

Short abstract
This article introduces an algorithmic method, termed modular decomposition, that defines the organization of protein-interaction networks as a hierarchy of nested modules.
Abstract
We introduce an algorithmic method, termed modular decomposition, that defines the organization of protein-interaction networks as a hierarchy of nested modules. Modular decomposition derives the logical rules of how to combine proteins into the actual functional complexes by identifying groups of proteins acting as a single unit (sub-complexes) and those that can be alternatively exchanged in a set of similar complexes. The method is applied to experimental data on the pro-inflammatory tumor necrosis factor-α (TNF-α)/NFκB transcription factor pathway.
Background
Protein complexes and their shared components
Most cellular processes are the result of a cascade of events mediated by proteins that act in a cooperative manner. Proteins combine into macromolecular complexes to execute essential tasks, such as replication, transcription, protein transport or metabolic reaction catalysis. Proteins can therefore be viewed as elementary building blocks of these molecular machines. Moreover, protein complexes can share components: proteins can be reused and participate to several complexes. Identifying protein complexes and the way they share components hence appears as an essential step in describing cellular biology on a molecular basis.
Several technologies for detecting protein interactions such as yeast two-hybrid (Y2H) and protein-complex purifications (PCP) have recently been scaled-up to high-throughput level and have generated large-scale protein-protein interaction datasets [1-4]. Up to now, methods for analyzing such datasets have mainly been based on clustering techniques. They have been applied to assign protein function by inference from the biological context as given by their interactors [5], and to identify complexes as dense regions of the network [6,7]. Such approaches, in general, do not aim to reveal the detailed structure within and between the detected regions. The logical organization into shared and specific components, and its representation, remains elusive.
The phenomenon of shared components, that is, proteins or groups of proteins occurring in different complexes, is fairly common. A shared component may be a small part of many complexes, acting as a unit that is constantly reused for its function. It may also be the main part of the complex, for example in a family of variant complexes that differ from each other by distinct proteins that provide functional specificity. It is important to capture and properly represent the modularity of protein-protein interaction networks by identifying the shared components and the way they are arranged to generate complexes.
Protein-protein interaction networks are classically represented by graphs with proteins as nodes and physical interactions represented by edges connecting the nodes. Here, we introduce a novel method to elucidate and represent the logical organization of protein-protein interaction networks by using the graph-theory notion of module and the related idea of modular decomposition. Following a brief description of the concept, we first verify the method on known complexes and then interpret a large-scale protein-protein interaction network around the transcription factor NFκB.
Modular decomposition of graphs
Modules
A graph is a formal framework for representing elements and their relations. Elements are represented as nodes and a link connects two nodes of elements in relation. Nodes connected by a link are said to be neighbors. In graph theory, a module is a set of nodes that have the same neighbors outside the module (Figure 1).
Figure 1.
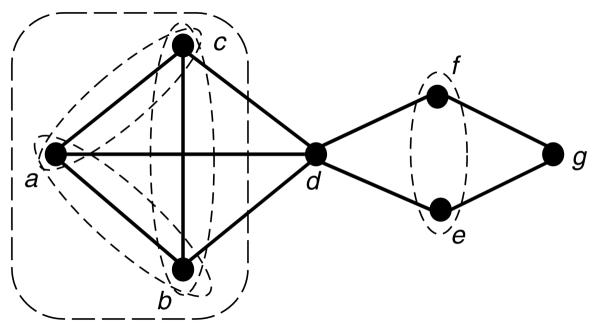
A graph and its modules. By definition, a module is a set of nodes that have the same neighbors outside the module. In addition to the trivial modules {a},{b},...,{g} and {a,b,c,..,g}, this graph contains the modules {a,b,c}, {a,b},{a,c},{b,c} and {e,f}.
Quotient
Because elements of a module have exactly the same neighbors outside the module, one can substitute all of them for a representative node. In a quotient, all elements of the module are replaced by the representative node, and the edges with the neighbors are replaced by edges to the representative.
Quotients can be iterated until the entire graph is merged into a final representative node. Iterated quotients can be captured in a tree, where each node represents a module, which is a subset of its parent and the set of its descendant leaves.
Modular decomposition
The modular decomposition is a unique, canonical tree of iterated quotients. Formal proofs as well as generalizations to structures other than graphs have been described by Möhring et al. [8,9]. The nodes of the modular decomposition are labeled in three ways (Figure 2): as series when the direct descendants are all neighbors of each other; as parallel when the direct descendants are all non-neighbors of each other; and by the structure of the module otherwise (the so-called prime module case). Modular decomposition derives an exact alternative representation of a graph as a tree of labeled nodes.
Figure 2.
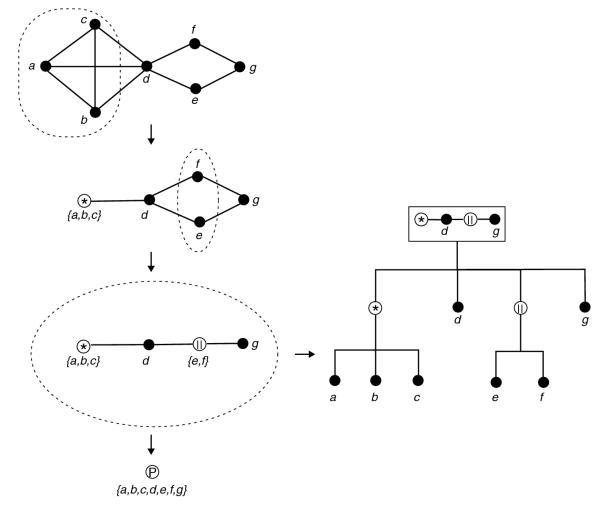
Modular decomposition of the example graph in Figure 1. Modular decomposition gives a labeled tree that represents iterations of particular quotients, here the successive quotients on the modules {a,b,c} and {e,f}. Series are labeled by an asterisk within a circle, parallel by two parallel lines within a circle, and prime by a P within a circle. The prime is advantageously labeled by its structure. The graph can be retrieved from the tree on the right by recursively expanding the modules using the information in the labels. Therefore, the labeled tree can be seen as an exact alternative representation of the graph.
Protein-protein interaction networks
The relationship between proteins identified via PCP and yeast two-hybrid (Y2H) methods is of a different nature (PCP in this instance comprises both the TAP-MS method (tandem affinity purification with mass spectrometric identification) used by Gavin et al. [10] and HMS-PCI (high-throughput mass spectrometric protein complex identification) as defined by Ho and colleagues [3]). TAP, for example, identifies multiprotein complexes, whereas Y2H systems [11] detect direct physical interactions between two proteins. Hence, edges in the corresponding networks symbolize different physical relationships between proteins. Therefore, with a different semantic given to the graph, modular decomposition has a different meaning. As we are interested in protein complexes, we focus our analysis on the PCP context.
A PCP experiment starts with the selection of a protein, called the bait. Purification of the bait results in the co-purification of proteins that co-occur in at least one complex with the bait protein. We assume in a first approach the datasets to be complete, that is, with all proteins in a given network systematically selected as baits. The dataset can be represented by a graph with proteins as nodes and an edge between proteins A and B if, and only if, there is at least one complex containing both A and B (Figure 3a).
Figure 3.
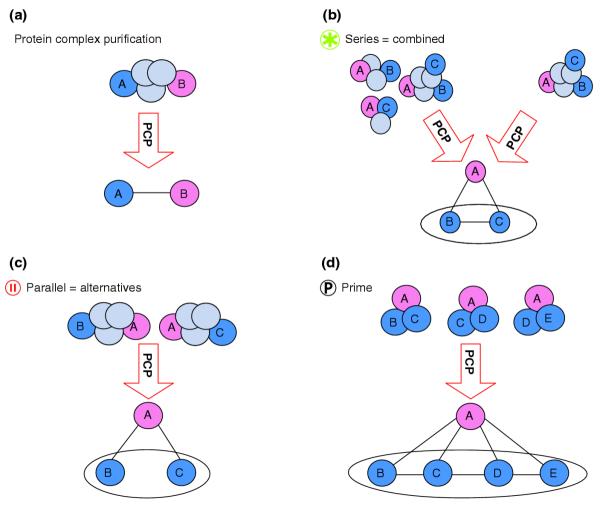
Interpretation of graph and module labels for systematic PCP experiments. (a) Two neighbors in the network are proteins occurring in a same complex. (b) Several potential sets of complexes can be the origin of the same observed network. Restricting interpretation to the simplest model (top right), the series module reads as a logical AND between its members. (c) A module labeled parallel corresponds to proteins or modules working as strict alternatives with respect to their common neighbors. (d) The prime case is a structure where none of the two previous cases occurs. Symbols are as in Figure 2.
It is important to note that a complex appears as a clique, that is, a fully connected sub-part of the network (Figure 4). However, the converse is not true: not every clique in the network necessarily derives from an existing complex. For example, three connected proteins can be the outcome of a single trimer, three heterodimers or combinations thereof (Figure 3b). On the basis of network analysis one cannot discriminate between these theoretical options. Moreover, the stoichiometry of complex constituents, that is, the respective number of copies of the same protein in the assembly cannot be inferred from PCP experiments. We therefore disregard stoichiometry issues and deal with the multiple options by adopting a parsimonious solution that embeds all possibilities: we consider the largest possible complex, which appears as a maximal clique in the graph. Finding maximal cliques is the basis for algorithms of protein-complex computation based on protein-protein interaction networks [6,7].
Figure 4.
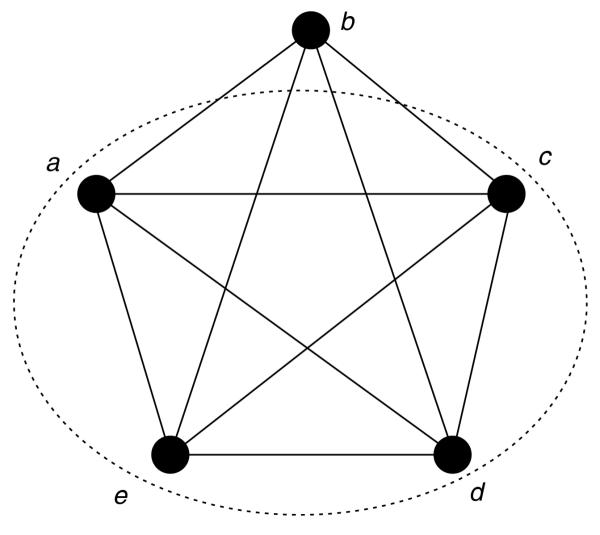
Cliques and maximal clique. A clique is a fully connected sub-graph, that is, a set of nodes that are all neighbors of each other. In this example, the whole graph is a clique and consequently any subset of it is also a clique, for example {a,c,d,e} or {b,e}. A maximal clique is a clique that is not contained in any larger clique. Here only {a,b,c,d,e} is a maximal clique.
Modular decomposition provides an instruction set to deliver all maximal cliques of a graph. In particular, when the decomposition has only series and parallels, the maximal cliques are straightforwardly retrieved by traversing the tree recursively from top to bottom. When encountered, a series module acts as a product: the maximal cliques are all the combinations made up of one maximal clique from each 'child' node. A parallel module acts as a sum: the set of maximal cliques is the union of all maximal cliques from the 'child' nodes. In the following, particular examples illustrate the application of this method.
Results
In this section, we demonstrate the application of modular decomposition for interpreting PCP networks of protein interactions. We find that modules have a functional interpretation, and that the labeling of the modules into prime, series and parallel corresponds to typical biological strategies of protein reuse.
Interpretation for PCP protein-protein interaction networks
Modular decomposition provides a comprehensive representation of the logical rules in the cooperation of the component proteins. In the tree the leaves are proteins; the root represents the whole network. In between, each node of the tree is a module that is a sub-part of its parent. The label of a node gives the nature of the relationship between its direct children.
Proteins or modules in a parallel module can be seen as alternatives (Figure 3c). If A is neighbor of B and C, which are not neighbors of each other, then A can belong to a complex together with either B or C, but not with both at the same time. B and C define a parallel module and thus are alternative partners in a complex with their common neighbor A. This situation corresponds to a logical 'exclusive OR' (also noted XOR) between B and C. Proteins or modules in parallel do not interact but can perform closely related biological functions.
Proteins or modules in a series module can be seen as potentially combined in any way (Figure 3b). If A is neighbor of B and C, which in turn are also neighbors of each other, then A can belong to a complex together with B or C, or with both at the same time. This situation corresponds to a logical 'OR' between B and C. The parsimonious solution restricts to the simplest model where the three proteins combine into a single complex. With a parsimonious solution, the series module interprets as a logical 'AND' between B and C. One can think of a series module as a unit: a set of proteins or modules that function together. A prime is a graph where neither of these cases occurs (Figure 3d).
Examples of established protein complexes in yeast
The following examples illustrate how modular decomposition can reveal the combinatorial assembly of complexes from interaction networks.
Protein phosphatase 2A
Parallel modules typically occur when related complexes exist in combinatorial variants. Such a case is represented by protein phosphatase 2A, which is a family of distinct, yet related, serine/threonine phosphatase complexes. Each complex is composed of a trimer that consists of the structural scaffold Tpd3, either of the two regulatory B subunits Rts1 or Cdc55, and one of the two catalytic subunits Pph21 or Pph22 [12] (Figure 5a).
Figure 5.
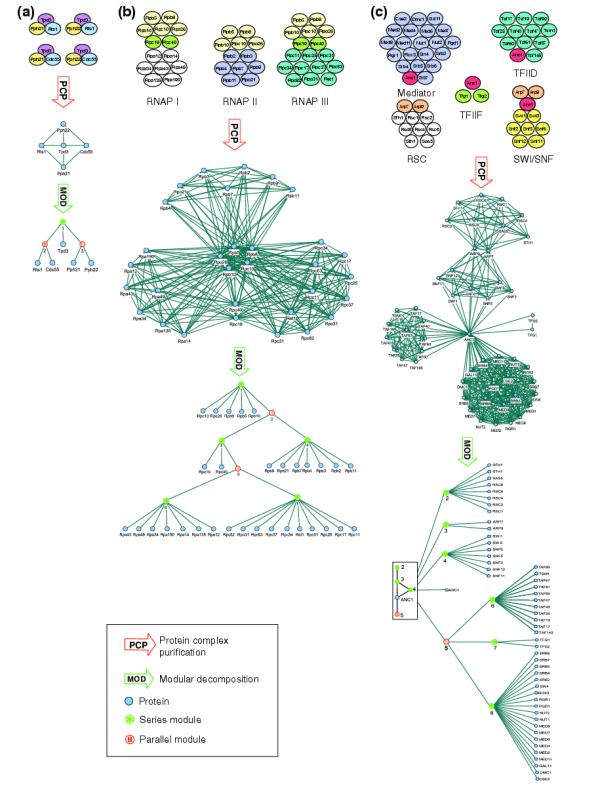
Three examples of modular decomposition of protein-protein interaction networks. In each case from top to bottom: schema of complexes, the corresponding protein-protein interaction network as determined from PCP experiments, and its modular decomposition (MOD). (a) Protein phosphatase 2A. Parallel modules group proteins that do not interact but are functionally equivalent. Here these are the catalytic Pph21 and Pph22 (module 2) and the regulatory Cdc55 and Rts1 (module 3). (b) RNA polymerases (RNAP) I, II and III. A good layout of the corresponding network gives an intuitive idea of what the constitutive units of the complexes are. Modular decomposition extracts them and makes their logical combinations explicit. (c) Transcriptional regulator complexes (see text for details). Modular decomposition condenses the network to its backbone prime structure (root of the tree) and identifies its constitutive units.
Modular decomposition of a simulated PCP experiment revealed the logical organization of the complex family. To derive the individual complexes as maximal cliques, the tree shows that Tpd3 combines (module 1, series) with either (module 2, parallel) Rts1 or Cdc55 and either (module 3, parallel) Pph21 or Pph22.
Modular decomposition groups together proteins with a similar function: the catalytic subunits Pph21 and Pph22 as alternatives in a parallel module and the regulatory subunits Cdc55 and Rts1 in another parallel module. Such a functional relationship is not obvious from the initial network of interactions.
RNA polymerases
Series modules reveal the presence of sub-complexes, that is, groups of proteins that function as single units in several complexes. Examples of such modules are found in RNA polymerases, which are protein complexes that synthesize RNA on DNA templates. RNA polymerase I synthesizes rRNA, polymerase II mRNA, and polymerase III many small RNAs such as tRNA.
The three polymerases involve a total of 31 proteins (Figure 5b). Modular decomposition defines the organization into shared and specific subunits and the logical rule set to derive the three enzyme complexes. Like PP2A, the catalytic unit is represented by alternative variants. For RNA polymerases, however, these alternative units consist of a multiprotein sub-complex (modules 4, 6 or 7). The two proteins Rpc19 and Rpc40 in module 3, shared by polymerase I and III, correspond to alternative variants of Rpb3 and Rpb11 in polymerase II, a relationship that is detected by sequence homology. The two proteins also form a sub-complex in the three-dimensional structure [13-15].
A series module containing five proteins at the root of the tree captures proteins that are common to all RNA polymerases. Interestingly, these proteins are scattered over the surface of the catalytic complex. For one of them, Rpc10, there is evidence that it acts as a bridging component between the sub-complex Rpb3/Rpb11 and the catalytic sub-complex. It is conceivable that those shared proteins all serve a comparable adaptor role to other cellular structures and might therefore be under too strong evolutionary constraints to allow divergence.
Transcriptional regulator complexes
The organization of a network cannot always be summarized by series and parallel modules only. In complex interaction arrangements, prime modules emerge in the decomposition and can be interpreted as irreducible backbones of the network, as we illustrate with a selection of complexes involved in chromatin remodeling and the transcriptional machinery.
We consider a network of 50 proteins that define the chromatin-remodeling complexes RSC and SWI/SNF; the general transcription factor complex TFIIF; the general transcription factor complex TFIID, which is responsible for promoter recognition; and the mediator complex that mediates signals to RNA polymerase II [16-19].
Modular decomposition (Figure 5c) of the network identifies six series modules as elementary units of the networks: the proteins specific to the RSC, SWI/SNF, TFIID, TFIIF, and the mediator complex (modules 2, 4, 6, 7 and 8 respectively), and Arp7 and Arp9 (module 3), which are common to the two chromatin-remodeling complexes. The three series modules specifically interacting with Anc1 are then embedded as alternatives into a parallel module (module 5). The root of the tree is a prime module, which summarizes the rather complex picture of the network.
Anc1, whose specific role in the respective complexes remains unclear in the literature, appears as the common point in the SWI/SNF complex and the transcription-related complexes. As with the shared components of the RNA polymerases, Arp7, Arp9 and Anc1 are rather peripherally arranged in the individual complexes. Arp7 and Arp9 have recently been proposed to compose a heterodimeric sub-complex that cooperates with DNA-bending proteins to facilitate chromatin remodeling and complex-complex interactions [20].
The structure of the prime reflects the progression from chromatin remodeling to transcription. Chromatin remodeling appears to come in two contexts: with mRNA transcription (module 4, SWI/SNF-specific proteins); and not reflected in this network (module 2, RSC-specific proteins that have no further connections here except for the shared components in module 3). Module 5 contains the elements responsible for promoter recognition (module 6), the mediator (module 8), and a general transcription factor (module 7). Anc1 links the chromatin-remodeling part to the transcriptional machinery.
As illustrated by these three examples, modular decomposition defines modules in a molecular-interaction network. Acting as a factorization principle, it helps to represent protein-protein interaction networks in a condensed and structured manner. Although the module representation can be helpful to derive hypotheses from interaction networks, interpretation requires particular attention for experimental datasets. Nodes in the network are not necessarily equivalent, as usually not all of the corresponding proteins have also been tested as baits in the underlying experiments.
Analysis of a high-throughput dataset: the TNF-α/NFκB pathway
We applied modular decomposition to the analysis of a large experimental dataset based on a systematic study of the human TNF-α/NFκB signal transduction pathway [21,22]. In this study, 32 proteins implicated in TNF-α/NFκB signaling had been selected as baits. Purification of these bait proteins resulted in a high confidence protein interaction network displaying 221 interactions involving 131 proteins.
In this experimental setup, not all proteins have been selected as baits. In such a network a pair of two non-neighbor proteins occurs in two situations. If at least one protein is bait, there is experimental evidence for the absence of the interaction. If none is bait, there is just no information about their interaction. We decided to apply the stringent spoke model convention [23], that is, not to infer an edge between two proteins if they are not bait, even if they occur in the same purification. To distinguish the case of unobserved interactions we flag the proteins in the network as well as in the tree differently for baits and non-baits. Because of the consequent lack of edges, the modular decomposition holds fewer series and more parallel modules. The interpretation for series still holds; however, the interpretation for prime and parallel modules is not as stringent as described above if containing unbaited proteins. Alternatives, in particular, are not exclusive if the interaction between two proteins has never been tested. The tree shows a current view of the network that can change with experimental evidence for additional interactions.
Modular decomposition has been applied to the TNF-α/NFκB dataset [21], resulting in a tree with 20 modules (see Additional data file 1). The root is labeled parallel, joining the different isolated parts of the network. Most of the proteins selected for purification are direct descendants of a prime module together with 16 parallel modules that group mainly new interactors that had consistently and specifically been identified by the same bait proteins. Using the known annotations of the proteins, we observed that those modules group proteins of common biological processes. For instance, the majority of the proteins of the module specific to relB are members of the SWI/SNF chromatin-remodeling complex. The HSP90/CDC37 chaperone complex is grouped in a module sharing nine common interactors of the pathway. Hence, the modules derive a consistent grouping of the newly identified interactors as a basis for biological understanding and interpretation.
Modular decomposition of the whole network contains a large prime module, reflecting the epistatic order of functionally distinct units in the cascade that cannot be further compressed simply by ANDs and ORs (see Additional data file 1). Nevertheless, modular decomposition can be used to further investigate local zones of functionally related proteins in the transduction pathway. We illustrate this strategy on the central knot of the pathway: the NFκB system.
The transcription factor NFκB is the convergence point for several signal transduction pathways activated by various stimuli, including TNF-α, IL-1β, bacterial lipopolysaccharide (LPS) and the T-cell receptor. The prevailing model is that NFκB is a dimer composed of a DNA-binding subunit and a transcriptional activator subunit. In the absence of stimulation, NFκB dimers are sequestered in the cytoplasm by inhibitors called IκB. After stimulation, an activated IKK complex phosphorylates IκBs, earmarking them for proteasome-mediated degradation. Following IκB degradation, NFκB translocates to the nucleus to activate gene transcription. Each of the proteins and complexes mentioned above exists in several variants. The existence of combinatorial variants could explain why this central knot can transmit different upstream signals resulting in distinct downstream outputs. In the following we use modular decomposition of NFκB members to investigate this hypothesis.
The NFκB family consists of five structurally related members: relA, relB, c-rel, NFκB1/p50 and NFκB2/p52. To analyze the NFκB variants, we first considered the networks of interactions among those five proteins selected as baits. Modular decomposition of the experimental TAP network characterizes the existence of complex variants (Figure 6). In line with the prevailing hypothesis we detect mutually exclusive usage of NFκB1/p50 and NFκB2/p52, which occur in a parallel module. Surprisingly, we detect the transcriptional activator subunit relA in all purifications, indicating that relA is complexed with all other members. As noted above, we cannot infer a direct physical interaction or the stoichiometry from such PCP experiments. Therefore, this finding suggests either that relA can form dimeric interactions with c-rel and relB, or the presence of higher-order tetrameric complexes involving at least two transcriptional activator subunits that are conjoined via either of the two DNA-binding subunits. Modular decomposition gives the rule of all potential tetramers, namely any combination where neither NFκB1/p50 and NFκB2/p52 nor c-rel and relB coexist. RelA appears with a central role in the tree. It is a necessary component of any potential tetramer formed of different NFκB dimers.
Figure 6.
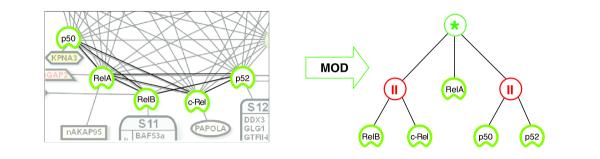
Investigating NFκB variants. Modular decomposition of NFκB members relB, c-rel, p50 and p52 delivers the potential NFκB dimers and tetramers. All combinations are possible (series) except those including both relB and c-rel (parallel), and those including both p50 and p52.
Next we analyzed the direct neighbors of NFκB members to obtain functional insight into the role of the various distinct NFκB complexes (Figure 7a). Here, the added proteins are not baits and therefore parallel modules are alternative, but not necessarily exclusive alternatives. Some proteins are specific to individual NFκB members. For instance, distinct members of the importin family of nuclear facilitator proteins have been co-purified with distinct NFκB subunits. In resting cells, KPNA2 and KPNA6 are identified only with relB. This is in line with the observation that relB is constitutively nuclear. Upon stimulation by TNF-α, a further member, KPNA3, co-purifies specifically with p50 [21], reflecting that p50 only translocates in response to TNF-α. Beside modules of interactors that are specific to individual proteins, others consist of shared interactors. In this context two modules are worth mentioning. One contains two IκB proteins (IκB-α and IκB-β) and ABIN2, which have been identified with all NFκB members except relB. ABIN2 had previously been implicated in TNF-α signaling [24]. Interestingly, overexpression of ABIN2 also inhibits TNF-α-induced activation of NFκB. However, the molecular mechanism has remained elusive. As ABIN2 appears in a module with IκB-α and IκB-β this suggests that ABIN2 exerts a function in directly modulating distinct NFκB complexes that do not contain relB, either by facilitating IκB-mediated retention in the cytosol or alternatively through an IκB-independent retention mechanism. The other module worth mentioning contains IKKα and Cot/Tpl2, which are common interactors of NFκB1/p50, NFκB1/p52 and relA. Cot/Tpl2 is a MAP kinase known as an agonist of NFκB, in particular by being implicated in the proteolysis of the precursor of p50, NFκB1/p105 [25].
Figure 7.
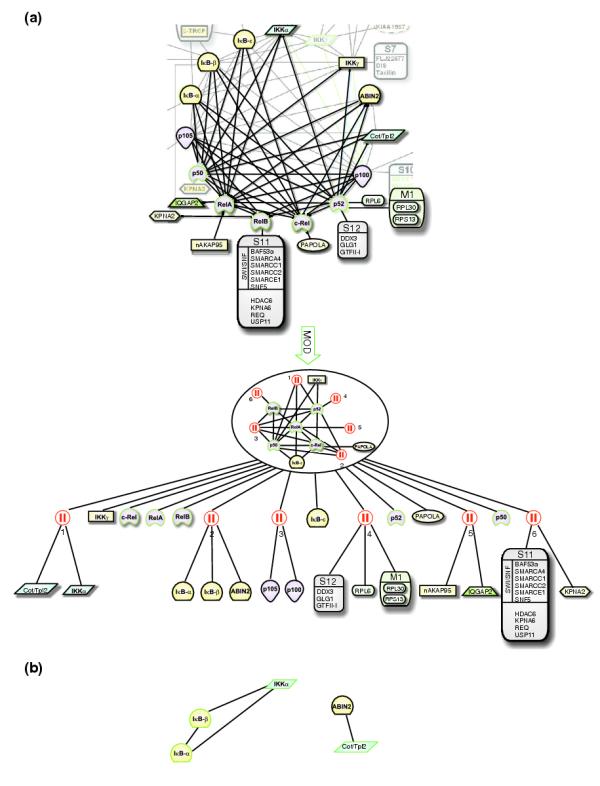
Analysis of the partners of NFκB members in resting cells. (a) Modular decomposition of the network of NFκB members and their partners. The network is composed of the NFκB members as defined in Figure 6 and their interactors. In this step, interactions among the interactors are disregarded. Symbols for the proteins are as defined in [21]. Baits are outlined in green. Modular decomposition organizes the interactors into modules. The root is a prime whose structure is shown in the encircled network. Module 1 and module 2, respectively, group the new interactors into activators and inhibitors of NFκB. (b) Further purifications using IKKα, IκB-α, IκB-β and Cot/Tpl2 as baits resolve the interactions between module 1 and module 2 members and suggest a complex composed of ABIN2 and Cot/Tpl2 as a NFκB modulation mechanism alternative to IKKα, IκB-α, IκB-β.
Therefore, modular decomposition delivers one module of inhibitors and one module of activators of NFκB. Relations within and between those two modules can be further investigated by selecting their member proteins as baits to capture the pattern of interactions among those proteins (Figure 7b). Confirming previous reports, we show here that IKKα binds constitutively to IκB-α and IκB-β. Interestingly, Cot/Tpl2 as bait co-purifies with ABIN2, but we did not observe any interactions between IκB-α or IκB-β and ABIN2, nor between IKKα and Cot/Tpl2. From this observation we hypothesize that Cot/Tpl2 may perhaps modulate ABIN2 in a manner akin to the action of IKK on the IκBs. As no directionality can be derived from interaction networks, ABIN2 may alternatively be a modulator of Cot/Tpl2.
Discussion
We introduced a definition of a module in the context of protein-protein interaction networks as a group of proteins that share the same interactors outside the module. Networks can be decomposed into a hierarchy of nested modules in a canonical way: the modular decomposition. More than just a hierarchical decomposition [26-28], the modular decomposition also states the logical relation between the members of the identified modules. Within a series module the members are all interacting with each other and can be considered from the outside as a single unit. Within a parallel module the members are all disconnected and can be considered as exclusive alternatives for the rest of the network. Prime modules appear as the most condensed structures of the network, where alternatives and units have been factored out. The whole tree provides a comprehensive representation of the logical organization of the network into modules. If primes are labeled by their structure, the tree is an exact alternative representation of the network.
We applied the method of modular decomposition to established examples of protein complexes and retrieved a consistent modular description of the composition that groups proteins with common biological role. The labeling of modules captures their relationship. In particular, series correspond to cooperative proteins whereas parallel modules correspond to alternative proteins for fulfilling the same function. Alternative proteins can be supposed to be paralogs. From a structural point of view, proteins in a parallel module most probably have the same or overlapping binding sites, whereas proteins in a series module are likely to have non-shared, non-overlapping binding sites.
We used modular decomposition to analyze a large PCP protein-protein interaction network of experimental data. It helped to structure the whole network in a consistent way but was also applied locally to reveal the variant complexes of NFκB members and their interactors. Application to experimental interaction data of the TNF-α/NFκB pro-inflammatory pathway requires appropriate distinction of bait proteins and retrieved interactors. For the whole dataset, bait proteins happen to stay in a large prime structure. Interactors, however, are more systematically grouped in modules on the basis of the baits they co-purified with. Those groups do typically define functionally related proteins. Starting from a selected set of proteins, here the NFκB proteins, modular decomposition of their interactions provided a clear rule set for their combinatorial assembly into complexes. Expanding the network to the direct interactors with subsequent modular decomposition refines their interactors' role with respect to NFκB, characterizing alternative modulation routes.
Two particular graph features had been recognized in previous reports to be helpful in reducing the complexity of biological networks. First, disconnected sub-graphs, termed connected components, are readily identified and treated independently (see, for example [29]). Second, groups of nodes that share exactly the same neighbors were described [30]. Both those features correspond to parallel modules, and both kinds of groups are consistently reflected as special cases in the modular decomposition tree. Modular decomposition offers a unified framework where connected components define a module at the root and the nodes with identical neighbors define bottom-level modules. Consequently, modular decomposition can be successfully applied to experimental datasets presenting large numbers of these modules, as it is the case in the TNF-α/NFκB pathway dataset.
A related question to be explored is the definition of the system of study. Indeed, the definition of a module is relative and depends strongly on the network being considered. This question comes up when facing large-scale experimental datasets and has been investigated, for example, for metabolic networks [31]. Our study of the TNF-α/NFκB pathway illustrates that this question goes hand in hand with the definition of the scope of the biological question, here the investigation of NFκB variants. Modular decomposition is not appropriate to define subsystems on its own, and should be applied in combination with strategies dedicated to this purpose.
It is tempting to apply modular decomposition to networks derived from Y2H experiments. Modules can be interpreted in this context as proteins with the same direct binding partners, that can all (series) or not (parallel) physically bind to each other, but we could not come up with a functional interpretation. However, Y2H gives information on the geometrical structure of complexes, whereas PCP informs on their compositions. Combining these two data sources can help in reducing the ambiguities intrinsic to each technique.
A modular description of molecular biology has been demanded and the lack of clear module definition stressed [32,33]. Two main directions have been followed to specify this notion, one toward dense parts of the network [7,27], the other one looking at repeated motifs [34]. We propose an alternative direction: a module is a group of elements that are indistinguishable from the rest of the network. This definition for graphs is general in nature and thus could be applied to and reinterpreted for other graphs in biology. Some biological relationships, like gene regulatory networks or metabolic networks, are better represented by different graph variants such as directed graphs or hypergraphs. Fortunately, the notion of module and its related decomposition has been defined in these cases [9] and can be used there. Another example is the state graph, a directed graph that describes the states of gene regulatory networks and the transitions between these states. The state graph can get exponentially large when all potential states are considered. However, to provide a more compact description that entirely captures the system behavior, groups of equivalent states can be condensed [35] which relates to the notion of modules of this directed graph. Similarly, the regulatory modules described by Segal and collaborators [36] are groups of genes obeying the same regulatory program, and hence are indistinguishable from the rest of the gene regulatory network. We assume that those examples are only a fraction of the potential types of biological study that would benefit from the concept of modular decomposition. Therefore, we foresee modular decomposition as a general and fundamental tool for network-based research and systems biology.
Materials and methods
Complex purifications
Experimental procedures for TAP-tagged purification of complexes in the TNFα/NFκB signaling pathway are described in [21].
Modular decomposition implementation
We followed the description of a practical algorithm for modular decomposition of graphs [37]. We give free access to our implementation [38].
Additional data files
A PDF file (Additional data file 1) showing the modular decomposition of the filtered dataset for the TNF pathway is available with the online version of this article. It contains annotations of the modules with the names of their common interactors and reference to the modules described in [21].
Supplementary Material
The modular decomposition of the filtered dataset for the TNF pathway
Acknowledgments
Acknowledgements
This work is supported by Cellzome AG. We thank Nathalie Chabrier for revealing to us the notion of modules in graphs and Ross McConnell for encouraging support in the implementation phase. We are grateful to Anne-Claude Gavin, Paola Grandi and Gerard Drewes for stimulating discussions and biological insight. We thank anonymous referees for their constructive comments.
References
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Deng M, Sun F, Chen T. Assessment of the reliability of protein-protein interactions and protein function prediction. Pac Symp Biocomput. 2003:140–151. [PubMed] [Google Scholar]
- Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. Proc Natl Acad Sci USA. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhring RH. Algorithmic aspects of the substitution decomposition in optimization over relations, set systems and boolean functions. Annls Operations Res. 1985;4:195–225. [Google Scholar]
- Möhring RH, Radermacher FJ. Substitution decomposition for discrete structures and connections with combinatorial optimization. Annls Disc Math. 1984;19:257–356. [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Yang H, Jiang W, Gentry M, Hallberg RL. Loss of a protein phosphatase 2A regulatory subunit (Cdc55p) elicits improper regulation of Swe1p degradation. Mol Cell Biol. 2000;20:8143–8156. doi: 10.1128/MCB.20.21.8143-8156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Bushnell DA, Fu J, Gnatt AL, Maier-Davis B, Thompson NE, Burgess RR, Edwards AM, David PR, Kornberg RD. Architecture of RNA polymerase II and implications for the transcription mechanism. Science. 2000;288:640–649. doi: 10.1126/science.288.5466.640. [DOI] [PubMed] [Google Scholar]
- Rubbi L, Labarre-Mariotte S, Chedin S, Thuriaux P. Functional characterization of ABC10alpha, an essential polypeptide shared by all three forms of eukaryotic DNA-dependent RNA polymerases. J Biol Chem. 1999;274:31485–31492. doi: 10.1074/jbc.274.44.31485. [DOI] [PubMed] [Google Scholar]
- Lalo D, Carles C, Sentenac A, Thuriaux P. Interactions between three common subunits of yeast RNA polymerases I and III. Proc Natl Acad Sci USA. 1993;90:5524–5528. doi: 10.1073/pnas.90.12.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry NL, Campbell AM, Feaver WJ, Poon D, Weil PA, Kornberg RD. TFIIF-TAF-RNA polymerase II connection. Genes Dev. 1994;8:2868–2878. doi: 10.1101/gad.8.23.2868. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Bjorklund S, Li Y, Sayre MH, Kornberg RD. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell. 1994;77:599–608. doi: 10.1016/0092-8674(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, Kornberg RD. RSC, an essential, abundant chromatin-remodeling complex. Cell. 1996;87:1249–1260. doi: 10.1016/S0092-8674(00)81820-6. [DOI] [PubMed] [Google Scholar]
- Cairns BR, Kim YJ, Sayre MH, Laurent BC, Kornberg RD. A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc Natl Acad Sci USA. 1994;91:1950–1954. doi: 10.1073/pnas.91.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szerlong H, Saha A, Cairns BR. The nuclear actin-related proteins Arp7 and Arp9: a dimeric module that cooperates with architectural proteins for chromatin remodeling. EMBO J. 2003;22:3175–3187. doi: 10.1093/emboj/cdg296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, et al. A physical and functional map of the human TNF-alpha/NF-kappaB signal transduction pathway. Nat Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- A physical map of the human TNF-NFkappaB signal transduction pathway http://tnf.cellzome.com
- Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nat Biotechnol. 2002;20:991–997. doi: 10.1038/nbt1002-991. [DOI] [PubMed] [Google Scholar]
- Van Huffel S, Delaei F, Heyninck K, De Valck D, Beyaert R. Identification of a novel A20-binding inhibitor of nuclear factor-kappa B activation termed ABIN-2. J Biol Chem. 2001;276:30216–30223. doi: 10.1074/jbc.M100048200. [DOI] [PubMed] [Google Scholar]
- Belich MP, Salmeron A, Johnston LH, Ley SC. TPL-2 kinase regulates the proteolysis of the NF-kappaB-inhibitory protein NF-kappaB1 p105. Nature. 1999;397:363–368. doi: 10.1038/16946. [DOI] [PubMed] [Google Scholar]
- Holme P, Huss M, Jeong H. Subnetwork hierarchies of biochemical pathways. Bioinformatics. 2003;19:532–538. doi: 10.1093/bioinformatics/btg033. [DOI] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- Gagneur J, Jackson DB, Casari G. Hierarchical analysis of dependency in metabolic networks. Bioinformatics. 2003;19:1027–1034. doi: 10.1093/bioinformatics/btg115. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. The identification of functional modules from the genomic association of genes. Proc Natl Acad Sci USA. 2002;99:5890–5895. doi: 10.1073/pnas.092632599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BH, Han K. Complexity management in visualizing protein interaction networks. Bioinformatics. 2003;19 (Suppl 1):I177–I179. doi: 10.1093/bioinformatics/btg1022. [DOI] [PubMed] [Google Scholar]
- Schuster S, Pfeiffer T, Moldenhauer F, Koch I, Dandekar T. Exploring the pathway structure of metabolism: decomposition into subnetworks and application to Mycoplasma pneumoniae. Bioinformatics. 2002;18:351–361. doi: 10.1093/bioinformatics/18.2.351. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Alon U. Biological networks: the tinkerer as an engineer. Science. 2003;301:1866–1867. doi: 10.1126/science.1089072. [DOI] [PubMed] [Google Scholar]
- Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- Remy E, Mosse B, Chaouiya C, Thieffry D. A description of dynamical graphs associated to elementary regulatory circuits. Bioinformatics. 2003;19 (Suppl 2):II172–II178. doi: 10.1093/bioinformatics/btg1075. [DOI] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- McConnell RM, Spinrad JP. Ordered vertex partitioning. Disc Math Theor Comp Sci. 2000;4:45–60. [Google Scholar]
- Modular decomposition of protein-protein interaction networks. Supplementary material. http://www.mas.ecp.fr/labo/equipe/gagneur/module/module.html [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The modular decomposition of the filtered dataset for the TNF pathway