

Abstract
Background
MKEY, a synthetic cyclic peptide inhibitor of CXCL4–CCL5 heterodimer formation, has been shown to protect against atherosclerosis and aortic aneurysm formation by mediating inflammation, but whether it modulates neuroinflammation and brain injury has not been studied. We therefore studied the role of MKEY in stroke‐induced brain injury in mice.
Methods and Results
MKEY was injected into mice after stroke with 60 minutes of middle cerebral artery occlusion. Infarct volume and neurological deficit scores were measured. Protein levels of CCL5 and its receptor CCR5 were detected by Western blot and fluorescence‐activated cell sorting (FACS), respectively. Numbers of microglia‐derived macrophages (MiMΦs) and monocyte‐derived MΦs (MoMΦs) in the brain, and their subsets, based on the surface markers CD45, CD11b, CCR2, CX3CR1, and Ly6C, were analyzed by FACS. MΦs and neutrophil infiltration in the ischemic brain were stained with CD68 and myeloperoxidase (MPO), respectively, and assessed by immunofluorescent confocal microscopy. The results showed that expressions of CCL5 and its receptor CCR5, were increased in the ischemic brain after stroke. MKEY injection significantly reduced infarct sizes and improved neurological deficit scores measured 72 hours after stroke. In addition, MKEY injection inhibited the number of MoMΦs, but not MiMΦs, in the ischemic brain. Furthermore, MKEY inhibited protein expression levels of Ly6C,CCR2, and CX3CR1 on MoMΦs. Lastly, the confocal study also suggests that the number of CD68‐positive MΦs and MPO‐positive neutrophils was inhibited by MKEY injection.
Conclusions
MKEY injection protects against stroke‐induced brain injury, probably by inhibiting MoMΦ‐mediated neuroinflammation.
Keywords: cerebrovascular disease/stroke, immunology, infarct or infarction, inflammation
Subject Categories: Ischemic Stroke, Neuroprotectants
Introduction
Stroke is a leading cause of death worldwide, but there are limited effective therapeutic strategies available.1 Neuroinflammation plays a key role in stroke‐induced brain injury,2, 3 as the neuroinflammatory response exists in all stages of the ischemic cascade, from early arterial occlusion to late brain injury.4 It is well known that macrophages (MΦs) prominently aggravate brain infarction,5 but the underlying mechanisms remain unknown.
MΦ activities in the ischemic brain are regulated by many inflammatory factors, including chemokines.6, 7, 8 Chemokines are small polypeptide molecules that vary in molecular weight from 8 to 15 kDa and are classified into 4 subgroups: CC, CXC, XC, and CX3C.6, 9, 10 CXCL4 in the CXC subgroup plays important roles in neuroinflammation.11, 12, 13, 14 In addition, the CC chemokine CCL5 regulates leukocyte migration. The interaction between CCL5 and its receptor CCR5, can produce many biological effects, such as the inflammatory response, cytophylaxis, and signal transcription.15 Importantly, CXCL4 and CCL5 can form a heterodimer, CXCL4‐CCL5, which significantly enhances the effect of CCL5 in mediating the adhesion and migration of monocytes during inflammation. How CXCL4‐CCL5 is involved in stroke‐induced brain injury and neuroinflammation has not been studied.
CXCL4‐CCL5 heterodimer formation can be reversed by MKEY, a mouse CCL5‐based synthetic cyclic peptide, which competes with CXCL4 for CCL5 binding sites, thus releasing CXCL4 from existing heterodimers.16 The efficacy of MKEY interrupting CXCL4‐CCL5 heterodimer formation has been confirmed.16 It has been shown that MKEY suppresses abdominal aortic aneurysm formation by modulating inflammation.17 We therefore tested the hypothesis that MKEY also protects against stroke in mice by affecting MΦ activity.
MΦs derive from brain resident microglia and circulating monocytes. Resident microglia are activated immediately and transformed into MΦs after stroke onset Blood monocytes are then recruited a few hours after stroke onset and differentiate to MΦs. Microglia‐derived MΦs (MiMΦs) and monocyte‐derived MΦs (MoMΦs) are morphologically similar but distinguishable by flow cytometric analyses using the MΦ surface markers CD45intCD11b+ and CD45hiCD11b+, respectively. Moreover, MΦ activity can be further analyzed by other markers, including Ly6C, CCR2, and CX3CR1. We thus studied how MKEY injection affects the accumulation of MΦs and their subsets.
Materials and Methods
All experimental procedures were approved by the Stanford University Administrative Panel on Laboratory Animal Care, and experiments were conducted in accordance with the guidelines of Animal Use and Care of the National Institutes of Health and Stanford University. All efforts were made to minimize the number of animals used, stress, and pain.
Focal Cerebral Ischemia Model
Male C57BL/6J mice, aged 10 weeks and weighing nearly 25 g, were purchased from The Jackson Laboratory (Bar Harbor, ME). Anesthesia was introduced with 5% isoflurane and maintained at 1% to 2% during surgery. The surgeon then exposed the left common carotid artery, internal carotid artery, and external carotid artery by making a midline neck incision, as described in our previous studies.5, 18 A 6‐0 silicone‐coated nylon suture (Doccol Corporation, Sharon, MA) was introduced into the common carotid artery through an incision, and the suture was advanced about 9 mm beyond the carotid bifurcation to occlude the middle cerebral artery (MCA) for 60 minutes.19, 20, 21 A rectal probe was inserted to monitor body core temperature, which was maintained at 37±0.5°C with a surface heating pad during the entire procedure. The animals were allowed to wake up and returned to their cages during MCA occlusion. They were then reanesthetized to remove the suture, ligate the artery, and close the incision. In the sham group, the same surgery was conducted but without inserting the suture into the MCA. A total of 85 mice, including 14 mice with sham surgery and 71 mice with stroke, were used for the whole experiment.
Drug Injection
In the treatment group, the mice were injected intravenously with MKEY (molecular weight 2.9 kDa. Formula CT‐2009ca, Carolus Therapeutics, La Jolla, CA) immediately and 3, 24, and 48 hours after reperfusion, for a total of 4 injections. The dosage was 20 mg/kg per injection, which was selected based on a previous study treating abdominal aortic aneurysm (AAA) disease.17 In that study, 2 dosages were compared, 10 and 20 mg/kg, and it was found that the latter showed stronger protection, which significantly inhibited leukocyte migration. In the control group, vehicle alone (CT‐2009 “MKEY” vehicle, Carolus Therapeutics, La Jolla, CA) with a volume equal to MKEY was administered at the identical time points.
Neurological Deficit Scores and Infarct Size Measurements
The neurological deficit score of the mice was estimated 72 hours after stroke using a neurological grading score from 0 (no observable neurological deficit) to 4 (unable to walk spontaneously and with a depressed level of consciousness) by an evaluator who was blinded to the experimental treatments, as previously described.5
The experimental mice were deeply anesthetized with isoflurane and decapitated 72 hours after cerebral ischemia. Brains were cut into 4 coronal sections and stained with 2% 2,3,5‐triphenyltetrazolium chloride (TTC; Sigma‐Aldrich, St. Louis, MO) at 37°C for 15 minutes and fixed in 4% paraformaldehyde for 24 hours. Infarct size was measured using Image J software (1.37v; Wayne Rasband, available through National Institutes of Health), normalized to the volumes of the nonischemic hemisphere to exclude the effect of cerebral edema induced by ischemia on the infarct size, and expressed as percentages of the hemisphere.22
Western Blot Analysis
To detect the expression levels of CCL5 in stroke mice, Western blot was performed as previously described.23 Samples were harvested 72 hours after ischemia, gently homogenized in cold lysis buffer (10 mmol/L 4‐2‐hydroxyethyl‐1‐piperazine‐ethanesulfonic acid [pH 7.9], 1.5 mmol/L MgCl2, 10 mmol/L KCl, 1 mmol/Ldithiothreitol) and protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), and then centrifuged. The supernatants were collected and used for analysis. Protein concentrations were measured using bicinchoninic acid (Pierce Chemicals, Rockford, IL). Proteins (36 μg) were loaded into each lane, separated with 4% to 15% polyacrylamide gels (Bio‐Rad, Hercules, CA), and electrotransferred to Immobilon polyvinylidene fluoride membranes (Millipore Corp, Billerica, MA). Membranes were blocked with 5% nonfat dry milk in phosphate‐buffered saline (PBS) with 0.1% Tween 20 for 1 hour, and incubated with 1:250 anti–mouse CCL5 antibody (R&D Systems Inc, Minneapolis, MN) overnight at 4°C. They were then washed 3 times with 0.1% Tween in PBS, then incubated with 1:1000 secondary antibody (Cell Signaling, Danvers, MA) for 2 hours at room temperature, and washed 3 times with 0.1% Tween in PBS. Signals were detected with ECL reagent (Amersham, Piscataway, NJ). β‐Actin was used as an internal control. Densitometric analysis of bands was performed with Image J software.
Fluorescence‐Activated Cell Sorting (FACS) Analysis
Mice were euthanized with an overdose of isoflurane 72 hours after stroke onset and perfused with 50 mL cold PBS. Their brains were collected and homogenized on ice using FACS buffer (PBS containing 1% fetal bovine serum) up to 7 mL, mixed with 3 mL of 90% Percoll (GE Healthcare, Little Chalfont, UK), and loaded with 1 mL 70% Percoll under cell suspension. Then the samples were centrifuged at 500g for 30 minutes at 4°C. Leukocytes at the interphase were collected and washed with FACS buffer. Then cells were resuspended for FACS analysis.18 Cell suspensions were labeled with antibodies against CD45, CD11b, Ly6C (BioLegend Inc.), CCR2, and CX3CR1 (R&D Systems Inc) on ice for 30 minutes in the dark. Cell surface molecule expression measurement was performed on the BD LSR II flow cytometer using Diva software (v6.1.2, Becton Dickinson, Franklin Lakes, NJ) and analyzed using FlowJo (Ashland, OR) software (v7.6.2, Tree Star).
Tissue Immunofluorescence Staining
Mice were euthanized with an overdose of isoflurane and perfused with icy PBS and 4% PFA at 72 hours after stroke as previously described.5 The brains were cut into 6 2‐mm blocks from rostral to caudal. Blocks 3 to 5, which were within the infarct area, were selected and sectioned into 50 μm slices on a cryostat. One slice from each block was randomly chosen; thus, a total of 3 slices from each animal were selected for immunostaining. Immunofluorescence staining was carried out with moderate shaking. Sections were washed with 0.1 mmol/L PBS (pH 7.4) and incubated for 1 hour with blocking solution (0.1 mmol/L PBS, 5% equine serum). After washing, sections were incubated overnight at 4°C with a rat anti–mouse CD68 antibody (diluted 1:200; MCA1957GA; AbD Serotec, Raleigh, NC), a marker for reactive MΦs/microglia, or a rabbit anti–human myeloperoxidase (MPO) antibody (cross‐reacted with mouse MPO, diluted 1:50; Cat# A0398; Dako North America, Inc, Carpinteria, CA). Sections were then rinsed and incubated for 2 hours at room temperature with an Alexa 488‐conjugated goat anti‐rabbit (for MPO) or Alexa 488‐conjugated goat anti‐rat (for CD68‐positive MΦs/microglia, diluted 1:200; Invitrogen, Carlsbad, CA) antibodies. The sections were then washed and covered using Vectashield mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). A negative control without primary antibodies was performed in parallel.
The expression of CD68 or MPO was investigated using the optical fractionator method on epifluorescent photomicrographs (Zeiss axiovert inverted scanning microscope; Zeiss, Jena, Germany). On each slice stained with antibodies, a region that was 400 μm away from the ischemic border was defined as the ischemic core. Positively stained cells (for both CD68 and MPO) in the predefined infarct area were counted using Image J software, and a mean number of positive cells was calculated from the 3 selected slices. All counts were performed on coded sections to blind the investigator to the treatment group.
Statistical Analysis
All values were expressed as mean±SE, and were analyzed using GraphPad Prism 5 (GraphPAD Software for Science, La Jolla, CA). Nonparametric tests were used for comparing infarct sizes, neurological scores, CCL‐5 expressions, and immunostaining results between vehicle and MKEY groups. Two‐way analysis of variance (ANOVA) (vehicle versus MKEY and stroke versus sham) was performed for FACS results, followed by a Bonferroni post‐hoc test. A P<0.05 was considered statistically significant.
Results
MKEY Injection Reduced Infarct Sizes and Improved Neurological Scores After Stroke in Mice
We first examined the protective effect of MKEY injection on stroke outcomes. The results showed that infarction was significantly reduced in the MKEY group (Figure 1A), and the neurological scores were also significantly improved 3 days after stroke in mice (Figure 1B).
Figure 1.
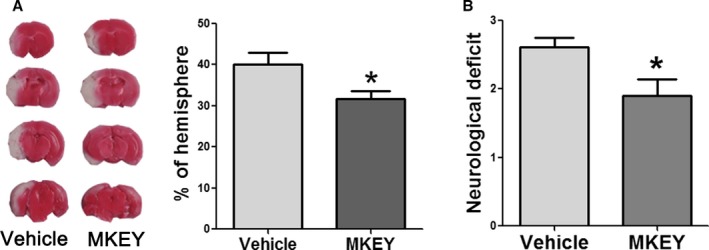
The effect of MKEY injection on infarction and neurological scores measured 72 hours after stroke. A, Left: Representative TTC staining images for infarction in mice treated with vehicle and MKEY. Right: Infarct volumes expressed as a percentage of the nonischemic hemisphere. B, Neurological scores in mice treated with vehicle and MKEY. n=9 to 11/group. *P<0.05 compared to vehicle.
CCL5 and CCR5 Expression was Increased in the Ischemic Hemisphere
As the major role of MKEY is to prevent formation of the CXCL4–CCL5 heterodimer, it was essential to examine whether CCL5 expression was increased after stroke. The Western blot results showed that CCL5 expression was significantly increased in the ischemic brain measured 3 days after stroke (Figure 2).
Figure 2.
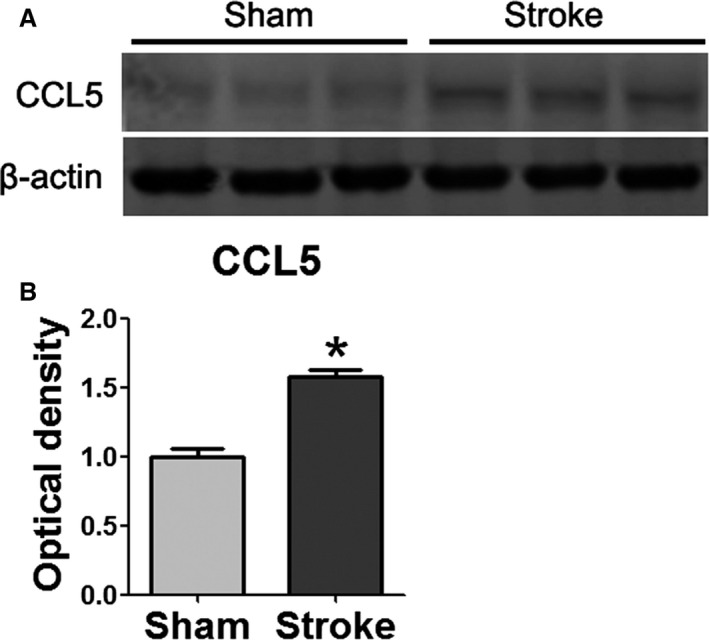
CCL5 expression was increased and measured 72 hours poststroke. A, Representative Western blots for CCL5 at 10.2 kDa in sham surgery and stroke brain. B, Quantification of CCL5 protein levels in sham and stroke brain. n=3/group. *P=0.05 vs sham.
In addition, as CCL5 executes its effect by stimulating its receptor CCR5, we also quantified surface CCR5 expression on MΦs using flow cytometry. We found that CCR5 expression was robustly increased in both MiMΦs and MoMΦs in the ischemic brain measured 3 days after stroke (Figure 3).
Figure 3.
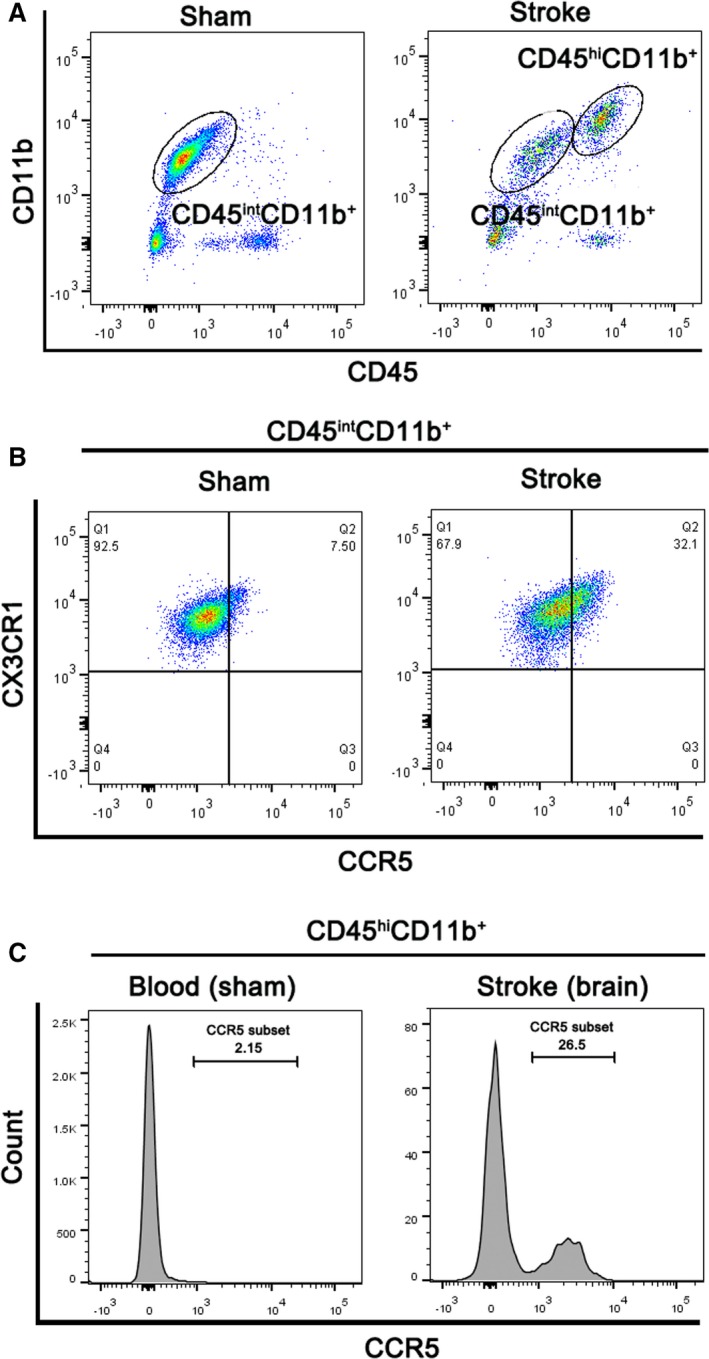
CCR5 expression was increased in both MiMΦs and MoMΦs. A, Representative FACS gates for MiMΦs (CD45int CD11b+ cells) and MoMΦs (CD45hi CD11b+) are shown. B, Representative FACS plots for CCR5 and CX3CR1 expression on gated MiMΦs isolated from sham and stroke brain are shown. C, Representative FACS histograms for CCR5 expression on peripheral blood or brain CD45hi CD11b+ cells in mice with sham surgery or stroke.
MKEY Significantly Attenuated the Number of Infiltrating MoMΦs
We further analyzed how MKEY injection affected MΦs and their subtypes in the ischemic brains 3 days after stroke. As discussed, MΦs can be classified into MiMΦs and MoMΦs as CD45intCD11b+ and CD45hiCD11b+, respectively. In addition, their subgroups can be identified by surface CCR2 and Ly6C expression.24 CX3CR1 is expressed on monocytes, dendritic cells, natural killer cells, and cytotoxic effector T cells in the periphery, and is involved in inflammatory processes.25, 26, 27 In the brain, CX3CR1 is mostly expressed on microglia and is important to maintain normal microglial activity.27, 28 Our results show that numbers of both MiMΦs and MoMΦs were significantly increased in the ischemic brains (Figure 4). MKEY injection significantly attenuated the number of MoMΦs, but not MiMΦs. Accordingly, MKEY injection also significantly inhibited the number of CCR2, Ly6C, and CX3CR1 subsets of MoMΦs, but not those of MiMΦs (Figure 4).
Figure 4.
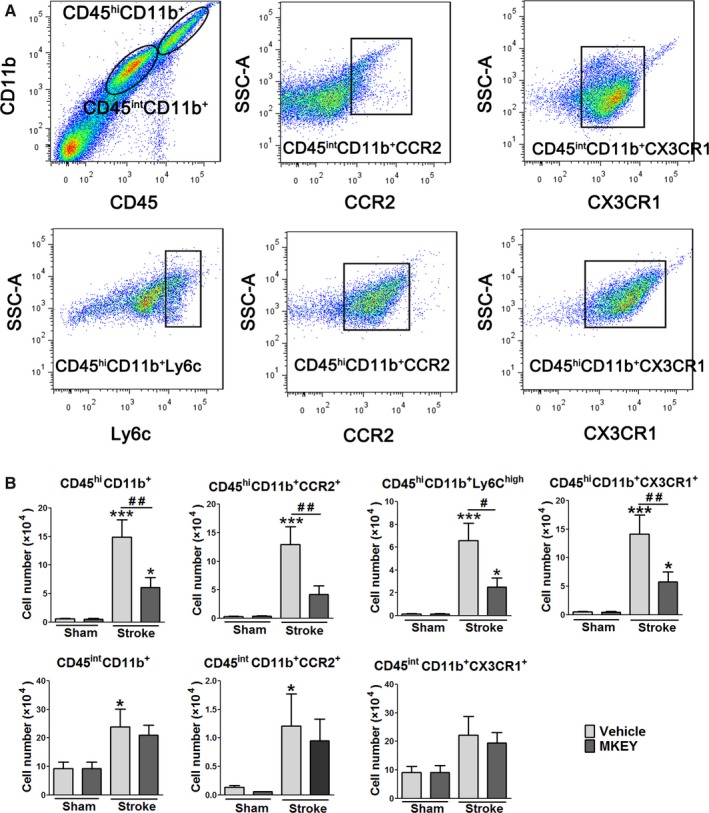
MKEY injection reduced numbers of MoMΦs and their subsets, but not those of MiMΦs, in the ischemic brain 72 hours after stroke. A, Gating strategies to identify MiMΦs and MoMΦs, as well as their subsets, CD45hi CD11b+ CCR2+, CD45hi CD11b+Ly6C+, CD45hi CD11b+ CX3CR1+, CD45int CD11b+ CCR2+, and CD45int CD11b+ CX3CR1+ in the ischemic brain. B, Quantification of MΦ numbers and their subsets in mice treated with vehicle or MKEY, with sham or stroke surgery. *, **, ***, vs sham surgery, P<0.05, 0.01, 0.001, respectively; #, ##, between the 2 indicated groups, P<0.05, 0.01, respectively.
MKEY Injection Reduces the Infiltration of CD68‐ and MPO‐Expressing Cells
Lastly, we also examined CD68‐labeled MΦs and MPO‐expressing neutrophils using confocal microscopy. Our results showed that a large number of MΦs and neutrophils were detected in the ischemic brain, and MKEY injection significantly inhibited their infiltration (Figure 5).
Figure 5.
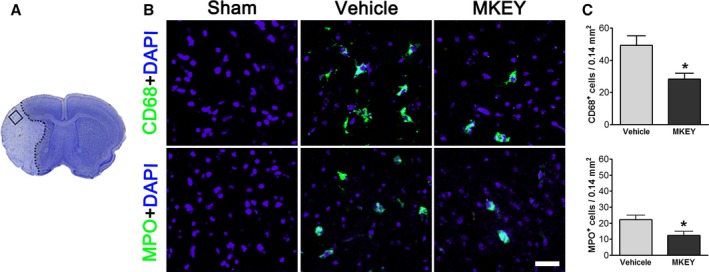
MKEY treatment poststroke reduced the infiltration of CD68+ and MPO + cells. A, A square area in a coronal brain section stained with cresyl violet was predefined and used for counting immunostained cells. B, Representative immunofluorescence staining for CD68, MPO, and DAPI (control) from vehicle and MKEY‐treated animals euthanized 72 hours after stroke. C, Quantification of CD68‐ and MPO‐positive cells in the predefined area. N=5/group. Bar: 20 μm. *P<0.05 compared to vehicle.
Discussion
We demonstrate the first evidence that MKEY, the inhibitor of CXCL4‐CCL5 heterodimer formation, can significantly decrease infarct size and attenuate neurological scores after stroke in mice, suggesting that CXCL4‐CCL5–mediated neuroinflammation contributes to brain injury. Indeed, we have confirmed that the expression of CCL5 was increased in the ischemic hemisphere, and its receptor CCR5 was increased in both MiMΦs and MoMΦs, suggesting that CCL5 and its receptor CCR5 are involved in ischemic brain injury. In addition, we found that MKEY robustly reduced the number of infiltrating MoMΦs, but not MiMΦs, indicating that inhibition of MoMΦs may play a crucial role in the protective effects of MKEY. Lastly, we further confirmed that MKEY attenuated the number of total CD68‐positive MΦs and MPO‐positive neutrophils as measured by confocal microscopy.
Previous studies have demonstrated that CCL5 is a proinflammatory chemokine with an important role in recruiting leukocytes to inflammatory sites.29 However, the role of CCL5 in ischemic stroke remains controversial. CCL5 is involved in cerebral microvascular dysfunction, inflammation, and tissue damage produced by cerebral ischemia and reperfusion.30 Dénes et al have shown that chronic systemic infection exacerbated ischemic brain injury through a CCL5‐mediated proinflammatory response in mice.31 In contrast, Tokami et al have shown that CCL5 has the potential to protect neurons by producing neurotrophic factors.32 In addition, CCL5 levels were reported by Montecucco et al to be increased in the serum of stroke patients,33 and significant levels of CCL5 were shown by Canouï‐Poitrine et al to be independent predictors of ischemic stroke,34 whereas Zaremba et al reported no elevation of CCL5 serum levels in the first 3 days after ischemic stroke.35 In the current study, we found that expression of CCL5 was increased in the ischemic brain, as was CCR5 expression in MΦs, suggesting that CCL5 and CCR5 may play pathogenic roles in brain damage after stroke.
CCL5 forms a heterodimer with CXCL4, which increases its efficacy in stimulating CCR5. Grommes et al have shown that CXCL4‐CCL5 heterodimers can cause lung edema, neutrophil infiltration, and tissue damage, all of which were inhibited by MKEY, the inhibitor of CXCL4‐CCL5 heterodimer formation.36 We previously found that MKEY treatment strongly decreased aortic monocyte/MΦ infiltration and thus suppressed formation and progression of experimental abdominal aortic aneurysms.17 These previous studies suggest that MKEY acts as an inflammation inhibitor. Consistent with these findings, our current results demonstrate that injection of MKEY can also reduce infarction by attenuating neuroinflammation.
MΦs in the ischemic brain consist of MiMΦs and MoMΦs, which can be identified as CD45intCD11b+ and CD45hiCD11b+e. In addition, MoMΦs can be further classified as pro‐ and anti‐inflammatory based on the expression levels of CCR2, CX3CR1, and Ly6C. Our results showed that MKEY injection significantly inhibited numbers of MoMΦs and all of their subsets labeled by CCR2, Ly6C, and CX3CR1, but it had no effect on these markers of MiMΦs. These results indicate that inhibition of MoMΦs plays a more important role than that of MiMΦs in ischemic injury. The overall inhibitory effect of MKEY injection against the inflammatory response was confirmed by the confocal results, which suggests that MKEY inhibited the infiltration of CD68‐positive MΦs and MPO‐positive neutrophils in the ischemic brain.
We are aware that there are some limitations in our study. First, we did not study detailed therapeutic time windows for the protective effect of MKEY. Second, we did not test whether MKEY injection has long‐term protection against stroke. Third, we did not analyze CXCL4‐CCL5 formation with and without MKEY treatment. Fourth, we have no information regarding its capability of crossing the blood‐brain barrier (BBB). Nevertheless, the issue of BBB might not be critical, as the drug was injected after stroke, when BBB is open. In addition, MKEY exerts its effects mainly in the intraluminal space. In any case, these issues should be clarified in future studies.
In conclusion, our experiments indicate that MKEY protects against brain injury induced by stroke, mainly by preventing infiltration of circulating MoMΦs, and that inhibition of the CXCL4‐CCL5 heterodimer is a potential target for therapeutic intervention in stroke.
Sources of Funding
This study is supported by NIH R01 grant 2R01NS064136 (Zhao).
Disclosures
None.
Acknowledgments
The authors wish to thank Cindy H. Samos for her assistance with manuscript preparation.
(J Am Heart Assoc. 2016;5:e003615 doi: 10.1161/JAHA.116.003615)
References
- 1. Nabavi SF, Li H, Daglia M, Nabavi SM. Resveratrol and stroke: from chemistry to medicine. Curr Neurovasc Res. 2014;11:390–397. [DOI] [PubMed] [Google Scholar]
- 2. Dziedzic T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev Neurother. 2015;15:523–531. [DOI] [PubMed] [Google Scholar]
- 3. Basic Kes V, Simundic AM, Nikolac N, Topic E, Demarin V. Pro‐inflammatory and anti‐inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clin Biochem. 2008;41:1330–1334. [DOI] [PubMed] [Google Scholar]
- 4. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gu L, Xiong X, Zhang H, Xu B, Steinberg GK, Zhao H. Distinctive effects of T cell subsets in neuronal injury induced by cocultured splenocytes in vitro and by in vivo stroke in mice. Stroke. 2012;43:1941–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mirabelli‐Badenier M, Braunersreuther V, Viviani GL, Dallegri F, Quercioli A, Veneselli E, Mach F, Montecucco F. CC and CXC chemokines are pivotal mediators of cerebral injury in ischaemic stroke. Thromb Haemost. 2011;105:409–420. [DOI] [PubMed] [Google Scholar]
- 7. Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14:3574–3589. [DOI] [PubMed] [Google Scholar]
- 8. Cavaillon JM. Cytokines in inflammation. C R Seances Soc Biol Fil. 1995;189:531–544. [PubMed] [Google Scholar]
- 9. Murphy PM, Baggiolini M, Charo IF, Hébert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 10. Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 11. Vandercappellen J, Van Damme J, Struyf S. The role of the CXC chemokines platelet factor‐4 (CXCL4/PF‐4) and its variant (CXCL4L1/PF‐4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011;22:1–18. [DOI] [PubMed] [Google Scholar]
- 12. Daly TJ, LaRosa GJ, Dolich S, Maione TE, Cooper S, Broxmeyer HE. High activity suppression of myeloid progenitor proliferation by chimeric mutants of interleukin 8 and platelet factor 4. J Biol Chem. 1995;270:23282–23292. [DOI] [PubMed] [Google Scholar]
- 13. Gengrinovitch S, Greenberg SM, Cohen T, Gitay‐Goren H, Rockwell P, Maione TE, Levi BZ, Neufeld G. Platelet factor‐4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J Biol Chem. 1995;270:15059–15065. [DOI] [PubMed] [Google Scholar]
- 14. Crisi GM, Katz IR, Zucker MB, Thorbecke GJ. Induction of inhibitory activity for B cell differentiation in human CD8 T cells with pokeweed mitogen, dimaprit, and cAMP upregulating agents: countersuppressive effect of platelet factor 4. Cell Immunol. 1996;172:205–216. [DOI] [PubMed] [Google Scholar]
- 15. Katayama H, Yokoyama A, Kohno N, Sakai K, Hiwada K, Yamada H, Hirai K. Production of eosinophilic chemokines by normal pleural mesothelial cells. Am J Respir Cell Mol Biol. 2002;26:398–403. [DOI] [PubMed] [Google Scholar]
- 16. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. [DOI] [PubMed] [Google Scholar]
- 17. Iida Y, Xu B, Xuan H, Glover KJ, Tanaka H, Hu X, Fujimura N, Wang W, Schultz JR, Turner CR, Dalman RL. Peptide inhibitor of CXCL4‐CCL5 heterodimer formation, MKEY, inhibits experimental aortic aneurysm initiation and progression. Arterioscler Thromb Vasc Biol. 2013;33:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joo SP, Xie W, Xiong X, Xu B, Zhao H. Ischemic postconditioning protects against focal cerebral ischemia by inhibiting brain inflammation while attenuating peripheral lymphopenia in mice. Neuroscience. 2013;243:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiong X, Gu L, Zhang H, Xu B, Zhu S, Zhao H. The protective effects of T cell deficiency against brain injury are ischemic model‐dependent in rats. Neurochem Int. 2013;62:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao H, Yenari MA, Sapolsky RM, Steinberg GK. Mild postischemic hypothermia prolongs the time window for gene therapy by inhibiting cytochrome C release. Stroke. 2004;35:572–577. [DOI] [PubMed] [Google Scholar]
- 21. Han RQ, Quyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG. Postischemic brain injury is attenuated in mice lacking the beta2‐adrenergic receptor. Anesth Analg. 2009;108:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin‐4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42:2026–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A role for spleen monocytes in post‐ischemic brain inflammation and injury. J Neuroinflammation. 2010;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arai M, Ikawa Y, Chujo S, Hamaguchi Y, Ishida W, Shirasaki F, Hasegawa M, Mukaida N, Fujimoto M, Takehara K. Chemokine receptors CCR2 and CX3CR1 regulate skin fibrosis in the mouse model of cytokine‐induced systemic sclerosis. J Dermatol Sci. 2013;69:250–258. [DOI] [PubMed] [Google Scholar]
- 26. Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. [DOI] [PubMed] [Google Scholar]
- 27. Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. [DOI] [PubMed] [Google Scholar]
- 28. Luhmann UF, Carvalho LS, Robbie SJ, Cowing JA, Duran Y, Munro PM, Bainbridge JW, Ali RR. Ccl2, Cx3cr1 and Ccl2/Cx3cr1 chemokine deficiencies are not sufficient to cause age‐related retinal degeneration. Exp Eye Res. 2013;107:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014;2014:292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terao S, Yilmaz G, Stokes KY, Russell J, Ishikawa M, Kawase T, Granger DN. Blood cell‐derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia‐reperfusion. Stroke. 2008;39:2560–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dénes A, Humphreys N, Lane TE, Grencis R, Rothwell N. Chronic systemic infection exacerbates ischemic brain damage via a CCL5 (regulated on activation, normal T‐cell expressed and secreted)‐mediated proinflammatory response in mice. J Neurosci. 2010;30:10086–10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tokami H, Ago T, Sugimori H, Kuroda J, Awano H, Suzuki K, Kiyohara Y, Kamouchi M, Kitazono T. RANTES has a potential to play a neuroprotective role in an autocrine/paracrine manner after ischemic stroke. Brain Res. 2013;1517:122–132. [DOI] [PubMed] [Google Scholar]
- 33. Montecucco F, Lenglet S, Gayet‐Ageron A, Bertolotto M, Pelli G, Palombo D, Pane B, Spinella G, Steffens S, Raffaghello L, Pistoia V, Ottonello L, Pende A, Dallegri F, Mach F. Systemic and intraplaque mediators of inflammation are increased in patients symptomatic for ischemic stroke. Stroke. 2010;41:1394–1404. [DOI] [PubMed] [Google Scholar]
- 34. Canouï‐Poitrine F, Luc G, Mallat Z, Mallat Z, Machez E, Bingham A, Ferrieres J, Ruidavets JB, Montaye M, Yarnell J, Haas B, Arveiler D, Morange P, Kee F, Evans A, Amouyel P, Ducimetiere P, Empana JP. Systemic chemokine levels, coronary heart disease, and ischemic stroke events: the PRIME study. Neurology. 2011;77:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zaremba J, Ilkowski J, Losy J. Serial measurements of levels of the chemokines CCL2, CCL3 and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006;44:282–289. [PubMed] [Google Scholar]
- 36. Grommes J, Alard JE, Drechsler M, Wantha S, Mörgelin M, Kuebler WM, Jacobs M, von Hundelshausen P, Warkart P, Wygrecka M, Preissner KT, Hackeng TM, Koenen RR, Weber C, Soehnlein O. Disruption of platelet‐derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med. 2012;185:628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]