

Short abstract
This study presents two novel normalization schemes for cDNA microarrays. They are based on iterative local regression and optimization of model parameters by generalized cross-validation.
Abstract
In this study we present two novel normalization schemes for cDNA microarrays. They are based on iterative local regression and optimization of model parameters by generalized cross-validation. Permutation tests assessing the efficiency of normalization demonstrated that the proposed schemes have an improved ability to remove systematic errors and to reduce variability in microarray data. The analysis also reveals that without parameter optimization local regression is frequently insufficient to remove systematic errors in microarray data.
Background
Microarrays have been widely used for the study of gene expression in biological and medical research. They allow the simultaneous measurement of the expression of thousands of genes in cells. However, microarrays do not assess gene expression directly, but only indirectly by monitoring fluorescence intensities of labeled target cDNA hybridized to probes on the arrays [1]. The first step in the analysis of microarray data is, therefore, the transformation of fluorescence signals into quantities of gene expression. This includes several data pre-processing procedures - for example, excluding artifacts and correcting for background intensities. The signals also have to be adjusted for differences in dye labeling, fluorescence yields, scanning amplification and other systematic variability in the measurement. Although this so-called normalization procedure is only an intermediate step in the analysis, it has a considerable influence on the final results [2]. Assessment of the efficiency of a chosen normalization method should therefore be an integral part of every normalization procedure.
Important and widely used microarray platforms are spotted cDNA microarrays consisting of probes that are spatially ordered on a rigid surface. Probes for cDNA arrays are generally the polymerase chain reaction (PCR) products derived from cDNA clone sets and are spotted on the array using a set of pins [1]. To measure gene expression by cDNA microarrays, RNA samples are reverse transcribed to cDNA and labeled with fluorescent dyes. The labeled target cDNA is then hybridized to the microarray probes. To control variability due to variable spot size and concentration of arrayed PCR product, cDNA microarrays arrays are generally co-hybridized with two samples, one of which serves as the reference sample. The two samples for a cDNA array are labeled by different dyes (for example, Cy5, Cy3) with distinct optical properties. Pairing the signal intensities of both samples for each spot aims to eliminate the variability of the spotting procedure. The calculated ratio of signal intensities for each spot delivers a measure for fold changes in gene expression. However, raw fluorescence ratios are frequently misleading. The corresponding fold changes might reflect experimental biases rather than changes in gene expression.
A well known experimental bias for cDNA arrays is the so-called dye bias, referring to the systematic error that originates from using two different dyes. Dye bias is most apparent in self-self hybridization experiments, in which identical samples are labeled by two different dyes and hybridized on the same array. It could be expected that ratios of spot signal intensities vary around one. However, intensity-dependent deviations from such behavior have frequently been observed [3,4]. These deviations can be related to a variety of experimental factors such as differing labeling efficiencies, fluorescence quantum yields, background intensities, scanning sensitivity, signal amplification and total amount of RNA in the samples [1,4,5]. Besides intensity-dependent dye bias, other types of dye bias have been reported [5-8].
Normalization aims to correct for systematic errors in microarray data. A variety of normalization methods have been proposed for two-color arrays (for a recent review see [9]). One of the first methods proposed to correct for dye bias was global linear normalization, which assumes that the total fluorescence in both channels is equal [10]. On the basis of this assumption, a normalization constant can be derived and used to adjust the fluorescence intensities of the two channels. However, recent reports have shown that this procedure is insufficient to correct for nonlinear dependence of spot intensities and fluorescence ratios [4,6,11]. Several normalization methods have been developed to overcome this shortcoming of global normalization [6-8,11]. They commonly regress fluorescence ratios with respect to spot intensities in a nonlinear fashion. Some of these local regression methods have been further extended to correct for spot location-dependent dye bias [6,7].
Although nonlinear normalization procedures have been able to reduce systematic errors, an optimal adjustment of these normalization models to the data has not been discussed. Current methods are based on default parameter values and leave it up to the researcher to adjust the normalization parameters. Instructions on how to optimize parameter settings are generally not given. Optimization of parameters is, however, crucial for the normalization process. We show in our study that systematic errors in cDNA microarray data exhibit a large variability between, and even within, experiments. This requires an adjustment of the model parameters to the data. A set of normalization parameters of fixed value is frequently insufficient to correct experimental biases.
In this study we introduce two normalization schemes based on iterative local regression and model selection. The underlying relations between experimental variables and gene-expression changes were derived from an explicitly formulated hybridization model. Both normalization schemes aim to correct for intensity- and location-dependent dye bias in cDNA microarray data. For model selection, we applied generalized cross-validation (GCV), which has computational advantages compared to standard cross-validation. The efficiencies of correction for dye bias of different normalization schemes were compared using permutation tests for two independently generated cDNA microarray datasets. Several statistical measures were used to assess the variability and reproducibility of results obtained by different normalization methods. Finally, the normalized fold changes of multiple genes were compared to externally validated fold changes for a third microarray experiment.
Results
Hybridization model
A first step in the analysis of microarray data is the development of a hybridization model relating intensity of fluorescent signals to mRNA abundance. The model should describe the influence of experimental parameters on the data variability and include error terms. Explicitly modeling the relation between signal intensities and changes in gene expression can separate the measured error into systematic and random errors. Systematic errors may be corrected in the normalization procedure, whereas random errors cannot be corrected, but have to be assessed by replicate experiments. Removal of systematic errors is important, as they limit the accuracy of the measurement, whereas random errors limit its precision.
Our hybridization model applies to two-color arrays commonly consisting of a red (Cy5) and green (Cy3) fluorescence channel. The model relates the measured spot fluorescence intensity to changes in the labeled transcript abundances in which we are interested. Its explicit derivation can be found in Materials and methods. Specifically, the model relates the ratios Ir/Ig of spot signal intensities (Ir/g, spot fluorescence intensity in red/green channel) with the ratios Tr/Tg of labeled transcript abundance (Tr/g, abundance of transcript labeled by red/green dye). The relation has the following form:
M - κ(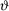 ) = D + ε (1)
) = D + ε (1)
where M is the log fluorescence intensity ratio (M = log2Ir/Ig), D is the logged ratio of transcript abundance (D = log2Tr/Tg) and ε represents the random error. The term κ is an additive factor that can depend on a set of experimental variables , for example, spot intensity and location. In our model, κ() can be seen as a term for systematic errors. Using Equation (1), we can derive D from M up to the random error term ε once we know κ(). The factor κ() is generally calibrated by exploiting Equation (1). Depending on the assumptions about the experiment, we can proceed with different normalization methods.
Assuming κ is constant and the majority of assayed genes are not differentially expressed, the ratios can be linearly scaled to an average value of one. This leads to linear normalization. If κ depends on the fluorescence intensity, it may be derived from the signal ratios assuming symmetry of the logged fold changes D and error term ε. The factor κ() can then be calculated by a local regression of M with respect to the fluorescence intensity. This procedure can be performed using all or a selected subset of genes and is frequently called intensity-dependent normalization. In the experiments analyzed, we found that the measured spot intensity ratios showed not only intensity-dependent, but also spatial bias across the array. We introduce, therefore, two normalization schemes that simultaneously correct for dye bias due to intensity and spatial location.
Normalization schemes
Two normalization schemes were developed to determine the normalization factor κ() in the hybridization model (Equation (1)). They are based on iterative local regression and incorporate optimization of model parameters. Local regression is performed using LOCFIT, which is based on the same computational ideas as the popular lowess method [12,13]. However, it differs from lowess in that its implementation offers more flexibility to the user. For local fitting, LOCFIT (as well as lowess) requires the user to choose a smoothing parameter α that controls the neighborhood size h. The parameter α specifies the fraction of points that are included in the neighborhood and thus has a value between 0 and 1. Larger α values lead to smoother fits. In addition, the setting of scale parameter s is necessary for a local regression with two or more predictor variables. These parameters provide the scales of the predictor variables for the fitting procedures. The parameter s can be of arbitrary value.
For normalization by LOCFIT, therefore, model parameters α and s have to be chosen. The choice of model parameters for local regression is crucial for the efficiency and quality of normalization. To optimize the model parameters, we use a cross-validation procedure. Because conventional leave-one-out cross-validation becomes computationally prohibitive for this task, we used GCV, which approximates the leave-one-out method [14]. GCV is computationally less expensive to perform as it does not require multiple constructions of regression models based on partial data, as standard cross-validation does.
Both normalization schemes aim to correct for systematic errors linked to spot intensity and location. The first procedure leaves the scale of log intensity ratios M unchanged, whereas the second procedure includes an adjustment of the scale of M. The notation is as follows: A = 0.5 (log2 Ir + log2Ig), which is the geometric mean of the fluorescent intensities of both channels; X is the spot location on the array in the X direction; Y is the spot location on the array in the Y direction; αA is the smoothing parameter for local regression of M with respect to A; αXY is the smoothing parameter for local regression of M with respect to spatial coordinates X and Y; sY is the scale parameter allowing a different amount of smoothing in the Y direction compared to smoothing in the X direction.
Optimized local intensity-dependent normalization (OLIN)
For a set of smoothing parameter αA, local regression of M with respect to A is performed, generating a set of regression models. Then, the regression models are compared by GCV. The model with 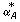 resulting in the minimum GCV criterion is chosen. The optimal fit
resulting in the minimum GCV criterion is chosen. The optimal fit 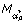 (A) corresponds to a normalization factor κ(A) in Equation (1). (A) is subtracted from M, generating an intensity normalized M: M ← M - (A). For a set of smoothing parameter αXY and a set of scale parameter sY, local regression of M with respect to X and Y is performed. The resulting models are compared by GCV. The optimal fit
(A) corresponds to a normalization factor κ(A) in Equation (1). (A) is subtracted from M, generating an intensity normalized M: M ← M - (A). For a set of smoothing parameter αXY and a set of scale parameter sY, local regression of M with respect to X and Y is performed. The resulting models are compared by GCV. The optimal fit 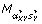 (A) corresponds to a normalization factor κ(X,Y) in Equation (1). (A) is subtracted from M generating a spatially normalized M: M ← M - (A). The steps above are repeated, unless the maximal number of iterations N is reached. If the maximal number of iterations is reached, M is the normalized log intensity ratio.
(A) corresponds to a normalization factor κ(X,Y) in Equation (1). (A) is subtracted from M generating a spatially normalized M: M ← M - (A). The steps above are repeated, unless the maximal number of iterations N is reached. If the maximal number of iterations is reached, M is the normalized log intensity ratio.
Optimized scaled local intensity-dependent normalization (OSLIN)
First, OLIN is performed. Then, for a set of smoothing parameter α and a set of scale parameter s, local regression of abs(M) with respect to X and Y is performed. The resulting models are compared by GCV. The model with α* and s* producing the minimum GCV criterion is chosen and an optimal fit 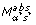 produced. M is locally scaled by : M' ← M/ . The global scale of M' is adjusted, so that total variation of M remains constant:
produced. M is locally scaled by : M' ← M/ . The global scale of M' is adjusted, so that total variation of M remains constant:
![]()
M" is the normalized log intensity ratio.
We applied our hybridization model and normalization schemes to data from two independent spotted cDNA microarray experiments. In the first experiment, gene expression in two colon cancer cell lines (SW480/SW620) was compared [15]. The SW480 cell line was derived from a primary tumor, whereas the SW620 cell line was cultured from a lymph-node metastasis of the same patient. Sharing the same genetic background, these cell lines serve as an in vitro model of cancer progression [16]. The comparison was direct, that is without using a reference sample. cDNA derived from SW480 cells was labeled by Cy3; cDNA derived from SW620 was labeled by Cy5. The SW480/620 experiment consisted of four technical replicates. In the second experiment (apo AI), gene expression in tissue samples from eight apo AI knock-out and eight control mice was studied [17,18]. Cy5-labeled cDNA from each tissue sample was co-hybridized with a Cy3-labeled reference sample consisting of pooled cDNA from the control mice. Hence, a total of 16 cDNA microarrays comprise the apo AI experiment. Technical replicates were missing. Further information regarding the experiments can be found in the Materials and methods section.
The effects of the normalization schemes are illustrated here for a chosen microarray (slide 3) of the SW480/620 experiment. The first step of normalization is, however, the identification of systematic experimental variability in the data.
Identification of systematic errors: intensity-dependent and location-dependent dye bias
Visual inspection of different plot representations of the data pointed to two major types of systematic errors: intensity-dependent and location-dependent dye bias. Although visual inspection lacks the stringency of statistical analysis, it provides an important first tool to detect artifacts in microarray data.
Popular representations are plots of Cy5 (Ir) versus Cy3 (Ig) intensities on linear or log scale. To illustrate the effect of the normalization procedures, however, the use of transformed log intensities is preferable [4]. In so-called MA-plots the log ratio M = log2(Ir/Ig) = log2(Ir) -log2(Ig) is plotted against the mean log intensities A = 0.5(log2(Ir) + log2(Ig)) Although MA-plots are basically only a 45° rotation with a subsequent scaling, they reveal intensity-dependent patterns more clearly than the original plot. MA-plots also introduce a measure for the spot intensity A, which was used in our normalization schemes. Figure 1a presents the MA-plot for the raw data of slide 3. Clearly, it shows a general nonlinear dependence of log ratios M on spot intensity A. For low intensities, M is biased towards negative values, which is generally the case for arrays of the SW480/620 experiment. This is contrasted by MA-plots of the apo AI experiment, where log ratios are generally biased towards positive values for low spot intensities (see Figure 3 in additional data file 1). The differing characteristics of this dye bias may be caused by differences in labeling or scanning protocol used in the two experiments. In addition to standard MA-plots, we found that it can be favorable to smooth the MA-plot by calculating the average value of M within a moving window along the intensity scale. Such  A-plots frequently display the dependence of M on A more clearly (see Figure 2 in additional data file 1). As well as intensity-dependent bias, the MA-plot in Figure 1 also reveals saturation effects for spots of high intensities. A substantial saturation is indicated by arrow-shaped distributions of spots in MA-plots. Such effects are caused by the limited dynamical range of M due to the scanner detection limit of 216 - 1 units. A more detailed analysis of saturation and how it affects MA-plots can be found in [19]. Ratios corresponding to spots with saturation in one or both channels should be treated with care, as a recovery of the unsaturated intensities is generally not possible (see the Hybridization model section in Materials and methods). Therefore, normalization cannot correct for such saturation effects. To avoid this difficulty, saturation should be prevented by adjustment of scanning parameters. Alternatively, a multiple scanning procedure can be applied [20].
A-plots frequently display the dependence of M on A more clearly (see Figure 2 in additional data file 1). As well as intensity-dependent bias, the MA-plot in Figure 1 also reveals saturation effects for spots of high intensities. A substantial saturation is indicated by arrow-shaped distributions of spots in MA-plots. Such effects are caused by the limited dynamical range of M due to the scanner detection limit of 216 - 1 units. A more detailed analysis of saturation and how it affects MA-plots can be found in [19]. Ratios corresponding to spots with saturation in one or both channels should be treated with care, as a recovery of the unsaturated intensities is generally not possible (see the Hybridization model section in Materials and methods). Therefore, normalization cannot correct for such saturation effects. To avoid this difficulty, saturation should be prevented by adjustment of scanning parameters. Alternatively, a multiple scanning procedure can be applied [20].
Figure 1.
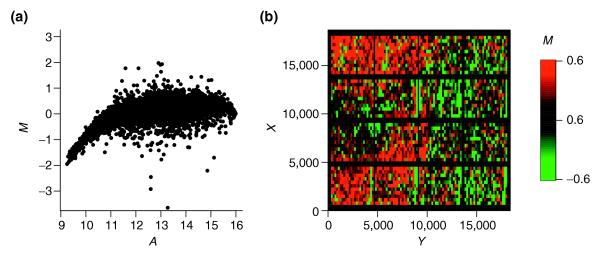
Intensity and spatial distribution of raw log intensity ratios M of slide 3 of the SW480/620 experiment. (a) The MA-plot indicates a strong bias towards the Cy3 channel for low spot intensities A. (b) The spatial MXY-plot shows uneven distribution of positive M (red squares) and negative M (green squares). The columns with consistently negative M correspond to empty control spots. The axis labels X and Y refer to the spot location as determined by the QuantArray scanning software.
Less frequent than the Ir - Ig-plots or MA-plots is the representation of log ratios based on the corresponding spot location. This type of plot, termed here a MXY-plot, offers, however, a valuable tool for assessing the quality of hybridization as well as the subsequent normalization. MXY-plots show the log ratios M with respect to the spot location on the array. Positive M values are represented as red squares, whereas negative M values are shown as green squares. The MXY-plot for the raw data of slide 3 is shown in Figure 1b. Large areas show a tendency towards positive M (for example, the lower left side). For slides of both experiments, MXY-plots point to the existence of spatial bias. Whereas spatial bias was variable across different slides of the SW480/620 experiment, it was more consistent for slides of the apo AI experiment (see Figures 4 and 5 in additional data file 1). As an alternative to MA-plots, the average value  of neighboring spots can again be used instead of M for plotting. These XY-plots frequently display spatial artifacts more clearly than MXY-plots (see Figure 2 in additional data file 1).
of neighboring spots can again be used instead of M for plotting. These XY-plots frequently display spatial artifacts more clearly than MXY-plots (see Figure 2 in additional data file 1).
In contrast to intensity-dependent dye bias, the origin of spatial bias is less clear. Possible reasons for the observed spatial bias might be spatial inhomogeneities of hybridization, uneven slide surfaces or unbalanced scanning procedures [1]. Schuchhardt et al. [5] and Yang et al. [6] suggested a ratio bias linked to the use of different pins. In this case, a block-wise bias would be apparent, which we did not observe. In our experiment, the spatial dye bias seemed to be continuous across arrays. Of course, one explanation for the uneven spatial distribution is that it reflects actual biological variability. For example, the lower left side of the array in Figure 1b could be enriched with spots corresponding to upregulated genes. This, however, seems to be unlikely, as the print order of spots in the SW480/620 experiment did not follow functional categories of genes. Even if genes are grouped on the used microtiter plates on the basis of their functions, the spotting procedure used for cDNA arrays leads to an even distribution of those genes across the array. Moreover, the spatial patterns of log ratios M differed between replicate arrays of the SW480/620 experiment. If they were specific for the print layout of the probes, similar patterns in all arrays would be expected. Other arguments also point against a biological source of the observed intensity-dependent and spatial dye bias for the experiments analyzed here. First, log ratios close to zero can be expected for empty control spots in the SW480/620 experiment. However, a large number of empty control spots with low fluorescence signals due to nonspecific hybridization had consistently large negative log ratios. They would be falsely detected as significant if no data normalization was applied [15]. Second, only a small number of genes are expected to be differentially expressed in the apo AI experiment [6]. Therefore, both MA- and MXY-plots should show log ratios close to zero for the vast majority of spots.
As well as visual inspection, we used permutation tests to detect intensity-dependent and spatial dye bias. The tests determined the significance of observing a median log ratio within a spot intensity or location neighborhood as introduced in Materials and methods. The number of neighborhoods with significant for the false discovery rate (FDR) = 0.01 is shown in Tables 1 and 2. For spot-intensity neighborhoods, a symmetrical window of 50 spots was chosen, whereas a 5 × 5 window was chosen as the spot-location neighborhood. For slide 3 of the SW480/620 experiment, testing the dependency of log ratio M on spot intensity A revealed that 1,138 spot neighborhoods (or 27% of all neighborhoods) had a significantly large positive or negative median log ratio. Testing for location-dependent dye bias, 837 neighborhoods (20%) were detected as significant.
Table 1.
Number of significant spot neighborhoods on the intensity scale for arrays of the experiments analyzed
| Microarray | No normalization | Linear normalization | Global lowess | P-lowess | S-lowess | LIN | OLIN | OSLIN |
| SW 1 | 596 | 580 | 2 | 5 | 7 | 39 | 12 | 0 |
| SW 2 | 1,090 | 1,080 | 0 | 0 | 0 | 52 | 0 | 0 |
| SW 3 | 1138 | 988 | 0 | 0 | 0 | 0 | 0 | 0 |
| SW 4 | 745 | 655 | 0 | 9 | 4 | 0 | 12 | 0 |
| Apo 1 | 1,748 | 1,810 | 18 | 0 | 0 | 26 | 0 | 0 |
| Apo 2 | 2,739 | 2,683 | 16 | 17 | 26 | 24 | 0 | 0 |
| Apo 3 | 3,479 | 3,559 | 52 | 30 | 15 | 0 | 1 | 0 |
| Apo 4 | 2,122 | 2,184 | 1 | 17 | 22 | 0 | 0 | 11 |
| Apo 5 | 3,885 | 3,886 | 100 | 13 | 11 | 94 | 0 | 0 |
| Apo 6 | 3,540 | 3,555 | 0 | 0 | 0 | 114 | 0 | 0 |
| Apo 7 | 3,725 | 3,724 | 0 | 0 | 7 | 0 | 0 | 0 |
| Apo 8 | 3,253 | 3,296 | 66 | 0 | 0 | 507 | 0 | 0 |
| Apo 9 | 1,040 | 1,044 | 114 | 0 | 0 | 455 | 0 | 0 |
| Apo 10 | 1,554 | 1,575 | 0 | 0 | 0 | 45 | 0 | 0 |
| Apo 11 | 2,903 | 2,889 | 5 | 34 | 35 | 2 | 0 | 0 |
| Apo 12 | 3,536 | 3,543 | 14 | 0 | 0 | 47 | 0 | 0 |
| Apo 13 | 2,819 | 2,901 | 132 | 12 | 3 | 354 | 4 | 0 |
| Apo 14 | 2,291 | 2,342 | 313 | 0 | 12 | 484 | 64 | 4 |
| Apo 15 | 3,050 | 3,034 | 95 | 36 | 0 | 164 | 0 | 0 |
| Apo 16 | 1,338 | 1,374 | 159 | 0 | 12 | 918 | 0 | 0 |
Spot neighborhoods are found significant if the median log ratio M has larger positive and negative value than expected from random order of spots along the intensity scale. The level of significance was defined by FDR = 0.01. The spot neighborhood was defined by a symmetrical window of 50 spots.
Table 2.
Number of significant spot neighborhoods across the spatial layout of the arrays analyzed
| Microarray | No normalization | Linear normalization | Global lowess | P-lowess | S-lowess | LIN | OLIN | OSLIN |
| SW 1 | 1,500 | 1,483 | 1,625 | 214 | 220 | 106 | 0 | 0 |
| SW 2 | 808 | 831 | 1,068 | 218 | 113 | 67 | 0 | 0 |
| SW 3 | 874 | 815 | 723 | 126 | 96 | 78 | 1 | 0 |
| SW 4 | 741 | 757 | 846 | 76 | 43 | 49 | 0 | 0 |
| Apo 1 | 1,173 | 1,196 | 1,276 | 913 | 491 | 755 | 100 | 1 |
| Apo 2 | 521 | 518 | 801 | 176 | 74 | 221 | 3 | 0 |
| Apo 3 | 562 | 576 | 706 | 334 | 79 | 258 | 0 | 0 |
| Apo 4 | 770 | 771 | 1,058 | 177 | 14 | 176 | 2 | 0 |
| Apo 5 | 670 | 648 | 844 | 357 | 222 | 381 | 5 | 0 |
| Apo 6 | 432 | 432 | 1,003 | 129 | 106 | 296 | 10 | 0 |
| Apo 7 | 516 | 526 | 1,258 | 186 | 88 | 194 | 17 | 0 |
| Apo 8 | 850 | 833 | 1,202 | 684 | 342 | 458 | 61 | 9 |
| Apo 9 | 1,596 | 1,621 | 1,780 | 1,105 | 644 | 798 | 21 | 5 |
| Apo 10 | 707 | 711 | 896 | 261 | 108 | 279 | 3 | 0 |
| Apo 11 | 504 | 484 | 1,306 | 166 | 87 | 258 | 11 | 0 |
| Apo 12 | 1,313 | 1,323 | 1,144 | 425 | 288 | 370 | 12 | 17 |
| Apo 13 | 1,357 | 1,368 | 1,155 | 653 | 394 | 568 | 41 | 0 |
| Apo 14 | 862 | 1,005 | 987 | 273 | 108 | 272 | 82 | 0 |
| Apo 15 | 733 | 743 | 1,004 | 588 | 241 | 502 | 97 | 0 |
| Apo 16 | 942 | 985 | 1,347 | 786 | 333 | 470 | 47 | 0 |
The level of significance was defined by FDR = 0.01. The spot neighborhood was defined by a window of 5 × 5 spots.
A simple but popular method for normalizing cDNA microarray data is global linear normalization. However, linear normalization leads only to a vertical shift along the M-axis in the plots (see Figure 1 in additional data file 1). Thus, the intensity- and location-dependent bias remained apparent. This was confirmed by the results of the permutation tests: 988 spot-intensity and 815 spot-location neighborhoods were detected as significant. This shows that linear normalization was insufficient to remove the observed dye and spatial bias
Local intensity-dependent normalization
Inspection of the MA- and MXY-plots showed that the relations between log ratio M and spot intensity A and between log ratio M and spot location (X, Y) are nonlinear. In our hybridization model, the normalization factor κ should therefore be a function of A as well as X and Y:
Mi-κ(A,X,Y) = Di + εi (2)
If we combine the logged fold change Di and error term εi to a random variable ζi which is assumed to be symmetrical distributed around zero, we get
Mi = κ(A,X,Y) + ζi (3)
Since this relation is of the same form as Equation (10) (see below), we can apply a local regression model to capture the intensity and location dependence of M. The residuals of the regression provided the logged fold changes D up to an error term and were used for the MA- and MXY-plots. The assumption that variable ζi is symmetrically distributed has to taken with caution, as it is based on two requirements: most genes arrayed are not differentially expressed or the numbers of up- and downregulated genes are similar; and the spotting procedure did not generate an spatial accumulation of up- or downregulated genes in localized areas on the array. Both requirements have to be assessed for each experiment individually. From the discussion in the previous section, we believe that both requirements are fulfilled for the datasets analyzed in this study.
To examine the influence of model selection on final normalization results, we first conducted the same normalization procedure as OLIN but without parameter optimization by GCV. Instead, we used default values for the model parameters. This provides a 'baseline' model termed LIN, which we compared to the optimized models OLIN and OSLIN. A default value of 0.5 was used for fitting parameters αA and αXY. The scaling parameter sY was set to 1. The iterative procedure was maintained for LIN to ensure self-consistency of results, as we regress stepwise with respect to intensity A and location (X,Y). The number of iterations was set to three. The results are displayed in Figure 2. The MA-plot data normalized by LIN showed that the residuals are centered around zero (Figure 2a). The considerable bias of log ratios M for low spot intensities A was removed. This was confirmed by testing normalized log ratios for intensity-dependent bias. No spot-intensity neighborhood with a significant median log ratio was detected (Table 1).
Figure 2.
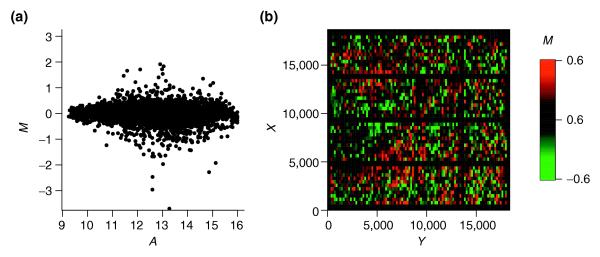
Intensity and spatial distribution of log ratios for local intensity-dependent normalization (LIN) with default model parameters. (a) The residuals of the local regression are well balanced around zero in the MA-plot. (b) Patterns of spatial bias are still apparent in the MXY-plot, whereas the lines of negative M corresponding to empty spots disappeared as a result of the intensity-dependent normalization.
However, careful inspection of the MXY-plot shows that the spatial bias was only partially removed, as spatial patterns were still visible (Figure 2b). The permutation tests also revealed that the distribution of M is not balanced across the array. Seventy-eight spots had neighborhoods with a significant large median log ratio (Table 2). The result indicates that local (spatial) features exist in the data, which were not appropriately fitted by LIN. This points to the importance of model parameter optimization, especially for location-dependent normalization.
Optimized local intensity-dependent normalization
To improve the efficiency of normalization, we conducted OLIN with model optimization by GCV. Three parameters (αA, αXY, sY) had to be optimized during each iteration. Parameters (αA, αXY) determine the proportion of data used for local intensity-dependent and spatial regression of log ratio M, respectively. They control the smoothness of fits. Choosing accurate parameter αA and αXY is crucial for the quality of the regression. Too large parameter values result in a poor fit, where local data features are missed; too small values lead to overfitting of the data. Two extreme cases illustrate the importance of parameters αA and αXY. If we choose a value of 1, all data points are included in the local regression. Although the weight function tricube W used by LOCFIT forces larger weights to be put on neighboring points, the fit becomes increasingly linear. The other extreme case is the use of a diminutive parameter value, which leads to fitting of every point independently of its neighborhood. Overfitting of the data occurs and the residuals are subsequently underestimated. Besides smoothing parameters αA and αXY, OLIN demands the setting of scaling parameter sY. This is especially important if spatial patterns of log ratio M vary on differing scales across the array. GCV was used to determine the optimal setting of model parameters. For αA and αXY, a parameter range of 0.1 to 1 was tested. For sY, values between 0.05 and 20 were compared. The number of iterations was set again to three. If more iterations were performed only minor changes in the outcome of normalization were observed, indicating that self-consistency of normalization was reached.
Inspection of the MXY-plot revealed that the optimized local intensity-dependent normalization was able to correct for the spatial bias (Figure 3b). Spots with positive and negative log ratio M were evenly distributed across the slide. The patterns of spatial bias across the array were no longer apparent. Similarly, the residuals were well balanced around zero in the MA-plot (Figure 3a). The results of the statistical tests underlined these findings. No significant neighborhoods were found on testing for intensity-dependent dye bias and only one neighborhood remained significant on testing for spatial bias (Tables 1 and 2).
Figure 3.
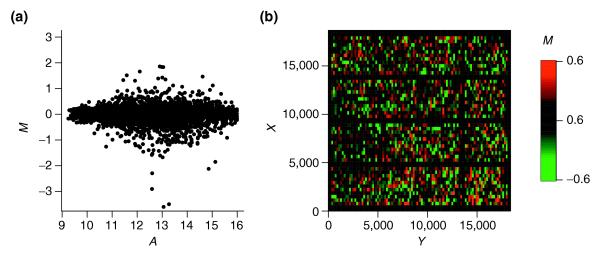
Intensity and spatial distribution of log ratios for optimized local intensity-dependent normalization. (a) MA-plot; (b) MXY-plot. Both plots indicate no apparent bias for log ratio M with respect to the intensity A or the spot location (X,Y). Note, however, that the MXY-plot shows areas of differing brightness corresponding to areas of differing variability of M. Regions with large abs(M) appear, therefore, brighter than regions with small abs(M). For example, the variance of M seems to be larger around spot location (X = 2,500, Y = 16,000) than round the location (X = 7,000, Y = 3,000).
The GCV procedure only approximates the prediction error of standard cross-validation. To test if this approximation is accurate for the microarray data analyzed, we compared the GCV estimates with the estimates produced by fivefold cross-validation. Although GCV is considerably less computationally demanding, it reproduces estimates of the computationally intensive fivefold cross-validation generally well (see Figure 4). The αA values selected by GCV ranged from 0.1 to 0.7 for the SW480/620 experiment and between 0.2 and 0.7 for the apo AI experiment. Smaller values produced overfitting of data; larger values yielded underfitting. For the third iteration, an αA value of 1 was generally selected, resulting in an approximately linear fit. Optimization of spatial regression parameters αXY and sY showed a more complex behavior and varied between experiments and slides.
Figure 4.
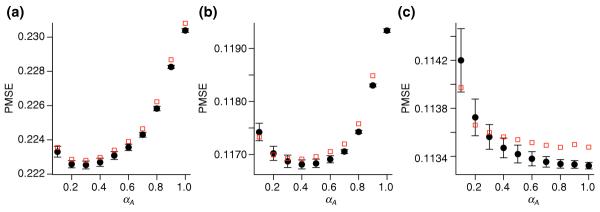
Comparison of GCV and fivefold cross-validation. The relation between prediction mean square error (PMSE) and smoothing parameter αA is shown for the three iterations (a-c) in the OLIN procedure applied to slide 16 of the apo AI experiment. The fivefold cross-validation was conducted for five random splits of the data. Mean values and standard errors of PMSE estimates are represented as black circles and error bars. PMSE estimates by GCV are represented by red squares. Generally, these estimates lie within the error margin of PMSE produced by fivefold cross-validation. The GCV-optimized value of αA was 0.3 for the first (a), 0.4 for the second (b), and 1.0 for the third (c) iteration.
Although OLIN leads to an even spatial distribution of positive and negative log ratios M, visual inspection of Figure 3b indicates that the variability of log ratios might be unbalanced across the array. This can also be assessed by permutation tests. In the same manner as for spatial bias detection, we derived the number of neighborhoods with significant median abs(M) values. The results can be found in Table 1 of additional data file 1. For slide 3 of SW480/620 experiment, 25 spot neighborhoods were detected as significant using FDR = 0.01. Therefore, it may be favorable to adjust the scale of log ratios M locally.
Optimized scaled local intensity-dependent normalization
If we can assume that the variability of log ratios M should be equal across the array, local scaling of M can be performed. As in the previous section, the validity of these assumptions has to be carefully checked for each array analyzed. The underlying requirement is again random spotting of arrayed genes. As we believe this requirement is fulfilled for our experiments, we applied optimized local scaling within the OSLIN scheme. The local scaling factors are derived by optimized local regression of the absolute log ratio M. The range of regression parameters tested by GCV is (0.1,1) for smoothing parameter α and (0.05,20) for scaling parameter sY. The resulting MA- and MXY-plots for slide 3 are presented in Figure 5. The variability of log ratios M appears to be even across the array. This is consistent with the result of the corresponding permutation test: No significant spot neighborhood was detected (see Table 2 in additional data file 1).
Figure 5.
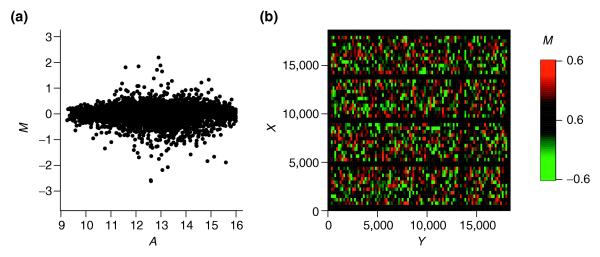
Intensity and spatial distribution of log ratios M for optimized scaled local intensity-dependent normalization. (a) MA-plot. (b) MXY-plot. This shows that the variability of log ratios is even across slide 3 of the SW480/620 experiments.
Slide-wise comparison of normalization schemes
The normalization methods proposed in this study yielded different results. To choose the optimal method, the efficiency of normalization in removing systematic errors has to be compared. As well as the methods presented above, we included three previously proposed normalization methods based on lowess regression and implemented in the Bioconductor software package [6,21]: global intensity-dependent normalization (global lowess) which regresses log ratios M with respect to spot intensity A; within print-tip group normalization (P-lowess) which regresses M with respect to A for every print-tip group independently; and scaled within-print-tip group normalization (S-lowess), which scales log ratios M for each print-tip group after P-lowess is applied. Note that the smoothing parameter α for these methods is constant and the default value of 0.4 was used.
The results of the comparison are examined in detail here for slide 1 of the apo AI experiment. The corresponding MA-plots can be found in Figure 3 in additional data file 1. Although linear normalization led to an overall balanced distribution of M, it was insufficient to remove the intensity-dependent dye bias. The nonlinear methods applied were generally able to correct for intensity-dependent bias. Figure 6 presents the MXY-plots for slide 1 of the apo AI experiment. For global linear normalization, the corresponding MXY-plot indicates that this method is insufficient to remove spatial artifacts on the array. Easily noticeable stripes of positive or negative log ratio remained. Spots near the right edge of slide 1 show a considerable bias towards positive log ratios. Note that these spatial patterns do not correlate with the sub-grid defined by the 4 × 4 print-tips. Application of global lowess normalization failed to remove these spatial artifacts. This can be expected, as the global lowess method does not incorporate any special normalization. A reduction in spatial bias can be seen for P-lowess, S-lowess and LIN which all include spatial normalization. However, spatial patterns remain prominent. For P-lowess and S-lowess, this indicates that they are not able to correct for spatial artifacts that are not correlated with print-tip groups. For LIN, it points to underfitting of the data, and thus the necessity of parameter optimization. Inspection of the MXY-plots for OLIN and OSLIN confirms that this was indeed the case: location-dependent dye bias was absent in both plots. In addition, the MXY-plot for OSLIN shows an even variability of log-ratios across the array.
Figure 6.
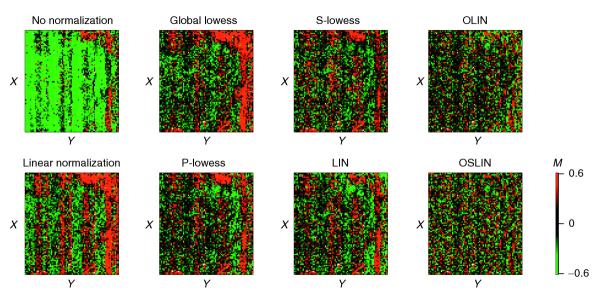
MXY-plots of slide 1 of the apo AI experiment for raw and normalized data. In this case, the X and Y coordinates correspond to rows and columns of the array, as exact spot locations are not given for the publicly available dataset.
To assess the validity of the findings based on visual inspection, the efficiency of normalization was also examined by permutation tests (Tables 1 and 2). For 1,750 spots, a significant intensity neighborhood was detected if no normalization was applied. Most significant spot neighborhoods could be found at low spot intensities. Global linear normalization even led to a slight increase in number of significant neighborhoods. All methods incorporating local intensity-dependent normalization performed with similar efficiency. For P-lowess, S-lowess, OLIN and OSLIN, no spots with significant neighborhoods were detected, whereas 18 remained for global lowess and 15 for LIN. Testing for spatial bias, we found 1,173 spot neighborhoods with significant large log ratios if no normalization was applied. Linear and global lowess normalization increased the number of spatially biased neighborhoods. P-lowess, S-lowess and LIN reduced the number of significant neighborhoods, although only with a limited efficiency (P-lowess, 913; S-lowess, 491; LIN, 755). A considerable reduction of spatial bias was achieved by OLIN: 100 neighborhoods were detected as significant after normalization. OSLIN showed the best performance. Only one spot neighborhood remained significant.
Besides giving an indication about the efficiency of normalization, the testing procedure applied also enabled us to identify regions of dye and spatial bias. This is illustrated in Figure 7. Spots are represented by red squares if their neighborhood has a significant positive median log ratio M. Correspondingly, spots are represented by green squares if their neighborhood has a significant negative median log ratio. By varying the level of FDR, the grade of significance can be assigned. This approach enables a stringent localization of significant experimental bias. Figure 6 shows, for example, that spots close to the right edge are especially affected by spatial artifacts.
Figure 7.
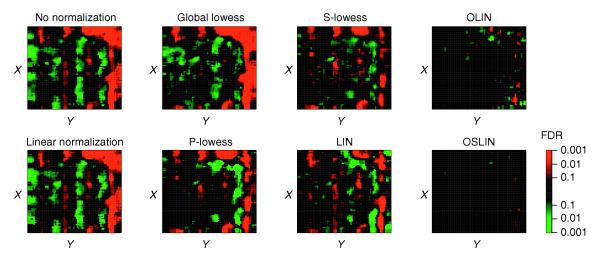
Significance of spatial bias for slide 1 of the apo AI experiment. Spots were represented by red or green squares if their neighborhood had a significant positive or negative median M value, respectively. The level of significance (FDR) is encoded by the brightness of the colors as shown in the color scale.
Although the number of significant neighborhoods varied between slides and experiments, the results of the comparison undertaken for slide 1 of the apo AI experiment remain generally valid for the other slides analyzed (Tables 1 and 2). Linear normalization was unable to remove intensity-dependent and location-dependent dye bias. Global lowess corrected for intensity-dependent, but not for spatial bias. P-lowess, S-lowess and LIN performed well in the correction for intensity-dependent bias, but were less efficient in correcting for spatial dye bias. For most slides, OLIN and OSLIN showed the highest efficiencies in removing both types of systematic error.
An alternative, and computationally less expensive, way to examine intensity- and location-dependent bias is the correlation of the log ratio M with average in the spot's neighborhood [5]. Assuming that log ratios of neighboring spots are uncorrelated, a correlation close to zero can be expected. A large positive correlation, however, indicates the existence of bias. Successful normalization, therefore, should remove the correlation of log ratios of neighboring spots. We conducted this type of correlation analysis for each array independently. Spot intensity and location neighborhoods were defined as before. The results can be found in Tables 2 and 3 of additional data file 1. We present and discuss here the average correlation coefficients for the two experiments analyzed (Table 3). For the SW480/620 experiment, the average Pearson correlation of a spot's log ratio M and the median log ratio of spots in its intensity neighborhood was 0.50. Whereas linear normalization lead to exactly the same correlation coefficient, the nonlinear methods compared yielded a correlation coefficient close to zero. Correlating the log ratio M of spots with the median log ratio of their spatial neighborhood resulted in a correlation of 0.53 for raw data. Linear normalization again yielded the same correlation. Global lowess slightly increased the correlation. P-lowess, S-lowess and LIN achieved a considerable, but limited, decorrelation. Only OLIN and OLIM resulted in correlation coefficients close to zero. The same analysis was applied to the apo AI experiment with a similar outcome. The coefficients for spatial correlation were, however, generally larger, indicating a more prominent spatial dye bias.
Table 3.
Statistical comparison of normalization schemes
| Experiment | No normalization | Linear normalization | Global lowess | P-lowess | S-lowess | LIN | OLIN | OSLIN | |
| Intensity-dependent cor(M, |
SW480/620 | 0.50 | 0.50 | 0.01 | -0.01 | -0.01 | 0.04 | 0.00 | 0.00 |
| Intensity-dependent cor(M, |
Apo AI | 0.47 | 0.47 | 0.06 | 0.05 | 0.05 | 0.09 | 0.00 | 0.00 |
| Spatial cor(M, |
SW480/620 | 0.53 | 0.53 | 0.56 | 0.34 | 0.32 | 0.27 | 0.07 | 0.08 |
| Spatial cor(M, |
Apo AI | 0.58 | 0.58 | 0.59 | 0.41 | 0.38 | 0.43 | 0.15 | 0.15 |
| Mean pairwise cor(M) | SW480/620 | 0.46 | 0.46 | 0.50 | 0.59 | 0.59 | 0.63 | 0.67 | 0.64 |
| Var(M) | SW480/620 | 927.1 | 658.7 | 455.4 | 216.3 | 213.0 | 205.7 | 163.0 | 186.5 |
| Number of significant genes | SW480/620 | 26 | 71 | 51 | 75 | 88 | 94 | 99 | 129 |
| Average cor(MqPCR, Mma) | Fibroblast | 0.82 | 0.81 | 0.82 | 0.82 | 0.81 | 0.81 | 0.84 | 0.88 |
Intensity-dependent correlation (intensity-dependent cor(M, ![]() )) describes the correlation between the log ratio M of the spot and the median value of M within a symmetrical neighborhood of 50 spots on the intensity scale. Spatial correlation (spatial cor(M,
)) describes the correlation between the log ratio M of the spot and the median value of M within a symmetrical neighborhood of 50 spots on the intensity scale. Spatial correlation (spatial cor(M, ![]() )) describes the correlation between the log ratio M of the spot and the median value of M within a neighborhood defined by a window of 5 × 5 spots. To ensure independence, M of the spot was not included in the median
)) describes the correlation between the log ratio M of the spot and the median value of M within a neighborhood defined by a window of 5 × 5 spots. To ensure independence, M of the spot was not included in the median ![]() of the neighborhood. For the calculation of mean pairwise correlation of slides, spots with intensity A < 11.6 were excluded. The significance of differential gene expression was examined by a one-sample t-test with the null hypothesis of mean log ratio M = 0. Duplicated spots on SW480/620 arrays were treated as independent measurements producing a maximum of eight observations per gene. Genes were detected as significantly differentially expressed if their Bonferroni adjusted p-values were smaller than 0.01. For the fibroblast experiment, the average Pearson correlation between qPCR-based logged fold changes MqPCR and microarray-based logged fold changes Mma of the genes for IL-8, COX2, Mast, B4-2 and actin is shown.
of the neighborhood. For the calculation of mean pairwise correlation of slides, spots with intensity A < 11.6 were excluded. The significance of differential gene expression was examined by a one-sample t-test with the null hypothesis of mean log ratio M = 0. Duplicated spots on SW480/620 arrays were treated as independent measurements producing a maximum of eight observations per gene. Genes were detected as significantly differentially expressed if their Bonferroni adjusted p-values were smaller than 0.01. For the fibroblast experiment, the average Pearson correlation between qPCR-based logged fold changes MqPCR and microarray-based logged fold changes Mma of the genes for IL-8, COX2, Mast, B4-2 and actin is shown.
Experiment-wide comparison of normalization schemes
In the ideal case, results derived by replicated arrays should be the same. In practice, however, variable experimental conditions lead to random and systematic changes in the outcome. Normalization aims to correct for systematic errors, and thereby to increase the consistency of outcome. To assess this capacity, we calculated total variation of log ratios M between replicated arrays for the SW480/620 experiment (Table 3). The total variance of raw log ratios M was var(M) = 927. This is reduced to 659 by linear normalization and to 455 by global lowess. P-lowess, S-lowess and LIN performed similarly, and further reduced the total variance. A minimum total variance of 163 was achieved using OLIN. This is a reduction of variance by over 80% compared to raw data. This analysis was not possible for the apo AI experiment, as only biological, but no technical, replicates were included. A reduction of variability between biological replicates by normalization, however, cannot be assumed.
A related measure of consistency is the overall correlation between arrays. Random error, however, may interfere with this analysis. Because log ratios of spots at low intensity can be expected to be highly affected by random error, spots in the lower third of the intensity distribution were excluded. On the basis of the remaining two thirds of the data, the average pairwise correlation  of log ratios M between all four slides was 0.46 for raw data as well as for linear normalization. A slight increase was achieved by global lowess ( = 0.50). Using methods incorporating spatial normalization, we obtained a considerable improvement. P-lowess and S-lowess produced the same correlation coefficients ( = 0.59). LIN and OSLIN yielded further increase in correlation. The highest correlation was achieved by OLIN with = 0.67.
of log ratios M between all four slides was 0.46 for raw data as well as for linear normalization. A slight increase was achieved by global lowess ( = 0.50). Using methods incorporating spatial normalization, we obtained a considerable improvement. P-lowess and S-lowess produced the same correlation coefficients ( = 0.59). LIN and OSLIN yielded further increase in correlation. The highest correlation was achieved by OLIN with = 0.67.
The main goal of the SW480/620 experiment was the identification of differentially expressed genes. Appropriate data normalization should facilitate detecting these genes. For means of comparison, we used a one-sample t-test. Because multiple tests are performed, p-values obtained were subsequently adjusted by Bonferroni correction. This produced a conservative estimate of significance. Normalization was found to have a considerable impact on this outcome of the significant test; the number of significant genes varied up to a factor of five between different methods (Table 3). Without normalization, only 26 genes were detected as significant. A maximum of 129 significant genes was found after application of OSLIN. Scaling generally had a positive effect on the number of significant genes. For both methods incorporating scaling (S-lowess, OSLIN) more genes were found to be significant compared with the corresponding method without scaling (P-lowess, OLIN). This may indicate that scaling facilitates the detection of differential expression.
A prominent example illustrating the impact of normalization on the significance of genes was given by the results for tissue inhibitor of metalloproteinases type 3 (TIMP-3). For raw data of the SW480/620 microarray experiment, the p-value was 0.52. The use of linearly normalized data resulted in a reduced p-value of 0.27. A borderline significance was achieved using global lowess, P-lowess and S-lowess (p = 0.022, 0.019, 0.015). The effect of parameter optimization was clearly demonstrated by the comparison of significance after application of LIN or OLIN/OSLIN. Whereas the p-value of TIMP-3 was 0.089 for LIN, it was considerably reduced for OLIN and OSLN (p = 0.009, 0.007). Consisting with the overall trend, scaling (by S-lowess or OSLIN) increased the significance. Downregulation of TIMP-3 in SW620 compared to SW480 cells was independently validated by northern plotting [22]. As TIMP-3 inhibits enzymes (metalloproteinases) required for invasion, reduced expression of TIMP-3 is conjectured to contribute to the invasive potential of SW620 cells [23].
Internal validation of normalization results by analysis of replicated control spots
As the previous sections revealed, selection of smoothing parameter is especially crucial for removing spatial artifacts in the experiments analyzed. The MXY-plots showed generally more complex patterns than corresponding MA-plots. This was reflected in the comparison of normalization results. Whereas all local regression methods applied in this study performed similarly in removing intensity-dependent bias of log ratios M, permutation tests indicated that methods without parameter optimization were insufficient to remove spatial bias. To validate this conclusion, we compared the variation of M of replicated spots for the SW480/620 experiment (Table 4). These control spots were spatially distributed across the array. Under ideal circumstances, the spatial location should not influence the corresponding value of M and thus variation of M should be minimal. However, a considerable effect of spot location was detected for all three types of replicated spots. Although all normalization schemes including spatial correction procedures could reduce the variability of M, their performance differed consistently for the three types of replicated spots. P-lowess reduced the variance of M on average by 39% compared to global lowess that does not incorporate spatial normalization. However, the corresponding OLIN procedure based on optimized parameter selection clearly outperformed P-lowess. Compared to global lowess, it yielded an average reduction of var(M) by over 60%. A similar result was obtained comparing normalization schemes that included scaling (S-lowess, OSLIN). The average reduction var(M) was, however, lower relative to the corresponding schemes without scaling. In the case of replicated Cot-1 control spots, S-lowess even increased the variability of M. Altogether, analysis of the included control spots supports the conclusion that parameter optimization can be crucial for the quality of normalization.
Table 4.
Comparison of variance of log ratios for control spots in SW480/620 experiments
| Control spot | Number of replicate spots per slide | Global lowess | P-lowess | S-lowess | OLIN | OSLIN |
| SS-DNA | 48 | 6.46 | 3.33 | 4.03 | 1.90 | 2.82 |
| Cot-1 DNA | 12 | 4.34 | 4.10 | 5.07 | 2.90 | 3.73 |
| Rice DNA | 12 | 12.0 | 4.34 | 5.03 | 2.35 | 2.79 |
The average within-slide variance (× 10-2) of log ratios M of control spots is shown after applying different normalization schemes. The three types of control spots derived from genomic DNA were used: salmon sperm (SS) DNA; Cot-1 DNA; rice DNA. Their intensities were above background as a result of nonspecific cross-hybridization. The locations of the replicated control spots were spatially distributed across the array. Comparison of corresponding log ratios M thus provides a measure for the spatial consistency of results produced by normalization.
External validation of normalization results by comparison of microarray and qPCR data
We showed in the previous section that model selection can considerably improve the consistency of data within a microarray experiment. However, the crucial question to ask (as one reviewer correctly pointed out) is whether the methods introduced can provide greater precision of the actual biological changes occurring. To address this valid point, we reanalyzed the microarray experiment of Iyer et al. [24]. In their study, the temporal response of gene expression in fibroblasts to serum was measured by spotted cDNA microarrays representing more than 8,600 human genes. The changes in expression were recorded for 12 time points ranging from 15 minutes to 24 hours after serum stimulation. Iyer and co-workers confirmed the temporal expression patterns of five genes (for IL-8, COX2, Mast, B4-2 and actin) by quantitative polymerase chain reaction (qPCR). This additional data enabled us to compare the results of normalization methods used with an external standard for multiple genes at multiple conditions. For this comparison, the correlation of qPCR-based logged fold changes with microarray-based logged fold changes was calculated. The use of the log-scale was motivated by the results of the fibroblast study, which showed a good overall correlation of logged fold changes derived by both methods (see Figure 3 of reference [24]). Any improvement in the correlation is especially desirable regarding time-series experiments where clustering is commonly used to identify coexpressed genes. As most clustering algorithms are based directly or indirectly on correlation as measure of similarity, the correlation of microarray data with actual biological transcriptional changes is of crucial importance.
We first normalized again all microarrays using the methods to be compared. MA- and MXY-plots indicated regions of intensity-dependent and spatial bias for the microarrays used in the study. Several of these plots are presented in Figure 8 in additional data file 1. After normalization of the data, the Pearson correlation between qPCR-based logged and microarray-based logged fold changes was calculated. The largest differences between normalization methods were obtained for COX2 (Figure 8). Whereas the correlation of logged fold-changes was only 0.56 for raw data, it increased to 0.60, 0.64 and 0.64 using lowess, P-lowess and S-lowess, respectively. The correlation was further improved to 0.70 by OLIN. However, the most considerable increase was observed for data normalized by OSLIN. A correlation coefficient of 0.86 was obtained. Remarkably, the relatively weak correlation for COX2 was already noted by Iyer and colleagues. They attributed this observation to a "localized area on the corresponding array scan resulting in an underestimation of the expression ratio" (see note 10 in reference [24]). The result indicates that optimized normalization methods can correct for such artifacts, at least partially. For the other genes, the differences between methods were less prominent, as the correlation of qPCR- and microarray-based fold changes was already strong (above 0.7) for raw data. The overall comparison, however, shows that only methods with model selection could improve the correlation of microarray data with the external standard (Table 3). Methods without model selection did not yield an increase in correlation compared the correlation obtained for raw data. The comparison demonstrates that optimized normalization can lead to greater precision of microarray data and to a better correlation of measured fold changes with the actual biological changes in expression.
Figure 8.
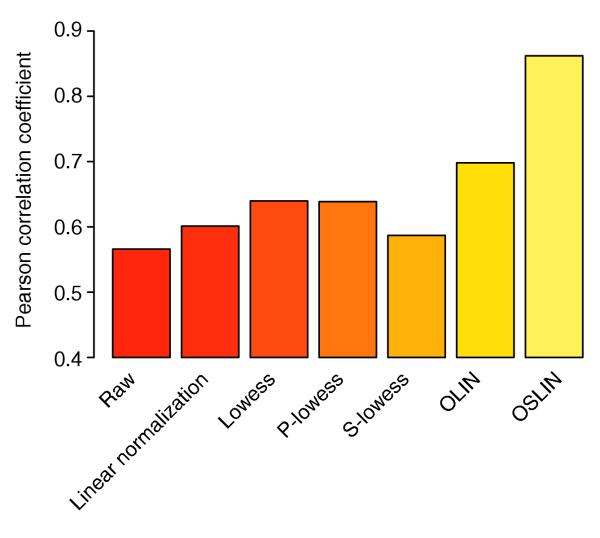
Histogram of Pearson correlation between logged qPCR- and microarray-based fold changes of COX2 expression for the fibroblast microarray experiment of Iyer et al. [24] after application of various normalization methods.
Discussion
Microarray measurements are affected by a variety of systematic experimental errors that limit the accuracy of the data produced. Such errors have to be identified and removed before further data analysis. Several approaches to the identification of intensity-dependent and spatial dye bias were developed in this study. The most basic is the visual inspection of MA- and MXY-plots. Alternatively, A- and XY-plots can be examined. Statistically more stringent, but also computationally more expensive, are permutation tests detecting regions of significant bias in microarray data. Although permutation tests have frequently been used to assess the significance of differential gene expression, to our knowledge, their use to detect artifacts in cDNA microarray data has not previously been proposed. The analysis showed, however, that they can be a valuable tool for identifying regions of dye bias.
Normalization aims to correct for experimental bias. A popular class of normalization methods is based on local regression, as they are flexible and straightforward to use. They have become the method of choice for many researchers and have been implemented in numerous freely available or commercial microarray data-analysis systems, for example Bioconductor [21], MIDAS [25], SNOMAD [26] and GeneTraffic [27]. Other methods, such as ANOVA models, often require statistical expertise in their interpretation and construction [28,29]. One unresolved challenge in using local regression methods has been, however, the choice of regression parameters. This has generally been left to the user, with only default values given. For example, a variety of smoothing parameters α have been suggested without further evaluation of their effects on normalization, for example 0.4 by Yang et al. [6], 0.5 by Kepler et al. [11], 0.5/0.7 by Colantuoni et al. [7], and 0.7 by Yuen et al. [30]. As our analysis shows, however, the use of such default parameters can severely compromise the efficiency of normalization.
To improve the quality of normalization, we developed two schemes incorporating iterative local regression and model selection. We based our normalization schemes on an explicitly formulated hybridization model linking the amount of labeled RNA to the observed fluorescence intensities. The basic goal is modeling the relation between response variable and a set of predictor variables. In our case, the response variable is the log fluorescence intensity ratio M and the predictor variables are spot intensity A and spot location (X,Y). To determine the influence of experimental variables on the measurement results, we use an iterative procedure alternating between local regression of M with respect to A and local regression of M with respect to X and Y. The iterative scheme ensures self-consistency of the stepwise regression procedure. Residuals of the local regression were interpreted as corrected fold changes. This allows a separation of the systematic errors due to intensity and spatial effects from biological changes in expression. To increase the accuracy of the normalization model, we optimized the model parameters. GCV was applied for parameter optimization since it is computationally of advantage for large data sets compared with standard cross-validation. The regression parameters selected by GCV varied between slides and experiments analyzed, reflecting the variability of systematic dye bias, and this manifests the need for model selection for each array individually. Visual inspection of spatial distribution of absolute log ratio M suggested an uneven variability of M across slides. As the span of log ratios seemed to vary continuously across the array, a correction by locally optimized scaling was performed. This procedure yielded an even variability of M across the spatial dimension of the array after local normalization and scaling.
An important criterion for the quality of normalization is its efficiency in removing systematic errors. However, the assessment of normalization efficiencies has been neglected in previous studies. Using methods that we developed for error identification, we compared the efficiency of several normalization schemes for two independently generated cDNA microarray datasets. Statistical efficiency testing was based on permutation tests detecting spot neighborhoods affected by experimental bias. These tests allow a stringent identification of regions of significant bias in microarray data. We believe that this feature is especially valuable for the important assessment of data quality, as it facilitates rapid detection of artifacts and may help to improve the experimental procedures. Fold changes should be treated with care if the corresponding spots have significantly biased neighborhoods even after normalization. As an alternative to permutation tests, we also applied correlation analysis for comparison of normalization efficiencies. Correlation analysis is less computationally expensive and agrees well with the results of the permutation test, but cannot deliver localization of experimental bias in the data.
Besides the schemes presented, we tested several other variations of iterative local regression with parameter optimizations. As an alternative to the proposed normalization by OLIN, we conducted local regression of log ratios M with respect to spot intensity A and spot location (X,Y) simultaneously. The computational costs of parameter optimization increased considerably, as cross-validation has to be applied to a three-dimensional parameter space. The results of this procedure yielded, however, no improvement in efficiency and were frequently less stable. Reversing the order of intensity-dependent and spatial normalization in the OLIN procedure also yielded a decreased performance of normalization. Moreover, if the fold changes are asymmetrically distributed or a high background noise exists, the use of a more robust local regression procedure might be favorable. A robust version of LOCFIT is implemented by iterative fitting of the data with successive downweighting of outliers in the regression [13]. The application of robust LOCFIT to the datasets examined showed, however, only minimal difference in outcome compared with the results of the original algorithm.
We restricted our normalization approaches to correction for spot intensity- and location-dependent dye bias. However, the principal components of our schemes, iterative regression and model selection, can be applied to the correction for other types of bias in cDNA microarray, such as those linked to differing microtiter plates, print-runs, scanner settings, and so on. As well as better performance, such extended normalization systems may give researchers new insights about the sources of variability in cDNA microarray data and may support the optimization of experimental protocols.
Conclusions
Although several other studies have recently introduced normalization by local regression, none has addressed the selection of model parameters. On the basis of two independently generated microarray datasets, the major conclusions of our study are as follows.
First, our analysis shows that parameter selection is crucial for the efficiency of normalization and that the use of default parameters can severely compromise the quality of normalized data. This finding is important, as normalization by local regression has become the method of choice for many researchers and has been implemented in numerous software packages for microarray data analysis. The final choice of regression parameters, however, remains with the user. Accepting the default parameters of the software without further evaluation can easily lead to insufficient normalization interfering with the subsequent data analysis.
Second, extensive comparison of normalization efficiencies showed that schemes based on parameter optimization can considerably reduce systematic errors in microarray data. Using these schemes, researchers can avoid insufficient normalization of microarray data and improve the overall consistency of measured gene expression between replicated arrays. These schemes can also yield an improved correlation of microarray measurements with actual biological changes in expression and, thus, support the validity of results derived in follow-up gene-expression analysis.
Third, GCV was used successfully for model selection. To our knowledge, this procedure has not been applied in the field of microarray data analysis so far. However, we found that it is of considerable computational advantage compared to the popular standard cross-validation, and it may be useful for a wide range of tasks in the analysis of high-throughput data.
Fourth, we developed methods for stringent detection of systematic errors. Independently of the normalization schemes proposed, these new methods can rapidly identify artifacts and experimental variability that is obscuring changes of biological interest. They may also assist in the optimization of experimental protocols and will be useful for researchers, especially if they are new to microarrays. It should be noted that GCV is only one of many methods proposed for smoothing parameter selection (see Materials and methods). The careful comparison of these methods is therefore an important task for future study.
Finally, the core methods and procedures introduced in this study are not restricted to cDNA microarrays, but can be applied to other array platforms as well. We believe therefore that they will be helpful to many researchers using array technologies.
Materials and methods
Hybridization model
To relate fluorescence signals to changes in gene expression, we introduce a hybridization model on which we base our normalization methods. Explicitly modeling the relation between signal intensities and changes in gene expression can separate the measured error into systematic and random errors. The model is especially developed for two-color arrays consisting commonly of a red (Cy5) and a green (Cy3) fluorescence channel. The basic model might, however, be generalized to other types of microarrays. The fundamental variables in our hybridization model are the fluorescence intensities of spots in the red (Ir) and the green channel (Ig). These intensities are functions of the abundance of labeled transcripts (Tr/g).
Thus, we have
Ir/g = fr/g (Tr/g, ) (4)
with functions fr/g relating the abundance of the transcripts to the measured intensities and a set of parameters in the experiment. Note that the functions fr and fg might be different.
Under ideal circumstances, this relation of I and T is linear up to an additional experimental error ε:
I = N() T + ε (5)
where N is a normalization factor and a function of experimental parameters such as the laser power or amplification of the scanned signal.
Generally, this simple relation does not hold for microarrays because of effects such as intensity background and saturation. Including an additive background Ib leads to
![]()
The normalization factor N' now depends on transcript abundance T. We can obtain the original Equation (5) by subtracting the background intensity Ib, so that the background corrected intensity Ibc is derived by
Ibc = I - Ib = NT + Ib + ε - Ib = NT + ε (6)
This step is included in most normalization procedures where the background intensity is estimated by the local background fluorescence surrounding the spot. Frequently, saturation also affects the relation between intensity and abundance of labeled transcript. A possible model for these effects is
![]()
where N1,N2 and c are constants. Although the right-hand side of the Equation (7) has the same form as Equation (6), the normalization factor N' is not constant, but varies with the transcript abundance T. Because the saturation is generally of unknown form, the recovery of the original relation between I and T might not be possible.
In a two-color experiment, ratios of fluorescence intensities are generally used to represent fold changes of gene expression. This procedure has the advantage of controlling for several variations that are inherent to spotted arrays such as size and morphology of the spots and a variable amount of spotted DNA. Therefore, fold changes (or ratios) of gene expressions are the major quantities derived in two-color experiments. To relate the ratios for labeled transcript abundances (Tr/Tg) to the ratios of signal intensities by (Ir/Ig), we propose following hybridization model:
![]()
which is based on Equations (4-7). The normalization factors kr/g() are functions dependent on a set of experimental parameters . This gives the relation between the measured quantities (Ir/Ig) and the unknown quantities (Tr/Tg) in which we are interested. Equation (8) can be log2-transformed to facilitate the computational evaluation. This leads to
M = log2(R)
= log2 (kr()Tr + sr) - log2(kg()Tg + εg)
To simplify this equation, we use the Taylor expansion
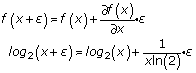
We can thus approximate the above Equation (6) by
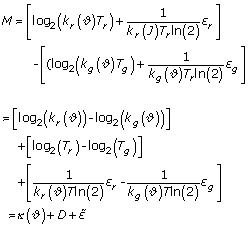
with κ() as additive normalization factor, D as logged fold changes and 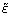 as the random error. This results in the final relation:
as the random error. This results in the final relation:
M - κ() D + (9)
Local regression
For the two schemes proposed in this study, a local regression method is used. Generally, regression methods aim to model the relation between a response variable Y and a set of predictor variables x. Regression models can be expressed as
Yi = μ(xi) + εi (10)
with a function μ of a chosen class and an error term εi. A standard procedure is the use of global regression methods. They, however, assume that the chosen global model holds over the whole range of x. A more flexible fitting approach is offered by local regression using polynomial functions, which are fitted at x based on data points in a neighborhood of chosen size h. The popular lowess method belongs to this type of local regression [12]. For our normalization schemes, we use local regression as performed by the LOCFIT method, as it is computationally more flexible. The main points of LOCFIT are outlined below. LOCFIT is described in further detail in [13].
LOCFIT algorithm
Evaluation points: LOCFIT does not perform local regression at every point of the dataset, but only at the vertex points z of a grid which spans the whole range of variable values of x.
Local polynomial fit: quadratic polynomials are locally fitted at the vertex points z. In a one-dimensional regression, for example, this leads to the approximation of μ by
M(z) ≈ a0 + a1(xi - z) + a2(xi - z)2
The neighboring points xi are weighted according to the tricube weight function
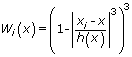
with h(x) as the bandwidth that defines the size of the smoothing window. The bandwidth h(x) is the minimal neighborhood size that includes the fraction α of the total number of points. By choosing α, the user of LOCFIT can determine the smoothness of the fit.
Multivariate regression: if the local regression is based on multiple predictor variables xj, multivariate local polynomials are used for fitting. The independent predictor variable xj are adjusted by a scaling factor sj:
![]()
Fitting criteria: the polynomial coefficients ai are determined by a local likelihood model. The response variable Yi is assumed to follow a chosen distribution function. The default distribution in LOCFIT is Gaussian. This leads to a local likelihood criterion that is equivalent to the local least square criterion.
Interpolation: after a local regression is performed for vertex points of the grid, the function μ for an arbitrary point xi is obtained by interpolation of the function approximations at the vertex points. To ensure that the function μ is globally differentiable, LOCFIT uses a cubic polynomial for interpolation, which includes estimates of the derivatives at the vertices.
Model selection
A standard approach for model selection is k-fold cross-validation. It splits the data into k segments of which k -1 segments are used for the model construction and one segment for the validation of the model. This is repeated k times, so that every segment is used for validation. Cross-validation estimates the prediction error by averaging the mean squared errors in the k runs. If different models are compared by cross-validation, the model yielding the lowest prediction error is generally selected. In the extreme case that k equals the number of data points, the cross-validation is also referred to as the leave-one-out method.
However, because of the large number of data points in microarray data, regression model selection by leave-one-out cross-validation becomes computationally prohibitive as the number of models constructed for cross-validation equals the number of data points. Even the computationally less expensive k-fold cross-validation is not practicable if a large number of models are compared for selection. As an alternative to standard cross-validation methods, we used, therefore, the generalized cross-validation (GCV), which approximates the leave-one-out method [14]. GCV is easier to perform, as this procedure does not include multiple constructions of regression models based on partial data. For the local regression model 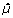 , the GCV criterion is
, the GCV criterion is
![]()
where n is the number of data points and ν is the degrees of freedom of the local fit and is given by the trace of the hat matrix linking data and fitted values [13]. Basically, the nominator term of the GCV criterion is the square error of the fit and thus favors models that fit well the data. The denominator term punishes models with large degrees of freedom compared to the number of data points, and thus aims to prevent over-fitting. For model selection, the cross-validation estimate of the prediction error is replaced by the GCV criterion. Thus, the model with the minimal GCV score is chosen.
GCV is an example of smoothing parameter selection, which is an intensively studied subject in nonparametric function estimation. A variety of selection methods have been proposed. They are commonly divided into two classes. Classical methods such as CV, GCV and Akaike's information criterion are extensions of approaches used in parametric function estimation. These methods are also called 'first generation methods'. Plug-in methods (or so-called 'second generation methods') have been primarily developed for kernel density estimation. They are generally based on the Taylor expansion of the bias of the estimation. A 'pilot bandwidth' is then plugged into the expansion to calculate the optimal smoothing parameter. A difficulty in using plug-in methods is, however, the selection of such pilot bandwidths. For an introduction to plug-in methods see [31]. The issue of which of these parameter selection methods is superior is highly controversial, as their performance seems to depend not only on the assumption about the fitted data, but also on the chosen criterion for the goodness of fit. Further information and discussion about smoothing parameter selection can be found in [32-35].
Significance of systematic errors
To examine dependencies between observed log ratios M and experimental variables, permutation tests were applied. Permutation (or randomization) tests have the advantage that a particular data distribution is not assumed. They rely solely on the observed data examples and can be applied with a variety of test statistics. A major restriction, however, is that permutation tests are computationally very intensive. The basic idea of a permutation test is simple: given labeled data, all permutations of the labels should be equally likely [36]. In evaluating a chosen test statistic for the permutations generated, an empirical distribution of the test statistic can be constructed. The significance of experimental observations can be determined by comparing the test statistic derived from permutated data with the test statistic of the original data.
In detail, the dependence of log ratios M on spot intensity A or spot location (X,Y) was tested for each slide independently. The null hypothesis states the independence of M and A or (X,Y). To test if log ratios M depend on spot intensity A, spots were ordered with respect to A. This defines a neighborhood of spots with similar A for each spot. Next, a test statistic was generated by calculating the spots' median log ratio within a neighborhood of chosen size. An empirical distribution of the test statistic was produced by calculating for 100 randomly permutated intensity orders of spots. Comparing the empirical distribution of with the observed distribution, we can evaluate the independence of M and A. If M is independent of A, is expected to be symmetrically distributed around its mean value. To assess the significance of observing positive deviations of , we used the false discovery rate (FDR), which indicates the expected proportion of false discoveries amongst rejected null hypotheses [37]. It is defined as FDR = n/s, where n is the number of neighborhoods with larger than a chosen threshold c for the empirical distribution of , and s is the number of neighborhoods with >c for the distribution derived from the original data. Varying threshold c delivers the number of significant for a set of chosen FDRs; that is, in our case FDR = 0.001, 0.005, 0.01, 0.05, 0.1. Correspondingly, the significance of observing negative deviations of can be determined based the number of values lower than a chosen threshold.
The same testing procedure was applied to test the dependence of log ratios M on spot location (X,Y). The null hypothesis states random spotting, that is, the independence of log ratio M and spot location. The neighborhood of a spot is defined here by a two-dimensional window of chosen size. The empirical distribution of was based on 100 random permutations of the spot locations on the array. The significance of was assessed using the FDR as above. In the same manner, the dependence of the absolute value of log ratio M and spot location can be tested.
Microarray data
The normalization models were applied to cDNA microarray data generated in two independent experiments.
Experiment 1: SW480/SW620 (SW) experiment
Gene expression in two cancer cell lines, SW480 and SW620, was compared. The SW480 cell line was derived from a colon tumor of a 50-year-old male patient. The second cell line (SW620) originated from a lymph-node metastasis of the same patient. Target cDNA from SW480 was labeled with Cy3 whereas cDNA from SW620 was labeled with Cy5 using the amino-allyl labeling method. Both cDNA pools were co-hybridized on glass slides with 8,448 spots. The spots consisted of 3,986 distinct sequence-verified human cDNA clones (Research Genetics, release GF211) printed in duplicates, 84 spots from non-human cDNA clones and a further 154 control spots. Spots were printed by 4 × 4 pins. The experiment consisted of four replicated arrays derived from separate labeling reactions. The slides were scanned using a Scanarray 5000 system. Local background spot intensities were extracted by QuantArray software (version2.1). Preliminary analysis showed that replicated spots were highly correlated (average Pearson correlation: 0.94). As this may interfere with the efficiency testing performed in this study, we excluded replicated spots to ensure the independence of spot intensities. Because all spots were printed in duplicate, only half of the spots (4,224) were included in the analysis. However, all normalization methods and statistical tests were also applied to the excluded spots, yielding very similar results (data not shown). Experimental details and further analysis can be found in Futschik et al. [15].
Experiment 2: apolipoprotein AI (apo AI) experiment
This experiment consists of cDNA microarray data from eight apo AI knock-out mice and eight control mice. Target cDNA from each of the 16 mice was indirectly labeled with Cy5 and was co-hybridized with a reference sample on glass slides. The reference sample was prepared by pooling cDNA from the eight control mice and was labeled with Cy3. Each of the 16 microarrays contained 6,384 cDNA probes. Spots were assayed by 4 × 4 pins. For imaging of slides, an Axon GenePix scanner was used. Fluorescence intensities of spots were extracted using the software package Spot. Further details can be found in Callow et al. [17]. The microarray data are publicly available [18]. This dataset was previously used by Yang et al. to present several normalization methods based on local regression by lowess [6].
Experiment 3: fibroblast experiment
To study growth control and cell-cycle progression, Iyer and co-workers measured the temporal response of fibroblasts to bovine fetal serum using cDNA microarrays [24]. Cultured fibroblasts were first induced to enter a quiescent state (G0) by serum deprivation. Subsequent addition of serum evoked fibroblasts to re-enter the cell cycle and to proliferate. To measure gene expression, Iyer and colleagues used cDNA microarrays representing 8,613 human genes. After serum stimulation, cells were sampled at 12 different time points ranging form 15 min to 24 h. The extracted mRNA was reverse transcribed and labeled with Cy5. All the samples were then separately co-hybridized with Cy3-labeled reference cDNA derived from cells in the quiescent state. A major finding of this experiment was that many transcriptional changes observed were related to wound healing. To validate the microarray measurements, the transcript levels of five genes (for IL-8, COX2, Mast, B4-2 and actin) were measured for the different time points using TaqMan 5' nuclease fluorigenic qPCR.
Comparing the logged fold changes based on PCR with those based on microarrays, Ivyer and co-workers found that these methods gave generally similar results. However, they also noted some exceptions from this overall similarity (see Figure 3 and note 10 of reference [24]). The data of the fibroblast experiment is publicly accessible at the Serum stimulation database [38].
Additional data files
Additional data file 1 contains figures and tables with more details of the data analysis presented and referred to in this paper. Additional data file 2 is a .zip file containing the implementation of the OLIN algorithm as add-on an R-package.
Supplementary Material
Gigures and tables with more details of the data analysis presented and referred to in this paper
The implementation of the OLIN algorithm as add-on an R-package
Acknowledgments
Acknowledgements
The normalization schemes presented in this study are implemented in the statistical language R using the LOCFIT and Bioconductor add-on packages [24,39]. The implementation as Bioconductor R-package is currently under development and will be freely available from the Bioconductor project [24]. A current version of the OLIN software and additional documentation are available at the OLIN webpage [40]. M.F. was supported by a PhD scholarship from the University of Otago. We thank Bronwyn Carlisle for proofreading and the referees for their critical and constructive comments.
References
- Holloway AJ, van Laar RK, Tothill RW, Bowtell DDL. Options available - from start to finish - for obtaining data from DNA microarrays. II. Nat Genet. 2002;32 Suppl:481–489. doi: 10.1038/ng1030. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Seidl T, Dugas M. Profound effect of normalization on detection of differentially expressed genes in oligonucleotide microarray data analysis. Genome Biol. 2002;3:research0033–0033.11. doi: 10.1186/gb-2002-3-7-research0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudoit S, Yang YH, Speed TP, Callow MJ. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Stat Sinica. 2002;12:111–139. [Google Scholar]
- Schuchhardt J, Beule D, Malik A, Wolski E, Eickhoff H, Lehrach H, Herzel H. Normalization strategies for cDNA microarrays. Nucleic Acids Res. 2000;28:e47. doi: 10.1093/nar/28.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed T. Normalization for cDNA microarray data: a robust composite method addressing single and multiple systematic variation. Nucleic Acid Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Henry G, Zeger S, Pevsner J. Local mean normalization of microarray element signal intensities across an array surface: quality control and correction of spatially systematic artifacts. Biotechniques. 2002;32:1316–1320. doi: 10.2144/02326mt02. [DOI] [PubMed] [Google Scholar]
- Finkelstein DB, Gollub J, Ewing R, Sterky F, Somerville S, Cherry J. Iterative linear regression by sector: renormalization of cDNA microarray data and cluster analysis weighted by cross homology. In: Lin SM, Johnson KF, editor. In Methods of Microarray Data Analysis. Boston: Kluwer Academic Publishers; 2001. http://www.stat.psu.edu/~chiaro/BioinfoII/NormMiss/finkelstein1.pdf [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002;32 Suppl:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler TB, Crosby L, Morgan KT. Normalization and analysis of DNA microarray data by self-consistency and local regression. Genome Biol. 2002;3:research0037–0037.12. doi: 10.1186/gb-2002-3-7-research0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland WS. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74:829–836. [Google Scholar]
- Loader C. Local Regression and Likelihood. New York: Springer; 1999. [Google Scholar]
- Craven P, Wahba G. Smoothing noisy data with spline functions. Numerische Math. 1979;31:377–403. [Google Scholar]
- Futschik M, Jeffs A, Pattison S, Kasabov N, Sullivan M, Merrie A, Reeve A. Gene expression profiling of metastatic and nonmetastatic colorectal cancer cell lines. Genome Lett. 2002;1:26–34. doi: 10.1166/gl.2002.005. [DOI] [Google Scholar]
- Leibovitz A, Stinson JC, McCombs WB, McCoy CE, Mazur KC, Mabry ND. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976;36:4562–4569. [PubMed] [Google Scholar]
- Callow MJ, Dudoit S, Gong EL, Speed TP, Rubin EM. Microarray expression profiling identifies genes with altered expression in HDL-deficient mice. Genome Res. 2000;10:2022–2029. doi: 10.1101/gr.10.12.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apo AI Data http://www.stat.berkeley.edu/users/terry/zarray/Html/apodata.html
- Sharov V, Kwong KY, Frank B, Chen E, Hasseman J, Gaspard R, Yu Y, Yang I, Quackenbush J. The limits of log-ratios. BMC Biotechnol. 2004;4:3. doi: 10.1186/1472-6750-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AM, Aach J, Steffen MA, Church GM. Measuring absolute expression with microarrays using a calibrated reference sample and an extended signal intensity range. Proc Natl Acad Sci USA. 2002;99:7554–7559. doi: 10.1073/pnas.112683499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcome to Bioconductor http://www.bioconductor.org
- Hewitt RE, Brown KE, Corcoaran M, Stetler-Stevenson WG. Increased expression of tissue inhibitor of metalloproteinases type I (TIMP-1) in a more tumourigenic colon cancer cell line. J Pathol. 2000;192:455–459. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH777>3.3.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Henriet P, Blavier L, Declerck YA. Tissue inhibitors of metalloproteinases (TIMP) in invasion and proliferation. APMIS. 1999;107:111–119. doi: 10.1111/j.1699-0463.1999.tb01533.x. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JCF, Trent JM, Staudt LM, Hudson J, Boguski MS, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- TM4: MIDAS http://www.tigr.org/software/tm4/midas.html
- SNOMAD - Standardization and NOrmalization of MicroArray Data http://pevsnerlab.kennedykrieger.org/snomad.htm [DOI] [PubMed]
- Iobion Informatics LLC | Products http://www.iobion.com/products/products.html
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Yuen T, Wurmbach E, Pfeffer RL, Ebersole BJ, Sealfon SC. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acid Res. 2002;30:e48. doi: 10.1093/nar/30.10.e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand MP, Jones MC. Kernel Smoothing. London: Chapman & Hall; 1995. [Google Scholar]
- Jones MC, Marron JS, Sheather SJ. A brief summary of bandwidth selection for density estimation. J Am Stat Assoc. 1996;91:401–407. [Google Scholar]
- Gu C. Model indexing and smoothing parameter selection in nonparametric regression (with discussion). Stat Sinica. 1998;8:607–646. [Google Scholar]
- Härdle W, Schimek MG, Eds . Statistical Theory and Computational Aspects of Smoothing. Heidelberg: Physica-Verlag; 1996. [Google Scholar]
- Loader CR. Bandwidth selection: classical or plug-in? Annls Stat. 1999;27:415–438. doi: 10.1214/aos/1018031200. [DOI] [Google Scholar]
- Fisher R. The Design of Experiments. Edinburgh: Oliver and Boyd; 1960. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- Serum stimulation database http://genome-www.stanford.edu/serum
- The R project for statistical computing http://www.r-project.org
- OLIN webpage http://itb.biologie.hu-berlin.de/~futschik/software/R/OLIN
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gigures and tables with more details of the data analysis presented and referred to in this paper
The implementation of the OLIN algorithm as add-on an R-package