowing laboratories in various clinical and nonclinical settings to design customized and fully automated assays as needed. In addition, such a system will also allow CLIA labs to remain compliant to the anticipated changes in LDT regulations, since the laboratories will not have to rely upon a manufacturer to prepare specific DNA arrays.
2. Materials and methods
2.1. Generic cartridges
The cartridge used in the present studies is an injection molded homopolymeric polystyrene structure (Fig. 1) similar to that previously described [25]. It contains all of the pumps, valves, microchannels, reaction and reagent reservoirs, and the integrated universal DNA array necessary to perform fully automated assays. Each of the cartridges can perform sample lysis, purification, amplification and hybridization steps simultaneously on four separate samples. When inserted into the workstation, the workstation's software controls all of the assay steps on up to a total of six cartridges in parallel, thus allowing the simultaneous analysis of up to 24 separate samples.
Fig. 1.

(Left) A schematic of the Rheonix CARD® cartridge depicting the four identical lanes used to automatically analyze four independent specimens. Within each lane, cells are lysed, DNA extracted, purified, and the resulting DNA subjected to PCR. The amplicons generated in the PCR reaction chambers are subsequently detected on the four separate DNA microarrays (MAR) shown. The various microchannels, diaphragm pumps and valves have been previously described in detail [25]. (Right) A side view of the cartridge showing the underside microchannel layer and suspended PCR tubes.
2.2. Universal probe design and synthesis
The twenty probes immobilized on the substrate were designed according to the methods previously described by Gerry et al. [28] with the exception that only four tetramers, instead of six, were used to design 16mer oligonucleotide probes. The 5′ aminoterminated probes were obtained from Integrated DNA Technologies (Coralville, IA) and the lyophilized oligonucleotides were then re-suspended in 0.5 M NaHCO3 at a concentration of 200 μM for covalent linkage to activated Biodyne membranes obtained from Pall Corporation (Port Washington, NY) via 1-ethyl-3-(3-dimethylaminopropyl) carbodiimde (EDAC) chemistry [29].
2.3. Generic cartridge and universal DNA microarrays
To achieve detection of specific PCR amplicons, two different types of probes were utilized (Fig. 2). One set of 16mer probes, independent of the amplicon sequences being detected, served as “universal” probes, and were immobilized on the surface of the DNA microarray (Table 1). A second set of “reporter” probes were designed with 34–45 nucleotides of their 3′ termini complementary to the 3′ portion of particular immobilized universal probes (Table 1) and 18–25 nucleotides at their 5′ termini complementary to the 3′ end of individual target amplicon sequences. To investigate this approach, the universal microarray filters were spotted with the universal probes (Table 1) at concentrations of 2 μM, 20 μM, or both concentrations, in spotting buffer containing 0.25 M NaHCO3, 25% polyethylene glycol (MW 8000), 0.1% Tween 20 and 150 μM Rhodamine-B. Once in solution, the universal probes were applied to the activated Biodyne membranes that were fixed on a plastic support.
Fig. 2.
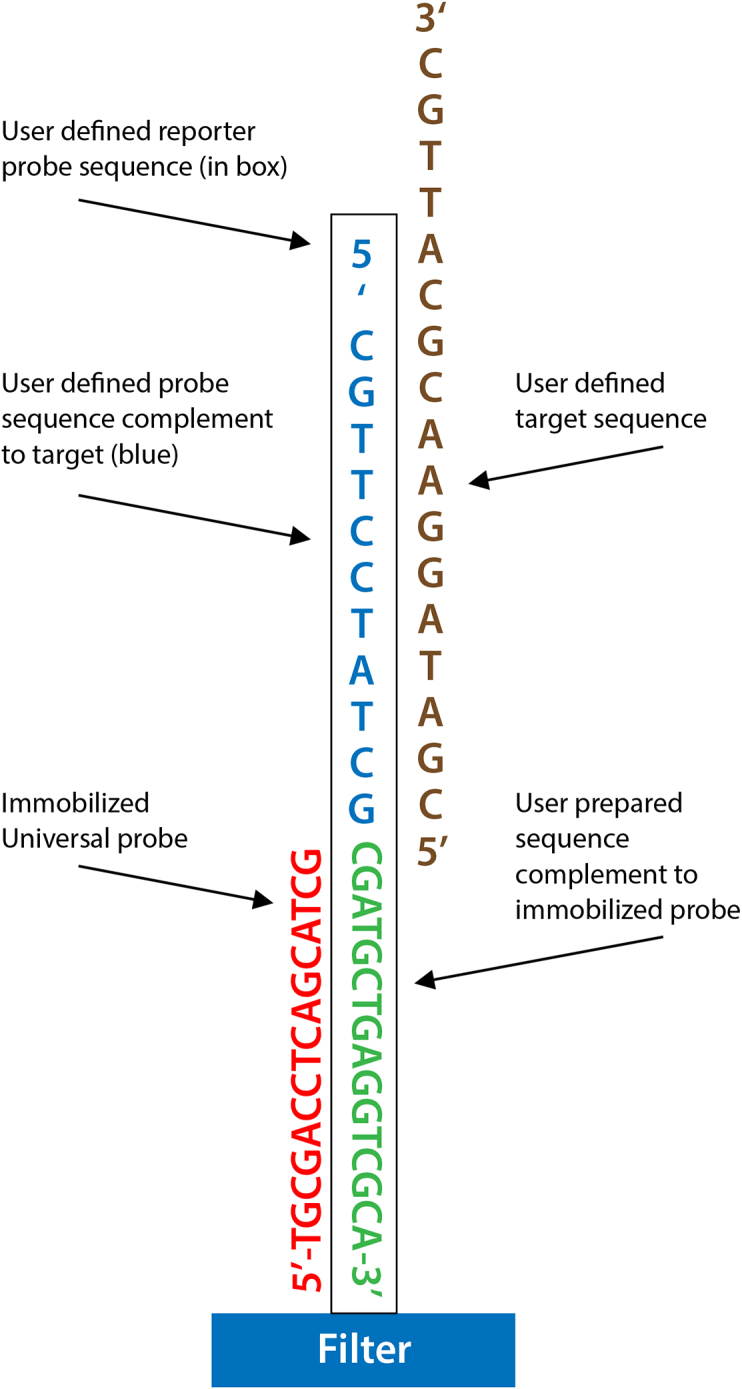
A schematic representation of a particular immobilized probe adhered at its modified 5’end to the filter substrate. A reporter probe is shown hybridized at its 3′ end to the immobilized probe and hybridized to the target amplicon at its 5′ end.
Table 1.
Nucleotide sequences of 20 different universal probes immobilized on the cartridge's integrated DNA array. All sequences are modified at their 5′ termini to facilitate binding to the activated membrane as described in Materials and Methods. The reporter probes were designed to be complementary, at their respective 3’termini, to the universal probes immobilized on the DNA array. As described in Materials and Methods, the reporter probes were further modified at their 5′ termini to include sequences complementary to the target amplicons (see also Fig. 2).
| Probe ID Number | Nucleotide sequence (5′ → 3′) | |
|---|---|---|
| Universal Probes |
Complementary Reporter Probes |
|
| 1 | /5AmMC6/TGCGACCTCAGCATCG | CGATGCTGAGGTCGCA |
| 2 | /5AmMC6/CAGCACCTGACCATCG | CGATGGTCAGGTGCTG |
| 3 | /5AmMC6/GACCACCTTGCGATCG | CGATCGCAAGGTGGTC |
| 4 | /5AmMC6/TGCGGGTACAGCACCT | AGGTGCTGTACCCGCA |
| 5 | /5AmMC6/CAGCGGTAGACCACCT | AGGTGGTCTACCGCTG |
| 6 | /5AmMC6/GACCGGTATGCGACCT | AGGTCGCATACCGGTC |
| 7 | /5AmMC6/TGCGATCGCAGCGGTA | TACCGCTGCGATCGCA |
| 8 | /5AmMC6/CAGCATCGGACCGGTA | TACCGGTCCGATGCTG |
| 9 | /5AmMC6/GACCATCGTGCGGGTA | TACCCGCACGATGGTC |
| 10 | /5AmMC6/AGCCCTTGCACGTCTG | CAGACGTGCAAGGGCT |
| 11 | /5AmMC6/CACGCTTGGTGCTCTG | CAGAGCACCAAGCGTG |
| 12 | /5AmMC6/GTGCCTTGAGCCTCTG | CAGAGGCTCAAGGCAC |
| 13 | /5AmMC6/AGCCGATGCACGCTTG | CAAGCGTGCATCGGCT |
| 14 | /5AmMC6/CACGGATGGTGCCTTG | CAAGGCACCATCCGTG |
| 15 | /5AmMC6/GTGCGATGAGCCCTTG | CAAGGGCTCATCGCAC |
| 16 | /5AmMC6/AGCCTCTGCACGGATG | CATCCGTGCAGAGGCT |
| 17 | /5AmMC6/CACGTCTGGTGCGATG | CATCGCACCAGACGTG |
| 18 | /5AmMC6/TCCCCTGTGGACTGTC | GACAGTCCACAGGGGA |
| 19 | /5AmMC6/ACGGCTGTTCCCTGTC | GACAGGGAACAGCCGT |
| 20 | /5AmMC6/GGACTGTCACGGAGTG | CACTCCGTGACAGTCC |
2.4. Samples used to isolate DNA
To demonstrate the versatility of the system, DNA was isolated from a variety of samples exactly as previously described [25, 26]. Detection of the infectious agents, Chlamydia trachomatis, Neisseria gonorrhoeae and Trichomonas vaginalis was demonstrated by spiking the organisms into phosphate buffered saline either individually or combined prior processing on the CARD cartridge. The detection of single nucleotide polymorphisms (SNPs) was demonstrated by analyzing buccal swabs collected from volunteers after IRB-approved informed consent forms were signed. The buccal swab samples were obtained using FLOQSwabs™ purchased from Copan Diagnostics, Inc. (Murrieta, CA) and suspended in phosphate buffered saline. DNA was then automatically isolated on the CARD cartridge from the samples and PCR amplified exactly as previously described [25, 26]. Finally, in order to demonstrate a higher multiplex detection of multiple targets, 5′ biotinylated DNA sequences, corresponding to multiple human papilloma virus (HPV) subtypes and other sexually transmitted infection (STI) agents were purchased from Integrated DNA Technologies and served as surrogate amplicons.
2.5. PCR amplification
The PCR amplification of individual sequences was first optimized for the particular target(s) of interest using standard bench top methods. Once optimized, the workstation was programmed to automatically perform each of the individual sample preparation, PCR and detection steps for the various assays. Since the assay conditions varied, particularly the levels of stringency required to detect SNPs versus the STI targets, the PCR amplification and detection conditions were separately optimized and then programmed into the workstation to conduct the fully automated assays.
The STI targets were amplified in a PCR master mix formulated for multiple targets containing 10 mM Tris-HCl pH 9.00, 50 mM KCl, 2.5 mM MgCl2, 400 μM dNTPs, 0.5 μM forward and biotinylated reverse primers for C.trachomatis, N. gonorrhoeae and T.vaginalis and 0.2 Units of Taq polymerase (Promega) in a final volume of 15 μl. The specimens were denatured at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing and extension at 71 °C for 30 s. The final extension was carried out for 3 min at 72 °C.
A previously published manual method [30] for detection of Mycoplasma genitalium (MG) was also migrated to the cartridge. The PCR assay conditions were exactly as previously described, with the exception that endpoint detection of the amplicons replaced the real time PCR process. Three separate reporter probes for the MG amplicon were designed that had identical sequences complementary to the gap sequence [30] on their 5′ termini, while the 3′ termini of the reporter probes were complementary to three different universal probes on the DNA array.
The PCR amplification of SNPs associated with warfarin sensitivity (Cyp2C9*2, Cyp2C9*3 and VKORC1) was carried out in a final volume of 15 μl containing 1.5 mM MgCl2, 200 μM dNTPs, 0.4 μM forward and reverse primers. Since the amplification of the CYP2C9 and VKORC1 targets required different master mixes, the cartridge design included two separate PCR chambers per lane. Therefore one of the master mixes was automatically introduced into one of the PCR chambers to amplify the CYP2C9 targets while the second master mix was introduced into the second PCR chamber to amplify the VKORC1 target. At the conclusion of the thermocycling, the amplicons from both PCR chambers were automatically combined on the cartridge for further analysis on the integrated universal DNA microarray. For both PCR reaction vessels, the thermal cycling conditions included an initial denaturation at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 48 °C for 30 s, and extension at 72 °C for 30 s. The final extension was carried out for an additional 5 min at 72 °C. For SNP PCR reactions, the primer pairs were biotinylated at both termini [26] in order to facilitate detection via streptavidinylated horse radish peroxidase.
2.6. Two-step versus sequential microarray hybridization
The methods used to detect the specific amplicons on the universal array were optimized according to the types of targets being analyzed. A two-step hybridization process, whereby the “reporter” probe was first allowed to hybridize to the immobilized “universal” probe and then the available reporter probe sequences allowed to hybridize to the PCR amplicons was ideal for detection of STI targets. On the other hand, a sequenced hybridization process was found to be effective for SNP detection. To accomplish detection of SNPs, the “reporter” probes designed to detect the wild type alleles were first allowed to hybridize to the immobilized “universal” probes, followed by hybridization of the denatured PCR amplicon mix to the reporter probes. Then, the “reporter” probes designed to detect the mutant alleles were subsequently introduced to hybridize to their appropriate “universal” probes, followed by hybridization of another aliquot of the same denatured PCR amplicon mix to the second set of reporter probes. As described below in more detail, although the sequence of events slightly differed in the two different hybridization approaches, they could easily be programmed into the workstation. In both cases, the ultimate detection of the various amplicons was achieved by incubation with streptavidinylated horse radish peroxidase (HRP), followed by 3, 3′,5, 5′-tetramethylbenzydine (TMB) for color development. The signal generated by incubation of HRP with TMB was detected after six minutes by allowing the workstation's onboard Cognex (Natick, MA) CMOS camera to capture the images which were then processed by the instruments imaging software [26].
2.7. Two-step hybridization
The integrated universal DNA microarray filters were first washed with 100 μl of hybridization buffer (150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA and 0.1% SDS) at 40 °C for 2 min after which the buffer was pumped out to the cartridge's waste reservoir. Fresh hybridization buffer (100 μl) was then pumped into the microarray chambers and 5 μl of the appropriate reporter probe mix allowed to flow over each of the microarrays. Hybridization was carried out at 40 °C for 10 min. While the first step of hybridization was underway 15 μl of amplicon was diluted to 100 μl with hybridization buffer and denatured at 95 °C for 5 min. Then 70 μl of the denatured amplicon solution was pumped into the microarray chamber along with 30 μl of hybridization buffer from the reagent reservoir. Hybridization was allowed to proceed for 10 min at 40 °C. After pumping the sample out of the microarray chamber, the filters were then washed with 100 μl of hybridization buffer at 40 °C for 5 min and a second wash carried out at room temperature for 30 s. Following these washes, the filters were incubated with 100 μl of streptavidinylated HRP for 5 min at room temperature followed by four washes of 30 s each with hybridization buffer at room temperature. One hundred μl of TMB was then dispensed and pumped into each of the microarray chambers and incubated for 7 min at room temperature to generate the color reaction which was stopped by washing 20% Ethanol through the microarray chamber. The filters were finally imaged by the workstation's optics system in the 20% Ethanol solution.
2.8. Sequenced hybridization for SNPs
Buccal cells were collected from volunteers whose genotypes for CYP2C9*2, CYP2C9*3 and VKORC-1 were determined by bidirectional Sanger Sequencing as previously described [26]. The microarray filters were initially washed in hybridization buffer for 2 min prior to reporter probe hybridization. The hybridization buffer was then pumped out of the microarray chamber to the waste reservoir and 100 μl of fresh hybridization buffer pumped into the microarray chamber, followed by 5 μl of the wild type reporter probe mixture (0.125 μM of Cyp2C9*2 and 0.5 μM of Cyp2C9*3 and VKORC-1) in Buffer A (300 mM NaCl, 20 mM NaH2PO4, 2 mM EDTA and 0.1% SDS). The reporter probe mixture was then allowed to hybridize with the universal probes in the microarray chamber at 50 °C for 10 min. After the final extension of the PCR reaction, the entire volume of the PCR amplicons (5 μl) was diluted in 100 μl of Buffer A and denatured at 95 °C for 5 min. Thirty μl of the diluted amplicons were then pumped into the microarray chamber along with 70 μl of Buffer A simultaneously pumped from reagent reservoir. Hybridization was then performed at 50 °C for an additional 10 min. Immediately following the first hybridization, a second hybridization was performed with the mutant reporter probes mix (0.125 μM of Cyp2C9*2 and VKORC-1, 0.5 μM of Cyp2C9*3) to which another aliquot of the same denatured amplified target was pumped into microarray chamber with hybridization buffer exactly as described above. After both hybridizations were completed, the microarray chambers were washed with 100 μl of Buffer B (75 mM NaCl, 5 mM NaH2PO4, 0.5 mM EDTA and 0.1% SDS) at 50 °C for 5 min, followed by a second wash carried out for 30 s. After these washes, the microarray filters were processed for color development by adding HRP and TMB exactly as described above for the two step hybridization process. All of the custom reporter probes used for the SNP assay were designed with locked nucleic acids (Table 2) to facilitate higher stringency melting temperatures and were purchased from Exiqon (Woburn, MA).
Table 2.
Six different reporter probes were designed to detect the wildtype (WT) and mutant (Mut) alleles of single nucleotide polymorphisms (SNP) defined by CYP2C9*2, CYP2C9*3 and VKORC1 loci. The 5′ termini of the reporter probes (shown in red) contain locked nucleic acid sequences (underlined) while the 3′ ends (shown in black) contain sequences complementary to the universal probes.
| Probe ID Number | Target SNP | Reporter Probe Sequence (5′ → 3′) |
|---|---|---|
| 1 | CYP2C9*2_WT | TGAGGA+C+C+GTGTTCA CGATGCTGAGGTCGCA |
| 2 | CYP2C9*2_Mut | T+GA+AC+AC+AG+TC+CT+CA CGATGGTCAGGTGCTG |
| 3 | CYP2C9*3_WT | A+GG+TC+AA+TG+TA+TC+TC CGATCGCAAGGTGGTC |
| 4 | CYP2C9*3_Mut | G+AGA+TA+CCT+TGA+CCTT AGGTGCTGTACCCGCA |
| 7 | VKORC-1_WT | CATCGA+C+C+CTTGGAC TACCGCTGCGATCGCA |
| 6 | VKORC1_Mut | GT+CCA+AGA+GT+CGA+TGA AGGTCGCATACCGGTC |
3. Results
The use of a universal microarray provides a powerful means to easily and rapidly detect single or multiplex PCR amplicons of interest. In order to allow an end user to functionalize the disposable microfluidic cartridge, we utilized 16mer “universal” probes (Table 1) that were first immobilized onto nylon membranes to serve as the anchors to which specific, user-defined, “reporter probes” could subsequently be hybridized. Once the reporter probes were in position via hybridization, sequences at their 5′ end could be used to hybridize to the PCR amplicons (Fig. 2). A total of twenty different 16mer universal probes were evaluated in this study (Table 1).
In order to optimize the hybridization steps on the microfluidic cartridge, we first evaluated multiple methods using standard “bench top” methods (data not shown). During the bench top studies, we determined that, depending upon the types of amplicons being detected, one of two different hybridization methods could be used to detect the targets of interest. A “two-step” hybridization process was developed whereby the user-defined reporter probes were first hybridized to the filter-bound universal probes, followed by hybridization of the PCR amplicons to the reporter probes. This approach was optimum for the detection of singleplex and multiplex amplicons resulting from PCR amplification of defined DNA sequences. For the detection of single nucleotide polymorphisms, however, a “sequenced hybridization” process provided better results. Since the wild type and mutant alleles were detected by reporter probes directed against opposite strands of the same target region and they differed at only a single nucleotide at the site of the SNP, allowing them to simultaneously flow over the filter-bound universal probes resulted in the reporter probes hybridizing to each other and thus reducing the overall signal intensity on the microarray. Therefore, to overcome this, the wild type reporter probes were allowed to hybridize to the filter-bound universal probes and then the denatured amplicons flowed over the DNA array. Then, immediately following this and without any intervening washing steps, the mutant reporter probes were flowed over the same array and allowed to hybridize to another set of universal probes on the array. After this hybridization step was completed, another aliquot of the same denatured amplicons was flowed over the array and hybridized to the reporter probes. Detection was then achieved by the introduction of streptavidinylated horse radish peroxidase and TMB substrate.
To demonstrate the ability to detect specific PCR amplicons from targets of interest, we first designed probes against three separate sexually transmitted infection (STI) agents, N. gonorroheae (NG), C. trachomatis (CT) and T. vaginalis (TV). A total of nine individual reporter probes complementary to the 3′ termini of the anchored universal probes were designed. Two were designed to detect NG amplicons, four were designed to detect CT amplicons, and three were designed to detect TV amplicons. Two hundred microliters of individual or combined cultures of the three STIs were introduced at 1000 cells/ml to the sample reservoir of the cartridge and automatically processed through the entire sample preparation, PCR amplification and detection. While the samples were being prepared on the cartridge, the reporter probes were simultaneously being hybridized to the universal probes on the universal DNA array. Using the two step hybridization method, detection of either singly amplified or multiplex amplified targets was easily distinguished on the DNA array (Fig. 3A). In a parallel study, the limit of detection for STI microbes was determined by introducing 200 μl of culture samples at 25, 50, and 75 copies/ml in phosphate buffer saline either individually or in combination. While the singleplex assay results yielded consistent detection limits of 25 copies/ml, inconsistency was observed for concentrations below 25 copies/ml (data not shown). Therefore, the limit of detection on the fully automated cartridge is 25 cells/ml (Fig. 3B).
Fig. 3.

(A) Image of microarray showing hybridization of target amplicons generated from DNA automatically isolated and PCR amplified from Chlamydia trachomatis (CT), Neisseria gonorrhoeae (NG) and Trichomonas vaginalis (TV). The sample preparation and PCR amplification was automatically performed using the cartridge by adding 200 μl of cultured targets (1 × 103copies/ml) to the sample reservoir either individually or all three mixed together. The PCR amplicons were detected using the two step hybridization method described in Materials and Methods. Each universal probe was immobilized at two concentrations (20 μM, 2 μM as described in Materials and Methods). (B) To estimate the limit of detection, the target microorganisms were diluted to 25, 50 and 75 copies per ml and 200 μl of the specimens added to the cartridge either individually or all three combined. The location and concentration of the various STI target probes as well as the Spotting Controls (SC) on the DNA array are shown in the filter key.
To investigate the ability to detect upwards of 20 individual targets, we employed synthetic biotinylated nucleotide sequences to mimic PCR amplicons. Twenty synthetic targets, ranging from 18 to 25 nucleotides long, were processed exactly as described for cell cultured targets. To mimic our standard approach, we designed 20 different “reporter” probes that were first allowed to hybridize to the 20 immobilized “universal” probes, followed by introduction of the biotinylated oligonucleotides that simulated authentic amplicons. As noted (Fig. 4A, B), all 20 targets, consisting of 19HPV subtypes and 1 STI target, were easily detectable and distinguishable.
Fig. 4.

(A) Bench top analysis of 20 different surrogate targets biotinylated nucleotide sequences corresponding to the L-1 region of various HPV subtypes and Chlamydia trachomatis (CT) was performed by allowing the various reporter probes to flow over the universal DNA array and after hybridization with the immobilized universal probes, the biotinylated surrogate sequences flowed over the array and then subsequently detected by incubation with horse radish peroxidase and tetramethyl benzidine as described in Materials and Methods. The biotinylated target sequences were introduced either individually or combined together. (B) The same approach that was optimized on the benchtop was automatically performed in the cartridge under the software control of the Encompass Optimum workstation. The location of the various probes and spotting controls (SC) are shown in the filter key.
Switching our attention to the detection of single nucleotide polymorphisms (SNPs), we first evaluated various approaches using bench top experiments (data not shown). Previous work by our group demonstrated that locked nucleic acid (LNA) reporter probes provided specific detection of SNPs in a standard array assay in which the various reporter probes were directly immobilized on the membrane [26]. Although the previous work demonstrated excellent correlation between the automated cartridge assay and Sanger sequencing results, that approach required that the reporter probes complementary to the targets had to first be immobilized on the membrane prior to the initiation of the assay. In the current universal array format, only universal probes were immobilized, thus allowing any user to introduce their own specific reporter probes to detect SNP targets of interest. Since the LNA reporter probes have increased Tm's as compared to corresponding reporter probes designed without locked nucleotides, the detection of the SNPs in the universal array required a different hybridization buffer and temperatures to facilitate detection of the SNP mutations. The sequential hybridization process described in Materials and Methods was used to perform the SNP detection. Our preliminary bench top method evaluations demonstrated that, in contrast to the two step hybridization methods used to detect the STI targets, higher salt concentrations and more stringent washes were necessary for the sequenced hybridization method to successfully detect the SNPs. The cartridge assay was first optimized using an in-house panel of samples of known genotype that included homozygous, heterozygous and mutant alleles for CYP29C*2, CYP29C*3 and VKORC-1 (Fig. 5A). Genomic DNA (1 × 103copies/μl) was amplified from these samples using bench top manual methods. Due to differing PCR conditions required for optimum amplification [26], the CYP2C9 targets (*2 and *3) were amplified in one PCR reaction and the VKORC-1 target in a separate PCR reaction. At the completion of the reactions, however, the amplicons from the two PCR reactions were pooled prior to hybridization. The amplicons were verified on agarose gel (Fig. 5A) and the sequenced hybridization carried out using manual methods (Fig. 5B). The results confirmed the known genotypes of the SNP panel samples. The same denatured amplicons were then applied to the universal array and the sequenced hybridization method also yielded correct results (Fig. 5C). Once we were certain that the sequenced hybridization approach could detect the correct SNPs in the panel samples, we then collected fresh buccal swabs from volunteers that had previously been screened by bidirectional Sanger sequencing for genotypes at the CYP2C9 and VKORC-1 loci. A total of 19 specimens and 2 negative (i.e., no target DNA provided) were analyzed in a blind manner (Fig. 6). All sample preparation, PCR amplification and detection on the universal microarray were automatically performed using the workstation with the results correlating perfectly with the bidirectional Sanger sequencing results.
Fig. 5.

DNA obtained from four samples of known genotype at the CYP2C9*2, CYP2C9*3, and VKORC1 loci (inset, as determined by Sanger Sequencing) were PCR amplified and run on an agarose gel (A) to confirm amplification. Then, using bench top methods, the amplicons were analyzed with the Universal DNA array using the sequenced hybridization method described in Materials and Methods. The filter key for the Universal DNA array is shown. (B) Universal DNA array results obtained when amplicons generated in (A) were analyzed using manual methods on the bench top. (C) PCR amplicons generated on the bench top were analyzed on the cartridge using the sequenced hybridization method described in Materials and Methods.
Fig. 6.

Universal DNA arrays resulting from fully automated analysis of 21 different specimens obtained from volunteers that had previously been genotyped using bi-directional Sanger Sequencing. Starting with buccal swabs, all assay steps were automatically performed on the Rheonix CARD® cartridge under the control of the Encompass Optimum workstation. The location of the probes against the various wild type and mutant alleles is identical to the filter key shown in Fig. 5.
Once parameters were established to convert existing benchtop assays to the fully automated platform, we then converted an existing manual, bench top PCR assay for detection of Mycoplasma genitalium (MG) onto the cartridge. Three different “reporter” probes (Table 3) were designed that had identical nucleotides at their 5′ ends complementary to the gap target amplicons [30] and different nucleotides at their 3′ ends complementary to three different “universal” probes (Table 1) immobilized on the DNA array. Two hundred microliters of thimerosol-inactivated MG, diluted in PBS at concentrations of 25, 50 and 100 cfu/ml were evaluated in the cartridge assay under the control of the Encompass Optimum workstation. As noted (Fig. 7), strong signals were obtained for all three concentrations on the universal cartridge's arrays.
Table 3.
Three different reporter probes were designed to detect the gap gene found within M. genitalium. Each probe had identical sequences at its 5′ terminus (shown in red), corresponding to the complement of the gap amplicon, while the 3′ termini had differing sequences, corresponding to the complements of the immobilized universal probes.
| Universal Probe ID | Reporter probe sequence (5′→ 3′) |
|---|---|
| MG-ID_8 | TGTTGTTCCAGAAGCAAATGGCAAACTT-TACCGGTCCGATGCTG |
| MG-ID_9 | TGTTGTTCCAGAAGCAAATGGCAAACTT-TACCCGCACGATGGTC |
| MG-ID_21 | TGTTGTTCCAGAAGCAAATGGCAAACTT-GAC AGG GAA CAG CCG T |
Fig. 7.

Universal DNA array results obtained by introducing 200 μl of cultures of Mycoplasma genitalium (MG) to the cartridge. All steps of the assay, including cell lysis, DNA extraction, DNA purification, PCR amplification, and detection on the integrated universal DNA array, were automatically performed on the cartridge under the control of the Encompass Optimum software. The location of the three probes directed against the MG target is shown in the filter key.
4. Discussion
Despite the power of nucleic acid assays to provide valuable information in multiple fields, laboratories seeking to use commercially available, end point detection of multiplex PCR amplicons in fully automated platforms must work with the respective manufacturers to assure that the proper detection probes are integrated into the systems’ DNA arrays. Since many laboratories would benefit from a system that will allow the individual users to modify the DNA arrays at will, the approach described herein provides a means to develop, alter and optimize their own assays in an easy-to-use format. In addition, for those clinical laboratories seeking to develop LDTs, such a system will allow those laboratories to remain compliant with the anticipated changes in FDA's enforcement of regulations pertaining to LDTs [23].
Another advantage of the described system is that since the introduction of the specific reporter probes can be automatically controlled by the workstation while the remainder of the assay is underway, no time is lost while the DNA array is being functionalized using user-defined reporter probes. This has the advantage of more efficient assay development and added versatility for the end user.
Since the various “universal” probes immobilized on the microfluidic cartridge's DNA array have a fixed nucleic acid sequence, laboratories seeking to develop their own assays only need to design the various “reporter” probes required to hybridize to their own PCR amplicons by designing the 3′ end of the reporter probe to be complementary to the immobilized universal probe and the 5′ end of the reporter probe to be complementary to the target sequence. Moreover, the reporter probes should be designed to assure that they do not anneal to themselves or to other reporter probes in the system. We have taken care to provide sufficient variability in the universal probe sequences to facilitate such a design. Since all hybridizations take place under identical conditions, laboratories should also take care to assure that the various reporter probes and PCR amplicons will favorably anneal to their respective complements under these identical conditions.
Depending upon the design of the detection scheme, various hybridization conditions can be implemented. As described above, detection of single or multiple infectious disease targets can be accomplished by use of a “two step” hybridization process whereby the reporter probes are first allowed to hybridize to the immobilized universal probes, followed by a second hybridization step of the PCR amplicons with the reporter probes now available. In the case of SNP detection, however, we found it necessary to employ an equally effective, but alternate “sequenced hybridization” whereby reporter probes, differing by one nucleotide, were designed against the opposing strands of the amplicons. One reporter probe was directed against the wild type allele while the other reporter probe designed to detect the mutant allele was directed against the complementary strand. Since those two reporter probes would be expected to hybridize to each other if employed simultaneously, we found that effective detection of SNPs could be achieved by first delivering the wild type reporter probes and allowing the PCR amplicons to hybridize, followed by the delivery of the mutant allele reporter probes and hybridization with a second aliquot of the same PCR amplicons. This approach allowed the detection and distinction of the various wild type and mutant alleles while avoiding the potential pitfall of self-annealing of the various reporter probe combinations.
To demonstrate the ease of transferring an existing “bench top” assay to the automated system, we were able to successfully detect Mycoplasma genitalium using assay parameters developed by another laboratory [30]. The ability to automatically process untreated specimens through cell lysis, DNA purification and PCR amplification demonstrates proof of principal and provides a convenient method to streamline existing assays or assays under development by laboratories developing their own user-defined assays.
Taken together, the proposed system will provide the convenience and ease-of-use for laboratories to develop their own fully automated molecular assays. A further advantage of the system when used in clinical settings is it will provide a means for clinical laboratories to develop their own user defined assays in a manner that will allow the labs to remain compliant with anticipated changes in FDA's oversight of LDTs.
Declarations
Author contribution statement
Rubina Yasmin, Hui Zhu, Zongyuan Chen, Richard A. Montagna: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Funding statement
This study was supported by Rheonix, Inc.
Competing interest statement
The authors declare the following conflict of interests: All authors were employed by Rheonix, Inc. while the studies were performed. All authors except for Zongyuan Chen remain currently employed. All authors own either stock or stock options in Rheonix, Inc.
Additional information
No additional information is available for this paper.
References
- 1.Muldrew K.L. Molecular diagnostics of infectious diseases. Curr. Opin. Pediatr. 2009;21:102–111. doi: 10.1097/MOP.0b013e328320d87e. [DOI] [PubMed] [Google Scholar]
- 2.Macgregor P.F., Squire J.A. Application of Microarrays to the Analysis of Gene Expression in Cancer. Clin. Chem. 2002;48:1170–1177. [PubMed] [Google Scholar]
- 3.Gage B.F., Eby C., Johnson J.A., Deych E., Rieder M.J., Ridker P.M., Milligan P.E., Grice G., Lenzini P., Rettie A.E., Aquilante C.L., Grosso L., Marsh S., Langaee T., Farnett L.E., Voora D., Veenstra D.L., Glynn R.J., Barrett A., McLeod H.L. Use of Pharmacogenetic and Clinical Factors to Predict the Therapeutic Dose of Warfarin. Clin. Pharmacol. Ther. 2008;84(3):326–331. doi: 10.1038/clpt.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ochiai T., Nishimura K., Watanabe T., Kitajima M., Nakatani A., Marusasa T., Hashiguchi T., Uchida T., Sakuyama N., Sato T., Kishine K., Futagawa S., Nagaoka I. Identification of responders/non-responders to 5-fluorouracil based on individual 50% inhibitory area under the concentration curve of 5-fluorouracil obtained with collagen gel droplet-embedded culture-drug sensitivity test in colorectal cancer. Oncol. Lett. 2011;2(2):309–313. doi: 10.3892/ol.2011.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathew C. Postgenomic technologies: hunting the genes for common disorders. BMJ. 2001;322:1031–1034. doi: 10.1136/bmj.322.7293.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brusa V., Galli L., Linares L.H., Ortega E.E., Lirón J.P., Leotta G.A. Development and validation of two SYBR green PCR assays and a multiplex real-time PCR for the detection of Shiga toxin-producing Escherichia coli in meat. J. Microbiol. Methods. 2015;119:10–17. doi: 10.1016/j.mimet.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 7.Mandappa I.M., Joglekar P.I., Manonmani H.K. Application of a molecular beacon based real-time isothermal amplification (MBRTIA) technology for simultaneous detection of Bacillus cereus and Staphylococcus aureus. J. Food Sci. Technol. 2015;52(7):4642–4646. doi: 10.1007/s13197-014-1525-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saiki R.K., Walsh P.S., Levenson C.H., Erlich H.A. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc. Natl. Acad. Sci. USA. 1989;86(16):6230–6234. doi: 10.1073/pnas.86.16.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linacre A., Graham D. Role of molecular diagnostics in forensic science. Expert Rev. Mol. Diagn. 2002;2(4):346–353. doi: 10.1586/14737159.2.4.346. [DOI] [PubMed] [Google Scholar]
- 10.Patidar M., Agrawal S., Parveen F., Khare P. Molecular insights of saliva in solving paternity dispute. J. Forensic Dent. Sci. 2015;7(1):76–79. doi: 10.4103/0975-1475.150325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luis J. Molecular diagnosis of microbial contamination in cosmetic and pharmaceutical products. J. AOAC Int. 2001;84(3):671–675. [PubMed] [Google Scholar]
- 12.Curtis K.A., Rudolph D.L., Owen S.M. Rapid detection of HIV-1 by reverse-transcription, loop-mediated isothermal amplification (RT-LAMP) J. Virol. Methods. 2008;151(2):264–270. doi: 10.1016/j.jviromet.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Haakensen M., Dobson C.M., Deneer H., Ziola B. Real-time PCR detection of bacteria belonging to the Firmicutes Phylum. International Journal of Food Microbiology. 2008;125(3):236–241. doi: 10.1016/j.ijfoodmicro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Molenkamp R., van der Ham A., Schinkel J., Beld M. Simultaneous detection of five different DNA targets by real-time Taqman PCR using the Roche LightCycler480: Application in viral molecular diagnostics. J. Virol. Methods. 2007;141(2):205–211. doi: 10.1016/j.jviromet.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Sanjuan-Jimenez R., Colmenero J.D., Bermúdez P., Alonso A., Morata P. Amplicon DNA Melting Analysis for the Simultaneous Detection of Brucella spp and Mycobacterium tuberculosis Complex. Potential Use in Rapid Differential Diagnosis between Extrapulmonary Tuberculosis and Focal Complications of Brucellosis. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0058353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou S., Han J., Wen L., Shanjuan H.Y.L.K.C., Dong L.L.G.J., Guo Y.Z.Y., Shu Y. Human Influenza A Virus (H5N1) Detection by a Novel Multiplex PCR Typing Method. J. Clin. Microbiol. 2007;45(6):1889–1892. doi: 10.1128/JCM.02392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y.J., Kim D., Chun J.Y. Single-channel multiplexing without melting curve analysis in real-time PCR. Sci. Rep. 2014;4 doi: 10.1038/srep07439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Xu Z., Zhang Q., Jin M., Yu J., Li J., Liu N., Cui S., Kong X., Wang H., Li H., Cheng W., Ma X., Duan Z. Simultaneous Detection of Seven Enteric Viruses Associated with Acute Gastroenteritis by a Multiplexed Luminex-Based Assay. J. Clin. Microbiol. 2012;50(7):2384–2389. doi: 10.1128/JCM.06790-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riccelli P.V., Merante F., Leung K.T., Bortolin S., Zastawny R.L., Janeczko R., Benight A.S. Hybridization of single-stranded DNA targets to immobilized complementary DNA probes: comparison of hairpin versus linear capture probes. Nucleic Acids Res. 2001;29(4):996–1004. doi: 10.1093/nar/29.4.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archer M.J., Lin B., Wang Z., Stenger D.A. Magnetic bead-based solid phase for selective extraction of genomic DNA. Anal. Biochem. 2006;355(2):285–297. doi: 10.1016/j.ab.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 21.Cho M., Oh S.S., Nie J., Stewart R., Eisenstein M., Chambers J., Marth J.D., Walker F., Thomson J.A., Soha H.T. Quantitative selection and parallel characterization of aptamers. Proc. Natl. Acad. Sci. USA. 2013;110(46):18460–18465. doi: 10.1073/pnas.1315866110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glushakova L.G., Bradley A., Bradley K.M., Alton B.W., Hoshika S., Hutter D., Sharma N., Yang Z., Kim M.J., Benner S.A. High-thoughput multiplexed xMAP Luminex array panel for detection of twenty two medically important mosquito-borne arboviruses based on innovations in synthetic biology. J. Virol. Methods. 2015;214:60–74. doi: 10.1016/j.jviromet.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.US Food Drug Administration Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories. Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs) October 3, 2014 [Google Scholar]
- 24.US Food and Drug Administration Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories. FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs) October 3, 2014 [Google Scholar]
- 25.Spizz G., Young L., Yasmin R., Chen Z., Lee T., Mahoney D., Zhang X., Mouchka G., Thomas B., Honey W., Roswech T., McGuire C., Montagna R., Zhou P. Rheonix CARD® Technology: an innovative and fully automated molecular diagnostic device. Point Care. 2012;11:42–51. doi: 10.1097/POC.0b013e318222e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spizz G., Chen Z., Li P., McGuire C., Klimkiewicz P., Zysling D., Yasmin R., Hungerford W., Thomas B., Wilding G., Mouchka G., Young L., Zhou P., Montagna R.A. Determination of Genotypes Using Fully Automated Molecular Detection System. Arch. Path. Lab Med. 2015;139:805–811. doi: 10.5858/arpa.2014-0059-OA. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z., Zhu H., Malamud D., Barber C., Ongagna Y.Y.S., Yasmin R., Modak S., Janal M.N., Abrams W.R., Montagna R.A. A rapid, self-confirming assay for HIV: Simultaneous detection of anti-HIV antibodies and viral RNA. J. AIDS Clin. Res. 2016;7:540. doi: 10.4172/2155-6113.1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerry N.P., Witowski N.E., Day J., Hammer R.P., Barany G., Barany F. Universal DNA Microarray Method for multiplex detection of low abundance point mutation. JMB. 1999;292:251–262. doi: 10.1006/jmbi.1999.3063. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y., Coyne M.Y., Will S.G., Levenson C.H., Kawasaki E.S. Single-base mutational analysis of cancer and genetic diseases using membrane bound modified oligonucleotides. Nucleic Acids Res. 1991;19(14):3929–3933. doi: 10.1093/nar/19.14.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svenstrup H.F., Jensen J.S., Bjornelius E., Lidbrink P., Birkelund S., Christiansen G. Development of a Quantitative Real-Time PCR Assay for Detection of Mycoplasma genitalium. J. Clin Microbiol. 2005;43(7):3121–3128. doi: 10.1128/JCM.43.7.3121-3128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]