

Abstract
Introduction
In patients with Alzheimer's disease (AD) and mild cognitive impairment, structural changes in the retina (i.e., reduced thicknesses of the ganglion cell and retinal nerve fiber layers and inclusion bodies that appear to contain beta-amyloid protein [Ab]) have been previously reported. We sought to explore whether anatomic retinal changes are detectable in the preclinical stage of AD.
Methods
A cross-sectional study (as part of an ongoing longitudinal cohort study) involving 63 cognitively normal adults, all of whom have a parent with AD and subjective memory complaints. We compared neocortical amyloid aggregation (florbetapir PET imaging) to retinal spectral domain optical coherence tomography (SD-OCT) markers of possible disease burden. Retinal biomarkers, including the number and surface area of retinal inclusion bodies and the thickness of retinal neuronal layers, were compared across groups with high vs. low neocortical beta-amyloid load.
Results
The surface area of inclusion bodies increased as a function of cortical amyloid burden. Additionally, there was a trend toward a selective volume increase in the inner plexiform layer (IPL; a layer rich in cholinergic activity) of the retina in Aβ+ relative to Aβ− participants, and IPL volume was correlated with the surface area of retinal inclusion bodies.
Discussion
These initial results suggest that retinal imaging may be a potential cost-effective and noninvasive technique that can be used to identify those at-risk for AD. Layer-specific changes in the IPL and their association with surface area of inclusion bodies are discussed as a possible reflection of early inflammatory processes associated with cholinergic disruption and concurrent Ab accumulation in the neocortex.
Keywords: Retina, Alzheimer's disease, Biomarkers, Preclinical AD, Inner plexiform layer, Acetylcholine, Cholinergic hypothesis, Optical Coherence Tomography, OCT
Alzheimer's disease (AD) is a complex neurodegenerative process with multiple known and suspected etiologies, with the disease having variable phenotypic presentations [1]. Notwithstanding, there is now consensus that abnormally high levels of cerebral beta-amyloid (Aβ) indicate AD pathology in individuals with clinically classified dementia or mild cognitive impairment (MCI) [2], [3], [4], [5]. Recent advances have allowed for in vivo detection of levels of Aβ using positron emission tomography (PET) scans [6]. These studies have prospectively documented that cognitively normal (CN) older adults who have high levels of cerebral beta-amyloid (Aβ+) have an increased rate of decline in cognitive function, particularly in episodic memory, when compared with CN older adults with low or normal levels of cerebral beta-amyloid (Aβ−) [7], [8], [9], [10]. This decline in cognitive function is often subtle (e.g., approximately 0.3–0.4 standard deviations per year on standard tests of episodic memory), such that individuals, their caregivers, or both are often unaware of the progressive deterioration in memory [11], [12]. This finding has led to consensus from researchers and regulators for the classification of a preclinical AD disease stage that precedes MCI or AD by several years, with this period becoming a target for therapeutic interventions (i.e., secondary prevention trials) designed to intervene earlier in the disease progression by slowing the aggregation of Aβ [13].
Recently, the US Food and Drug Administration approved three fluorine-18 labeled PET radiopharmaceuticals (florbetapir, flutemetamol, and florbetaben) for identification of cerebral amyloid burden in clinical settings. Although the cost of PET imaging remains very high (≈$5000 USD), an increasing number of CN older adults are motivated to participate in current and planned secondary prevention trials if they meet diagnostic criteria for preclinical AD [13]. Moreover, as novel therapeutics are advanced to slow progression of the disease, it will become increasingly important to identify point-of-care clinical markers of early disease burden, to determine (at lower cost) which individuals should be referred for Aβ PET imaging [14]. Although age and apolipoprotein ε4 (APOE ε4) are reliable risk factors for Aβ+, there remain no clinical or behavioral markers that can reliably detect risk for Aβ+ within periods of <18 months.
There is growing evidence that anatomic changes in the retina may provide an opportunity for identifying and quantifying central nervous system (CNS) amyloid levels. When compared with healthy age-matched controls, patients with AD had reduced numbers of ganglion cell axons and were three times more likely to have increased optic nerve cup-to-disc ratio, a potential consequence of ganglion cell and nerve fiber loss [15]. Furthermore, peripapillary retinal nerve fiber layer (RNFL) thickness has been documented to be significantly thinner (suggestive of optic atrophy) in patients with MCI and mild-to-moderate AD when compared with age-matched controls [16], [17], [18]. In addition to thinning of the peripapillary RNFL, individuals with AD have corresponding thinning of the macular RNFL [16]. Berisha et al. also have observed narrowing of retinal veins, with decreased blood flow, in patients with early AD as compared to healthy age-matched controls [19].
Age-related macular degeneration (AMD) and AD are both age-related neurodegenerative diseases, which share several risk factors including cigarette smoking, hypertension, and hyperlipidemia, and histopathologic features involving oxidative stress, inflammation, and complement activation [20], [21], [22], [23]. Like AD, which has Aβ deposition in the senile plaques, AMD has characteristic Aβ deposition in retinal drusen. Retinal drusen are typically located in the outer retina, at the level of the retinal pigment epithelium (RPE) and Bruch's membrane [21], [22]. It is important to ensure that the Aβ deposition in AD can be differentiated from the retinal drusen of AMD that may also contain amyloid. Accordingly, all multicolor and OCT images were evaluated by a neuro-ophthalmologist (L.N.J.) to screen for hyperflourescent drusen at the RPE level.
Most studies to date have been cross-sectional comparisons of healthy controls and those with MCI or AD burden. Given that peripapillary and macular RNFL thinning are accelerated with chronological age, there could be considerable overlap and difficulty in differentiating between pathologic changes in RNFL thickness changes from normal age-related decline in RNFL thickness [24]. Moreover, there is a paucity of data describing longitudinal changes over time within the same subjects with respect to the retinal layers, the retinal microvasculature, and the aggregation of inclusion bodies that may contain fibrillar Aβ from the earliest, preclinical stage of AD [25], [26]. Herein, we describe baseline spectral domain optical coherence tomography (SD-OCT) retinal imaging results from an ongoing longitudinal natural history study of individuals who are at high risk for the earliest, preclinical stage of AD.
1. Methods
1.1. Participants
A total of 63 adults, ranging in age from 55 to 75 years, with two well-established risk factors for AD, namely, a self-reported first-degree family history of the disease, as well as self-identification of subjective memory concerns, were recruited using a selection process described previously [27]. All participants underwent a detailed medical screening interview. Exclusion criteria were a diagnosis of MCI or AD, history of neurological or psychiatric disorder, any significant systemic illness or unstable medical condition (e.g., active cardiovascular disease), and current use of any medications known to affect cognition (e.g., use of sedative narcotics). Subjects with histories of cataract surgery, corneal LASIK surgery, AMD, glaucoma, or subjects with known ophthalmic pathology were excluded. In addition, multicolor imaging (topographical three-wavelength; available through the SPECTRALIS system used in this study) was obtained on all subjects, and these images were reviewed by a neuro-ophthalmologist with ophthalmology board certification (L.N.J.) at the time that all BluePeak autofluorescence (BAF) imaging data were scored.
Inclusion criteria were a score on the mini-mental state examination (MMSE) > 27 and performance within normal limits on a battery of cognitive tests described previously (listed in Table 1) [9], [28]. All participants were living independently, most were engaged in full-time or part-time employment, and many were caretakers for a parent with AD. Aβ status and APOE genotype were unknown at the time of assessment and were not used to determine enrollment. Although Aβ status and APOE genotyping were conducted as a part of the study protocol, researchers remained blinded to these results throughout testing. The study was approved by and complied with the regulations of Rhode Island Hospital's Institutional Review Board, and all participants provided written informed consent in accordance with the Declaration of Helsinki. The study complied with HIPAA regulations.
Table 1.
Demographic and clinical characteristics
| Variable | Main outcome | Full sample (n = 63) |
Aβ+ (n = 10) |
Aβ− (n = 53) |
P | Cohen's d (95% CI) |
|---|---|---|---|---|---|---|
| N (%) | N (%) | N (%) | ||||
| Sex | No. of female | 39 (61.9%) | 31 (58.5%) | 8 (80.0%) | .199 | — |
|
APOE |
No. of ε4 carriers |
29 (46.8%) |
22 (41.5%) |
7 (70.0%) |
.108 |
— |
| Mean (SD) |
Mean (SD) |
Mean (SD) |
P |
|||
| Age | No. of years | 62.79 (5.35) | 62.28 (5.15) | 65.50 (5.87) | .081 | −0.56 (−1.23 to 0.13) |
| Education | No. of years | 17.21 (2.77) | 17.18 (2.47) | 17.40 (4.17) | .819 | −0.06 (−0.73 to 0.62) |
| SUVr (Neocortex) | Standardized uptake value ratio | 0.98 (0.16) | 1.27 (0.22) | 0.93 (0.07) | .000 | 3.20 (2.28–4.03) |
| GDS | Total score | 1.86 (2.16) | 1.91 (2.27) | 1.60 (1.51) | .685 | 0.19 (−0.49 to 0.86) |
| DASS depression subscale | Total depression subscale score | 3.56 (6.70) | 3.73 (7.24) | 2.70 (2.58) | .660 | 0.28 (−0.40 to 0.96) |
| DASS anxiety subscale | Total anxiety subscale score | 2.73 (4.53) | 2.87 (4.75) | 2.00 (3.27) | .585 | 0.25 (−0.43 to 0.92) |
| DASS stress subscale | Total stress subscale score | 6.73 (6.77) | 6.65 (7.18) | 7.10 (4.23) | .850 | −0.09 (−0.77 to 0.58) |
| MAC-Q | Total score | 24.92 (3.57) | 24.92 (3.64) | 24.90 (3.31) | .984 | 0.01 (−0.67 to 0.68) |
| MMSE | Total score | 29.05 (1.02) | 29.13 (0.96) | 28.60 (1.26) | .132 | 0.43 (−0.25 to 1.11) |
| GMLT (moves/second) | Total correct moves per second | 0.81 (0.20) | 0.78 (0.20) | 0.81 (0.19) | .540 | −0.16 (−0.83 to 0.52) |
| GMLT (total errors) | Total no. of errors | 61.33 (18.38) | 59.87 (23.35) | 61.79 (16.80) | .726 | −0.10 (−0.78 to 0.57) |
| GMLT composite | Standardized z-score | 0.00 (0.80) | 0.02 (1.00) | −0.01 (0.75) | .919 | 0.04 (−0.64 to 0.71) |
| ISLT total recall | Total words recalled | 25.59 (4.22) | 24.73 (4.46) | 25.85 (4.16) | .374 | −0.26 (−0.94 to 0.41) |
| One-card learning task | Accuracy of performance | 1.01 (0.10) | 0.95 (0.09) | 1.02 (0.10) | .011 | −0.72 (−1.39 to −0.01) |
| One back task | Accuracy of performance | 1.45 (0.11) | 1.40 (0.14) | 1.46 (0.10) | .063 | −0.54 (−1.24 to 0.13) |
Abbreviations: APOE, apolipoprotein; SUVr, standardized uptake value ratio; GDS, Geriatric Depression Scale; DASS, Depression, Anxiety, and Stress Scale; MAC-Q, memory complaints questionnaire; MMSE, mini mental state examination; GMLT, Groton maze learning test; ISLT, International Shopping List Task.
NOTE. Bolded values are significant at the P < .001 level.
1.2. Procedure
Participants completed baseline clinical and neuropsychological assessment involving neuropsychological tests (Table 1) which assessed visuospatial learning and reasoning (Groton maze learning test [GMLT]) [29], [30], verbal memory (International Shopping List Test) [31], visual memory (Cogstate One-Card Learning [OCL] task) [28] and working memory (Cogstate One Back [OBK] task) [28]. The MMSE was used to assess general cognitive function, and participants' mood was measured using the 15-item Geriatric Depression Scale and the Depression, Anxiety and Stress Scale (DASS). Subjective memory impairment was determined using the memory complaint questionnaire. All measures were performed by trained staff supervised by a neuropsychologist.
Enrolled participants then completed SD-OCT imaging, APOE genotyping from collected saliva samples, and Aβ PET imaging within 6 weeks of their baseline entry visit.
1.3. Measures
1.3.1. APOE genotyping
A 2-mL saliva sample was collected from all participants and sent for APOE genotyping at the Providence Veterans Administration Hospital laboratory. Although this was completed as part of the study, the resulting group sample sizes were too small for subsequent statistical analysis. Therefore, APOE genotype was not included as a factor in subsequent analyses.
1.3.2. Aβ PET imaging
To assess neocortical amyloid burden, all participants had an Aβ PET scan. A 370MBq (10 mCi +/− 10%) bolus injection of 18F-florbetapir was administered intravenously. Approximately 50 minutes post-injection, a 20-minute PET scan was performed with head CT scan for attenuation correction purposes. Images were obtained using a 128 × 128 matrix and reconstructed using iterative or row action maximization likelihood algorithms. PET standardized uptake value (SUV) data were summed and normalized to the whole cerebellum SUV, resulting in a region-to-cerebellum ratio termed SUV ratio (SUVr). An SUVr threshold of ≥1.1 was used to discriminate between Aβ− and Aβ+. Table 1 provides between-group differences on the established measure of total neocortical SUVr (averaged over six regions of interest) [32]. These SUVr calculations were performed using the MIMneuro software, with a normative database of 74 healthy normal individuals (48 males, 26 females), aged between the ages of 18–50 years, who all had negative amyloid scans on visual assessment [32]. For all cases, Aβ positivity was confirmed by consensus over-read by two board-certified radiologists who were also board certified in nuclear medicine.
1.3.3. SD-OCT & BAF imaging
Participants were administered two drops of tropicamide (Mydriacyl 1%) per eye for pupil dilation before OCT imaging. The Heidelberg SPECTRALIS SD-OCT was used to acquire retinal OCT scans of the optic nerve head and the macula. Multicolor (MC) imaging of modified five field sections of the retina (central, nasal, superior, temporal, and inferior) in addition to retinal BAF was completed for the right and left eyes of both participants, using the Heidelberg confocal scanning ophthalmoscope. Outcome measures for the SD-OCT imaging sequences included peripapillary RNFL and macular retinal segmentation volumes for RNFL, ganglion cell layer, inner plexiform layer (IPL), inner nuclear layer, outer plexiform layer, and outer nuclear layer. For each individual participant, the volume and thickness of all retinal neuronal layers for each eye were expressed as a ratio of the total retinal volume and thickness for each eye. Because we suspected that volumetric RNFL changes in our cohort would reflect systemic changes of preclinical AD, we averaged the ratio for each retinal neuronal cell layer across both eyes. Additionally, we conducted preliminary analyses to examine potential asymmetry between eyes and found no asymmetry across retinal layers (all P < .171).
Additionally, the number count and summed surface area (μm2) of retinal inclusion bodies identified via BAF were compared between Aβ− and Aβ+ groups. These inclusion bodies differ from the age-related extracellular drusen that typically accumulate at the level of Bruch's membrane and the retinal pigment epithelium. Research in both animal and human models suggest that these inclusion bodies contain fibrillar Aβ [25], [26], [33], which is characteristic of the neocortical Aβ plaque deposits found in preclinical AD [34]. The number and surface area of inclusion bodies was summed across both eyes for each participant. The precise locations of these inclusion bodies were primarily identified via BAF (en face orientation). Multicolor imaging, which uses individual laser colors to characterize anatomic and pathologic detail at different retinal depths, was used specifically to identify and exclude microbleeds and to ensure that we did not count artifacts within the α and β crescents surrounding the optic nerve head. However, the inclusion bodies that we identified with BAF are not easily visualized with multicolor imaging. Similarly, these inclusion bodies are very difficult to identify using SD-OCT imaging (coronal sections), appearing as faint white spots with poor delineation of borders. Our independent raters reviewed SD-OCT images to the best of their ability, relying on major vessels as landmarks, and noting their observations on visual inspection. On BAF analysis, we excluded the 360-degree region within 1 disc diameter from the center of the optic disc and the 360-degree macula region within 1 disc diameter from the foveal center. This was done to avoid including the area of hypopigmentation around the optic nerve in participants with zone α and/or β crescents and to avoid including age-related macular drusen. All other remaining sections across the standard 1, 2.22, 3.45 mm Early Treatment of Diabetic Retinopathy Study grid were included in these analyses. Individuals with background diabetic retinopathy and retinal microbleeds (identified via multicolor imaging) were excluded. Inclusion bodies were identified via 100% consensus ratings from a post-doctoral research fellow and board-certified ophthalmologist. Both raters were blinded to participant amyloid status. See Fig. 1 for an example of identified retinal inclusion bodies.
Fig. 1.

Inclusion bodies (circled in red ellipses) identified via BluePeak autofluorescence (BAF) imaging in a high neocortical amyloid-beta load (Aβ+) cognitively normal older adult.
Surface area of inclusion bodies was measured using the semi-automated Heidelberg Region Finder tool on the BAF images obtained for each eye [35]. This tool allows the operator to place a ‘seed’ in the area of interest, and the software automatically delineates the boundary of the object (in this case, the inclusion bodies as shown in Fig. 1). On visual inspection, the operator has the ability to modify the boundaries, and then surface area is computed automatically and recorded.
1.4. Data analysis
For each neuropsychological outcome measure, performance between Aβ+ and Aβ− groups was compared using a series of one-way analyses of variance (ANOVAs), and the magnitude of differences was quantified using Cohen's d. Although age was not significantly different between groups, it is a key predictor of volumetric loss in the retina. As such, the volumes of retinal layers were compared between the Aβ+ and Aβ− groups with a series of analyses of covariance (ANCOVAs) with age as the only covariate. We applied the false discovery rate (FDR) approach to correct for multiple comparisons [36]. Additionally, a linear regression model covarying for age was used to examine the relationships between surface area of inclusion bodies and cortical Aβ burden (SUVr). Correlations between number and surface area of inclusion bodies and retinal layer volumes were computed using Pearson's r.
2. Results
2.1. Demographic and clinical characteristics
Table 1 summarizes the demographic and clinical characteristics of the study sample. Of the 63 participants, 10 participants were Aβ+, and 53 were Aβ−. There were no statistically significant differences between the Aβ− and Aβ+ groups on any demographic variables or any neuropsychological assessments, with the exception of a small but statistically significant difference (Aβ+ < Aβ−) on the One-Card Learning task, a test of working memory (Table 1).
2.2. Effect of Aβ on retinal layer volumes
Differences between Aβ− and Aβ+ groups on the volume of each retinal layer are summarized in Table 2. The Aβ+ group had higher volume in the inner plexiform layer (IPL; mean [SD] = 0.371 [0.028] μm3) in comparison with the Aβ− group (mean [SD] = 0.354 [0.028] μm3; Table 2, Fig. 2). However, this difference was not statistically significant after controlling for multiple comparisons using FDR correction (see Table 2). The Aβ+ group also showed moderate increases in the volume of the retinal nerve fiber layer (RNFL), but this was not significantly different from the Aβ− group (Fig. 2). Although we endeavored to compare our volume measurements for the IPL to a comparable normative sample, we were unable to identify a published normative database that reports isolated IPL segmentation in an older adult sample of healthy control subjects. Hence, to increase confidence that our measurements are valid, we compared our measures of RNFL thickness to well-established published normative data. A recent meta-analysis of 17 OCT studies compared RNFL thicknesses between patients with AD and older adult control subjects (with measurements from 790 control eyes) [18]. Across these 17 studies, the mean RNFL thickness ranged between 75 μm (SD = 3.8) and 113.16 μm (SD = 6.72). For our total sample of subjects, we found an overall mean RNFL thickness (both eyes) of 111.65 μm (SD = 4.3), which falls within the range of reported values for these other studies [18]. Hence, we have reasonable confidence in our measures for other retinal layers reported herein, including the IPL.
Table 2.
Differences in each retinal neuronal layer between low neocortical amyloid-beta load (Aβ−) and high neocortical amyloid-beta load (Aβ+) cognitively normal older adults
| Retinal layer | Age, df (F) | P | Amyloid, df (F) | P | Pf | Aβ− (n = 53), mean (SD) | Aβ+ (n = 10), mean (SD) |
|---|---|---|---|---|---|---|---|
| RNFL volume | (1.60) 0.06 | .811 | (1.60) 1.56 | .216 | .504 | 0.073 (0.001) | 0.077 (0.003) |
| GCL volume | (1.60) 0.60 | .441 | (1.60) 0.29 | .595 | .694 | 0.188 (0.003) | 0.193 (0.008) |
| INL volume | (1.60) 0.05 | .828 | (1.60) 0.11 | .745 | .745 | 0.166 (0.051) | 0.124 (0.119) |
| IPL volume | (1.60) 0.25 | .622 | (1.60) 4.98 | .029 | .203 | 0.115 (0.001) | 0.122 (0.003) |
| ONL volume | (1.60) 0.01 | .924 | (1.60) 1.03 | .315 | .551 | 0.218 (0.003) | 0.226 (0.007) |
| OPL volume | (1.60) 0.82 | .369 | (1.60) 0.69 | .410 | .574 | 0.097 (0.002) | 0.100 (0.004) |
| Inclusion bodies volume | (1.55) 2.17 | .146 | (1.55) 2.59 | .113 | .400 | 2.850 (0.618) | 5.485 (1.493) |
Abbreviations: Pf, P value after adjustment for false discovery rate (FDR; Benjamini & Hochberg, 1995); OPL, outer plexiform layer; INL, inner nuclear layer; GCL, ganglion cell layer; ONL, outer nuclear layer; IPL, inner plexiform layer; RNFL, retinal nerve fiber layer.
Note. Volume/thickness of all layers has been normed to total retinal volume/thickness for each individual participant. Values reported are ratios.
Fig. 2.

Magnitude of difference between low neocortical amyloid-beta load (Aβ−) and high neocortical amyloid-beta load (Aβ+) cognitively normal older adults on each retinal neuronal layer and the number of inclusion bodies (“0” line represents Aβ− cognitively normal older adults group) (error bars represent 95% confidence intervals).
2.3. Effect of Aβ on inclusion bodies
Relative to the Aβ− group, the Aβ+ group showed a larger surface area of inclusion bodies identified with BAF imaging; however, although this difference was moderate in magnitude, it was not large enough to reach statistical significance (Fig. 2). When Aβ burden was treated continuously in the complete sample, there was a significant relationship of a moderate magnitude between SUVr and surface area of inclusion bodies (Fig. 3). Linear regression with age as a covariate showed that surface area of inclusion bodies was a significant predictor of SUVr (F(2, 31) = 4.83, P < .05), accounting for 22.5% of SUVr variance.
Fig. 3.
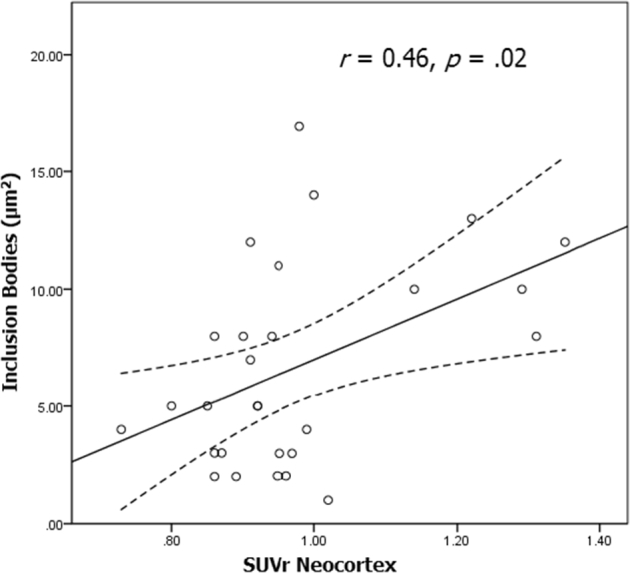
Relationship between surface area of inclusion bodies and amyloid-beta (Aβ) burden (dotted lines represent 95% confidence intervals).
2.4. Relationship between inclusion bodies and retinal layer volumes
A significant relationship (r = 0.41, P = .03) was identified between surface area of inclusion bodies and the volume of the IPL (Fig. 4A). This relationship existed despite the trend toward decreased IPL volume with increasing age (r = −0.30, P = .08). There were no significant relationships between surface area of inclusion bodies and the volume of any other retinal layer (Fig. 4B; all P's > .17). Moreover, using the HE Region Finder tool with the cross-sectional OCT imaging, the majority of these inclusion bodies were located within the IPL or adjacent to the IPL, and the vast majority of these also appeared to be in close proximity to microvessels.
Fig. 4.

Relationship between surface area of inclusion bodies and the volume of the inner plexiform layer (A) and the outer plexiform layer (B) (dotted lines represent 95% confidence intervals).
3. Discussion
This study used SD-OCT with BAF to examine retinal biomarkers of preclinical AD. Our results show that, in CN older adults who are at-risk for AD due to high levels of CNS amyloid that: (1) surface area of retinal inclusion bodies increased as a function of neocortical Aβ accumulation; (2) Aβ+ older adults show increased thickness selective to the IPL relative to Aβ− older adults. Although the effect size difference between these two groups is large (Cohen's d = 0.80), this difference did not reach statistical significance due to small sample size for the Aβ− group (N = 10); and (3) IPL volume is associated with increased total surface area of retinal inclusion bodies.
The association between neocortical Aβ deposition and the surface area of retinal inclusion bodies adds to an expanding body of literature demonstrating that pathologic changes associated with early AD can be visualized in the retina. From as early as the fourth week of gestation, the eyes, particularly the RNFL and the optic nerve, are direct sensory extensions of the central nervous system, as their axons synapse directly with several brain regions. The insoluble aggregates of Aβ plaques and neurofibrillary tangles in the brain that underlie the pathologic cascade of AD can also be detected in the retinae of both living and post mortem AD patients [37], [38]. Accordingly, it has been posited that the presence of Aβ plaques and neurofibrillary tangles may contribute to ocular changes such as RNFL thinning and associated optic nerve head cupping, and changes within the lens and blood vessel that have been observed in AD patients [15], [19], [39], [40], [41]. Kayabasi et al. used OCT to examine retinal changes in Aβ in patients with MCI. They visualized retinal Aβ by injecting curcumin, a ligand that putatively binds selectively to fibrillar Aβ, and found more inclusion bodies in the plexiform retinal layers of MCI patients than healthy controls [41]. This study builds on these findings using SD-OCT with BAF (a noninvasive technique) to visualize inclusion bodies. In this study, we have extended these findings from symptomatic patients with MCI [41] or AD [37], [38] to our participants with subjective memory complaint and a first-degree family history of AD, that is, a population at-risk for AD. It is important to note that findings of retinal Aβ in symptomatic AD patients have been mixed, with several postmortem studies failing to reveal significant retinal amyloid deposition in AD patients [42], [43]. However, our findings support the hypothesis that retinal biomarkers could be a useful screening tool to distinguish individuals at risk for developing AD and could be helpful in identifying ideal candidates for secondary prevention trials.
Our principal finding is that the surface area (μm2) accounted for by the retinal inclusion bodies that largely appear within (or adjacent to) the IPL, and are mostly observed for the Aβ+ group, is moderately correlated with neocortical amyloid burden across the entire series of 63 subjects. Moreover, there was an additional positive correlation observed between the surface area of these inclusion bodies and the thickness of the IPL layer (and this relationship was not observed for any other neuronal layer). Finally, it was only the IPL that showed a robust increase in thickness for the Aβ+ group relative to the Aβ− group, although this difference failed to reach statistical significance due to a small sample size in the Aβ− group (N = 10). The IPL is where bipolar and amacrine cells synapse with the dendrites of the ganglion cells. In several species, markers for cholinergic synapses are concentrated in the IPL, and acetylcholine is released by distinct assemblies of amacrine and possibly bipolar cells [44], [45]. Moreover, immunohistochemistry using acetylcholinesterase and choline acetyltransferase (ChAT) as markers has revealed discrete bands of cholinergic activity within the IPL in lampreys [46], amphibians [47], rodents [48], [49], nonhuman primates [49], and humans [49]. Although we acknowledge that symptomatic AD patients have shown a thinning of the RNFL [19], [41], in contrast to our current findings in the preclinical population, we hypothesize that the finding that the cholinergic-rich IPL seems to be the central neuronal layer affected in our population is important and specific to a preclinical AD population. A disruption to the cortical cholinergic system is one of the most reliable and earliest neurochemical changes in AD [50]. Recent imaging studies of the cortical cholinergic system in living AD patients have supported an early cholinergic disruption [51], [52]. Additionally, in vivo and in vitro animal work has shown that decreased activation of cholinergic receptors leads to increased Aβ burden [53], [54], [55], [56]. Finally, postmortem histopathological studies have revealed decreased ChAT activity in nonsymptomatic participants in areas with high Aβ burden [53], [57]. Taken together, this work indicates that cholinergic disruption and Aβ deposition in the neocortex co-occur in preclinical AD. The finding of increased volume in the cholinergic-rich IPL in the Aβ+ group associated with increased surface area of retinal inclusion bodies (thought to contain Aβ) suggests that a similar process could be occurring in the retina in this group of at-risk AD patients. It is possible that these retinal changes represent an early inflammatory process occurring in the major cholinergic center of the retina that parallels the early pathologic cascade of AD in the neocortex, in which early cholinergic changes co-occur with Aβ deposition in the major cholinergic centers of the brain. However, this hypothesis is tentative and requires replication and further investigation in larger cohorts to ensure greater statistical power and confidence.
Our study has several limitations. First, there is no conclusive postmortem histopathology confirming the presence of fibrillar Aβ in the inclusion bodies identified in this study. Although there are histopathologic studies confirming the presence of Aβ in inclusion bodies [25], [26], confirmation by within-subjects study design has not been completed to date. Future longitudinal investigations should attempt to include postmortem histopathology to confirm the presence of fibrillar Aβ in the same type and conformation of inclusion bodies that we found in the majority of our Aβ+ subjects (Fig. 1). Furthermore, because the SPECTRALIS system uses new technology and an advanced segmentation algorithm, normative volumetric data on finely segmented retinal layers, specifically the IPL, were not available at the time of publication. Development of these normative data as this technology becomes more widely available will be critical in the assessment of retinal correlates of preclinical AD. Additionally, our sample sizes are relatively small, with 10 Aβ+ and 53 Aβ− participants. Despite the small sample of Aβ+ participants, these initial results are promising and provide foundation for future detailed exploration of the relationship between neocortical and retinal disease burden in preclinical AD. We plan to follow these participants longitudinally to visualize within-subjects changes in retinal and cortical biomarkers associated with disease progression. Along with longitudinal analysis, replication of this study will be the next step in validating these retinal biomarkers of preclinical AD and further examining the retinal cholinergic hypotheses generated from these results.
These limitations notwithstanding, our results show that SD-OCT imaging of the retina may allow for the prediction of neocortical amyloid burden. This is the first study to demonstrate these results in a preclinical AD population, using a noninvasive imaging technique. Additionally, the preclinical stage of AD may be associated with increased volume of the inner plexiform layer of the retina, and this volume increase is correlated with increased presence of neocortical Aβ. This IPL volume increase could reflect an early inflammatory process related to disruption of cholinergic transmission that co-occurs with Aβ deposition in the neocortex and/or be a result of these inclusion bodies occupying space within (and adjacent to) this retinal layer. These preliminary results indicate that SD-OCT imaging of these early changes in the retina in preclinical AD patients could provide a more cost-effective method of examining biomarkers that are indicative of disease burden and reflective of time-specific changes in the neocortex.
Research in Context.
-
1.
Systematic review: The authors review current state of knowledge with respect to nonvascular retinal imaging markers of early Alzheimer's disease (AD) and describe the conduct of a cross-sectional study of individuals at high risk for AD, with a subset of these individuals identified as falling within the preclinical stage of the disease. This study compares neocortical beta-amyloid aggregation (PET imaging) to retinal spectral domain optical coherence tomography (SD-OCT) markers of possible disease burden in the preclinical stage of AD.
-
2.
Interpretation: Using blue-light autoflourescence imaging, inclusion bodies were found in individuals with significant neocortical amyloid burden, and these were absent in the retinal imaging of individuals without substantial beta-amyloid aggregation. The surface of these inclusion bodies increased as a function of cortical amyloid burden. Additionally, there was a trend toward a selective volume increase in the inner plexiform layer (IPL; a layer rich in cholinergic activity) of the retina, in amyloid-positive participants, and IPL volume was correlated with the surface area of the same retinal inclusion bodies.
-
3.
Future directions: If these findings are replicated, these retinal imaging markers may eventually lead to a potential cost-effective and noninvasive technique that can be used to identify those at-risk for AD. Layer-specific changes in the IPL and their association with surface area of inclusion bodies are discussed as a possible reflection of early inflammatory processes associated with cholinergic disruption and concurrent Aβ accumulation in the neocortex.
Acknowledgments
This study was funded in part by an unrestricted educational grant to P.J.S. from Pfizer Inc. Internal funding was provided for the study from Lifespan Clinical Research Center, Providence, RI, USA. The funding organization had no role in the conduct or design of this research. The Heidelberg SPECTRALIS SD-OCT imaging system was placed on loan to P.J.S., for research use, by Heidelberg Engineering GmbH, Heidelberg, Germany. The authors thank Ms. Christine Getter for her coordination of visits of our study subjects to the Lifespan Clinical Research Center, Rhode Island Hospital, Providence, RI. We also thank all of our research participants for their generosity, enthusiasm, and participation. This article is dedicated to the loving memory of Edward L. Etkind, M.D. (1936–2015).
Footnotes
All co-authors contributed to the design, execution, data analyses and manuscript preparation for this report. B.F. is a full-time employee of Heidelberg Engineering, Inc. The authors have no other relevant conflicts to disclose.
References
- 1.Au R., Piers R.J., Lancashire L. Back to the future: Alzheimer's disease heterogeneity revisited. Alzheimers Dement (Amst) 2015;1:368–370. doi: 10.1016/j.dadm.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Kawas C.H. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute of Aging and the Alzheimer's Association workgroup. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villemagne V.L., Burnham S., Bourgeat P., Brown B., Ellis K.A., Salvado O. Amyloid β deposition, neurodegeneration and cognitive decline in sporadic Alzheimer's disease: A prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 4.Uchida K., Shan L., Suzuki H., Tabuse Y., Nishimura Y., Hirokawa Y. Amyloid-β sequester proteins as blood-based biomarkers of cognitive decline. Alzheimers Dement (Amst) 2015;1:270–280. doi: 10.1016/j.dadm.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashton N.J., Kiddle S.J., Graf J., Ward M., Baird A.L., Hye A. Blood protein predictors of brain amyloid for enrichment in clinical trials? Alzheimers Dement (Amst) 2015;1:48–60. doi: 10.1016/j.dadm.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeo J.M., Waddell B., Khan Z., Pal S. A systematic review and meta-analysis of 18 F-labeled amyloid imaging in Alzheimer's disease. Alzheimers Dement (Amst) 2015;1:5–13. doi: 10.1016/j.dadm.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doraiswamy P.M., Sperling R.A., Johnson K., Reiman E.M., Wong T.Z., Sabbagh N. Florbetapir F 18 amyloid PET and 36-month cognitive decline: A prospective multicenter study. Mol Psychiatry. 2014;19:1044–1051. doi: 10.1038/mp.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mormino E.C., Betensky R.A., Hedden T., Schultz A.P., Ward A., Huijbers W. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer's disease. Neurology. 2014;82:1760–1767. doi: 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim Y.Y., Ellis K.A., Pietrzak R.H., Ames D., Darby D., Harrington K. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012;79:1645–1652. doi: 10.1212/WNL.0b013e31826e9ae6. [DOI] [PubMed] [Google Scholar]
- 10.Small G.W., Siddarth P., Kepe V., Ercoli L.M., Burggren A.C., Bookheimer S.Y. Prediction of cognitive decline by positron emission tomography of brain amyloid and tau. Arch Neurol. 2012;69:215–222. doi: 10.1001/archneurol.2011.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckley R., Saling M.M., Ames D., Rowe C.C., Lautenschlager N.T., Macaulay S.L. Factors affecting subjective memory complaints in the AIBL aging study: biomarkers, memory, affect, and age. Int Psychogeriatr. 2013;25:1307–1315. doi: 10.1017/S1041610213000665. [DOI] [PubMed] [Google Scholar]
- 12.Fyock C.A., Hampstead B.M. Comparing the relationship between subjective memory complaints, objective memory performance, and medial temporal lobe volumes in patients with mild cognitive impairment. Alzheimers Dement (Amst) 2015;1:242–248. doi: 10.1016/j.dadm.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim Y.Y., Maruff P., Getter C., Snyder P.J. Disclosure of PET Amyloid Imaging Results: A Preliminary Study of Safety and Tolerability. Alzheimers Dement. 2016;12:454–458. doi: 10.1016/j.jalz.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Lim Y.Y., Maruff P., Schindler R., Ott B.R., Salloway S., Yoo D.C. Disruption of cholinergic neurotransmission exacerbates Aβ-related cognitive impairment in preclinical Alzheimer's disease. Neurobiol Aging. 2015;36:2709–2715. doi: 10.1016/j.neurobiolaging.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Danesh-Meyer H.V., Birch H., Ku J.Y., Carroll S., Gamble G. Reduction of optic nerve fibers in patients with Alzheimer disease identified by laser imaging. Neurology. 2006;67:1852–1854. doi: 10.1212/01.wnl.0000244490.07925.8b. [DOI] [PubMed] [Google Scholar]
- 16.Iseri P.K., Altina O., Tokay T., Yuskel N. Relationship between cognitive impairment and retinal morphological and visual abnormalities in Alzheimer disease. J Neuroophthamol. 2006;26:18–24. doi: 10.1097/01.wno.0000204645.56873.26. [DOI] [PubMed] [Google Scholar]
- 17.Paquet C., Boissonnot M., Roger F., Dighiero P., Gil R., Hugon J. Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer's disease. Neurosci Lett. 2007;420:97–99. doi: 10.1016/j.neulet.2007.02.090. [DOI] [PubMed] [Google Scholar]
- 18.Thomson K.L., Yeo J.M., Waddell B., Cameron J.R., Pal S. A systematic review and meta-analysis of retinal nerve fiber layer change in dementia, using optical coherence tomography. Alzheimers Dement (Amst) 2005;1:136–143. doi: 10.1016/j.dadm.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berisha F., Feke G., Trempe C., Wallace McMeel J., Schepens C. Retinal abnormalities in early Alzheimer's disease. Invest Ophthamol Vis Sci. 2007;48:2285–2289. doi: 10.1167/iovs.06-1029. [DOI] [PubMed] [Google Scholar]
- 20.Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer's disease. Prog Retin Eye Res. 2011;30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 21.Howlett D.R., Bate S.T., Collier S., Lawman S., Chapman T., Ashmeade T. Characterisation of amyloid-induced inflammatory responses in the rat retina. Exp Brain Res. 2011;214:185–197. doi: 10.1007/s00221-011-2819-4. [DOI] [PubMed] [Google Scholar]
- 22.Keenan T.D., Goldacre R., Goldacre M.J. Associations between age-related macular degeneration, Alzheimer disease, and dementia record linkage study of hospital admissions. JAMA Ophthalmol. 2014;132:63–68. doi: 10.1001/jamaophthalmol.2013.5696. [DOI] [PubMed] [Google Scholar]
- 23.Xu H., Chen M., Forrester J.V. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz D.S., Castilla M., Rodríguez O., i Rovira M.B. Alzheimer disease and mild cognitive impairment assessment using optical coherence tomography. Poster presented at: 8th Annual Clinical Trials in Alzheimer's Disease (CTAD) Conference; November, 2015; Barcelona, Spain. J Prev Alzheimers Dis. 2015;2:368. [Google Scholar]
- 25.Dentchev T., Milam A.H., Lee V.M., Trojanowski J.Q., Dunaief J.L. Amyloid-β is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis. 2003;9:184–190. [PubMed] [Google Scholar]
- 26.Isas J.M., Luibl V., Johnson L.V., Kayed R., Wetzel R., Glabe G. Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthamol Vis Sci. 2010;51:1304–1310. doi: 10.1167/iovs.09-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snyder P.J., Lim Y.Y., Schindler R., Ott B.R., Salloway S., Daiello L. Microdosing of Scopolamine as a ‘Cognitive Stress Test’: Rationale and Test of a Very Low Dose in an At-Risk Cohort of Older Adults. Alzheimers Dement. 2014;10:262–267. doi: 10.1016/j.jalz.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Maruff P., Lim Y.Y., Darby D., Ellis K.A., Pietrzak R.H., Snyder P.J. Clinical utility of the Cogstate Brief Battery in identifying cognitive impairment in mild cognitive impairment and Alzheimer's disease. BMC Psychol. 2013;1:1–11. doi: 10.1186/2050-7283-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snyder P., Bednar M.M., Cromer J.R., Maruff P. Reversal of scopolamine-induced deficits with a single dose of donepezil, an acetylcholinesterase inhibitor. Alzheimers Dement. 2005;1:126–135. doi: 10.1016/j.jalz.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Fredrickson A., Snyder P.J., Cromer J., Thomas E., Lewis M., Maruff P. The use of effect sizes to characterize the nature of cognitive change in psychopharmacological studies: An example with scopolamine. Hum Psychopharmacol. 2008;23:425–436. doi: 10.1002/hup.942. [DOI] [PubMed] [Google Scholar]
- 31.Lim Y.Y., Prang K.H., Cysique L., Pietrzak R., Snyder P.J., Maruff P. A method for cross-cultural adaptation of a verbal memory assessment. Behav Res Methods. 2009;41:1190–1200. doi: 10.3758/BRM.41.4.1190. [DOI] [PubMed] [Google Scholar]
- 32.Clark C.M., Schneider J.A., Bedell B.B., Beach T.G., Bilker W.B., Mintun M.A. Use of Florbetapir-PET for imaging amyloid-β pathology. JAMA. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson L.V., Leitner W.P., Rivest A.J., Staples M.K., Radeke M.J., Anderson D.H. The Alzheimer's Aβ peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jack C.R., Wiste H.J., Lesnick T.G., Weigand S.P., Knopman D.S., Vemuri P. Brain β-amyloid load approaches a plateau. Neurology. 2013;80:1–7. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Region Finder Use Manual . Heidelberg Engineering GmBH; Carlsbad, CA: 2011. Software Version 2.4. [Google Scholar]
- 36.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 37.Frost S., Kanagsingam Y., Sohrabi H., Vignarajan J., Bourgeat P., Salvado O. Retinal vascular biomarkers for early detection and monitoring of Alzheimer's disease. Transl Psychiatry. 2013;3:e233. doi: 10.1038/tp.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koronyo-Hamaoui M., Koronyo Y., Ljubimov A., Miller C.L., Ko M.K., Black K.L. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54:S204–S217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein L.E., Muffat J.A., Cherny R.A., Moir R.D., Ericsson M.H., Huang X. Cystolic β-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet Neurol. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 40.Koronyo Y., Salumbides B., Black K., Koronyo-Hamaoui M. Alzheimer's disease in the retina: Imaging retinal Aβ plaques for early diagnosis and therapy treatment. Neurodegener Dis. 2012;10:285–293. doi: 10.1159/000335154. [DOI] [PubMed] [Google Scholar]
- 41.Kayabasi U., Sergott R., Rispoli M. Retinal examination for the diagnosis of Alzheimer's disease. Int J Ophthamalic Pathol. 2014;3:4–7. [Google Scholar]
- 42.Ho C.Y., Troncoso J.C., Knox D., Stark W., Eberhart C.G. Beta-amyloid, phospho-tau, and alpha-synuclein deposits similar to those in the brain are not identified in the eyes of Alzheimer's and Parkinson's disease patients. Brain Pathol. 2014;24:25–32. doi: 10.1111/bpa.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schon C., Hoffman N.A., Ochs S.M., Burgold S., Filser S., Steinbach S. Long-term in-vivo imaging of fibrillary tau in the retina of P310S transgenic mice. PLoS One. 2012;7:e53547. doi: 10.1371/journal.pone.0053547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masland R.H., Mills J.W. Autoradiographic identification of acetylcholine in the rabbit retina. J Cell Biol. 1979;83:159–178. doi: 10.1083/jcb.83.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Massey S.C., Neal M.J. The light evoked release of acetylcholine from the rabbit retina in vivo and it's inhibition by γ-aminobutyric acid. J Neurochem. 1979;32:1327–1329. doi: 10.1111/j.1471-4159.1979.tb11062.x. [DOI] [PubMed] [Google Scholar]
- 46.Pombal M., Abalo X., Rodicio M., Anadon R., Gonzalez A. Choline acetyltransferase-immunoreactive neurons in the retina of adult and developing lampreys. Brain Res. 2003;993:154–163. doi: 10.1016/j.brainres.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Lopez J., Moreno N., Gonzalez A. Localization of choline acetyltransferase in the developing and adult retina of Xenopus laevis. Neurosci Lett. 2002;330:61–64. doi: 10.1016/s0304-3940(02)00739-5. [DOI] [PubMed] [Google Scholar]
- 48.Ivanova E., Toychiev A., Yee C., Sagdullaev B. Intersublaminar vascular plexus: The correlation of retinal blood vessels with functional sublaminae of the inner plexiform layer. Invest Ophthamol Vis Sci. 2014;55:78–86. doi: 10.1167/iovs.13-13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zarbin M., Wamsley J., Palacios J., Kuhar M. Autoradiographic localization of high affinity GABA, benzodiazepine, dopaminergic, adrenergic, and muscarinic cholinergic receptors in the rat, monkey, and human retina. Brain Res. 1986;374:75–92. doi: 10.1016/0006-8993(86)90396-3. [DOI] [PubMed] [Google Scholar]
- 50.Bowen D.M., Benton J.S., Spillane J.A., Smith C.C.T., Allen S.J. Choline acetyltransferase activity and histopathology of frontal neocortex from biopsies of demented patients. J Neurol Sci. 1982;57:191–202. doi: 10.1016/0022-510x(82)90026-0. [DOI] [PubMed] [Google Scholar]
- 51.Pike K.E., Savage G., Villemagne V.L., Ng S., Moss S.A., Maruff P. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 52.Reiman E.M., Chen K., Liu X., Bandy D., Yu M., Lee W. Fibrillar amyloid-β burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beach T.G., Kuo Y.M., Spiegel K., Emmerling M.R., Sue L.I., Kokjohn K. The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer's disease. J Neuropathol Exp Neurol. 2000;59:308–313. doi: 10.1093/jnen/59.4.308. [DOI] [PubMed] [Google Scholar]
- 54.Beach T.G., Walker D., Sue L., Scott S., Layne K.J., Newell A.J. Immunotoxin lesion of the cholinergic nucleus basalis causes Aβ deposition: Towards a physiologic animal model of Alzheimer's disease. Curr Med Chem. 2003;3:57–75. [Google Scholar]
- 55.Davis A.A., Fritz J.J., Wess J., Lah J.J., Levey A.I. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci. 2010;30:4190–4196. doi: 10.1523/JNEUROSCI.6393-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seo H., Ferree A.W., Isacson O. Cortico-hippocampal APP and NGF levels are dynamically altered by cholinergic muscarinic antagonist or M1 agonist treatment in normal mice. Eur J Neurosci. 2002;15:498–506. doi: 10.1046/j.0953-816x.2001.01884.x. [DOI] [PubMed] [Google Scholar]
- 57.Potter P.E., Rauschkolb P.K., Pandya Y., Sue L.I., Sabbagh M.N., Walker D.G. Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer's disease. Acta Neuropathol. 2011;122:49–60. doi: 10.1007/s00401-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]