

Short abstract
A classification of Arabidopsis gene products on the basis of their sequence similarity to other organisms has identified a set of potential plant specific genes.
Abstract
Background
The availability of the complete genome sequence of Arabidopsis thaliana together with those of other organisms provides an opportunity to decipher the genetic factors that define plant form and function. To begin this task, we have classified the nuclear protein-coding genes of Arabidopsis thaliana on the basis of their pattern of sequence similarity to organisms across the three domains of life.
Results
We identified 3,848 Arabidopsis proteins that are likely to be found solely within the plant lineage. More than half of these plant-specific proteins are of unknown function, emphasizing the general lack of knowledge of processes unique to plants. Plant-specific proteins that are membrane-associated and/or targeted to the mitochondria or chloroplasts are the most poorly characterized. Analyses of microarray data indicate that genes coding for plant-specific proteins, but not evolutionarily conserved proteins, are more likely to be expressed in an organ-specific manner. A large proportion (13%) of plant-specific proteins are transcription factors, whereas other basic cellular processes are under-represented, suggesting that evolution of plant-specific control of gene expression contributed to making plants different from other eukaryotes.
Conclusions
We identified and characterized the Arabidopsis proteins that are most likely to be plant-specific. Our results provide a genome-wide assessment that supports the hypothesis that evolution of higher plant complexity and diversity is related to the evolution of regulatory mechanisms. Because proteins that are unique to the green plant lineage will not be studied in other model systems, they should be attractive priorities for future studies.
Background
Plants have played a major role in the geochemical and climatic evolution of our planet. Today, in addition to their fundamental ecological importance (plants account for more than 99% of the terrestrial biomass and support most of the biodiversity on Earth), plants are essential for humans as the main source of food, provide raw materials for many types of industry and chemicals for medical applications. It is thus daunting to realize how little we understand about them. For example, only approximately 10% of the genes of Arabidopsis thaliana, the best explored model system for plant biologists, have been characterized experimentally [1].
What makes plants different from other organisms? This is a central question in plant biology that has a complex, multilayered answer. The genomic sequence of Arabidopsis, published in December 2000, allows us to begin answering this fundamental question from the perspective of genetic information. By identifying the similarities and the differences in the gene content of this model plant as compared with organisms in other phylogenetic lineages, one can begin to differentiate ancient processes that are shared by cells in plants and other organisms, from those that evolved independently in the plant lineage and contributed to determining plant form and function.
The vast majority of the genes in Arabidopsis are encoded in the nuclear genome. Most of the original genetic content of the cyanobacterial ancestor of the chloroplast and the proteobacterial ancestor of the mitochondrion have been transferred to the plant cell nucleus. The remaining 79 chloroplast-encoded proteins are mostly components of the photosystem and electron-transport chain or are involved in protein synthesis [2]. Most of the 58 genes encoded in the mitochondrial genome are devoted to components of the respiratory chain and tRNAs [3].
The initial analysis of the sequenced Arabidopsis genome suggested that 70% of the Arabidopsis gene products can be assigned to functional categories on the basis of sequence similarity to genes of known function in other organisms [1]. However, the proportion of Arabidopsis genes with related counterparts in other organisms with completed genome sequences (Escherichia coli, Synechocystis sp. PC6803, Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster and Homo sapiens) vary greatly depending on the functional category: from 8-23% in transcription to 48-60% in protein synthesis when using a stringent BLASTP cutoff value of E < 10-30. These results suggest that plant transcription factors have evolved independently, but that proteins that participate in other cellular processes are highly conserved in the plant lineage [1].
Determining the function of proteins deduced from genomic sequences is a central goal in this post-genomic era. The importance of this pursuit is emphasized by the observation that even for some of the most extensively studied organisms, such as E. coli, a high proportion of the predicted genes are as yet uncharacterized [4]. How do we prioritize the characterization of thousands of genes with only hints of what their cellular roles might be, based on sequence similarity? Comparative genomics is a powerful approach that has been used extensively to predict protein function. This technique also promises to help us identify lineage-specific functions or processes that can tell us about the unique aspects of plants and other organisms. One way to promote discoveries that are of special significance to plants or other lineages is to focus attention on those genes that are found preferentially in that lineage. However, few comparative genomic studies have been carried out that focus on lineage-specific aspects of the sequenced genomes [5]. The recent availability of several complete genome sequences from all three domains of life (Eukarya, Bacteria, Archaea) makes comparative genomic strategies particularly attractive to tackle this issue.
As a first step towards identifying plant-specific genes, we classified the nuclear-encoded protein-coding genes of Arabidopsis on the basis of their pattern of sequence similarity to protein sequences of organisms that belong to Eukarya, Bacteria and Archaea. We then used this initial classification to identify 3,848 Arabidopsis proteins that are likely to be found only in green plants. We believe that these plant-specific proteins may play important roles in processes that are unique to and of significance to green plants. We found that many plant-specific proteins are known or putative transcription factors, indicating that evolution of plant-specific mechanisms of regulating gene transcription was important for the evolution of the plant lineage. However, many plant-specific proteins have no known function. From our analysis of the latter proteins, we suggest that plant-specific processes that occur in chloroplasts or mitochondria, and/or that are associated with membranes, are the most understudied. Interestingly, plant genes encoding plant-specific proteins were often expressed in an organ-specific manner. To facilitate the functional characterization of plant-specific proteins, we have compiled and integrated information from multiple public databases (for example, TIGR, MIPS, TAIR, SIGNAL) and computer prediction programs into a searchable database that is accessible from the worldwide web [6].
Results
Arabidopis protein-coding genes exhibit diverse phylogenetic profiles
To try and understand what makes plants different from other organisms at the genetic level, we sought to identify and characterize the Arabidopsis protein-coding genes that are present in plant species but not in organisms outside the Plantae. As a first step we investigated the pattern of similarity of Arabidopsis protein sequences among organisms that belong to the three domains of life (Eukarya, Bacteria and Archaea). We determined the phylogenetic profile (PP) of each Arabidopsis protein sequence by recording the presence or absence of similar sequences in protein sets from other organisms. This is similar to the "phylogenetic profiles" defined by Pellegrini et al. [7] to hypothesize protein function in E. coli; or the "binary vectors" used by Peregrin-Alvarez et al. [8] to study the phylogenetic distribution of metabolic enzymes in the same bacterium. In contrast to these studies, however, we applied a more conservative two-way cutoff to generate the PPs of plant proteins. A 'presence of similar sequence' call was made when the BLASTP E-value of the best match between a given Arabidopsis protein query and one of the nine protein sets utilized (below) was 10-10 or less. 'Absence of similar sequence' calls were made when the BLASTP E-values were greater than 0.01. Protein coding genes that exhibit BLAST E-values between 0.01 and 10-10 against any of the protein sets were not considered further (13,469 protein sequences). Although 49% of Arabidopsis proteins could not be assigned a PP with this conservative criterion, it allowed us to focus on the proteins with the best-defined pattern of conservation throughout the phylogeny. This resulted in 13,819 Arabidopsis proteins being associated to a vector of nine characters that indicated whether similar sequences were found or not found in the following protein sets: Homo sapiens, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Mus musculus, Schizosaccharomyces pombe, Saccharomyces cerevisiae, a combined set of 88 species of Bacteria, and a combined set of 16 species of Archaea.
PPs are not true phylogenies. Nevertheless, we argue that the PP is a trait of the protein sequence that reflects its evolutionary origin. In this study, we were most interested in identifying the protein sequence innovations of plants. Therefore, we did not register or consider domain architecture or other structural features beyond primary structure in the PP definition. As a consequence, proteins that are unique to the plant lineage because of innovative arrangements of ubiquitous domains will not be included in our analyses. For example, proteins with a novel arrangement of kinase domains will be classified according to the uniqueness of the primary structure alone, and kinase-domain-containing proteins are likely to have PPs indicative of presence in all organisms. Similarly, some members of gene families that are mostly plant specific might be classified in other lists if they have acquired one of these ubiquitous domains.
The current Arabidopsis genome is composed of protein-coding genes with different evolutionary histories (see, for example [5,9,10]). Consequently, it is expected that Arabidopsis protein sequences would have different PPs. Although 512 PPs are combinatorially possible with nine protein sets, genome-wide differences should be most marked when comparing across kingdoms or domains of life. Indeed, 12,521 (91%) of the Arabidopsis proteins that were assigned PPs exhibited one of the eight possible PPs defined by the presence or absence in other eukaryal, bacterial or archaeal genomes (sum of the black circle and the three intersecting circles in Figure 1a). For example, 2,436 Arabidopsis proteins were found to have sequence similarity to proteins in all the protein sets utilized (intersection of the Bacteria, Archaea and Eukarya circles in the Venn diagram in Figure 1a); 1,152 Arabidopsis proteins were found to have sequence similarities to proteins in all the eukaryotic sets examined; and 576 proteins had sequence similarity with proteins in the bacterial domain exclusively (Figure 1a). In addition, 434 proteins exhibited PPs suggestive of patterns of conservation across kingdoms (included in the circle labeled 'Other' in Figure 1a). For example, 163 Arabidopsis proteins were found only in the animal proteomes, while 27 were found only in the two fungal proteomes. PPs not readily classified accounted for 838 proteins (also included in the circle labeled 'Other' in Figure 1a). These numbers are not solely the result of gene-family expansion (for example, a few Arabidopsis proteins are similar to metazoan proteins but have given rise to large protein families). The size of gene families is similar in all PP groups and trends observed in the numbers are maintained even when looking at proteins that are unique in the Arabidopsis genome (as determined by BLASTCLUST analysis; see Figure 4 in [6]).
Figure 1.
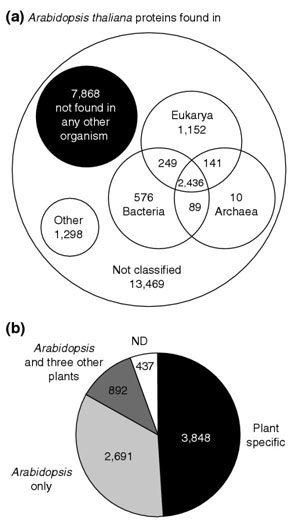
Identification of plant-specific proteins. (a) Classification of Arabidopsis proteins based on their pattern of sequence similarity to other organisms. The 27,288 Arabidopsis proteins were classified on the basis of their phylogenetic profiles (PP). Each PP recorded whether similar sequences were found or not found in the protein sets from the following organisms: Homo sapiens, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Mus musculus, Schizosaccharomyces pombe, Saccharomyces cerevisiae, a combined set of 88 species of Bacteria, and a combined set of 16 species of Archaea. Not drawn to scale. (b) Identification of putative plant-specific proteins. The Arabidopsis proteins that lack similarity to any other organism (7,868 proteins represented in the black circle in (a)) were compared against sequences in the expressed sequence tag (EST) database of Arabidopsis and 13 other plant species. A total of 3,848 Arabidopsis proteins were identified as plant specific because they showed sequence similarity to proteins in the Arabidopsis EST database and to proteins in EST databases of at least four other plant species (at E-value ≤ 10-10). In addition, 892 other Arabidopsis proteins show similarity to the Arabidopsis and one to three other plant EST databases, 2,691 Arabidopsis proteins exhibit similarity to sequences in the Arabidopsis EST database only, and 437 lack similarity to any sequence in the EST databases used.
Predominant PP corresponds to putative plant-specific proteins
Notably, the most abundant PP, exhibited by 7,868 protein sequences, corresponded to Arabidopsis proteins that showed no detectable similarity to any sequence in the protein sets used in this study (black circle in Figure 1a). This class contains proteins that are likely to be plant specific. Characterization of the plant-specific protein functions and identification of the processes in which they participate should help us understand the molecular features that distinguish plants from other organisms.
Absence of Arabidopsis proteins in all other protein sets examined could be explained in two major ways: these Arabidopsis proteins are generally present in plants but are absent in non-plant lineages (plant innovation, divergence, gene loss); or these Arabidopsis proteins are spurious or incorrect gene predictions. If these are real plant-specific proteins, we reasoned that similar proteins should be expressed in other plant species. Therefore, to distinguish these two possibilities, we compared the 7,868 protein sequences against the Arabidopsis expressed sequence tag (EST) database and the EST databases of 13 other vascular plant species, including both monocotyledonous and dicotyledonous species (see the list of species in Materials and methods). ESTs comprise the largest pool of sequence data for many plant species and contain portions of transcripts from many uncharacterized genes. Because ESTs have no annotated coding sequences, a TBLASTN search was performed. We considered as plantspecific any of the 7,868 proteins identified previously that showed significant sequence similarity (E ≤ 10-10) against protein sequences in the Arabidopsis EST database and the databases of at least four other plant species. After manually excluding proteins encoded in retroelements or transposable elements, 3,848 Arabidopsis proteins were selected and classified as putative plant-specific proteins (Figure 1b). Because of the stringency of our criteria, it is likely that we have missed many true plant-specific genes. In addition, it is clear that even with a relaxed cutoff, the list of plant-specific proteins will still not be exhaustive. ESTs are fragments of genes and EST populations often do not include rarely expressed genes. In addition, we analyzed only protein-coding genes, and it is known that many noncoding RNAs are likely to be kingdom specific ([11], and J. Kastenmayer and P.J.G., unpublished work). Although non-exhaustive, this approach allowed us to identify a set of sequences that are strong candidates for expressed proteins that are plant specific.
Many plant-specific proteins are of unknown function
To gain insight into the cellular functions of the plant-specific proteins defined above, we examined their annotation. As shown in Table 1, approximately 38% of the plant-specific proteins are annotated only as 'expressed protein' in version 4 of the TIGR Arabidopsis genome (from April 2003). This is almost twice the proportion of expressed proteins currently annotated in the whole genome (Table 1). Another 14% of plant-specific proteins were annotated as 'hypothetical protein', a proportion comparable to that observed in the whole genome. Together, 2,017 (52%) of plant-specific proteins are without known cellular function. In stark contrast, only 2.5% of the proteins that are conserved in all protein sets are annotated as hypothetical or expressed proteins (Table 1). Because of this, we consider this list of plant-specific proteins a good starting point for future reverse-genetic strategies. Understanding the function of uncharacterized plant-specific proteins is likely to provide a rich source of novel insights about plant biology.
Table 1.
Proteins with poor annotation are abundant among plant-specific proteins
| Plant-specific proteins | Arabidopsis proteins found in all protein sets | Whole Arabidopsis protein set* | |
| Hypothetical protein | 557 (14%) | 12 (0.5%) | 4,363 (15%) |
| Unknown proteins | 3 (0.1%) | 0 (0.0%) | 26 (0.1%) |
| Expressed protein | 1,457 (38%) | 50 (2.0%) | 6,683 (23%) |
| Total | 2,017 (52%) | 62 (2.5%) | 11,072 (38%) |
| Total in class | 3,848 | 2,436 | 28,581 |
Comparison of the number of proteins annotated as 'expressed protein', 'hypothetical protein' or 'unknown protein' in the list of plant-specific proteins, proteins conserved throughout the phylogeny and in the whole Arabidopsis proteome. Total number of protein sequences per category (percentage relative to the total in the class) are shown. *Although the Arabidopsis TIGR genome release v3.0 (2002) was used to make the classification (plant-specific or other groups) and for all the other data analysis in this study, the numbers in this table reflect the latest protein annotation available from TIGR (genome release v4.0, April 2003).
In an effort to provide clues about the cellular roles and facilitate prioritization of the study of proteins without known function, we analyzed their predicted subcellular localization and some other properties (all predictions and information gathered are available in our database [6]). Subcellular localization and membrane-spanning predictions were carried out with TargetP [12] and TMHMM [13], respectively, as described in Materials and methods. We predict that 614 plant-specific proteins with unknown function are targeted to chloroplasts or mitochondria (Table 2). This suggests that many plant-specific proteins of unknown function have roles in processes that occur in these plant organelles. Interestingly, a statistically significant difference in the predicted subcellular localization was found for plant-specific proteins of unknown function compared with those with known functions (p < 0.0001). Plant-specific proteins of unknown function are more likely to be found in organelles (mitochondria and chloroplasts) and less likely to be in the secretory pathway compared with those with known function. This suggests that the secretory pathway has been the focus of greater research attention than have the other two organelles in plants. While this conclusion is less surprising for chloroplasts, because their metabolism cannot be studied in non-plant systems, it is somewhat surprising for mitochondria, which have been well studied in yeast and mammalian systems. On the other hand, the high proportion of plant-specific mitochondrial proteins of unknown function may reflect the extent to which plant mitochondrial metabolism differs from that of mitochondria of other organisms [14].
Table 2.
Prediction of subcellular localization and transmembrane helices
| Unknown plant-specific proteins | Known plant-specific proteins | Whole Arabidopsis protein set | |
| TargetP predictions | |||
| Any other location | 1,073 (53%) | 1,006 (55%) | 15,706 (58%) |
| Chloroplast | 360 (18%) | 253 (14%) | 3,972 (15%) |
| Mitochondria | 254 (13%) | 178 (10%) | 2,963 (11%) |
| Secretory pathway | 330 (16%) | 394 (22%) | 4,647 (17%) |
| TMHMM predictions | |||
| Membrane associated | 220 (11%) | 89 (5%) | 2,075 (8%) |
| Total | 2,017 | 1,831 | 27,288 |
Total number of protein sequences per category (percentage relative to the total in the group).
Similarly, a significantly higher proportion of plant-specific proteins of unknown function were predicted to be membrane associated when compared with the known plant-specific proteins (p < 0.0001) or with the entire proteome of Arabidopsis (p < 0.0001) (Table 2). This suggests that membrane association also contributes to a poorer understanding of protein function. Most of the plant-specific proteins of unknown function were represented by ESTs. In fact, 74 genes that encode plant-specific proteins were represented by 20 or more ESTs, suggesting that at least some of them are highly or moderately expressed (see [6]). Together, these data suggest that putative membrane-associated plant-specific proteins that are predicted to be targeted to the mitochondria or chloroplasts, and in particular those judged from the EST frequency to be highly expressed, are attractive candidates for future studies.
Plant-specific proteins with annotated function are often transcription factors, while other basic cellular processes are underrepresented
We found 1,831 (45%) plant-specific proteins with annotations suggestive of various functional roles. Conspicuous by their prevalence, 494 (13%) of the total plant-specific proteins are known or putative transcription factors. This is more than twice as many as expected, based on the current estimate of 5% of the total number of plant transcription factors encoded in the Arabidopsis nuclear genome [15], and is by far the most prevalent functional group among plant-specific proteins. We surmise that evolution of proteins specifically involved in the control of gene transcription was an important factor in the evolution of vascular plants. This finding further supports the hypothesis that the evolution of plant form was in large part determined by the evolution of regulatory mechanisms [16]. As expected, none of the plant-specific proteins is an obvious component of the conserved basal transcriptional machinery (RNA polymerase subunits, for example) or general transcription factors (such as the TATA-box binding protein or subunits of the transcription factors TFIIB, IIE, IIF, IIH and so on). Instead, most plant-specific proteins related to transcription appear to be proteins that can bind DNA and can presumably activate or repress transcription in response to specific developmental, environmental or physiologic cues.
The biggest group belongs to the AP2/ERF domain transcription factor family. In fact, 124 of the 142 annotated AP2/ERF transcription factors are plant-specific on the basis of our criteria. AP2/ERF is one of the largest families of Arabidopsis transcription factors. Members of this family are involved in a wide range of processes, in development as well as in response to biotic or abiotic stress [17]. The second largest group of plant-specific transcription factors (73 proteins) given by our analysis belongs to the NAC superfamily. NAC transcription factors are specific to plants, in which they are known to play a role in developmental processes [18]. The third largest group of plant-specific transcription factors comprised 44 proteins from the WRKY superfamily. This represents about 55% of the total number of WRKY transcription factors currently annotated in the Arabidopsis genome [19]. WRKY transcription factors are involved in a variety of processes such as pathogen defense, trichome development and senescence [19]. Other plant-specific proteins involved in transcription belong to various families of transcription factors - bHLH (31), GRAS (28), C2C2 family (42), MYB (25), TCP (21) and others (Table 3) - and to transcriptional regulator families such as the AUX/IAA family (24) and others (10).
Table 3.
Arabidopsis plant-specific proteins with known or hypothetical function and that are involved in central cellular processes
| Gene family | Number of plant-specific proteins |
| Known or putative transcription factors | |
| AP2/ERF | 124 |
| ARF | 16 |
| B3 | 14 |
| bHLH | 31 |
| bZIP | 12 |
| C2C2 | 42 |
| C2H2 | 2 |
| EIN/EIL | 5 |
| GRAS | 28 |
| HD | 13 |
| Leafy | 1 |
| MADS | 2 |
| MYB | 25 |
| NAC | 73 |
| SBP | 11 |
| TCP | 21 |
| Trihelix | 1 |
| VP1/ABI3 | 1 |
| WRKY | 44 |
| Other | 28 |
| Other transcriptional regulators | |
| AUX/IAA | 24 |
| Other | 10 |
| Pre-mRNA processing and transport | 1 |
| Translation | 7 |
| Protein degradation | |
| F-box proteins | 115 |
In contrast to transcription, other basic cellular processes were poorly represented in the plant-specific category (Table 3). Only one plant-specific protein was found that could be related to pre-mRNA processing (At5g19480). In addition, no known components of the mRNA transport and degradation machineries could be found, although some plant-specific proteins contained domains (such as LRP1, RRM) that have been associated with RNA metabolism in a previous study [6,20]. A few plant-specific proteins were involved in translation. The two Arabidopsis acidic 60S ribosomal proteins of the P3 type - Rpp3a (At4g25890) and Rpp3b (At5g57290) - were classified as plant-specific. Acidic ribosomal proteins form a characteristic complex or 'stalk' on the side of the large subunit of eukaryotic ribosomes [21]. Though the functional role of this complex is unclear, it is thought to participate in the elongation phase of translation [21]. Animals, fungi and protozoans possess three classes of acidic ribosomal P proteins: P0, P1 and P2. However, consistent with our results, plants are known to possess an extra class of acidic P proteins termed P3 [22]. The eukaryotic initiation factor 4B (At3g26400, At4g38710, At1g13020) was also classified as plant specific. This classification is consistent with previous studies that indicate that eIF-4B in plants is different from its counterparts in other organisms [23]. Finally, the ribosomal L5 protein (At2g07725) and a ribosomal-related protein (At2g10980) were also classified as plant specific. Further characterization of the roles of these proteins in translation may uncover novel plant-specific aspects of protein synthesis.
Proteins involved in protein degradation were also poorly represented in the plant-specific category, with the exception of F-box proteins. The F-box is a structural motif about 50 amino acids in length [24]. It is present in F-box proteins and is involved in protein-protein interaction of the F-box proteins with the other members of the SCF complex [24,25]. The SCF complexes form one of the largest and best understood families of E3-ligases present in eukaryotic genomes. The function of these complexes is to facilitate the formation of a linkage between the ubiquitin peptide and a protein substrate [26]. A polyubiquitin chain is formed afterwards, thus targeting the protein for proteasome-mediated degradation. F-box proteins confer the substrate specificity on the SCF complex, and in plants have an important role in processes such as cell-cycle control, floral development, circadian rhythms and responses to the hormones auxin and jasmonic acid [27]. Of the proteins that appear to contain an F-box domain, 115 are unique to plants (Table 3). F-box proteins, as defined by the presence of the F-box domain but otherwise unrelated in primary structure, are found in other organisms [25]. This is also one of the protein families that shows the biggest expansion in the plant lineage [5]. Thus, together with transcriptional regulatory mechanisms, protein degradation may have also played a role in vascular plant evolution.
Protein phosphorylation/dephosphorylation is a major mechanism used by both plants and animals to regulate many cellular processes. It is estimated that about 4% of Arabidopsis genes encode proteins that belong to the eukaryotic kinase superfamily and 1% encode protein phosphatases (PlantsP database [28]). We found only one of the protein kinases annotated in the PlantsP database classified as plant-specific (At4g00340) and no protein phosphatases. We found that most protein kinases had PPs indicative of similar sequences in all the genomes analyzed. In contrast, phosphatases had PPs indicative of similar sequences in all eukaryote genomes. Protein phosphatases are highly conserved between plants and animals [29], but there are known groups of protein kinases that appear to be unique to plants (see, for example [30]). This novelty however, is based on plant-specific domain architecture and not the primary structure. Our study thus indicates that few signal transduction components involving phosphate transfer have been evolutionary novelties in plants; we have found that most, if not all, have been constructed by shuffling, addition or deletion of domains that are shared by all eukaryote genomes; similarly to other whole-genome sequences [31].
The role of plant-specific proteins in plant cell metabolism
Plants have an extraordinarily rich metabolic capacity, producing many thousands of structurally different compounds [32]. In an effort to understand the significance of plant-specific proteins for plant cell metabolism, we asked what proportion of plant-specific proteins contributed to this metabolic capability, and whether they define metabolic pathways that are innovations of the plant lineage. As one way to address these questions, we used the proteins classified in this study to query the AraCyc database of metabolic pathways [33]. AraCyc currently includes more than 170 metabolic pathways, covering primary and secondary metabolism as well as several plant-specific metabolic pathways [33]. Of the Arabidopsis proteins with PPs, 432 were assigned to reactions in one or more of the pathways in AraCyc for a total of 869 references in the metabolic database. Not surprisingly, 252 of these 432 proteins corresponded to proteins with conserved sequences throughout the phylogeny. These 252 proteins were found to catalyze several steps in catabolic pathways such as glycolysis, the tricarboxylic acid cycle, and fatty-acid degradation. Similarly, they also catalyzed reactions in anabolic pathways such as purine and pyrimidine biosynthesis, and participated in several amino-acid biosynthesis pathways, fatty-acid biosynthesis, gluconeogenesis and other pathways. In stark contrast, only 19 out of the 3,848 plant-specific proteins, catalyzing 11 different reactions, were found in AraCyc (Table 4). These results indicate that most plant-specific proteins do not participate in the metabolic pathways currently present in AraCyc. Furthermore, the few plant-specific proteins found catalyzed at most two steps in the same pathway, suggesting that most of the plant pathways in this database are not innovations of the plant lineage.
Table 4.
Plant-specific proteins that are found in the AraCyc database
| Locus | Protein description | Metabolic pathway | Enzyme name | Reaction* |
| At1g78240 | Similar to early-responsive to dehydration stress ERD3 protein | Carbon monoxide dehydrogenase pathway | Methyltransferase | 2.1.1.- |
| At1g08550 | Violaxanthin de-epoxidase precursor, putative | Carotenoid biosynthesis | Violaxanthin de-epoxidase | RXN-325 |
| At1g08550 | Violaxanthin de-epoxidase precursor, putative | Carotenoid biosynthesis | Violaxanthin de-epoxidase | RXN-314 |
| At1g78240 | Similar to early-responsive to dehydration stress ERD3 protein | CO2 formation from methanol | Methyltransferase | METHTRANSBARK-RXN |
| At1g53520 | Chalcone-flavanone isomerase-related | Flavonoid biosynthesis | Chalcone isomerase | 5.5.1.6 |
| At5g05270 | Chalcone-flavanone isomerase family | Flavonoid biosynthesis | Chalcone-flavonone isomerase | 5.5.1.6 |
| At5g66220 | Putative chalcone-flavanone isomerase (chalcone isomerase) (CHI) | Flavonoid biosynthesis | Chalcone isomerase | 5.5.1.6 |
| At1g27690 | Lipase -related | Glycerol biosynthesis | Lipase | 3.1.1.3 |
| At5g03980 | gdsl-motif lipase/hydrolase protein | Glycerol biosynthesis | Lipase | 3.1.1.3 |
| At1g13280 | Allene oxide cyclase family similar to ERD12 | Jasmonic acid biosynthesis | Allene oxide cyclase | 5.3.99.6 |
| At1g19640 | S-adenosyl-L-methionine:jasmonic acid carboxyl methyltransferase (JMT) | Jasmonic acid biosynthesis | S-adenosyl L-methionine:jasmonic acid carboxyl methyltransferase | RXN1F-28 |
| At1g13280 | Allene oxide cyclase family similar to ERD12 | Lipoxygenase pathway | Allene oxide cyclase | 5.3.99.6 |
| At4g21610 | Lsd1 like protein | L-serine degradation | LSD1 | 4.2.1.13 |
| At1g53520 | Chalcone-flavanone isomerase-related | Phytoalexin biosynthesis | Chalcone isomerase | 5.5.1.6 |
| At5g66220 | Putative chalcone-flavanone isomerase (chalcone isomerase) (CHI) | Phytoalexin biosynthesis | Chalcone isomerase | 5.5.1.6 |
| At5g05270 | Chalcone-flavanone isomerase family | Phytoalexin biosynthesis | Chalcone-flavonone isomerase | 5.5.1.6 |
| At1g03040 | bHLH protein component of the pyruvate dehydrogenase complex E3 | Pyruvate dehydrogenase | Pyruvate dehydrogenase (lipoamide) | 1.2.4.1 |
| At2g45880 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At3g23920 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At4g15210 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At4g17090 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At5g18670 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At5g45300 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At5g55700 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
| At2g32290 | Glycosyl hydrolase family 14 (beta-amylase) | Starch degradation† | Beta-amylase | 3.2.1.2 |
*EC number is given when available, otherwise the AraCyc [70] frame name for the reaction is given. †AraCyc designation for this metabolic pathway is 'Starch and cellulose biosynthesis'. However, as far as we know, genes in this family are only involved in starch degradation.
Consistent with the results obtained with AraCyc, visual inspection of protein annotations failed to detect obvious primary metabolic functions in the plant-specific category. In addition to this general observation, we consider in more detail lipid and carbohydrate metabolism.
A catalog of 600 Arabidopsis genes representing 210 cellular activities involved in acyl lipid metabolism was recently constructed based on annotations by experts in the field [34]. Because acyl lipid metabolism is a primary metabolic pathway common to all eukaryotic, and most prokaryotic, organisms, it might be expected that relatively few of these proteins would be classified as plant specific. This was the case. Although 82 of the 600 Arabidopsis proteins were plant specific, most of these were members of large gene families. For example, 52 of the plant-specific proteins are classified as lipid-transfer proteins, an abundant class of extracellular proteins possibly involved in defense responses or cuticle lipid production. Of the 210 enzymes or cellular activities involved in plant acyl lipid metabolism, only 15 were plant specific. Within this group of 15, in addition to lipid-transfer proteins, we found a lysophosphatidate acyltransferase, an allene oxide synthase (involved in jasmonic acid biosynthesis), oleosin, and several lipases or acyl hydrolases. However, with the exception of allene oxide synthase, oleosins and lysophosphatidate acyltransferase, the function of most of the plant-specific proteins that are involved in acyl lipid metabolism has not been confirmed, and they represent attractive targets for further study.
Another area of plant metabolism that may employ many plant-specific proteins is cell-wall metabolism. Because the walls surrounding plant cells are different from the cell walls of bacteria and fungi, it seems likely that many of the enzymes needed for the synthesis, reorganization and degradation of cell-wall components might be specific to plants. Although analysis of the plant-specific genes identified a number of proteins possibly involved in wall biosynthesis and reorganization (see below), many of the enzymes involved in wall degradation are shared with many other organisms, including bacteria and fungi. This is not surprising; many organisms use plants as a food source and therefore need the ability to degrade plant cell-wall components.
In contrast, proteins involved in wall biosynthesis and reorganization are well represented among the plant-specific genes, at least as far as they can be identified. For example, the expansins are a group of proteins involved in loosening the connections between wall components to allow growth and development [35], and most of them are present in the list of plant-specific proteins. The proteins involved in cellulose biosynthesis have been identified as plant homologs of bacterial cellulose synthases [36]. Thus, it is expected that these proteins would not be plant specific, but rather would be shared with bacteria. In contrast, some members of a family of proteins related to xyloglucan fucosyltransferase [37,38] are plant specific. This is somewhat unexpected, in that many organisms, including bacteria and animals, have fucosyltransferase enzymes. Although the evolution of this large family of enzymes is unclear, the genes encoding the plant enzymes either arose via convergent evolution or have diverged sufficiently that they no longer have significant sequence similarity.
Finally, because so few enzymes involved in cell-wall biosynthesis have been identified, it is likely that many of these enzymes are annotated as expressed proteins or hypothetical proteins. For example, many wall polysaccharides and glycoproteins contain arabinose, a sugar rarely found in the glycoconjugates of other organisms. Thus, the many different enzymes needed for the addition of arabinose to plant glycoconjugates have few, if any, homologs in better studied organisms. Because none of the plant arabinosyltransferases has yet been identified, it seems likely that they would be among the proteins annotated as expressed proteins or hypothetical proteins. Because they are expected to be integral membrane proteins and because it is expected that plants should contain a significant number of different arabinosyltransferases, this group of proteins would be a good candidate for functional genomic studies.
Many plant-specific genes have organ-specific expression
A phylogenetic mode of gene expression has been proposed for the development of C. elegans [39]. In this nematode, evolutionarily conserved genes are expressed early during development, whereas worm-specific genes are expressed preferentially during later developmental stages [39]. We posit that such a phylogenetic model of gene expression is also present in plants. A prediction from this model is that plant-specific genes are likely to show preferential expression in organs as compared to genes that are evolutionarily conserved. To test this prediction, we analyzed the expression of genes represented on publicly available microarray experiments. We found 1,071 genes that encode plant-specific proteins represented on glass slide microarrays from the Arabidopsis Functional Genomics Consortium (AFGC) (see Materials and methods). Interestingly, a high proportion of the plant-specific genes (600 of the 1,071) were differentially expressed in at least one organ comparison experiment (Figure 2a). Flowers and roots showed the strongest bias in the number of differentially expressed genes among the plant-specific group. However, a statistically significant bias was observed for all comparisons (Figure 2b). This bias suggests that many plant-specific proteins have roles in processes that occur in specific organs, particularly in roots and flowers. In contrast to plant-specific genes, genes encoding proteins that are conserved in other lineages show no greater preferential expression in organs than would a random group of genes (Table 5). Moreover, plant proteins with similarity to proteins encoded in other eukaryote genomes were less likely to be preferentially expressed in organs. Plant genes that code for proteins with similarity to bacterial proteins were also biased towards preferential expression in organs. However, most of the differentially expressed genes in this group encoded proteins with similarity to proteins in cyanobacteria and are predicted to function in the chloroplast (data not shown). Thus, these results are in agreement with a phylogenetic model of expression for plant genes.
Figure 2.
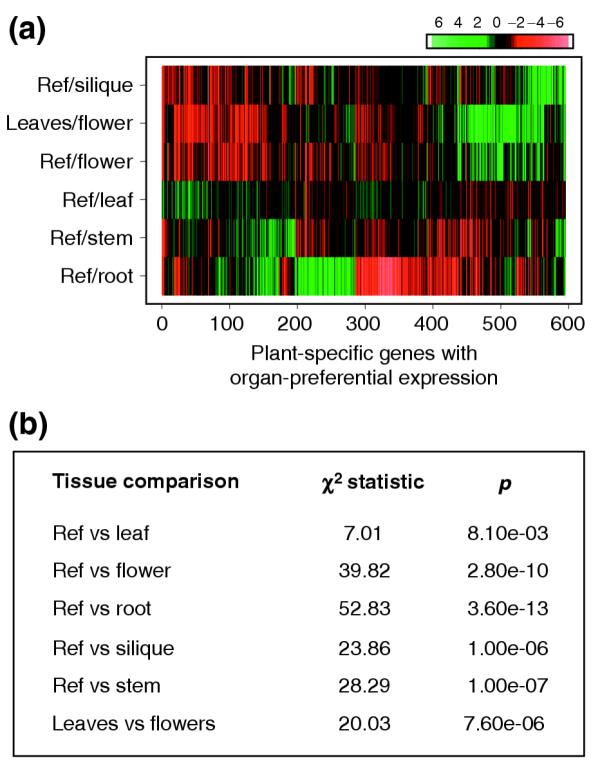
Arabidopsis genes encoding plant-specific proteins exhibit preferential expression in organs. (a) Heat map showing the 600 plant-specific genes that exhibited differential expression in at least one microarray experiment comparing RNA samples from different plant organs. Microarray experiments were obtained from the Stanford Microarray Database. The mean was calculated for the replicates. Organ preferential expression was defined as a twofold or higher ratio in the comparison. Gene expression is expressed as the log2(ratio). The bar at the top right indicates the magnitude of change. Green indicates induction and red indicates depression of gene expression. Ref, reference sample; see Materials and methods for details. (b) For all organ comparisons the number of differentially expressed genes in the plant-specific category was statistically higher than the number of differentially expressed genes that are not plant specific. Calculation of the statistical significance was done using the chi-square test for contingency tables.
Table 5.
Plant-specific genes are preferentially expressed in organs compared with genes that are evolutionarily conserved
| Classification | Differentially expressed | Percentage of total | χ2 | p | |
| Statistically significant organ-preferential expression | Plant specific | 600 | 56% | 51.27 | 8.1e-13 |
| Bacteria (includes cyanobacteria) | 122 | 67% | 32.74 | 1.1e-08 | |
| No statistically significant difference from a random sample | Eukaryotes and archaea | 37 | 61% | 4.82 | 2.8e-02 |
| Eukaryotes and bacteria | 370 | 46% | 0.22 | 6.4e-01 | |
| Archaea | 3 | 60% | 0.03 | 8.5e-01 | |
| Archaea and bacteria | 52 | 49% | 0.01 | 9.2e-01 | |
| Common to all | 15 | 48% | 0.00 | 9.6e-01 | |
| Statistically significant expression everywhere | Eukaryotes | 112 | 27% | 61.75 | 3.9e-15 |
The first column indicates the conclusion from the statistical test. The second column indicates the phylogenetic classification of the genes analysed in each row. The number of genes from each class (for example, plant-specific) that showed organ-preferential expression is indicated in the third column. The fourth column shows the percentage of genes that showed organ-preferential expression as compared to the total number of genes represented on Arabidopsis glass-slide microarrays for each class. The χ2 statistic and the p value are presented in the fifth and sixth columns, respectively.
We next asked whether the plant-specific group or some of the other groups defined by the PPs correlated with groupings derived from gene expression alone, thus identifying responses with a potentially common evolutionary history. In addition to the plant-specific proteins, the corresponding genes for 1,654 other proteins classified in this project (Figure 1a) were also represented on AFGC microarrays. We used hierarchical clustering analysis to group these 2,725 genes on the basis of their expression profiles. We only considered clusters with a 0.5 or higher correlation coefficient in the expression pattern among cluster members and clusters with 10 or more members. We found 34 such clusters and determined which of these showed 80% or higher enrichment in any of the PPs defined in this study. Interestingly, only two clusters were found enriched in any PP and they were enriched in plant-specific genes (Table 6). From the putative functions of the genes in the clusters found above, plant-specific processes may be related to defense or stress responses. These data suggest that these plant responses to biotic or abiotic stress have evolved entirely in the plant lineage.
Table 6.
Two groups of plant-specific genes exhibit common expression profiles
| PP* | Representative EST clone ID | Locus | Description |
| Cluster 1† | |||
| PS | 111O21XP | At1g19180 | Expressed protein |
| PS | 123B21T7 | At1g30755 | Expressed protein |
| PS | 209F11T7 | At1g63090 | F-box protein (SKP1 interacting partner 3-related) |
| PS | 181I16T7 | At1g72510 | Expressed protein |
| PS | 148B19T7 | At1g74950 | Expressed protein |
| PS | 40F4T7 | At2g23320 | Identical to WRKY DNA-binding protein 15 |
| EBA | 240G12T7 | At2g31880 | Putative leucine-rich repeat transmembrane protein kinase |
| EBA | 169J16T7 | At2g39660 | Putative protein kinase |
| PS | 172K21XP | At3g16860 | Expressed protein |
| PS | 94C19T7 | At3g25870 | Expressed protein |
| PS | 114O7T7 | At4g12070 | Expressed protein |
| PS | 250F15T7 | At4g19515 | Similar to disease resistance protein |
| PS | 137B1T7 | At4g30390 | Expressed protein |
| PS | 122N24T7 | At5g13180 | NAM-like protein; hypothetical senescence upregulated protein SENU5 |
| PS | 204H15T7 | At5g13200 | GRAM-domain-containing protein similar to ABA-responsive protein |
| E | 195M6T7 | At5g22250 | CCR4-associated factor-like protein |
| PS | 200J12T7 | At5g62520 | Expressed protein |
| Cluster 2‡ | |||
| PS | 169C12T7 | At1g05250 | Putative peroxidase |
| PS | 113H5XP | At1g52050 | Jacalin lectin family similar to myrosinase-binding protein homolog |
| EBA | 121N12T7 | At1g61590 | Putative serine/threonine protein kinase |
| PS | 40E4T7 | At1g74770 | Hypothetical protein; predicted by GenemarkHMM |
| B | 34E12T7 | At3g24670 | Polysaccharide lyase family 1 (pectate lyase) |
| PS | 122J15T7 | At4g14060 | Major latex protein (MLP)-related |
| PS | 194B13T7 | At4g15390 | Acyltransferase family |
| PS | 204N5XP | At4g26010 | Putative peroxidase |
| PS | 144C19T7 | At5g07080 | Transferase family similar to 10-deacetylbaccatin III-10-O-acetyl transferase |
| PS | 116F2T7 | At5g45070 | Putative disease resistance protein (TIR class) |
| PS | 110O2T7 | At5g57685 | Unknown protein; predicted by GenemarkHMM |
Experiments were ranked according to the proportion of genes in the cluster that were differentially expressed. The most important experiments for each cluster are indicated. *PP, phylogenetic profile. †Low expression in flowers compared to leaves, unstable and moderately unstable transcripts. ‡High expression in roots as compared to a reference made of the whole plant, repressed during shoot development from root explants. PS, plant specific; EBA, Arabidopsis protein with similarity to proteins in other eukaryotes, bacteria and archaea; B, Arabidopsis protein with similarity to proteins in bacteria.
Discussion
Comparative genome analysis has established that gene number alone is not sufficient to explain organismal complexity. Current estimates from the annotation of the Arabidopsis genome sequence indicate that there are around 27,000 protein-coding genes in this flowering plant [40]. Surprisingly, the sequence of the human genome indicates that the authors of this manuscript have only about 3,000 more protein-coding genes than the simple weed. It is clear that such a small number of genes cannot explain the vast differences in developmental, morphological and behavioral processes that separate the two species.
Several hypotheses have been developed to explain how organisms can increase greatly in complexity without major increases in numbers of genes (for example, alternative splicing [41], DNA rearrangement during differentiation [42], and transcriptional regulation [16,43]). Our data suggest that evolution of lineage-specific mechanisms of transcriptional control is one important factor in the evolution of plants. Among the 1,831 plant-specific proteins with known or putative function, transcription factors stand out as the most prevalent functional group. Four hundred and ninety-four transcription factors from various families were identified as plant specific. This result is consistent with the initial characterization of the Arabidopsis proteome, which indicated that many proteins in the transcription function category (including transcription factors and other aspects of mRNA metabolism) do not show sequence similarity with proteins in other sequenced model organisms [1]. This is also consistent with previous estimates that indicate that approximately half of Arabidopsis transcription factors are from families unique to plants [15]. Importantly, the predominance of this functional category among all plant-specific proteins, defined only on the basis of primary structure, indicates that the evolution of plant form was at least partly accomplished by the evolution of plant-specific mechanisms to control gene expression at the transcriptional level. This result also prompts us to speculate that the Arabidopsis genome contains an equally rich diversity of cis-acting regulatory elements. Thus, one could imagine that the combinations of plant-specific transcription factors and their cognate cis-acting sequences could provide a very large source of mechanistic alternatives and could easily bridge the gap in aspects of development, morphology, behavior, and processes that separate plants from organisms in other lineages. In fact, candidate plant-specific transcription factors involved in each of these processes can be identified from the list of plant-specific proteins. For example, members of the GRAS and homeodomain family are involved in developmental processes, members of the TCP family are needed for the body plan, and members of the AP2/ERF family are known to mediate responses to environmental stimuli. The hypothesis that evolution of complexity and diversity is related to the evolution of regulatory mechanisms over common sets of genes is not a new one [16,44,45]. Nevertheless, our results provide a quantitative assessment that supports this hypothesis and identifies the characteristics of the proteins involved.
F-box proteins are one of the most expanded gene families in plants [5]. Also, the Arabidopsis F-box protein family diversified in such a way that many members show no detectable sequence similarity to proteins in other organisms. Together, these observations suggest that regulation of protein turnover has also had an important role in plant evolution. Significantly, new connections between transcription and protein degradation [46] and the known importance of protein decay for the action of phytohormones such as auxin and other plant-specific processes [47], further suggests that the interplay between the evolution of protein degradation and plant-specific mechanisms to activate or repress gene transcription is an important theme in plant biology.
Plants produce an incredible diversity of chemical compounds [32]. In an effort to understand the significance of plant-specific proteins for plant cell metabolism, we used the AraCyc database of metabolic pathways [33] to investigate the metabolic pathways in which they participate. AraCyc currently includes a total of 170 metabolic pathways, approximately 100 of which have five or more reactions. AraCyc was constructed computationally and curated manually and is neither complete nor error-free. Nevertheless, it is one of the broadest compilations of plant metabolism currently available, covering many primary and secondary metabolic pathways. Only 19 out of the 3,848 plant-specific proteins, catalyzing 11 reactions, were found in AraCyc. In contrast, many steps in central pathways were catalyzed by proteins conserved throughout the phylogeny consistent with the ancient origin of primary metabolism. Thus, in accord with previous studies [32], our results suggest that the extremely rich metabolic capacity of plants has arisen largely from modification of metabolic pathways that were present in the plant ancestor, rather than by evolution of new pathways.
Interestingly, Arabidopsis genes that code for plant-specific proteins but not for proteins that are evolutionarily conserved were often expressed preferentially in plant organs. This result suggests that plant gene expression follows a phylogenetic model similar to that observed in C. elegans [39]. In this model, evolutionarily conserved genes that code for functions essential for all cell types tend to be expressed constitutively throughout the organism. In contrast, plant-specific genes, which evolved later to carry out regulatory or specialized functions, are expressed later during development and preferentially in certain cell types.
Although this study focused primarily on proteins that lack similar sequences in all other non-plant organisms, the frequency of occurrence of some other PPs can also inform us about the evolution of plant form. Arabidopsis proteins with a PP indicating conservation throughout all forms of life were the next most abundant group. Such plant sequences that are conserved in the genomes of Eukarya, Bacteria and Archaea are likely to belong to ancient protein domains that were created or acquired early in evolution, and are good candidates for proteins found in the last universal common ancestor [48]. Thus these proteins are likely to carry out basic cellular functions that are essential to cellular integrity in all organisms. Indeed, many of the proteins in this category are easily recognized as components of the translation machinery or involved in primary metabolic processes [6].
The distribution of PPs also suggests that plants have more in common with bacteria than with archaea (Figure 1a). This certainly reflects the rich bacterial heritage in plants. It is now widely accepted that extensive gene transfer has occurred from the cyanobacterial ancestor of the chloroplast to the plant nuclear genome [49]. Consistent with this, many Arabidopsis proteins that show sequence similarity to bacterial proteins are predicted to contain chloroplast transit peptides, suggesting plastid localization. In addition, about half of the Arabidopsis proteins with similarity to bacterial proteins are present in cyanobacterial proteomes; thus, as much as 55% of the current plant proteins shared with bacteria may come from the endosymbiotic event with a cyanobacterium (see supplementary information in [6]). Plants also seem to have retained metabolic capabilities of bacterial origin that are absent in other eukaryotic genomes (for example many plant glycoside hydrolases, pectinesterases and pectate lyases have similarity to bacterial proteins). In contrast to the commonality between plants and bacteria, only 10 Arabidopsis proteins were found to have similarity exclusively to archaeal proteins. This is somewhat surprising because many proteins encoded in archaeal genomes, such as components of the DNA replication, transcription and translation machineries, are more similar to proteins encoded in eukaryote genomes than to proteins from bacteria (reviewed in [50]). This result could be biased because of the higher number of bacterial genomes (88) over archaeal genomes (16) available for this study. However this bias could not be too significant because there are 141 plant proteins that are similar both in other eukaryotes and in archaea (Figure 1a). Many of the proteins in this last group are subunits of the ribosome or components of the basal transcriptional machinery. Consequently, these data suggest that the plant proteins shared with archaea have a deeper evolutionary origin than those shared with bacteria, as is observed through the analysis of clusters of orthologous groups [51].
Surprisingly, we found more Arabidopsis proteins in common with the five animal proteomes analyzed than with the two fungal proteomes (for example, 163 Arabidopsis proteins were found only in the animal proteomes, while 27 proteins were found only in the two fungal proteomes). Molecular phylogenetic studies based on multiple protein sequences often suggest a closer evolutionary relationship between fungi and animals (for example [52]), albeit the split into plants, animals and fungi is thought to have occurred in a relatively short period of time. The larger number of proteins in common between plants and animals as compared with fungi prompt us to speculate that the fungal ancestor might have diverged earlier than the ancestor that gave rise to plants and animals.
Although a large majority of PPs observed (more than 97%) were consistent for eukaryotic organisms (for example, if present in human, then also present in all other animal protein sets), a few PPs were unexpected. Unexpected patterns may reveal novel features about the biology of plants, but they can also point out problems with the genome information available. The latter was clearly the case for 18 Arabidopsis proteins, which were found in all eukaryotic proteomes and in the bacterial and archaeal protein sets but not in the proteome of Rattus norvegicus. These 18 proteins were distributed throughout the Arabidopsis genome, and showed no obvious relationship in terms of sequence or function. Similarly, 36 other proteins were present in all eukaryote proteomes analyzed except in Rattus norvegicus. Although it is formally possible that some of these genes are truly absent from the rat genome, this genome sequence is not complete and is less well annotated than most others. Therefore, a more plausible hypothesis is that similar genes are present in rat but in genome regions that have not yet been properly sequenced, assembled or annotated. Accordingly, PPs may be useful for evaluating and identifying potential problems with genomic sequences.
In any study of this type, the ultimate reasons for the observed plant-specific nature of protein sequences are unclear and require further detailed analysis. Beyond the technical issues described in previous sections, plant proteins without detectable similarity in other genomes can be explained in at least three ways: first, these proteins may have been present in the common ancestor but diverged early and/or rapidly after speciation such that similarity can no longer be detected; second, they may have been lost in other lineages; and third, they may be true plant innovations occurring after plants diverged from ancestors of the other organisms. In some cases, such as the AP2 domain transcription factors, it is plausible that this protein class evolved after the divergence of the major eukaryotic lineages. In other cases, such as transcription factors that are members of the GRAS family, they may be distant relatives of proteins that are present in other major eukaryotic lineages [53]. Regardless of the reason, it is clear that the presence of these sequences in plants but not in other forms of life leaves little doubt that they are important for plant functions and processes.
Because of their great economic importance, angiosperms have received disproportionately more attention than plant species from other phylogenetic groups. Although this has greatly advanced plant research, the relatively scarce genomic data on other green plant lineages (green algae, mosses, ferns, liverworts, hornworts, lycophytes and gymnosperms) preclude us from knowing the degree of conservation of the plant-specific proteins identified in this study and the general nature of our conclusions beyond the vascular plants. It will be of great interest in the future to extend this analysis to other phyletic groups inside and outside the Plantae as more genome sequences become available. A more extensive comparison of the proteins currently encoded in plant genomes (plant-specific and other) with other species should help us gain further insight into the evolutionary history of plants.
In contrast to evolutionarily conserved proteins, we know little about the function of most plant-specific proteins. The Arabidopsis genome annotation pipeline relies largely on sequence similarity to known genes in other species [40]. Thus, it is not surprising that a list of genes that have been selected on the basis of lack of similarity to other highly studied model systems (such as bacteria, yeast, and fly) have poor annotations. In addition, it is a common experimental strategy to look for homologs in plants for genes that have already been characterized in yeast, bacteria or animals. Because genes that are conserved in all organisms are more likely to have roles in processes that are fundamental for the function of all types of cells (such as basal transcription, translation, central metabolic pathways) it is also likely that these have received more attention. Nevertheless, this great difference emphasizes that proteins present only in plant species are much less studied than proteins that are also found in other model organisms. Predictions of subcellular localization and transmembrane helices suggest that plant-specific processes that occur in chloroplasts and mitochondria and membrane-associated plant-specific proteins are the least understood. In addition, some genes coding for unknown plant-specific proteins are highly expressed (as judged by high EST counts). Therefore, the study of proteins with unknown function that are highly expressed, membrane-associated, and/or that function in chloroplasts or mitochondria is likely to be a fruitful approach to discover novel aspects of plant processes.
With the completion of the genome sequence of this model plant, attention is shifting to functional studies. We believe plant-specific proteins of unknown function are great candidates for future study. Because these genes will not be studied in non-plant model systems, they represent attractive priorities for the plant community, especially in the context of the 2010 Project, the aim of which is to understand the function of every Arabidopsis gene by the end of the year 2010 [54].
Materials and methods
Sequences and generation of phylogenetic profiles
Arabidopsis thaliana proteins were obtained from The Institute for Genomics Research (TIGR) [55]. Drosophila melanogaster proteins were obtained from FlyBase [56]. All other protein sequences were obtained from the Genome section of the National Center for Biotechnology (NCBI) ftp server [57]. The complete expressed sequence tag (EST) databases utilized were obtained from TIGR [58]. A complete list of the organisms and EST databases used in this study is available from the Plant Specific Database website [6].
The complete set of predicted Arabidopsis proteins was used in BLASTP comparisons against each of the following protein sets: Homo sapiens, Rattus norvegicus, Drosophila melanogaster, Caenorhabditis elegans, Mus musculus, Schizosaccharomyces pombe, Saccharomyces cerevisiae, a combined set of 88 species of bacteria, and a combined set of 16 species of archaea. We constructed the phylogenetic profile (PP) of each Arabidopsis protein sequence from the BLASTP reports by recording the 'presence' or 'absence' in the other protein sets. A 'presence of similar sequence' call was made when the BLASTP E-value was 1e-10 or less. 'Absence of similar sequence' calls were made when the BLAST E-value was 0.01 or greater. Proteins that exhibit BLAST E-values between 0.01 and 1e-10 against any of the genomes were not considered any further.
The above procedure identified putative plant-specific genes by selecting PPs that indicated absence in all protein sets compared. These protein sequences were then further characterized by TBLASTN searches against EST databases from 13 species of monocotyledonous and dicotyledonous plants. Proteins with matches against the Arabidopsis EST database and EST databases of four other plant species (E-value cutoff <1e-10) were selected for further analysis. The following EST databases were utilized for this step: Arabidopsis Gene Index; Hordeum vulgare Gene Index; Cotton Gene Index; Ice Plant Gene Index; Maize Gene Index; Medicago truncatula Gene Index; Pinus Gene Index; Potato Gene Index; Rice Gene Index; Sorghum bicolor Gene Index; Soy Gene Index; Sunflower Gene Index; Tomato Gene Index; Wheat Gene Index.
At the outset of this study, we evaluated several E-value cutoff schemes to generate the PPs. We found that the PPs were not substantially affected by moderate changes to the E-value cutoff. Although the stringent E-values we chose may lead to some omissions, the number omitted is small and we felt that it was important to minimize false positives rather than allow too many false negatives. In this regard, previous studies have suggested a much larger number of proteins as plant specific, up to 10,000. The difference between this number and ours is due in part to the different E-value cutoff used, but largely because of the requirement we imposed for plant-specific genes to be expressed, as documented by their presence in EST databases. This expression requirement removed many gene annotations based solely on computational predictions and that were probably erroneous.
All BLAST report pages were parsed with the Bio::SearchIO module from the Bioperl project [59]. All other sequence manipulations and data analyses were done with custom-made Perl scripts.
Analysis of the annotation of plant-specific proteins
The following steps were performed to identify known or putative functions of plant-specific proteins. First, we used the automatic functional classification scheme developed by the Munich Information Center for Protein Sequences [60] to classify plant-specific proteins in functional groups (data not shown). We have implemented a web server that automates this analysis (available from the Bio-Webtools Server [61]). Second, for groups of proteins of interest we manually inspected the annotations made by TIGR. Known proteins were not corroborated further (for example, floral homeotic protein APETALA2 identical to SP:P47927). Proteins with putative annotations were manually inspected for the presence of corresponding protein domains using the Pfam database [62]. When available, the annotations in lists of proteins were also corroborated by comparing against expert databases. The transcription factors used in this study were corroborated by AGRIS [63]. Proteins involved in RNA metabolism were corroborated by a previous study [20]. Protein phosphatases and kinases were corroborated by the PlantsP database [28]. Metabolic enzymes were corroborated by AraCyc [33] and the Arabidopsis Lipid Gene Database [34]. Selected gene families were corroborated by the annotation of experts in the field (see 'Search by Gene Family' in [6]).
Prediction of subcellular localization and transmembrane helices
Subcellular localization predictions were carried out with TargetP [12] using the web server [64]. TargetP looks for amino-terminal sorting signals by feeding the outputs from SignalP, ChloroP and an analogous mitochondrial predictor into a neural network that makes the final choice between the different compartments. It provides a score and a reliability class (a measure of the difference between the winner and runner-up models) to evaluate the significance of the prediction. The TargetP web server size cutoff of 4,000 amino acids precluded analysis of the complete sequences of four Arabidopsis protein-coding genes (At1g48090.1, At1g67120.1, At3g02260.1 and At5g23110.1). In these cases, only the amino-terminal portion of the protein was utilized for the prediction.
Putative transmembrane helices were predicted using TMHMM [13] through the web server [65]. TMHMM uses a hidden Markov model to predict transmembrane helices from the amino-acid sequence.
The complete output of the TargetP and TMHMM programs for each protein sequence analyzed is available at [6].
Gene-expression analysis
To analyze the expression of genes in this study, we used a highly filtered dataset prepared from the publicly available two-color microarray experiments performed by the Arabidopsis Functional Genomics Consortium (AFGC, described in detail elsewhere and available upon request). Briefly, all microarray hybridizations, including a wide variety of experimental treatments available from the Stanford Microarray Database (SMD) [66] as of January 2002, were initially considered for the analysis. Hybridizations were discarded for technical reasons (partial microarrays, multiple scans of the same hybridization, control experiments of the AFGC) or because they corresponded to experiments done with RNA samples from other species. Spot quality parameters were applied to each hybridization to filter out sub-optimal data points. The parameters were: sum of raw channel intensities ≥ 1,000; channel-intensity values could not be saturated in more than one channel per hybridization; 50% or more of the pixels in the spot had to be greater than 1.5 times background (in at least one channel per hybridization); good values for qualitative indicators of spot quality (Flag = 0); we included only spots that were printed with DNA from good PCR reactions (SMD codes 0, 5 and 7). The lowess method by sector was then used to normalize each hybridization [67]. Slide quality parameters were then applied to filter out sub-optimal hybridizations: hybridizations with a strong gradient in the ratios after normalization were discarded [68]; experiments or hybridizations with low reproducibility were not considered further. Reproducibility was qualitatively assessed by scatter plots of the replicates. EST clones that had been printed several times were averaged and a final data table was generated by calculating the median of redundant EST clones (those that represent the same gene). Genes with data in 200 or more experiments were then selected to generate a final dataset that contained 7,513 rows (genes) × 338 columns (hybridizations).
For the analysis of gene expression in organs, the experiments with the following SMD identifiers were utilized: 7197, 7199, 7200, 7201, 7203, 7205, 21096, 21097, 21098, 21099, 2370, 2371. The mean was calculated for the replicates. Organ preferential expression was defined as a twofold or higher ratio between the two samples compared in the microarray experiments.
We used the uncentered correlation similarity metric to perform average linkage clustering. All data analysis and manipulation was done in S-PLUS.
Statistical analysis
Calculation of the statistical significance of over- or under-representation of categorical properties in the lists generated in this study was done using the chi-square test for contingency tables [69]. All data analysis and manipulation was done in S-PLUS.
Acknowledgments
Acknowledgements
We thank Matt Larson for expert technical assistance, Curtis Wilkerson for useful comments throughout this project, Ralph Taggart and Eric Brenner for critical reading of this manuscript, and Vivek Anantharaman and Eugene V. Koonin for providing the list of Arabidopsis proteins involved in RNA metabolism. We thank Karen Bird for editorial assistance and Lan Xue from the Michigan State University Statistical Consulting Service for statistical support. Funded by DOE (DE-FG02-91ER20021) to K.K. and P.G.; NSF (DBN987638) grant to P.G., K.K. and J.B.O.
References
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Sato S, Nakamura Y, Kaneko T, Asamizu E, Tabata S. Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 1999;6:283–290. doi: 10.1093/dnares/6.5.283. [DOI] [PubMed] [Google Scholar]
- Marienfeld J, Unseld M, Brennicke A. The mitochondrial genome of Arabidopsis is composed of both native and immigrant information. Trends Plant Sci. 1999;4:495–502. doi: 10.1016/S1360-1385(99)01502-2. [DOI] [PubMed] [Google Scholar]
- Koonin EV. Genome sequences: genome sequence of a model prokaryote. Curr Biol. 1997;7:R656–R659. doi: 10.1016/S0960-9822(06)00328-9. [DOI] [PubMed] [Google Scholar]
- Lespinet O, Wolf YI, Koonin EV, Aravind L. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002;12:1048–1059. doi: 10.1101/gr.174302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Plant-Specific Gene Database http://genomics.msu.edu/plant_specific
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peregrin-Alvarez JM, Tsoka S, Ouzounis CA. The phylogenetic extent of metabolic enzymes and pathways. Genome Res. 2003;13:422–427. doi: 10.1101/gr.246903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/S1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- Chothia C, Gough J, Vogel C, Teichmann SA. Evolution of the protein repertoire. Science. 2003;300:1701–1703. doi: 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- MacIntosh GC, Wilkerson C, Green PJ. Identification and analysis of Arabidopsis expressed sequence tags characteristic of non-coding RNAs. Plant Physiol. 2001;127:765–776. doi: 10.1104/pp.127.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Siedow JN, Day DA. Respiration and photorespiration. In: Buchanan B, Gruissem W, Jones R, editor. In Biochemistry and Molecular Biology of Plants. Rockville, MD: American Society of Plant Physiologists; 2000. pp. 676–728. [Google Scholar]
- Riechmann JL, Heard J, Martin G, Reuber L, Jiang CZ, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- Doebley J, Lukens L. Transcriptional regulators and the evolution of plant form. Plant Cell. 1998;10:1075–1082. doi: 10.1105/tpc.10.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–646. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- Xie Q, Frugis G, Colgan D, Chua NH. Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 2000;14:3024–3036. doi: 10.1101/gad.852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5:199–206. doi: 10.1016/S1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchorzewski M. The acidic ribosomal P proteins. Int J Biochem Cell Biol. 2002;34:911–915. doi: 10.1016/S1357-2725(02)00012-2. [DOI] [PubMed] [Google Scholar]
- Szick K, Springer M, Bailey-Serres J. Evolutionary analyses of the 12-kDa acidic ribosomal P-proteins reveal a distinct protein of higher plant ribosomes. Proc Natl Acad Sci USA. 1998;95:2378–2383. doi: 10.1073/pnas.95.5.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AM, Wong KC, Malmstrom SA, Browning KS. Eukaryotic initiation factor 4B from wheat and Arabidopsis thaliana is a member of a multigene family. Biochem Biophys Res Commun. 1999;266:314–321. doi: 10.1006/bbrc.1999.1814. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:reviews3002.1–3002.7. doi: 10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/S0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001;2:169–178. doi: 10.1038/35056563. [DOI] [PubMed] [Google Scholar]
- del Pozo JC, Estelle M. F-box proteins and protein degradation: an emerging theme in cellular regulation. Plant Mol Biol. 2000;44:123–128. doi: 10.1023/A:1006413007456. [DOI] [PubMed] [Google Scholar]
- PlantsP: Functional genomics of plant phosphorylation http://plantsp.sdsc.edu [DOI] [PMC free article] [PubMed]
- Rodriguez PL. Protein phosphatase 2C (PP2C) function in higher plants. Plant Mol Biol. 1998;38:919–927. doi: 10.1023/A:1006054607850. [DOI] [PubMed] [Google Scholar]
- Hrabak EM, Chan CW, Gribskov M, Harper JF, Choi JH, Halford N, Kudla J, Luan S, Nimmo HG, Sussman MR, et al. The Arabidopsis CDPK-SnRK superfamily of protein kinases. Plant Physiol. 2003;132:666–680. doi: 10.1104/pp.102.011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- De Luca V, St Pierre B. The cell and developmental biology of alkaloid biosynthesis. Trends Plant Sci. 2000;5:168–173. doi: 10.1016/S1360-1385(00)01575-2. [DOI] [PubMed] [Google Scholar]
- Mueller LA, Zhang P, Rhee SY. AraCyc: a biochemical pathway database for Arabidopsis. Plant Physiol. 2003;132:453–460. doi: 10.1104/pp.102.017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisson F, Koo AJ, Ruuska S, Schwender J, Pollard M, Thelen JJ, Paddock T, Salas JJ, Savage L, Milcamps A, et al. Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol. 2003;132:681–697. doi: 10.1104/pp.103.022988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove DJ. Loosening of plant cell walls by expansins. Nature. 2000;407:321–326. doi: 10.1038/35030000. [DOI] [PubMed] [Google Scholar]
- Pear JR, Kawagoe Y, Schreckengost WE, Delmer DP, Stalker DM. Higher plants contain homologs of the bacterial celA genes encoding the catalytic subunit of cellulose synthase. Proc Natl Acad Sci USA. 1996;93:12637–12642. doi: 10.1073/pnas.93.22.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RM, DeRocher AE, Bar-Peled M, Zeng W, Norambuena L, Orellana A, Raikhel NV, Keegstra K. Xyloglucan fucosyltransferase, an enzyme involved in plant cell wall biosynthesis. Science. 1999;284:1976–1979. doi: 10.1126/science.284.5422.1976. [DOI] [PubMed] [Google Scholar]
- Sarria R, Wagner TA, O'Neill MA, Faik A, Wilkerson CG, Keegstra K, Raikhel NV. Characterization of a family of Arabidopsis genes related to xyloglucan fucosyltransferase1. Plant Physiol. 2001;127:1595–1606. doi: 10.1104/pp.127.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL. Genomic analysis of gene expression in C. elegans. Science. 2000;290:809–812. doi: 10.1126/science.290.5492.809. [DOI] [PubMed] [Google Scholar]
- Wortman JR, Haas BJ, Hannick LI, Smith RK, Jr, Maiti R, Ronning CM, Chan AP, Yu C, Ayele M, Whitelaw CA, et al. Annotation of the Arabidopsis genome. Plant Physiol. 2003;132:461–468. doi: 10.1104/pp.103.022251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17:100–107. doi: 10.1016/S0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Eastman QM, Schatz DG. Transposition mediated by RAG1 and RAG2 and its implications for the evolution of the immune system. Nature. 1998;394:744–751. doi: 10.1038/29457. [DOI] [PubMed] [Google Scholar]
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Wilson AC, Maxson LR, Sarich VM. Two types of molecular evolution. Evidence from studies of interspecific hybridization. Proc Natl Acad Sci USA. 1974;71:2843–2847. doi: 10.1073/pnas.71.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D. Evolution of transcriptional regulation. Curr Opin Genet Dev. 2000;10:575–579. doi: 10.1016/S0959-437X(00)00130-1. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Brower CS, Conaway JW. Gene expression - emerging roles of ubiquitin in transcription regulation. Science. 2002;296:1254–1258. doi: 10.1126/science.1067466. [DOI] [PubMed] [Google Scholar]
- Hellmann H, Estelle M. Plant development: regulation by protein degradation. Science. 2002;297:793. doi: 10.1126/science.1072831. [DOI] [PubMed] [Google Scholar]
- Kyrpides N, Overbeek R, Ouzounis C. Universal protein families and the functional content of the last universal common ancestor. J Mol Evol. 1999;49:413–423. doi: 10.1007/pl00006564. [DOI] [PubMed] [Google Scholar]
- Douglas SE. Plastid evolution: origins, diversity, trends. Curr Opin Genet Dev. 1998;8:655–661. doi: 10.1016/S0959-437X(98)80033-6. [DOI] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Archaea and the prokaryote-to-eukaryote transition. Microbiol Mol Biol Rev. 1997;61:456–502. doi: 10.1128/mmbr.61.4.456-502.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Galperin MY. Sequence - Evolution - Function: Computational Approaches in Comparative Genomics. Boston: Kluwer Academic; 2003. [PubMed] [Google Scholar]
- Baldauf SL, Roger AJ, Wenk-Siefert I, Doolittle WF. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science. 2000;290:972–977. doi: 10.1126/science.290.5493.972. [DOI] [PubMed] [Google Scholar]
- Richards DE, Peng J, Harberd NP. Plant GRAS and metazoan STATs: one family? BioEssays. 2000;22:573–577. doi: 10.1002/(SICI)1521-1878(200006)22:6<573::AID-BIES10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Chory J, Ecker JR, Briggs S, Caboche M, Coruzzi GM, Cook D, Dangl J, Grant S, Guerinot ML, Henikoff S, et al. National Science Foundation-sponsored Workshop Report: 'The 2010 Project' functional genomics and the virtual plant. A blueprint for understanding how plants are built and how to improve them. Plant Physiol. 2000;123:423–426. doi: 10.1104/pp.123.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Institute for Genomic Research http://www.tigr.org
- FlyBase@flybase.net http://www.flybase.org
- Genome section of the National Center for Biotechnology (NCBI) ftp server ftp://ftp.ncbi.nih.gov/genomes
- TIGR gene indexes http://www.tigr.org/tdb/tgi
- bioperl.org - main page http://www.bioperl.org
- Frishman D, Mokrejs M, Kosykh D, Kastenmuller G, Kolesov G, Zubrzycki I, Gruber C, Geier B, Kaps A, Albermann K, et al. The PEDANT genome database. Nucleic Acids Res. 2003;31:207–211. doi: 10.1093/nar/gkg005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Bio-Webtools server http://128.122.133.135
- Pfam: Pfam home page http://www.sanger.ac.uk/Software/Pfam/index.shtml
- Davuluri R, Sun H, Palaniswamy S, Matthews N, Molina C, Kurtz M, Grotewold E. AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics. 2003;4:25. doi: 10.1186/1471-2105-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TargetP server v1.01 http://www.cbs.dtu.dk/services/TargetP
- TMHMM server, v. 2.0 http://www.cbs.dtu.dk/services/TMHMM
- SMD: home page http://genome-www5.stanford.edu
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez RA, Ewing RM, Cherry JM, Green PJ. Identification of unstable transcripts in Arabidopsis by cDNA microarray analysis: rapid decay is associated with a group of touch- and specific clock-controlled genes. Proc Natl Acad Sci USA. 2002;99:11513–11518. doi: 10.1073/pnas.152204099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels ML, Witmer JA. Statistics for Life Science. San Francisco: Pearson Education; 2003. [Google Scholar]
- TAIR biochemical pathways http://www.arabidopsis.org/tools/aracyc