

Abstract
Background
Myeloid cells play a central role in atherosclerosis. We investigated the associations between the plasma levels of growth factors and chemokines that regulate myeloid cell homeostasis and function and the risk of first‐time acute coronary events in middle‐aged persons.
Methods and Results
We measured baseline plasma levels of macrophage colony‐stimulating factor; monocyte chemotactic protein 1; C‐C motif chemokine ligands 3, 4, and 20; C‐X‐C motif chemokine ligands 1, 6, and 16; and C‐X3‐C motif chemokine ligand 1 in 292 participants who had a coronary event during follow‐up and 366 controls matched for age, sex, and time of inclusion who remained event free. Study participants were recruited from the Malmö Diet and Cancer Study population cohort and had no previous history of coronary artery disease. We found a strong independent negative association between macrophage colony‐stimulating factor and incident coronary events in a forward stepwise Cox proportional hazards model including all biomarkers alongside the classic Framingham risk factors (age, sex, smoking, total cholesterol, high‐density lipoprotein cholesterol, systolic blood pressure), diabetes mellitus, and medication. Conversely, monocyte chemotactic protein 1 had the strongest independent positive association with the outcome. The addition of macrophage colony‐stimulating factor and monocyte chemotactic protein 1 significantly improved the predictive ability of a model including traditional risk factors alone (C statistic 0.81 [95% CI 0.78–0.84] versus 0.67 [95% CI 0.63–0.71]; net reclassification index 0.52 [0.42–0.62]; P<0.001). The combined model led to a 54% net downclassification of participants who did not have a coronary event during follow‐up and was particularly effective in the intermediate‐risk group.
Conclusions
High levels of macrophage colony‐stimulating factor and low levels of monocyte chemotactic protein 1 in plasma characterize middle‐aged persons at low risk to develop clinically manifested coronary artery disease.
Keywords: coronary artery disease, inflammation, innate immunity, leukocytes, macrophage colony‐stimulating factor, monocyte chemotactic protein 1
Subject Categories: Biomarkers, Inflammation, Cardiovascular Disease, Risk Factors, Acute Coronary Syndromes
Introduction
Monocytes, macrophages, and neutrophils—myeloid effectors of innate immunity—play pivotal roles in atherosclerosis. Neutrophils have gained increasing interest in recent years, with the current consensus being that they have a proatherogenic role, especially in the initial stages of the disease.1, 2 Human monocytes can be divided into the “classical” CD14++CD16− and “nonclassical” CD14+CD16+ populations, which are considered to correspond to the “inflammatory” CCR2+CX3CR1−Ly‐6Chi and “steady state” CCR2−CX3CR1+Ly‐6Clo monocyte populations in mice.3 Classical monocytes predominate in human circulation and are preferentially recruited into inflammatory sites and atherosclerotic plaques compared with their nonclassical counterparts.3 We previously showed that blood counts of neutrophils and of the classical CD14++CD16− monocytes correlate with the incidence of acute cardiovascular events,4, 5 suggesting that factors governing myeloid cell homeostasis might play important roles in human cardiovascular disease.
Myeloid cell dynamics are tightly regulated through a complex network of growth factors, cytokines, and chemokines. In mice, macrophage colony‐stimulating factor (M‐CSF) promotes maturation of Ly‐6Chi into Ly‐6Clo monocytes,6 supports the survival of Ly‐6Clo monocyte and macrophages in blood and tissues,7 and promotes an anti‐inflammatory macrophage phenotype with important roles in tissue repair and homeostasis.8 In contrast, granulocyte and granulocyte‐macrophage colony‐stimulating factors induce proinflammatory activation of neutrophils, macrophages, dendritic cells, and eosinophils.8, 9 Under steady state conditions, only M‐CSF can be consistently measured in the circulation, whereas granulocyte and granulocyte‐macrophage colony‐stimulating factors increase and become measurable mainly during inflammatory reactions.9
Besides growth factors, a complex network of chemokines control myeloid cell production, trafficking, and function and have been found to be involved in the pathogenesis of cardiovascular disease.2, 10 Monocyte chemotactic protein 1 (MCP‐1; also known as C‐C motif chemokine ligand 2 [CCL2]) is the most important regulator of monocyte trafficking and has been shown to have potent proatherogenic properties.2, 11, 12 Similarly, fractalkine (also known as C‐X3‐C motif chemokine ligand 1 [CX3CL1]), macrophage inflammatory protein 1α (MIP‐1α; also known as CCL3), macrophage inflammatory protein 1β (MIP‐1β; also known as CCL4), CCL20, C‐X‐C motif chemokine ligand 1 (CXCL1) and CXCL16 promote monocyte and neutrophil recruitment and survival, supporting atherogenesis and plaque vulnerability in mice.2, 13, 14 The CXCR1 and CXCR2 ligand CXCL6 (granulocyte chemotactic protein 2) is also involved in neutrophil recruitment,15 but its role in cardiovascular disease has not been examined previously.
Much of the knowledge related to myeloid mediators and the roles they play in cardiovascular disease has been gained from animal studies, and human data are lacking to a large extent. The aim of our study was to identify whether plasma levels of M‐CSF, MCP‐1, CCL3, CCL4, CCL20, CXCL1, CXCL6, CXCL16, and CX3CL1 can predict the incidence of first‐time coronary events and improve the currently used models for cardiovascular risk prediction. To this end, we studied the correlations between baseline levels of these proteins and incident coronary events in plasma samples collected from 292 middle‐aged persons with no previous history of coronary artery disease who had an acute coronary event during follow‐up and 366 sex‐ and age‐matched controls who remained event free. Participants were recruited from the Malmö Diet and Cancer Study (MDC) cohort.
Materials and Methods
Study Cohort
The MDC has a population‐based prospective epidemiological cohort of 28 449 participants enrolled between 1991 and 1996.16 Between October 1991 and February 1994, every other participant was invited to take part in a substudy of the epidemiology of cardiovascular disease (MDC‐CV), yielding a cohort of 6103 participants.17 All participants were followed from the baseline examination until first hospitalization for an acute coronary event, death, emigration, or June 30, 2009. Incident cases of patients experiencing coronary events were retrieved by data linkage to the Swedish Hospital Discharge Register and the Cause of Death Registry of Sweden. Following prior exclusion of 102 participants with prevalent nonfatal myocardial infarction (1.7% of the MDC‐CV cohort), 402 incident coronary events (6.6% of the MDC‐CV cohort) were identified during the follow‐up period. A coronary event was defined as a nonfatal or fatal myocardial infarction on the basis of International Classification of Diseases, Ninth Revision (ICD9) code 410 and ICD10 code I21. Death due to ischemic heart disease was defined on the basis of codes 412 and 414 (ICD9) or I22, I23, and I25 (ICD10). We matched the incident coronary cases with 402 coronary event–free control participants of the same age, sex, and time of participation in the baseline examination (±6 months). These matching variables were selected because of their nonmodifiable nature. No paired analyses were used in the analysis of the data. We excluded 29 cases and 8 controls (4.6% of the case–control cohort) because of prevalent revascularization (percutaneous coronary intervention or coronary artery bypass grafting) or incident revascularization before the first coronary event in cases or before the end of the follow‐up period in controls. Furthermore, 81 cases and 28 controls (13.6% of the case–control cohort) were excluded because of incomplete clinical data or missing plasma samples, yielding a cohort consisting of 292 cases and 366 controls (81.8% of the case–control cohort). The study design and exclusion details are described in Figure 1. Although all participants were deemed to be apparently healthy at the time of inclusion, we cannot rule out the possibility that some participants might have had a potential history of chronic inflammatory conditions such as autoimmune disease, human immunodeficiency virus, cancer, or thrombosis. Because of unavailable information, we were unable to identify and exclude these participants from the study. The study was approved by the regional ethics review board and was conducted in accordance with the Declaration of Helsinki. All participants gave informed written consent.
Figure 1.
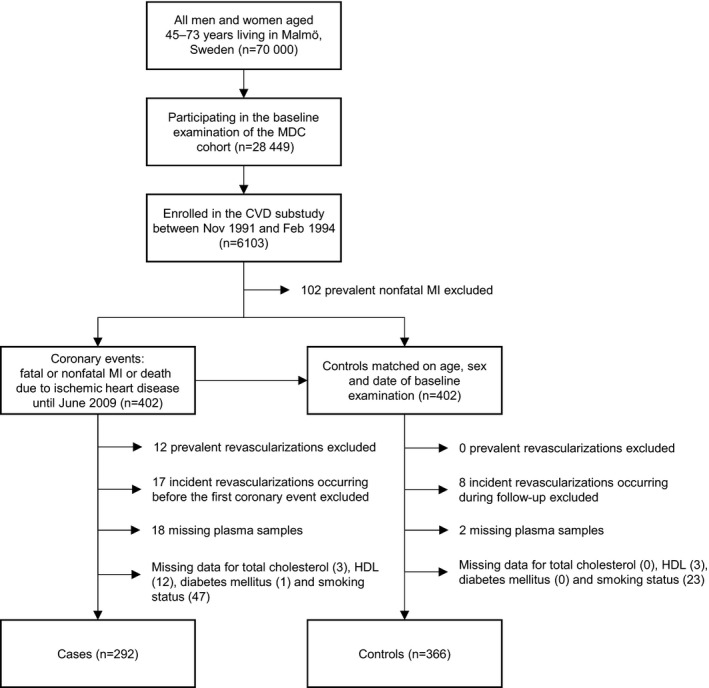
Study design. Diagram outlining how the matched sample of the case–control cohort was obtained. *Revascularization indicates coronary artery bypass grafting or percutaneous coronary intervention. CVD indicates cardiovascular disease; HDL, high‐density lipoprotein; MDC, Malmö Diet and Cancer Study; MI, myocardial infarction.
Baseline Assessment
Information on baseline characteristics was collected from self‐administered questionnaires and clinical examination. Smoking habits were categorized into never or former smokers (who quit smoking at least 1 year before the examination) and current smokers. Diabetes mellitus was defined as fasting whole blood glucose >6.1 mmol/L (corresponding to a threshold of 7.0 mmol/L in fasting plasma glucose), self‐reported physician diagnosis of diabetes mellitus, or use of antidiabetic medication. Blood pressure was measured twice in the right arm after a 10‐minute rest. The average of the 2 measurements was used. Hypertension was defined as systolic blood pressure ≥140 mm Hg or diastolic blood pressure ≥90 mm Hg or the use of blood pressure–lowering medication. Blood samples were drawn after overnight fasting. Fasting venous blood glucose, serum cholesterol, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, C‐reactive protein (CRP), and triglycerides were analyzed with standard methods at the clinical laboratory of Malmö University Hospital.
Analysis of Myeloid Markers in Plasma
Myeloid markers were analyzed in plasma by the proximity extension assay technique using the Proseek Multiplex CVD96x96 reagents kit (Olink Bioscience) at the Clinical Biomarkers Facility, Science for Life Laboratory, in Uppsala, Sweden. Briefly, oligonucleotide‐labeled antibody probe pairs were allowed to bind to their respective targets present in the plasma sample. Addition of DNA polymerase led to extension and joining of the 2 oligonucleotides and formation of a polymerase chain reaction template. Universal primers were used to preamplify the DNA templates in parallel. Finally, the individual DNA sequences were detected and quantified using specific primers in a microfluidic real‐time quantitative polymerase chain reaction chip (96.96, Dynamic Array integrated fluidic circuit, Fluidigm Biomark; Fluidigm Corp). The chip was run with a Biomark HD instrument.18 The respective intra‐ and interassay variations were 7% and 18% for MCP‐1, 7% and 12% for M‐CSF, 10% and 18% for CCL3, 8% and 12% for CCL4, 8% and 9% for CCL20, 6% and 13% for CXCL1, 8% and 12% for CXCL6, 10% and 14% for CXCL16, and 9% and 14% for CX3CL1. Data analysis was performed by a preprocessing normalization procedure using Olink Wizard for GenEx (Multid Analyses). All data are presented as arbitrary units. General calibrator curves to calculate the approximate concentrations are available on the Olink website (http://www.olink.com).
Statistics
Differences in baseline characteristics between the case and control groups were evaluated with Mann–Whitney nonparametric tests for nonnormally distributed continuous data and with Student t tests for normally distributed continuous data. Differences in categorical data were calculated with χ2 tests. Nonnormally distributed continuous data are presented as medians and interquartile ranges, and normally distributed continuous data are presented as mean±SD. Nonnormally distributed variables were natural logarithm transformed, and biomarker variables were standardized before inclusion into regression analyses.
Independent associations among myeloid biomarkers, baseline variables, and incident coronary artery disease were evaluated by calculating partial correlations between each pair of variables and constructing a partial correlation network that included correlations with a Bonferroni‐adjusted P<0.05. Calculations were performed in R v. 3.1.1 (R Foundation for Statistical Computing), and the partial correlation network was drawn using the yEd Graph Editor software v. 3.14 (yWorks GmbH), with a hierarchical layout algorithm.
Cox proportional hazards regression models were used to evaluate risk factor–adjusted hazard ratios and 95% CIs for each biomarker. We used 2 models for the Cox regression analyses. Model A included the traditional risk factors used in the Framingham risk score (age, sex, smoking, total cholesterol, high‐density lipoprotein cholesterol, and systolic blood pressure) as well as diabetes mellitus, blood pressure–lowering medication, and lipid‐lowering medication. Model B was additionally adjusted for potential confounders that differed significantly between cases and controls at baseline: diastolic blood pressure, body mass index, triglycerides, low‐density lipoprotein, creatinine‐based estimated glomerular filtration rate, CRP, and white blood cell count. The stepwise forward selection of variables also included MCP‐1, M‐CSF, CCL3, CCL4, CCL20, CXCL1, CXCL6, CXCL16, CX3CL1, and CRP in both models. P values were adjusted for multiple testing using the Bonferroni method. Plots of the hazard function in different groups over time did not indicate that the proportional hazards assumption was violated.
Metrics of risk discrimination were assessed using logistic regression analysis adjusted for matching variables to take the matched case–control design into account. To estimate the risk in the original population cohort, the risk model calculated from case–control data was adjusted for the incidence rate in the original cohort and for the case–control ratio as described by Huang et al19 and Pencina et al.20 Receiver operating curves and C statistics, net reclassification index, and integrated discrimination improvement were calculated and used to compare the performance of the model based on traditional risk factors with models including traditional risk factors and biomarkers for the prediction of incident coronary events. To be applicable to the case–control design, net reclassification index calculations were extended, as suggested by Pencina et al.20 Statistical differences in the areas under the receiver operating characteristic curves were calculated according to DeLong et al.21
Statistical analyses were carried out using IBM SPSS Statistics v. 22 (IBM Corp) and R v. 3.1.1 (and the R packages pROC, PredictABEL, Rcmdr, and qgraph).
Results
The clinical characteristics of the study cohort at baseline are summarized in Table 1. The case group contained a larger percentage of participants who were smokers, were overweight (body mass index ≥25), were diabetic, and had hypertension compared with the sex‐ and age‐matched control group. Cases also had higher total cholesterol, triglycerides, low‐density lipoprotein cholesterol, CRP, and white blood cell counts, whereas high‐density lipoprotein cholesterol levels were lower than in controls. A higher percentage of participants within the case group received antidiabetic medication at baseline. The median time from baseline to the occurrence of a coronary event was 10.6 years (interquartile range 6.4–13.5 years).
Table 1.
Baseline Characteristics of the Case–Control Cohort
| Cases (n=292)a | Controls (n=366)a | P Valueb | |
|---|---|---|---|
| Age at screening, y | 61.8 (57.3–64.9) | 61.4 (57.0–64.3) | 0.360 |
| Male sex (% male) | 172 (58.9%) | 217 (59.3%) | 0.920 |
| BMI ≥25 (%)c | 176 (60.5%) | 194 (53.2%) | 0.060 |
| Current smoker (%) | 102 (34.9%) | 93 (25.4%) | 0.008 |
| Diabetes mellitus (%)d | 49 (16.8%) | 27 (7.4%) | 1.8×10−4 |
| Hypertension (%)e | 229 (78.4%) | 249 (68.0%) | 0.003 |
| Medication | |||
| Antidiabetic (%) | 21 (7.2%) | 5 (1.4%) | 1.4×10−4 |
| Lipid‐lowering (%) | 3 (1.0%) | 5 (1.4%) | 0.694 |
| Blood pressure‐lowering (%) | 57 (19.5%) | 59 (16.1%) | 0.255 |
| Clinical parameters | |||
| Systolic blood pressure, mm Hg | 150±19 | 143±18 | 5×10−6 |
| Diastolic blood pressure, mm Hg | 91±10 | 88±9 | 2.8×10−4 |
| Cholesterol, mmol/L | 6.3±1.0 | 6.2±1.1 | 0.050 |
| Triglycerides, mmol/L | 1.3 (1.0–1.9) | 1.2 (0.9–1.7) | 0.037 |
| HDL, mmol/L | 1.2±0.3 | 1.4±0.4 | 8×10−6 |
| LDL, mmol/L | 4.4±1.0 | 4.2±1.0 | 0.005 |
| CRP, mg/L | 1.9 (0.9–3.4) | 1.3 (0.7–2.6) | 6.1×10−6 |
| White blood cell count, ×109/L | 6.4 (5.3–7.4) | 5.7 (5.0–6.8) | 7.7×10−6 |
| eGFR, mL/min/1.73 m2 | 76±15 | 75±16 | 0.894 |
| Myeloid markers | |||
| M‐CSF, au | 530 (440–600) | 580 (490–680) | 1.3×10−7 |
| MCP‐1, au | 22 (17–29) | 18 (15–22) | 9.9×10−5 |
| CXCL1, au | 71 (56–94) | 82 (64–114) | 1.1×10−14 |
| CCL4, au | 125 (96–171) | 105 (82–146) | 8.0×10−6 |
| CCL3, au | 3.2 (2.9–4.1) | 3.2 (2.8–3.9) | 0.939 |
| CXCL6, au | 119 (89–157) | 122 (94–167) | 0.308 |
| CXCL16, au | 13 (11–16) | 12 (10–16) | 0.007 |
| CX3CL1, au | 45 (37–58) | 50 (40–62) | 0.003 |
| CCL20, au | 52 (39–78) | 51 (36–74) | 0.453 |
au indicates arbitrary units; BMI, body mass index; CCL, C‐C motif chemokine ligand; CRP, C‐reactive protein; CXCL, C‐X‐C motif chemokine ligand; CX3CL1, C‐X3‐C motif chemokine ligand 1; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MCP‐1, monocyte chemotactic protein 1; M‐CSF, macrophage colony‐stimulating factor.
Data are presented as number (percentage of cases/controls) for categorical data, mean±SD for normally distributed continuous variables, and median (interquartile range) for nonnormally distributed variables.
Mann–Whitney test was used for nonnormally distributed data, t test was used for normally distributed data, and χ2 test was used for categorical data.
BMI was calculated as weight/height2 (kg/m2) and categorized as normal weight (BMI <25) and overweight/obese (BMI ≥25).
Positive questionnaire, medication, or glucose ≥6.1 (mmol/L).
Blood pressure ≥140/90 mm Hg or treatment.
Myeloid Markers in Plasma and Incidence of First‐Time Acute Coronary Events
To concomitantly explore the independent associations among myeloid biomarkers, baseline clinical variables, and incident coronary artery disease, partial correlations were calculated between each pair of variables, controlling for all of the others, and a partial correlation network was constructed (Figure 2). M‐CSF and CXCL1 showed independent negative associations with the case–control variable, whereas MCP‐1 and CCL4 displayed positive associations (Bonferroni‐adjusted P<0.05).
Figure 2.
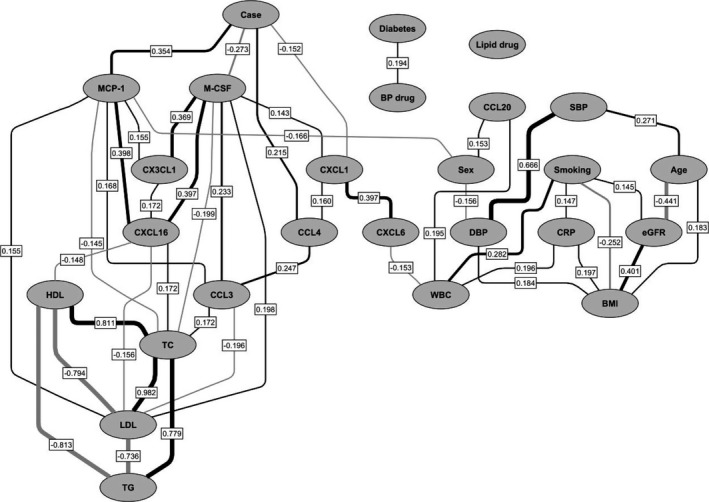
Partial correlation network of the relationships among clinical variables, myeloid biomarkers, and incident first‐time coronary events in the study group. Positive partial correlations are depicted in black, and negative partial correlations are shown in gray. Line thickness is proportional to the strength of the correlation. Only partial Pearson correlations with Bonferroni‐adjusted P values <0.05 are shown, with the partial correlation coefficients reported in boxes on the lines indicating correlations. Positive correlations for categorical variables indicate higher numbers of cases, patients with diabetes mellitus, current smokers, and women. BMI indicates body mass index; BP, blood pressure; CCL, C‐C motif chemokine ligand; CRP, C‐reactive protein; CXCL, C‐X‐C motif chemokine ligand; CX3CL1, C‐X3‐C motif chemokine ligand 1; DBP, diastolic blood pressure; DM, diabetes mellitus; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; MCP‐1, monocyte chemotactic protein 1; M‐CSF, macrophage colony‐stimulating factor; SBP, systolic blood pressure; TC, total cholesterol; TG, triglycerides; WBC, white blood cells.
When the 9 biomarkers were tested individually, M‐CSF, MCP‐1, and CXCL1 were significantly associated with incident coronary events in Cox proportional hazard models adjusted for the traditional cardiovascular risk factors included in the Framingham risk score (age, sex, smoking, total cholesterol, high‐density lipoprotein, systolic blood pressure) and for blood pressure–lowering medication, lipid‐lowering medication, and diabetes mellitus (model A) (Table 2). The associations remained significant following further adjustment for potential confounders that differed between cases and controls at baseline: diastolic blood pressure, body mass index, triglycerides, low‐density lipoprotein, estimated glomerular filtration rate, CRP, and white blood cell count (model B) (Table 2). The relationships between biomarkers and outcome were linear, as demonstrated by gradually increasing hazard ratios for each increasing MCP‐1 quartile and gradually decreasing hazard ratios for the M‐CSF and CXCL1 quartiles (P for linear trend <0.001) (Table 2).
Table 2.
Correlations Between Baseline Biomarker Levels and Incident Coronary Events During Follow‐up
| Biomarker | HRa | 95% CI | Nominal P Value | Corrected P Valueb | HR vs Q1 (95% CI) | Linear Trend P Value | Corrected P Valueb | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | |||||||
| MCP‐1 | ||||||||||
| Model Ac | 1.50 | 1.33–1.69 | 2.8×10−11 | 2.8×10−10 | 1 | 1.14 (0.78–1.65) | 1.57 (1.10–2.26) | 2.26 (1.62–3.16) | 1.1×10−7 | 1.1×10−6 |
| Model Bd | 1.49 | 1.32–1.69 | 2.2×10−10 | 3.7×10−9 | 1 | 1.12 (0.76–1.63) | 1.56 (1.08–2.27) | 2.22 (1.56–3.14) | 5.4×10−7 | 9.1×10−6 |
| M‐CSF | ||||||||||
| Model A | 0.67 | 0.59–0.76 | 2.5×10−10 | 2.5×10−9 | 1 | 0.61 (0.45–0.83) | 0.51 (0.37–0.69) | 0.31 (0.21–0.44) | 4.8×10−11 | 4.8×10−10 |
| Model B | 0.64 | 0.56–0.73 | 2.9×10−11 | 5.0×10−10 | 1 | 0.56 (0.41–0.78) | 0.46 (0.34–0.64) | 0.27 (0.19–0.40) | 8.9×10−12 | 1.5×10−10 |
| CXCL1 | ||||||||||
| Model A | 0.76 | 0.68–0.86 | 1.1×10−5 | 1.1×10−4 | 1 | 0.83 (0.62–1.13) | 0.64 (0.46–0.90) | 0.44 (0.31–0.62) | 1.2×10−6 | 1.2×10−5 |
| Model B | 0.76 | 0.67–0.86 | 1.5×10−5 | 2.6×10−4 | 1 | 0.84 (0.62–1.15) | 0.61 (0.43–0.86) | 0.44 (0.30–0.63) | 2×10−6 | 3.4×10−5 |
| CX3CL1 | ||||||||||
| Model A | 0.84 | 0.75–0.95 | 0.005 | 0.053 | 1 | 0.80 (0.58–1.09) | 0.69 (0.50–0.95) | 0.61 (0.44–0.84) | 0.002 | 0.016 |
| Model B | 0.83 | 0.73–0.94 | 0.004 | 0.063 | 1 | 0.74 (0.54–1.03) | 0.66 (0.48–0.92) | 0.58 (0.41–0.82) | 0.001 | 0.020 |
| CCL4 | ||||||||||
| Model A | 1.13 | 1.01–1.27 | 0.030 | 0.301 | 1 | 1.71 (1.19–2.45) | 1.77 (1.23–2.55) | 1.75 (1.21–2.53) | 0.006 | 0.060 |
| Model B | 1.14 | 1.01–1.28 | 0.028 | 0.476 | 1 | 1.65 (1.14–2.39) | 1.80 (1.24–2.62) | 1.74 (1.19–2.53 | 0.007 | 0.111 |
| CCL3 | ||||||||||
| Model A | 0.96 | 0.83–1.11 | 0.581 | 1 | 1 | 1.22 (0.84–1.78) | 0.65 (0.43–0.98) | 1.00 (0.68–1.49) | 0.331 | 1 |
| Model B | 0.94 | 0.80–1.11 | 0.474 | 1 | 1 | 1.17 (0.78–1.73) | 0.61 (0.39–0.94) | 0.93 (0.61–1.41) | 0.184 | 1 |
| CCL20 | ||||||||||
| Model A | 0.91 | 0.80–1.03 | 0.140 | 1 | 1 | 1.15 (0.82–1.60) | 0.87 (0.62–1.23) | 0.85 (0.60–1.21) | 0.158 | 1 |
| Model B | 0.91 | 0.80–1.04 | 0.165 | 1 | 1 | 1.17 (0.84–1.64) | 0.87 (0.61–1.24) | 0.88 (0.61–1.26) | 0.232 | 1 |
| CXCL6 | ||||||||||
| Model A | 0.93 | 0.83–1.05 | 0.244 | 1 | 1 | 0.88 (0.64–1.21) | 0.99 (0.72–1.37) | 0.77 (0.55–1.07) | 0.204 | 1 |
| Model B | 0.94 | 0.83–1.06 | 0.321 | 1 | 1 | 0.87 (0.63–1.22) | 0.99 (0.71–1.40) | 0.80 (0.56–1.13) | 0.328 | 1 |
| CXCL16 | ||||||||||
| Model A | 1.12 | 0.99–1.26 | 0.068 | 0.683 | 1 | 1.15 (0.82–1.62) | 1.05 (0.75–1.48) | 1.09 (0.77–1.53) | 0.789 | 1 |
| Model B | 1.10 | 0.97–1.25 | 0.144 | 1 | 1 | 1.14 (0.81–1.62) | 0.97 (0.68–1.39) | 1.04 (0.73–1.47) | 0.921 | 1 |
Number of participants included in the analysis: 292 cases and 366 controls. CCL indicates C‐C motif chemokine ligand; CXCL, C‐X‐C motif chemokine ligand; CX3CL1, C‐X3‐C motif chemokine ligand 1; HDL, high‐density lipoprotein; HR, hazard ratio; MCP‐1, monocyte chemotactic protein 1; M‐CSF, macrophage colony‐stimulating factor; Q, quartile.
Per 1‐SD increase in the respective variable.
After Bonferroni correction for multiple testing. Model A: 10 tests, critical cutoff P=0.005. Model B: 17 tests, critical cutoff P=0.003.
Model A: Cox regression analysis adjusted for age, sex, diabetes mellitus, current smoking, total cholesterol, HDL, systolic blood pressure, blood pressure–lowering medication, and lipid‐lowering medication.
Model B: Cox regression analysis adjusted for age, sex, diabetes, current smoking, total cholesterol, HDL, systolic blood pressure, blood pressure–lowering medication, lipid‐lowering medication, diastolic blood pressure, body mass index, triglycerides, low‐density lipoprotein, estimated glomerular filtration rate, C‐reactive protein, and white blood cell count.
Next, we included all biomarkers alongside traditional risk factors, diabetes mellitus, CRP, and medication in a Cox forward stepwise regression model to identify the strongest independent relationships between potential predictors and incident coronary events. Only the correlations for MCP‐1, M‐CSF, CRP, CXCL1, and CCL4 remained statistically significant in the final model when a Bonferroni correction for multiple testing was applied (model A) (Table 3). The distinctively strongest predictors of coronary events were M‐CSF (hazard ratio 0.49 [95% CI 0.41–0.6]; P=3.5×10−13) and MCP‐1 (hazard ratio 2.06 [95% CI 1.75–2.43]; P=5.1×10−18), confirming the results of the partial correlation network analysis. The Cox forward stepwise regression model including all potential confounders yielded similar results (model B) (Table 3). Among all considered biomarkers, MCP‐1 and M‐CSF remained the strongest predictors of coronary events when men, women, diabetic and nondiabetic participants, and those above and below the median age were analyzed separately (data not shown). Notably, the incidence of acute coronary events was negatively associated with M‐CSF and positively associated with MCP‐1, indicating that these 2 biomarkers might have complementary value as predictors of incident coronary events. To illustrate this relationship, we divided the patients into high and low groups based on median plasma levels of M‐CSF and MCP‐1 and created Kaplan–Meier survival curves for the combinations of high and low biomarker levels (Figure 3). We found that participants with high M‐CSF and low MCP‐1 had a markedly improved coronary event‐free survival compared with the rest of the cohort (log‐rank P for trend <0.001).
Table 3.
Independent Predictors of Incident Coronary Events in the Cohort
| Step | Model A | Model B | ||||||
|---|---|---|---|---|---|---|---|---|
| Variable | HR (95% CI)a | Nominal P Value | Corrected P Valueb | Variable | HR (95% CI)a | Nominal P Value | Corrected P Valueb | |
| 1 | MCP‐1 | 2.06 (1.75–2.43) | 5.1×10−18 | 9.8×10−17 | MCP‐1 | 1.93 (1.59–2.34) | 3.7×10−11 | 9.4×10−10 |
| 2 | M‐CSF | 0.49 (0.41–0.6) | 3.5×10−13 | 6.7×10−12 | M‐CSF | 0.41 (0.33–0.52) | 8.2×10−14 | 2.1×10−12 |
| 3 | CRP | 1.38 (1.19–1.6) | 2.9×10−5 | 5.5×10−4 | CRP | 1.33 (1.14–1.55) | 3.5×10−4 | 0.009 |
| 4 | CXCL1 | 0.73 (0.63–0.86) | 1.1×10−5 | 0.002 | CXCL1 | 0.76 (0.65–0.9) | 0.001 | 0.025 |
| 5 | CCL4 | 1.29 (1.11–1.49) | 7.0×10−4 | 0.013 | CCL4 | 1.3 (1.12–1.52) | 7.0×10−4 | 0.018 |
Final model of a Cox proportional hazards regression analysis with stepwise forward selection of variables. Only variables with an adjusted P value <0.05 after Bonferroni correction are shown. Number of participants included in the analysis: 292 cases and 366 controls. Variables not retained in the final model A: age, sex, diabetes, current smoking, systolic blood pressure, total cholesterol, HDL, blood pressure–lowering medication, lipid‐lowering medication, CCL3, CCL20, CX3CL1, CXCL6, and CXCL16. Variables not retained in the final model B: age, sex, diabetes mellitus, current smoking, systolic blood pressure, diastolic blood pressure, total cholesterol, blood pressure–lowering medication, lipid‐lowering medication, body mass index, low‐density lipoprotein, HDL, triglycerides, estimated glomerular filtration rate, white blood cells, CCL3, CCL20, CX3CL1, CXCL6, and CXCL16. CCL indicates C‐C motif chemokine ligand; CRP, C‐reactive protein; CXCL, C‐X‐C motif chemokine ligand; CX3CL1, C‐X3‐C motif chemokine ligand 1; HDL, high‐density lipoprotein; MCP‐1, monocyte chemotactic protein‐1; M‐CSF, macrophage colony‐stimulating factor.
Per 1‐SD increase in the respective variable.
After Bonferroni correction for multiple testing. Model A: 19 tests, critical cutoff P=0.003. Model B: 25 tests, critical cutoff P=0.002.
Figure 3.
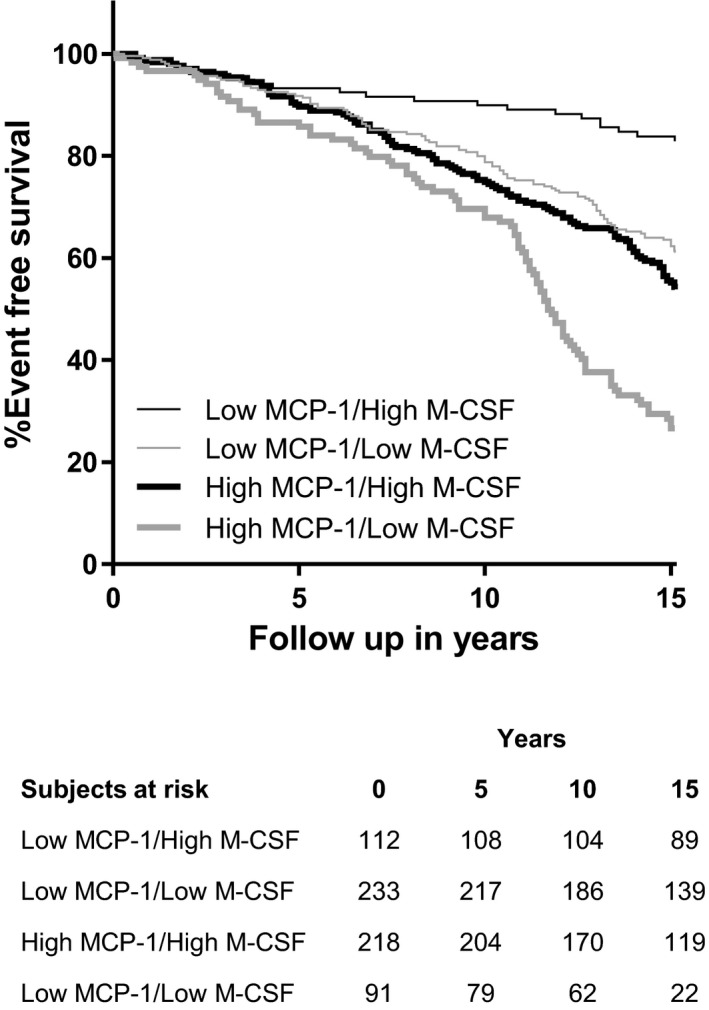
Kaplan–Meier curves illustrating coronary event‐free survival of participants with high or low plasma levels of MCP‐1 and M‐CSF. Thin lines indicate low MCP‐1 levels (below median), and thick lines indicate high MCP‐1, whereas gray lines indicate low M‐CSF and black lines indicate high M‐CSF. The x‐axis was curtailed when <10% of participants remained in follow‐up. MCP‐1 indicates monocyte chemotactic protein 1; M‐CSF indicates macrophage colony‐stimulating factor.
M‐CSF and MCP‐1 Improve the Discriminative Ability of a Traditional Risk Factor Model for Coronary Artery Disease
To test whether M‐CSF, MCP‐1, CXCL1, or CCL4 could improve the predictive value of the traditional risk factors for first‐time coronary events, receiver operating characteristic curves were constructed for binary logistic regression models including traditional risk factors (age, sex, smoking, total cholesterol, high‐density lipoprotein, systolic blood pressure, and diabetes mellitus), blood pressure–lowering medication, and lipid‐lowering medication, as well as M‐CSF, MCP‐1, CXCL1, CCL4, and CRP alone or in combination (Table 4 and Figure 4). The addition of M‐CSF or MCP‐1 alone significantly improved the discriminative ability of the traditional risk factor model for coronary artery disease risk, whereas addition of CXCL1, CCL4, or CRP alone did not improve discrimination. The addition of MCP‐1 to the model already including M‐CSF and traditional risk factors further improved discrimination (P=1.1×10−10). The model including M‐CSF and MCP‐1 alongside traditional risk factors had a significantly improved C statistic compared with traditional risk factors alone (0.81 [95% CI 0.78–0.85] versus 0.67 [95% CI 0.62–0.70]; P=6.6×10−13).
Table 4.
Comparison of Risk Prediction Models for Acute Coronary Events
| Model | C Statistic | 95% CI | P Valuea | Continuous NRI | 95% CI | P Value |
|---|---|---|---|---|---|---|
| Traditional risk factors | 0.67 | 0.63–0.71 | — | — | — | — |
| +MCP‐1 | 0.73 | 0.69–0.77 | 1.5×10−4 | 0.52 | 0.37–0.66 | <1×10−5 |
| +M‐CSF | 0.70 | 0.66–0.74 | 0.027 | 0.34 | 0.19–0.49 | 1×10−5 |
| +CRP | 0.67 | 0.63–0.72 | 0.268 | 0.16 | 0.006–0.31 | 0.042 |
| +CXCL1 | 0.69 | 0.64–0.73 | 0.069 | 0.25 | 0.09–0.40 | 0.001 |
| +CCL4 | 0.68 | 0.64–0.72 | 0.144 | 0.18 | 0.03–0.33 | 0.021 |
| +M‐CSF +MCP‐1 | 0.81 | 0.78–0.84 | 6.6×10−13 | 0.86 | 0.72–0.99 | <1×10−5 |
Comparative C statistic analysis of different predictive models for incident coronary events, based on traditional risk factors (age, sex, current smoking, total cholesterol, high‐density lipoprotein cholesterol, systolic blood pressure, blood pressure–lowering medication, lipid‐lowering medication, and presence of diabetes), with and without the addition of biomarkers. Number of participants included in the analysis: 292 cases and 366 controls. CCL indicates C‐C motif chemokine ligand; CRP indicates C‐reactive protein; CXCL, C‐X‐C motif chemokine ligand; MCP‐1, monocyte chemotactic protein 1; M‐CSF, macrophage colony‐stimulating factor; NRI, net reclassification index.
P value vs the model including traditional risk factors alone.
Figure 4.
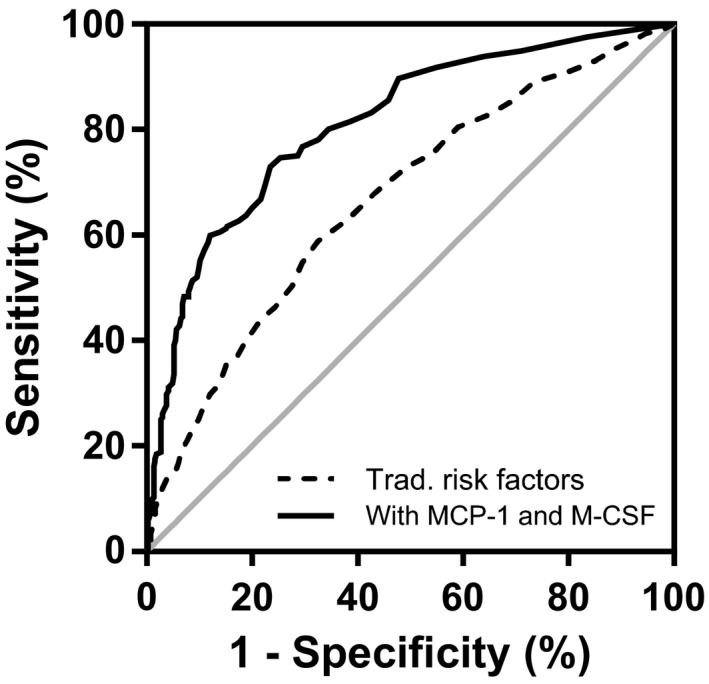
Receiver operating characteristic curves of binary logistic regression models for acute coronary event risk discrimination. The broken line represents the model including traditional risk factors (age, sex, total cholesterol, high‐density lipoprotein, systolic blood pressure, smoking, diabetes mellitus) as well as blood pressure–lowering medication and lipid‐lowering medication. The continuous line represents the model with traditional risk factors, medication, MCP‐1, and M‐CSF. MCP‐1 indicates monocyte chemotactic protein 1; M‐CSF indicates macrophage colony‐stimulating factor.
We also performed reclassification analyses for 3 predefined risk categories for incident coronary events: <10% (low risk), 10% to 20% (moderate risk), and >20% (high risk) (Table 5). The addition of M‐CSF and MCP‐1 significantly improved the predictive value of the traditional risk factor model, with a net reclassification index of 0.52 (95% CI 0.42–0.62; P<0.001) and integrated discrimination improvement of 0.18 (95% CI 0.15–0.21; P<0.001). Of 86 participants with low coronary event risk according to the traditional model, 8 (9.3%) were correctly upclassified and 6 (7%) were incorrectly upclassified after the addition of MCP‐1 and M‐CSF. Among participants in the intermediate‐risk group, 146 were correctly downclassified and 61 were correctly upclassified (total correct reclassification 207 of 292, 71%) compared with 26 incorrectly downclassified and 15 incorrectly upclassified (total incorrect reclassification 41 of 292, 14%). Within the high‐risk group, we recorded 73 of 280 (26%) with correct downclassification compared with 50 of 280 (18%) with incorrect downclassification. Taken together, these results suggest that addition of M‐CSF and MCP‐1 to the traditional cardiovascular risk factors for prediction of first‐time coronary events is useful mainly in the intermediate‐risk population, leading to correct net total reclassification of 166 of 292 (57%) participants in this category. Analysis of reclassification in cases and controls revealed that the addition of M‐CSF and MCP‐1 was particularly effective in the control group, leading to a net downclassification of 54% of participants who did not have a coronary event during follow‐up. In the coronary case group, the combined model led to a minimal net downclassification of 2% of participants (Table 5).
Table 5.
Reclassification of Study Participants Between Risk Categories for Incident Coronary Events After Addition of MCP‐1 and M‐CSF to a Traditional Risk Factor Model
| Without Biomarkers | With MCP‐1 and M‐CSF | Percentage Reclassified | |||||
|---|---|---|---|---|---|---|---|
| <10% | 10–20% | >20% | Total | Total Up | Total Down | Net | |
| Participants with coronary events | |||||||
| <10% | 15 (65%) | 3 (13%) | 5 (22%) | 23 | — | — | — |
| 10–20% | 26 (25%) | 17 (16%) | 61 (58%) | 104 | — | — | — |
| >20% | 20 (12%) | 30 (18%) | 115 (70%) | 165 | — | — | — |
| Total | 61 | 50 | 181 | 292 | 24% (69/292) | 26% (76/292) | 2% down |
| Participants without coronary events | |||||||
| <10% | 57 (90%) | 5 (8%) | 1 (2%) | 63 | — | — | — |
| 10–20% | 146 (78%) | 27 (14%) | 15 (8%) | 188 | — | — | — |
| >20% | 40 (35%) | 33 (29%) | 42 (37%) | 115 | — | — | — |
| Total | 243 | 65 | 58 | 366 | 6% (21/366) | 60% (219/366) | 54% down |
The number of participants and row percentage are shown. The model without biomarkers included age, sex, current smoking, total cholesterol, high‐density lipoprotein cholesterol, systolic blood. Net reclassification index was 0.52 (95% CI 0.42–0.62; P<1×10−5) and integrated discrimination improvement was 0.18 (95% CI 0.15–0.21; P<1×10−5) after addition of MCP‐1 and M‐CSF. MCP‐1 indicates monocyte chemotactic protein‐1; M‐CSF, macrophage colony‐stimulating factor.
Discussion
We are the first to compare the ability of several mediators related to myeloid cell homeostasis and function (M‐CSF, MCP‐1, CCL3, CCL4, CCL20, CXCL1, CXCL6, CXCL16, and CX3CL1) to predict first‐time acute coronary events in middle‐aged persons with no previous history of coronary artery disease. We identified, for the first time, a strong independent negative association between M‐CSF and coronary artery disease risk and confirmed the previously described positive relationship between MCP‐1 and coronary events in this population.22, 23 Participants at low risk of having acute coronary events were characterized by high levels of M‐CSF and low levels of MCP‐1 in plasma. Combining M‐CSF and MCP‐1 with the classic cardiovascular risk factors included in the Framingham risk score improved prediction accuracy in the intermediate‐risk group and led to correct net downclassification of approximately half of the participants that did not have a coronary event during follow‐up. Nevertheless, the model did not improve identification of participants who subsequently developed clinically manifested coronary artery disease.
M‐CSF and Coronary Artery Disease Risk
To our knowledge, this study is the first on the potential role of plasma M‐CSF as a biomarker of cardiovascular risk in persons with no clinical evidence of coronary artery disease. Interestingly, the negative correlation between the circulating levels of this myeloid growth factor and the incidence of coronary events was independent of traditional cardiovascular risk factors, CRP, and other myeloid mediators, including MCP‐1. Our findings raise the important question of whether M‐CSF plays a protective role in the pathogenesis of atherosclerotic plaque development and rupture. Interesting data recently published by Swirski and colleagues revealed that macrophage numbers in established atheroma are maintained mainly through local proliferation rather than constant replacement by recruited monocytes, whereas the development of early lesions relies on monocyte recruitment.24 Similar findings have been reported in other tissues, and M‐CSF has been identified to play an important role in both monocyte recruitment and macrophage proliferation.7, 25, 26 Macrophages generated in vitro in response to M‐CSF are being referred to as “M‐Mac” and are similar but not identical to M2 polarized macrophages.27 It has been proposed that M‐Mac/M2‐like macrophages represent the normal state of tissue macrophages and are hyporesponsive to inflammatory stimuli.9 M1 macrophages and their secreted mediators MCP‐1, tumor necrosis factor α, interleukin 1β, and interleukin 12 promote inflammation within the atherosclerotic plaques,12 whereas M2 macrophages and their signature cytokines interleukin 10 and transforming growth factor β inhibit inflammation, promote neovascularization and tissue repair, confer atheroprotection, and may be involved in plaque regression.12, 28, 29
Early studies have demonstrated immunosuppressive properties of M‐CSF in vitro and in vivo.30 M‐CSF or M‐CSF receptor blockade in mouse models of breast cancer led to the expansion of circulating neutrophils and inflammatory Ly‐6Chi monocytes, promoting enhanced metastasis.31 Conversely, M‐CSF administration before allogeneic hematopoietic cell transplantation in mice expanded the host macrophage pool, reduced donor T‐cell activation, and significantly inhibited the development of graft‐versus‐host disease.32 M‐CSF plays an important role in all stages of mononuclear phagocyte homeostasis, from bone‐marrow production to recruitment, proliferation, and survival in the tissues.33 Perhaps unsurprisingly, because monocytes and macrophages play a central role in atherogenesis, impaired monocyte/macrophage function and survival in M‐CSF–deficient Csf1 op/op mice and in hyperlipidemic mice treated with an M‐CSF receptor inhibitor impair monocyte recruitment and delay atherosclerosis development.34, 35 M‐CSF receptor blockade, however, had no effects on more advanced atherosclerotic plaques,36 and treatment with recombinant human M‐CSF suppressed atherogenesis in hyperlipidemic rabbits due to enhanced lipoprotein clearance and cholesterol metabolism.37, 38 Importantly, M‐CSF deficiency in LDLR−/−Csf1+/− mice is associated with increased macrophage apoptosis within the advanced atherosclerotic lesions, which could potentially trigger plaque instability and rupture.34
The ability of M‐CSF to predict incident coronary events has been investigated previously in patients with already established, clinically manifested coronary artery disease. Consecutive papers by Rallidis et al identified high plasma M‐CSF at hospital admission as a positive predictor of in‐hospital adverse events39 and high M‐CSF measured at the 6‐week follow‐up time point as a predictor of long‐term negative prognosis in patients with severe unstable angina.40 These results are supported by similar findings by Saitoh et al in a mixed cohort of 142 patients admitted with stable or unstable angina41 and by Ikonomidis et al in 100 stable angina patients.42 Notably, patients with acute or prevalent myocardial infarction or revascularization procedures were excluded from these studies, and the incident event rate during follow‐up was driven to a large extent by recurrent angina. In contrast, we reported a negative relationship between circulating M‐CSF and the incidence of hard coronary events, represented by myocardial infarction and death due to ischemic heart disease, in a medium‐sized cohort of persons with no previous history of coronary artery disease. The only other study to date to assess the predictive value of M‐CSF for first‐time cardiovascular events revealed a positive relationship between plasma M‐CSF and the incidence of stroke in a cohort of participants aged 70 years43; however, the relationship could not be confirmed after adjustment for traditional cardiovascular risk factors in the validation cohort consisting of men aged 77 years.43 Taken together with the conflicting experimental data, the results of the clinical studies performed so far, including ours, suggest that the role of M‐CSF in cardiovascular disease is complex and is probably dependent on the stage of the disease. If studies in other cohorts confirm our results, it is tempting to speculate that in healthy persons, M‐CSF might promote survival of the homeostatic anti‐inflammatory M2 macrophages, maintaining a stable plaque phenotype; however, these mechanisms could be disturbed by concurrent factors in patients with advanced atherosclerotic disease, leading to plaque destabilization. Whether M‐CSF plays a pathogenic role in this context or whether the elevated M‐CSF levels in these patients are the expression of an unsuccessful attempt to restore tissue homeostasis and plaque stability is unclear. Interestingly, previous experimental data demonstrating that M‐CSF treatment accelerates the healing process after myocardial infarction support the latter hypothesis.44, 45
MCP‐1 and Coronary Artery Disease Risk
MCP‐1 (also known as CCL2) mediates Ly‐6Chi monocyte release from the bone marrow and recruitment of neutrophils and Ly‐6Chi monocytes into inflammatory sites.2 MCP‐1 is upregulated in murine and human atherosclerotic lesions and has been shown to have a potent proatherogenic role in mice.11, 12, 46 In humans, polymorphisms in the MCP‐1 promoter are associated with increased plasma MCP‐1 and with a history of coronary events, suggesting active involvement of the chemokine in the pathology of coronary artery disease.47 In cross‐sectional studies of large patient cohorts, plasma MCP‐1 was directly correlated with the presence of peripheral artery disease23 and with the severity of coronary artery disease measured as coronary calcium score.48 Two independent prospective studies previously demonstrated that increasing plasma MCP‐1 concentrations are associated with elevated risk for first‐time acute coronary events.22, 23 In the work by Herder et al, the relationship between plasma MCP‐1 and incident coronary heart disease narrowly lost statistical significance after adjustment for age, sex, and other cardiovascular risk factors.22 Compared with our study, the authors used a case–cohort design based on a significantly younger population with a shorter follow‐up time and curtailed incident coronary event follow‐up at age 75 years,22 which can explain the difference in the results between the studies.
Study Limitations
Our study has some limitations that preclude direct translation of the data into clinical practice. The primary purpose of the study was to compare the value of several myeloid cell–related mediators as biomarkers with regard to incident coronary events. Consequently, we used a method able to measure a large number of parameters in the same plasma samples at the same time. The results of the analysis are expressed as relative arbitrary units and cannot be extrapolated directly to absolute plasma concentrations. Subsequently, we cannot propose a cutoff value for either biomarker that could be used in the clinic. Moreover, our study cohort was originally designed as a case‐control sample matched for sex, age, and time of inclusion; therefore, interpretation of the results with regard to unselected populations has to be done with due caution. To address this issue, we statistically compensated our calculations using the method proposed by Pencina et al,20 by taking into account event frequency in our cohort compared with event frequency among the 6103 participants in the MDC‐CV cohort, from which our cohort was selected. Last, matching cases and controls on age and sex may have lessened the influence of these strong risk predictors in the model based on traditional risk factors alone, and the C statistic of this model may have been underestimated compared with other studies. It is also possible that incremental improvements in risk prediction with the addition of MCP‐1 and M‐CSF to our traditional risk factor model may be overestimated.
Conclusions
In conclusion, we are the first to reveal a strong negative association between plasma M‐CSF and the incidence of coronary events in humans. We demonstrated that M‐CSF and MCP‐1 display opposing associations with the risk for first‐time coronary events in middle‐aged persons with no previous history of coronary artery disease. Those with high levels of M‐CSF and low levels of MCP‐1 in plasma are at low risk of developing acute coronary events. The negative relationship between M‐CSF and incident acute coronary events requires further confirmation in other cohorts and warrants further investigation into the potential protective role of M‐CSF against the development of acute coronary events in humans.
Sources of Funding
This work was supported by grants from the Swedish Research Council (Stockholm), the Swedish Heart–Lung Foundation (Stockholm), the Marianne and Marcus Wallenberg Foundation (Stockholm), Swedish Foundation for Strategic Research (Stockholm), the Albert Påhlsson Foundation (Stockholm), Skåne University Hospital Foundation (Lund), The Royal Physiographic Society in Lund, Bundy Academy Lund University (Lund), and the Lundström Foundation (Malmö).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002851 doi: 10.1161/JAHA.115.002851)
References
- 1. Doring Y, Drechsler M, Soehnlein O, Weber C. Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol. 2015;35:288–295. [DOI] [PubMed] [Google Scholar]
- 2. Zernecke A, Weber C. Chemokines in atherosclerosis: proceedings resumed. Arterioscler Thromb Vasc Biol. 2014;34:742–750. [DOI] [PubMed] [Google Scholar]
- 3. Gautier EL, Jakubzick C, Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1412–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, Nilsson J, Bjorkbacka H. Elevated CD14++CD16‐ monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122–131. [DOI] [PubMed] [Google Scholar]
- 5. Cotoi OS, Duner P, Ko N, Hedblad B, Nilsson J, Bjorkbacka H, Schiopu A. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle‐aged healthy individuals. Arterioscler Thromb Vasc Biol. 2014;34:202–210. [DOI] [PubMed] [Google Scholar]
- 6. Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–89. [DOI] [PubMed] [Google Scholar]
- 7. Jenkins SJ, Hume DA. Homeostasis in the mononuclear phagocyte system. Trends Immunol. 2014;35:358–367. [DOI] [PubMed] [Google Scholar]
- 8. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178:5245–5252. [DOI] [PubMed] [Google Scholar]
- 9. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. [DOI] [PubMed] [Google Scholar]
- 10. Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–1908. [DOI] [PubMed] [Google Scholar]
- 11. Coll B, Alonso‐Villaverde C, Joven J. Monocyte chemoattractant protein‐1 and atherosclerosis: is there room for an additional biomarker? Clin Chim Acta. 2007;383:21–29. [DOI] [PubMed] [Google Scholar]
- 12. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vistnes M. Macrophage inflammatory protein‐1beta: a novel prognostic biomarker in atherosclerosis? Cardiology. 2012;121:149–151. [DOI] [PubMed] [Google Scholar]
- 14. Yi GW, Zeng QT, Mao XB, Cheng M, Yang XF, Liu HT, Mao Y, Guo M, Ji QW, Zhong YC. Overexpression of CXCL16 promotes a vulnerable plaque phenotype in apolipoprotein E‐knockout mice. Cytokine. 2011;53:320–326. [DOI] [PubMed] [Google Scholar]
- 15. Gijsbers K, Gouwy M, Struyf S, Wuyts A, Proost P, Opdenakker G, Penninckx F, Ectors N, Geboes K, Van Damme J. GCP‐2/CXCL6 synergizes with other endothelial cell‐derived chemokines in neutrophil mobilization and is associated with angiogenesis in gastrointestinal tumors. Exp Cell Res. 2005;303:331–342. [DOI] [PubMed] [Google Scholar]
- 16. Berglund G, Elmstahl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. [DOI] [PubMed] [Google Scholar]
- 17. Rosvall M, Janzon L, Berglund G, Engstrom G, Hedblad B. Incidence of stroke is related to carotid IMT even in the absence of plaque. Atherosclerosis. 2005;179:325–331. [DOI] [PubMed] [Google Scholar]
- 18. Assarsson E, Lundberg M, Holmquist G, Bjorkesten J, Thorsen SB, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, Andersson AC, Lindstedt P, Stenvang J, Gullberg M, Fredriksson S. Homogenous 96‐plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014;9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Y, Pepe MS. A parametric ROC model‐based approach for evaluating the predictiveness of continuous markers in case‐control studies. Biometrics. 2009;65:1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pencina MJ, D'Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeLong ER, DeLong DM, Clarke‐Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 22. Herder C, Baumert J, Thorand B, Martin S, Lowel H, Kolb H, Koenig W. Chemokines and incident coronary heart disease: results from the MONICA/KORA Augsburg case‐cohort study, 1984–2002. Arterioscler Thromb Vasc Biol. 2006;26:2147–2152. [DOI] [PubMed] [Google Scholar]
- 23. Hoogeveen RC, Morrison A, Boerwinkle E, Miles JS, Rhodes CE, Sharrett AR, Ballantyne CM. Plasma MCP‐1 level and risk for peripheral arterial disease and incident coronary heart disease: Atherosclerosis Risk in Communities study. Atherosclerosis. 2005;183:301–307. [DOI] [PubMed] [Google Scholar]
- 24. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia‐Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tagliani E, Shi C, Nancy P, Tay CS, Pamer EG, Erlebacher A. Coordinate regulation of tissue macrophage and dendritic cell population dynamics by CSF‐1. J Exp Med. 2011;208:1901–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becker L, Liu NC, Averill MM, Yuan W, Pamir N, Peng Y, Irwin AD, Fu X, Bornfeldt KE, Heinecke JW. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One. 2012;7:e33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Gregoli K, Johnson JL. Role of colony‐stimulating factors in atherosclerosis. Curr Opin Lipidol. 2012;23:412–421. [DOI] [PubMed] [Google Scholar]
- 29. Randolph GJ. Immunology. No need to coax monocytes. Science. 2011;332:1268–1269. [DOI] [PubMed] [Google Scholar]
- 30. Sakurai T, Yamada M, Simamura S, Motoyoshi K. Recombinant human macrophage‐colony stimulating factor suppresses the mouse mixed lymphocyte reaction. Cell Immunol. 1996;171:87–94. [DOI] [PubMed] [Google Scholar]
- 31. Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, Hamilton JA. The promotion of breast cancer metastasis caused by inhibition of CSF‐1R/CSF‐1 signaling is blocked by targeting the G‐CSF receptor. Cancer Immunol Res. 2014;2:765–776. [DOI] [PubMed] [Google Scholar]
- 32. Hashimoto D, Chow A, Greter M, Saenger Y, Kwan WH, Leboeuf M, Ginhoux F, Ochando JC, Kunisaki Y, van Rooijen N, Liu C, Teshima T, Heeger PS, Stanley ER, Frenette PS, Merad M. Pretransplant CSF‐1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med. 2011;208:1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones CV, Ricardo SD. Macrophages and CSF‐1: implications for development and beyond. Organogenesis. 2013;9:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaposhnik Z, Wang X, Lusis AJ. Arterial colony stimulating factor‐1 influences atherosclerotic lesions by regulating monocyte migration and apoptosis. J Lipid Res. 2010;51:1962–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith JD, Trogan E, Ginsberg M, Grigaux C, Tian J, Miyata M. Decreased atherosclerosis in mice deficient in both macrophage colony‐stimulating factor (op) and apolipoprotein E. Proc Natl Acad Sci USA. 1995;92:8264–8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murayama T, Yokode M, Kataoka H, Imabayashi T, Yoshida H, Sano H, Nishikawa S, Nishikawa S, Kita T. Intraperitoneal administration of anti‐c‐fms monoclonal antibody prevents initial events of atherogenesis but does not reduce the size of advanced lesions in apolipoprotein E‐deficient mice. Circulation. 1999;99:1740–1746. [DOI] [PubMed] [Google Scholar]
- 37. Inoue I, Inaba T, Motoyoshi K, Harada K, Shimano H, Kawamura M, Gotoda T, Oka T, Shiomi M, Watanabe Y, Tsukada T, Yazaki Y, Takaku F, Yamada N. Macrophage colony stimulating factor prevents the progression of atherosclerosis in watanabe heritable hyperlipidemic rabbits. Atherosclerosis. 1992;93:245–254. [DOI] [PubMed] [Google Scholar]
- 38. de Villiers WJ, Fraser IP, Hughes DA, Doyle AG, Gordon S. Macrophage‐colony‐stimulating factor selectively enhances macrophage scavenger receptor expression and function. J Exp Med. 1994;180:705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rallidis LS, Zolindaki MG, Manioudaki HS, Laoutaris NP, Velissaridou AH, Papasteriadis EG. Prognostic value of C‐reactive protein, fibrinogen, interleukin‐6, and macrophage colony stimulating factor in severe unstable angina. Clin Cardiol. 2002;25:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rallidis LS, Zolindaki MG, Pentzeridis PC, Poulopoulos KP, Velissaridou AH, Apostolou TS. Raised concentrations of macrophage colony stimulating factor in severe unstable angina beyond the acute phase are strongly predictive of long term outcome. Heart. 2004;90:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saitoh T, Kishida H, Tsukada Y, Fukuma Y, Sano J, Yasutake M, Fukuma N, Kusama Y, Hayakawa H. Clinical significance of increased plasma concentration of macrophage colony‐stimulating factor in patients with angina pectoris. J Am Coll Cardiol. 2000;35:655–665. [DOI] [PubMed] [Google Scholar]
- 42. Ikonomidis I, Lekakis J, Revela I, Andreotti F, Nihoyannopoulos P. Increased circulating C‐reactive protein and macrophage‐colony stimulating factor are complementary predictors of long‐term outcome in patients with chronic coronary artery disease. Eur Heart J. 2005;26:1618–1624. [DOI] [PubMed] [Google Scholar]
- 43. Lind L, Siegbahn A, Lindahl B, Stenemo M, Sundstrom J, Arnlov J. Discovery of new risk markers for ischemic stroke using a novel targeted proteomics chip. Stroke. 2015;46:3340–3347. [DOI] [PubMed] [Google Scholar]
- 44. Yano T, Miura T, Whittaker P, Miki T, Sakamoto J, Nakamura Y, Ichikawa Y, Ikeda Y, Kobayashi H, Ohori K, Shimamoto K. Macrophage colony‐stimulating factor treatment after myocardial infarction attenuates left ventricular dysfunction by accelerating infarct repair. J Am Coll Cardiol. 2006;47:626–634. [DOI] [PubMed] [Google Scholar]
- 45. Okazaki T, Ebihara S, Asada M, Yamanda S, Saijo Y, Shiraishi Y, Ebihara T, Niu K, Mei H, Arai H, Yambe T. Macrophage colony‐stimulating factor improves cardiac function after ischemic injury by inducing vascular endothelial growth factor production and survival of cardiomyocytes. Am J Pathol. 2007;171:1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. [DOI] [PubMed] [Google Scholar]
- 47. McDermott DH, Yang Q, Kathiresan S, Cupples LA, Massaro JM, Keaney JF Jr, Larson MG, Vasan RS, Hirschhorn JN, O'Donnell CJ, Murphy PM, Benjamin EJ. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein‐1 levels and myocardial infarction in the Framingham Heart Study. Circulation. 2005;112:1113–1120. [DOI] [PubMed] [Google Scholar]
- 48. Deo R, Khera A, McGuire DK, Murphy SA, Meo Neto Jde P, Morrow DA, de Lemos JA. Association among plasma levels of monocyte chemoattractant protein‐1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44:1812–1818. [DOI] [PubMed] [Google Scholar]