

Abstract
Background
Mutations in the coding sequence of SCN5A, which encodes the cardiac Na+ channel α subunit, have been associated with inherited susceptibility to various arrhythmias. Variable expression of SCN5A is a possible mechanism responsible for this pleiotropic effect; however, it is unknown whether variants in the promoter and regulatory regions of SCN5A also modulate the risk of arrhythmias.
Methods and Results
We resequenced the core promoter region of SCN5A and the regulatory regions of SCN5A transcription in 1298 patients with arrhythmia phenotypes (atrial fibrillation, n=444; sinus node dysfunction, n=49; conduction disease, n=133; Brugada syndrome, n=583; and idiopathic ventricular fibrillation, n=89). We identified 26 novel rare variants in the SCN5A promoter in 29 patients affected by various arrhythmias (atrial fibrillation, n=6; sinus node dysfunction, n=1; conduction disease, n=3; Brugada syndrome, n=14; idiopathic ventricular fibrillation, n=5). The frequency of rare variants was higher in patients with arrhythmias than in controls. In the alignment with chromatin immunoprecipitation sequencing data, the majority of variants were located at regions bound by transcription factors. Using a luciferase reporter assay, 6 variants (Brugada syndrome, n=3; idiopathic ventricular fibrillation, n=2; conduction disease, n=1) were functionally characterized, and each displayed decreased promoter activity compared with the wild‐type sequences. We also identified rare variants in the regulatory region that were associated with atrial fibrillation, and the variant decreased promoter activity.
Conclusions
Variants in the core promoter region and the transcription regulatory region of SCN5A were identified in multiple arrhythmia phenotypes, consistent with the idea that altered SCN5A transcription levels modulate susceptibility to arrhythmias.
Keywords: arrhythmias, genetics, ion channels, sodium channels, transcription
Subject Categories: Arrhythmias, Genetics, Ion Channels/Membrane Transport
Introduction
Voltage‐gated sodium channels play a critical role in the generation and propagation of the cardiac action potential. Mutations in SCN5A, the gene encoding the major pore‐forming sodium channel α subunit in the heart (Nav1.5), are associated with inherited susceptibility to a wide variety of arrhythmia syndromes, the “cardiac sodium channelopathies.”1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 Gain‐of‐function mutations in SCN5A produce an enhanced sodium current during the action potential plateau and cause long QT syndrome (type 3),1, 2 whereas loss‐of‐function mutations produce a reduced sodium current and lead to various arrhythmias, including Brugada syndrome,3 early repolarization syndrome,4 idiopathic ventricular fibrillation,5 cardiac conduction disease,6 sinus node dysfunction,7 atrial standstill,8 and cardiomyopathy.12, 13 Both gain‐ and loss‐of‐function mutations are associated with atrial fibrillation.9, 10 Furthermore, sodium channel accessory subunit genes and sodium channel partner genes are also associated with arrhythmia syndromes.14, 15, 16, 17, 18, 19 Nevertheless, despite extensive efforts, most patients with arrhythmias suggestive of cardiac sodium channelopathies are genotype negative. Because reduced sodium channel expression is a major mechanism by which mutations in sodium channel genes alter sodium currents, leading to arrhythmia syndromes,1, 2, 3, 4, 5, 6, 7 genetic variants in regulatory regions of SCN5A may cause arrhythmias.
Increasing evidence shows that the regions regulating SCN5A transcription play a critical role in the electrophysiology of the heart.20, 21, 22, 23, 24, 25, 26, 27 A region around the noncoding exon 1 was initially identified as the core SCN5A promoter.21 Furthermore, it has been found that conserved noncoding sequences (CNSs) in intron 1 of SCN5A regulate promoter activity and cardiac conduction.20 Single‐nucleotide polymorphisms in the SCN5A core promoter region alter transcriptional activity, and a haplotype of the SCN5A promoter is associated with reduced transcriptional activity and the slowing of cardiac conduction.22, 23 A common haplotype of 2 single‐nucleotide polymorphisms in the promoter region has recently been associated with the severity of arrhythmia phenotypes, including conduction disease and ventricular tachyarrhythmias, in a family affected by a loss‐of‐function mutation in the coding region of SCN5A.24 Furthermore, evidence supporting the association of SCN5A transcriptional regulation and cardiac conduction includes the findings that genetic variation in a key cardiac transcription factor is associated with cardiac conduction in a genomewide association study and that the transcription factor regulates SCN5A expression.25, 26, 27 We tested the hypothesis that variants in the SCN5A core promoter region influence susceptibility to cardiac electrical diseases.
Methods
Study Participants
The study protocol was approved by the institutional review board of each institution. All participants provided written informed consent prior to the genetic and clinical investigations, in accordance with the standards of the Declaration of Helsinki and local ethics committees. This study included 1298 unrelated patients with arrhythmia phenotypes (atrial fibrillation, n=444; sinus node dysfunction, n=49; conduction disease, n=133; Brugada syndrome, n=583; idiopathic ventricular fibrillation, n=83; early repolarization syndrome, n=6) who did not have mutations in the coding sequences and flanking regions of SCN5A. Among the 1298 patients, 114 patients with Brugada syndrome and 376 patients with atrial fibrillation were white, and the remaining 808 patients were Japanese. We also resequenced the SCN5A promoter in 816 controls who were free from arrhythmias including 282 white and 534 Japanese participants. CNS23 and CNS28 in intron 1 of SCN5A regulate promoter activity,20 and we resequenced CNS23 and CNS28 in 405 patients with atrial fibrillation, 65 patients with cardiac conduction disease, and 664 controls.
Genetic Analysis
A genetic analysis was performed using genomic DNA extracted from peripheral white blood cells with standard methods. The core promoter region of SCN5A and the CNSs were amplified by polymerase chain reaction using primers described elsewhere, and direct DNA sequencing was performed.20 Variants found in controls, the 1000 Genomes Project data, the Tohoku Megabank Whole Genome data, or dbSNP (version 142) were excluded.28
Chromatin Immunoprecipitation Sequencing Analysis Using Adult Mouse Hearts
The association of the SCN5A promoter variants identified in this study with the key cardiac transcription factors was studied using data obtained from genomewide screens for binding sites of the key cardiac transcription factors in a previous study by our group (Figure 1).25 FASTQ files from the GEO data sets for Tbx3 (GSE44821), Tbx5 (GSE21529), Nkx2‐5 (GSE35151), and p300 and pol2 (GSE29184) as well as other markers of transcriptional activity were aligned to the mouse genome using the Galaxy server (http://galaxy.nbic.nl). Wiggle format data derived from these alignments were lifted over to the human genome (Hg18) for regions of interest and then uploaded as headed bedgraph track data to the University of California, Santa Cruz (UCSC) genome browser alongside positional data for the SCN5A promoter variants and other preselected tracks available for display on the UCSC browser.
Figure 1.
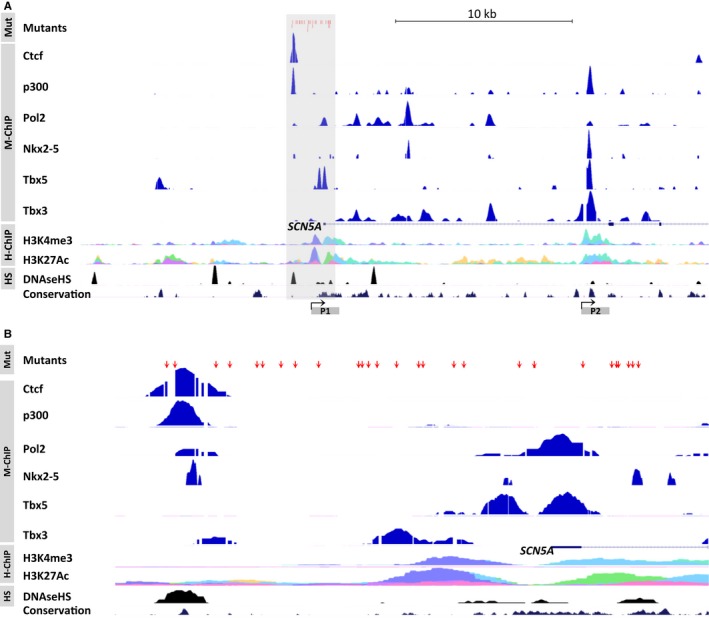
Human genomic promoter region of SCN5A. A, An overview of chromatin immunoprecipitation (ChIP) sequencing data sets including the SCN5A locus. B, A close‐up of the core promoter region of SCN5A marked in panel A with a gray box. The genomic positions of the identified mutants are marked with red arrows showing their location with respect to various transcription factors (Ctcf, Pol2, p300, Nkx2‐5, Tbx5 and Tbx3, lifted over to Hg18 from the mouse genome data sets) and other markers of transcriptional activity. The histone marks H3K4me3 and H3K27Ac and DNase hypersensitivity (HS) typically highlight regions of open active chromatin such as enhancers and promoters. Alternative promoters are marked P1 and P2. H‐ChIP indicates human heart ChIP sequencing; Mut, locations of mutants; M‐ChIP, mouse heart ChIP sequencing.
Functional Analysis of Rare Variants
Transient transfection analyses were performed, as previously reported.20, 21, 22, 23 Briefly, the full‐length human SCN5A promoter (haplotype A) was subcloned into the pGL3‐Basic plasmid (Promega, Madison, WI).21, 23 Mutant constructs were prepared using a QuikChange site‐directed mutagenesis kit (Stratagene), according to the manufacturer's instructions. The pGL3‐Basic plasmids carrying the wild‐type or mutant SCN5A promoter (1 μg) were transfected into HEK293 cells, Chinese hamster ovary cells, and the mouse cardiomyocyte cell line HL‐1 using Lipofectamine LTX (Invitrogen) or Fugene 6 transfection reagent (Roche Applied Science).21, 22, 23 To study the effects of variants in the CNSs on promoter activity, the pGL3‐Basic plasmids carrying wild‐type or mutant sequences with the SV40 promoter were also transfected into Chinese hamster ovary cells.20 In each experiment, the pRL‐TK plasmid (50 ng; Promega) encoding Renilla luciferase was cotransfected to normalize for experimental variability caused by differences in cell viability or transfection efficiency. Luminescence was measured 48 hours after transfection using the Dual‐Luciferase Reporter Assay System (Promega). The pGL3‐Basic plasmid without promoter was tested in each experiment, and its activity level served as the baseline. Potential muscle‐specific transcriptional regulatory modules in CNS28 were predicted using the M‐SCAN algorithm (http://www.cisreg.ca/cgi-bin/mscan/MSCAN).20 To study the affinities of a human heart protein for the transcription factors that were predicted to bind to sequences within CNS28, an electrophoretic mobility shift assay was performed using the standard protocol. Briefly, double‐stranded oligonucleotides containing the CNS28 sequence were incubated with a nuclear lysate from a human heart. A CNS28 unlabeled competitor probe was used as a control.
Statistical Analysis
The Mann–Whitney U test or Fisher exact test was used to evaluate significant differences. All statistical analyses were performed using SPSS version 20 (IBM Corp). A 2‐sided P<0.05 was considered statistically significant. Values are expressed as mean±SEM. The authors had full access to the data and take full responsibility for its integrity. All the authors have read and approved the paper as written.
Results
We identified 26 novel rare variants in the SCN5A promoter in 29 unrelated patients affected by various arrhythmias (atrial fibrillation, n=6; sinus node dysfunction, n=1; conduction disease, n=3; Brugada syndrome, n=14; idiopathic ventricular fibrillation, n=5) (Table). All variants were absent in controls and in the 1000 Genomes Project data.28 The patients carrying a rare variant in the SCN5A promoter included 23 men (79%) and had a mean age of 44±20 years. One patient with atrial fibrillation had 2 variants. The same variants were identified in multiple unrelated patients (Table): c.‐225‐849insTG was identified in 3 patients with Brugada syndrome (patients 17–19), and c.‐225‐115G>T was identified in 1 patient with conduction disease (patient 4) and 1 patient with Brugada syndrome (patient 23). Among patients carrying a rare variant in the SCN5A promoter, conduction abnormalities were present in 18 patients (62%), and early repolarization or J‐point elevation was present in 3 patients (12%). There was no abnormal QT interval in any patient. A family history of arrhythmias and/or sudden cardiac death was present in 11 patients (37%). Among patients with Brugada syndrome who were screened, rare variants in the SCN5A promoter were present in 14 of the 469 Japanese patients (3%) but were not present in any of the 114 white patients (0%).
Table 1.
Clinical Characteristics of Patients Carrying a Rare Variant in SCN5A Promoter
| Patient No. | Sex | Age at Onset, y | Disease | Promoter Variant | ECG Abnormalities | Family History of Arrhythmias | Family History of Sudden Death |
|---|---|---|---|---|---|---|---|
| 1 | F | 62 | SND | c.‐225‐820T>C | No | No | No |
| 2 | M | 71 | Conduction disease | c.‐53+147delG | Prolonged PR interval | No | No |
| 3 | F | 82 | Conduction disease | c.‐53+265_+269GGGTT | Prolonged PR interval | No | No |
| 4 | F | 9 | Conduction disease | c.‐225‐115G>T | Left bundle branch block | Conduction disease | No |
| 5 | M | 39 | AF | c.‐225‐1340G>T | Prolonged PR interval | SVT | No |
| 6 | F | 28 | AF | c.‐225‐1315G>T | No | AF | No |
| 7 | F | 47 | AF | c.‐225‐1161A>G, c.‐53+241C>A | No | No | No |
| 8 | M | 17 | AF | c.‐53+167G>T | No | AF | No |
| 9 | M | 64 | AF, SND | c.‐53+175delA | No | AF, SND | Yes |
| 10 | M | 58 | AF | c.‐53+222G>A | RBBB, prolonged PR interval | No | No |
| 11 | M | 21 | Brugada syndrome | c.‐225‐1763T>C | RBBB | No | No |
| 12 | M | 75 | Brugada syndrome | c.‐225‐1723C>T | RBBB, AF | No | No |
| 13 | M | 44 | Brugada syndrome | c.‐225‐1531C>T | No | No | Yes |
| 14 | F | 54 | Brugada syndrome | c.‐225‐1467G>A | No | No | No |
| 15 | M | 67 | Brugada syndrome | c.‐225‐782_779delGTTT | RBBB | No | No |
| 16 | M | 32 | Brugada syndrome | c.‐225‐866insTA | No | No | No |
| 17 | M | 67 | Brugada syndrome | c.‐225‐849insTG | RBBB, left anterior hemiblock | No | Yes |
| 18 | M | 54 | Brugada syndrome | c.‐225‐849insTG | Intraventricular block, inferior early repolarization | No | No |
| 19 | M | 34 | Brugada syndrome | c.‐225‐849insTG | RBBB | No | Yes |
| 20 | M | 22 | Brugada syndrome | c.‐225‐688T>C | No | No | No |
| 21 | M | 43 | Brugada syndrome | c.‐225‐587_‐584 del CAGT | Prolonged PR interval | No | Yes |
| 22 | M | 38 | Brugada syndrome | c.‐225‐565T>C | Inferior early repolarization | No | No |
| 23 | M | 57 | Brugada syndrome | c.‐225‐115G>T | No | No | No |
| 24 | M | 39 | Brugada syndrome | c.‐53+11G>A | RBBB | No | Yes |
| 25 | M | 13 | IVF | c.‐225‐1228A>G | RBBB | No | No |
| 26 | M | 26 | IVF | c.‐225‐1052G>A | No | No | No |
| 27 | M | 61 | IVF | c.‐225‐420G>C | Prolonged PR interval | No | No |
| 28 | M | 15 | IVF | c.‐225‐374 G>T | J‐point elevation | ERS | Yes |
| 29 | M | 41 | IVF | c.‐225‐51_‐42del CCGACCCCGC | RBBB | No | No |
| Male, n=23 (79%) | 44±20 | Conduction abnormalities, n=18 (62%) | n=7 (24%) | n=7 (24%) |
AF indicates atrial fibrillation; ERS, early repolarization syndrome; IVF, idiopathic ventricular fibrillation; RBBB, right bundle branch block; SND, sinus node dysfunction; SVT, supraventricular tachycardia.
In addition, 3 novel rare variants in the SCN5A promoter were identified in patients with arrhythmia phenotypes and in controls, but the frequency of the variants was not different. Similarly, the frequency of haplotype A/B, which has been associated with cardiac conduction,23 did not differ between patients with arrhythmia phenotypes and controls (allelic frequency 0.22 for both groups).
Two promoters of SCN5A have been reported previously in mice, and the human SCN5A promoter can be mapped to the first promoter of the mouse.29 The rare variants in the SCN5A promoter identified in patients affected by various arrhythmias were mapped to the mouse genome and aligned with data from genomewide screens for binding sites of the key cardiac transcription factors identified in a previous study from our group (Figure 1).25 All SCN5A promoter variants were successfully mapped. The majority of the variants were located in regions bound by transcription factors that are important for both the development and maintenance of cardiac function, suggesting that the variants disrupt the associations of these factors that have been shown to affect an enhancer involved in SCN5A expression.25 Using a luciferase reporter assay, 6 variants identified in patients with arrhythmias (Brugada syndrome, n=3; idiopathic ventricular fibrillation, n=2; conduction disease, n=1) were functionally characterized. Each mutant promoter consistently displayed decreased promoter activity compared with the wild‐type sequences (Figure 2).
Figure 2.
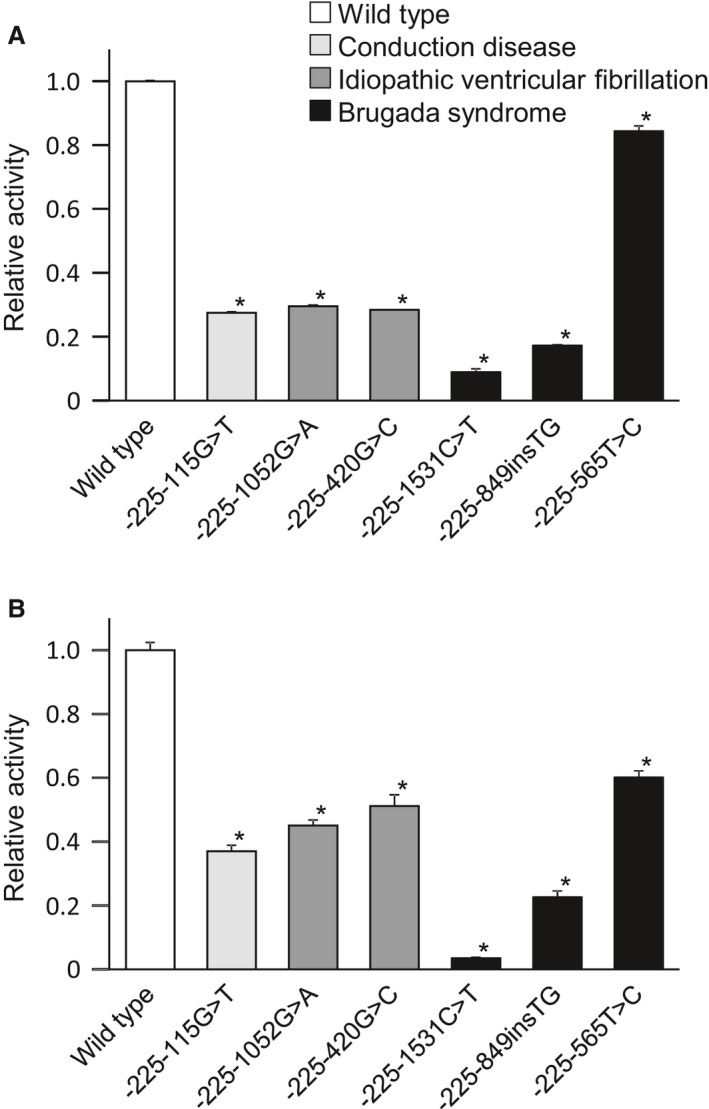
Promoter activity of rare variants in the SCN5A promoter region. In both HEK293 cells (A) and HL‐1 cardiomyocytes (B), each mutant promoter (n=4) displayed decreased activity compared with the wild‐type promoter (n=8). *P<0.01 vs the wild‐type promoter.
We identified a rare variant (c.‐53+15025C>T) in CNS28, which is important for the transcriptional regulation of SCN5A,20 in a patient with atrial fibrillation. This variant was absent in the controls and in the 1000 Genomes Project data. We also identified a variant (‐53+15307G>A) in CNS28 in 7 of the 405 patients with atrial fibrillation and 1 of the 664 controls and found a higher incidence rate in patients with atrial fibrillation than in controls (P=0.006) (Figure 3). This variant is located within 1 of the 3 tandem binding sites for transcriptional enhancer factor 1 (TEF‐1) in CNS28 and is predicted to disrupt the TEF‐1 binding site, which is well conserved across species.20 Electrophoretic mobility shift assays revealed a strong interaction between CNS28 and the nuclear protein isolated from the human heart, and the interaction was specifically blocked by anti‐TEF antibodies, indicating that TEF‐1 is associated with CNS28. Promoter reporter assays revealed that the variant was associated with decreased regulation of the promoter compared with the wild‐type sequence.
Figure 3.
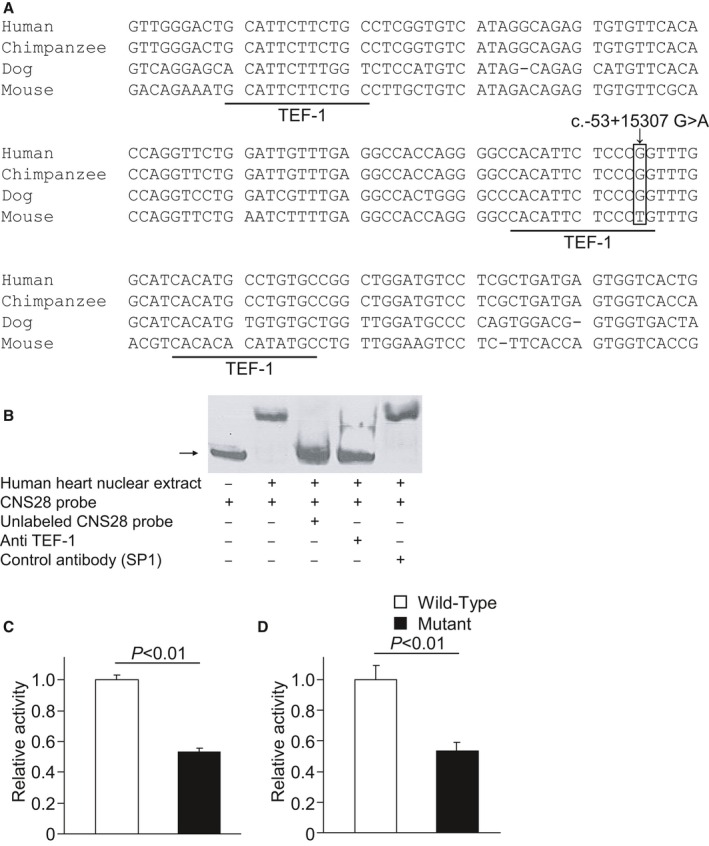
A variant in the conserved noncoding sequence 28 (CNS28) in intron 1 of SCN5A that regulates SCN5A transcription associated with atrial fibrillation. A, CNS28 includes 3 tandem binding sites for the transcriptional enhancer factor 1 (TEF‐1). A variant in CNS28 is predicted to disrupt the second binding site for TEF‐1. B, Electrophoretic mobility shift assays revealed a strong interaction between CNS28 and the nuclear protein isolated from the human heart. The interaction was specifically blocked by anti–TEF antibody. In both Chinese hamster ovary cells (C) and HL‐1 cardiomyocytes (D), the variant (n=4) decreased regulation of the promoter compared with the wild‐type sequence (n=4).
Discussion
We identified rare variants in the core promoter region of SCN5A in patients with multiple arrhythmia syndromes. Chromatin immunoprecipitation sequencing analysis revealed that the majority of the promoter variants were localized to regions bound by transcription factors and other factors that are important for transcriptional activity. In the functional analysis, the variants in the SCN5A promoter displayed decreased activity compared with the wild‐type sequences. We also identified a rare variant in the regulatory region of SCN5A transcription that was associated with decreased promoter activity. Our findings suggest that variability in SCN5A transcription affects susceptibility to a wide variety of arrhythmias.
The sodium channel plays an important role in normal cardiac function. The generation and propagation of electrical impulses throughout the atria, the ventricles, and the Purkinje network are critically dependent on normal sodium channel function; reductions in the sodium current slow the heart rate and cardiac conduction, as has been shown in heterozygous SCN5A knockout mice and during therapy with sodium channel blockers.30 Recent genomewide association studies have shown that the heritability of cardiac conduction is ≈30% to 40%, and these studies have consistently identified an association of the SCN5A and SCN10A loci with variation in cardiac conduction as one of the most significant association signals.27, 31, 32, 33, 34 Furthermore, loss‐of‐function mutations in SCN5A cause isolated conduction disease, and decreased sodium channel expression is a mechanism underlying such disease.6, 35, 36 In the present study, rare variants in the core promoter region of SCN5A were identified in patients with isolated conduction disease. Furthermore, 62% of patients carrying a rare variant in the SCN5A promoter had conduction abnormalities, further supporting the hypothesis that the sodium channel expression level is important for cardiac conduction. Similarly, the sodium channel controls cardiac excitability, and rare variants in the SCN5A promoter were identified in patients with sinus node dysfunction, which can be caused by mutations in the coding region of SCN5A.7, 8
To date, mutations in 20 different genes have been associated with Brugada syndrome, and sodium channel dysfunction is an important inherited mechanism of this disease.37, 38, 39, 40, 41, 42, 43 The causative genes of Brugada syndrome include sodium channel genes (α‐subunit gene SCN5A and β‐subunit genes) and sodium channel partner genes (GPD1L, MOG1, SLMAP, and PKP2).38, 39, 40, 42 Mutations in TRPM4 that can affect the resting membrane potential and thus reduce sodium channel availability have been identified recently in patients with Brugada syndrome.41 The present study added variants in the SCN5A promoter to the genetic cause of Brugada syndrome resulting in decreased sodium current. Evidence that common variants in 3 genes including SCN5A, SCN10A, and HEY2, all of which affect cardiac conduction, are associated with Brugada syndrome in a recent genomewide association study further supports this hypothesis.44 We screened for rare variants in the SCN5A promoter in Japanese and European patients with Brugada syndrome, but we identified unique variants only in the Japanese patients. The prevalence of Brugada syndrome is high in Southeast Asia compared with other areas, and our findings may explain, at least in part, this difference in prevalence.39, 45 Loss‐of‐function mutations in SCN5A are also associated with other forms of idiopathic ventricular fibrillation, including early repolarization syndrome characterized by J‐point elevation in the inferolateral leads and idiopathic ventricular fibrillation without J‐point elevation.4, 5 We identified rare variants in the SCN5A promoter in patients with these forms of idiopathic ventricular fibrillation. Furthermore, right bundle‐branch block has recently been associated with idiopathic ventricular fibrillation, and we also identified a rare variant in the SCN5A promoter in a patient with this disorder.46
Atrial fibrillation is the most common arrhythmia, with a high lifetime risk of 1 in 4 to 6.47 A number of studies have demonstrated a genetic contribution to atrial fibrillation susceptibility, and sodium channel dysfunction is an important pathogenic mechanism of atrial fibrillation.9, 10, 48, 49, 50 Mutations in SCN5A that result in either increases or decreases in sodium currents account for up to 6% of patients with atrial fibrillation, and we identified rare variants in the SCN5A promoter region in patients with atrial fibrillation.9, 10, 48, 49 Furthermore, a recent genomewide association study with a large cohort showed that polymorphisms in SCN5A are associated with a risk of atrial fibrillation.50 These polymorphisms in SCN5A have been associated with cardiac conduction in previous genomewide association studies, suggesting that the polymorphisms result in decreased sodium currents.27, 31 Our findings that a variant in the regulatory region of SCN5A transcription reduced promoter activity and was associated with atrial fibrillation are in accordance with previous reports.
Genetic screening is usually performed for nonsynonymous mutations in the coding exons and flanking intronic sequences, but genetic causes have not been sufficiently identified in patients with arrhythmia syndromes, especially those with arrhythmias that may result from loss‐of‐function mutations in sodium channel genes.51 In a recent study, 80% of the genome was shown to have biochemical functions, particularly portions outside of the protein‐coding regions, and increasing evidence suggests the importance of noncoding regions in cardiac electrophysiology.23, 24, 52 A haplotype of the SCN5A promoter that is commonly found in Asian persons affects transcriptional activity and modulates cardiac conduction in healthy persons, indicating that genetically determined variable transcription of the sodium channel occurs in the human heart.23 This haplotype has also modulated the extent to which sodium channel blockers slow conduction in patients with Brugada syndrome. Furthermore, another common haplotype of the SCN5A promoter is associated with the severity of phenotypes caused by a nonsynonymous mutation in SCN5A in a family affected with conduction disease and Brugada syndrome.24 In the present study, variants in the SCN5A promoter were associated with a wide variety of arrhythmia syndromes. Although variants in the SCN5A promoter may not be monogenic causes of arrhythmias, they may increase susceptibility to arrhythmias. This hypothesis is supported by evidence from a recent study that arrhythmia syndrome can be caused by multiple genetic factors.44 Future studies may elucidate an additional role of noncoding sequences in the pathogenesis of arrhythmia syndromes.
This study has several limitations. The frequency of variants was low, and thus the variants may not be responsible in a large number of patients. Although we used multiple control sets including direct sequencing data in control participants who were free from arrhythmias and in public data sets, the number of controls for white patients was relatively small. This study included only white and Japanese patients, and the results of this study may not be applied to other ethnicities. Linkage or segregation analysis was not conducted because DNA was not available in family members of probands included in this study. The same variants (c.‐225‐849insTG and c.‐225‐115G>T) were identified in multiple unrelated probands from different families. Although we did not check the relatedness of the probands, these 2 variants were absent in controls and in public databases and resulted in decreased promoter activity, suggesting that these 2 variants are associated with arrhythmia susceptibility. Although a relatively large number of rare variants in the SCN5A promoter region were identified, the number of variants that were functionally tested was limited. The functional analyses of variants were performed using HEK293 cells, Chinese hamster ovary cells, and mouse atrial myocytes (HL‐1) and the in vitro characteristics were consistent with the phenotype in the patients, but the environment was different from that in the native human cardiomyocyte, especially from that in ventricular myocytes.
Sources of Funding
This study was supported in part by grants from the Ministry of Health, Labour, and Welfare of Japan (H24‐033, Shimizu, Horie, Makita, Aiba, Kamakura, Nakano, Watanabe; 2012‐24591038, Watanabe); a research grant from Japan Agency for Medical Research and Development, AMED (15km0305015h0101) (Makita), a Grant‐in‐Aid for Scientific Research (B) 15H04823 (Makita), and a Grant‐in‐Aid for Project in Sado for Total Health (PROST) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Endo), the Dutch Heart Foundation (CVON2012‐10 Predict project), (Bezzina and Wilde), and NIH grant (HL R01092217) (Darbar).
Disclosures
None.
Acknowledgments
We thank Alfred George Jr, Cara Sutcliffe, Christiana Ingram, Gayle Kucera, and Tanya Stubblefield at Vanderbilt University; Naotaka Ohta at the National Cerebral and Cardiovascular Center; Saori Nakano at Nagasaki University; and Akinori Miyashita, Natsuyo Yahata, and Chieko Nishizawa, and Nobuyuki Takei at Niigata University for their assistance with performing and/or analyzing this work. The Vanderbilt DNA Resources Core provided technical assistance for this work.
(J Am Heart Assoc. 2016;5:e003644 doi: 10.1161/JAHA.116.003644)
References
- 1. Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. [DOI] [PubMed] [Google Scholar]
- 2. Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. [DOI] [PubMed] [Google Scholar]
- 3. Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz‐Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze‐Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. [DOI] [PubMed] [Google Scholar]
- 4. Watanabe H, Nogami A, Ohkubo K, Kawata H, Hayashi Y, Ishikawa T, Makiyama T, Nagao S, Yagihara N, Takehara N, Kawamura Y, Sato A, Okamura K, Hosaka Y, Sato M, Fukae S, Chinushi M, Oda H, Okabe M, Kimura A, Maemura K, Watanabe I, Kamakura S, Horie M, Aizawa Y, Shimizu W, Makita N. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–881. [DOI] [PubMed] [Google Scholar]
- 5. Akai J, Makita N, Sakurada H, Shirai N, Ueda K, Kitabatake A, Nakazawa K, Kimura A, Hiraoka M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000;479:29–34. [DOI] [PubMed] [Google Scholar]
- 6. Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–21. [DOI] [PubMed] [Google Scholar]
- 7. Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003;112:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AA. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003;92:14–22. [DOI] [PubMed] [Google Scholar]
- 9. Darbar D, Kucera G, Stubblefield T, Wang J, George AL, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T, Kita T, Horie M. A novel SCN5A gain‐of‐function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008;52:1326–1334. [DOI] [PubMed] [Google Scholar]
- 11. Balser JR. Sodium “channelopathies” and sudden death: must you be so sensitive? Circ Res. 1999;85:872–874. [DOI] [PubMed] [Google Scholar]
- 12. McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. [DOI] [PubMed] [Google Scholar]
- 13. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Medeiros‐Domingo A, Kaku T, Tester DJ, Iturralde‐Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales‐Quinteros S, Tusie‐Luna MT, Makielski JC, Ackerman MJ. SCN4B‐encoded sodium channel beta4 subunit in congenital long‐QT syndrome. Circulation. 2007;116:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kaab S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channel beta1 and beta2 subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F, Liao Y, Fang F, Chen Q, Tu X, Wang QK. Functional dominant‐negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem Biophys Res Commun. 2010;398:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr. Mutation in glycerol‐3‐phosphate dehydrogenase 1 like gene (GPD1‐L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kattygnarath D, Maugenre S, Neyroud N, Balse E, Ichai C, Denjoy I, Dilanian G, Martins RP, Fressart V, Berthet M, Schott JJ, Leenhardt A, Probst V, Le Marec H, Hainque B, Coulombe A, Hatem SN, Guicheney P. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011;4:261–268. [DOI] [PubMed] [Google Scholar]
- 20. Atack TC, Myers Stroud D, Watanabe H, Yang T, Hall L, Hipkens SB, Lowe JS, Leake B, Magnuson MA, Yang P, Roden DM. Informatic and functional approaches to identifying a regulatory region for the cardiac sodium channel. Circ Res. 2011;109:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang P, Kupershmidt S, Roden DM. Cloning and initial characterization of the human cardiac sodium channel (SCN5A) promoter. Cardiovasc Res. 2004;61:56–65. [DOI] [PubMed] [Google Scholar]
- 22. Yang P, Koopmann TT, Pfeufer A, Jalilzadeh S, Schulze‐Bahr E, Kaab S, Wilde AA, Roden DM, Bezzina CR. Polymorphisms in the cardiac sodium channel promoter displaying variant in vitro expression activity. Eur J Hum Genet. 2008;16:350–357. [DOI] [PubMed] [Google Scholar]
- 23. Bezzina CR, Shimizu W, Yang P, Koopmann TT, Tanck MW, Miyamoto Y, Kamakura S, Roden DM, Wilde AA. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation. 2006;113:338–344. [DOI] [PubMed] [Google Scholar]
- 24. Park JK, Martin LJ, Zhang X, Jegga AG, Benson DW. Genetic variants in SCN5A promoter are associated with arrhythmia phenotype severity in patients with heterozygous loss‐of‐function mutation. Heart Rhythm. 2012;9:1090–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, ‘t Hoen PA, Bakkers J, Barnett P, Christoffels VM. Genetic variation in T‐box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton‐Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller‐Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. Genome‐wide association study of PR interval. Nat Genet. 2010;42:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shang LL, Dudley SC Jr. Tandem promoters and developmentally regulated 5′‐ and 3′‐mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J Biol Chem. 2005;280:933–940. [DOI] [PubMed] [Google Scholar]
- 30. Royer A, van Veen TA, Le Bouter S, Marionneau C, Griol‐Charhbili V, Leoni AL, Steenman M, van Rijen HV, Demolombe S, Goddard CA, Richer C, Escoubet B, Jarry‐Guichard T, Colledge WH, Gros D, de Bakker JM, Grace AA, Escande D, Charpentier F. Mouse model of SCN5A‐linked hereditary Lenegre's disease: age‐related conduction slowing and myocardial fibrosis. Circulation. 2005;111:1738–1746. [DOI] [PubMed] [Google Scholar]
- 31. Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Lochen ML, Kong A, Thorsteinsdottir U, Stefansson K. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. [DOI] [PubMed] [Google Scholar]
- 32. Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El‐Hamamsy I, Wass MN, Dekker LR, de Jong JS, Sternberg MJ, McKenna W, Severs NJ, de Silva R, Wilde AA, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet. 2010;42:149–152. [DOI] [PubMed] [Google Scholar]
- 33. Smith JG, Magnani JW, Palmer C, Meng YA, Soliman EZ, Musani SK, Kerr KF, Schnabel RB, Lubitz SA, Sotoodehnia N, Redline S, Pfeufer A, Muller M, Evans DS, Nalls MA, Liu Y, Newman AB, Zonderman AB, Evans MK, Deo R, Ellinor PT, Paltoo DN, Newton‐Cheh C, Benjamin EJ, Mehra R, Alonso A, Heckbert SR, Fox ER. Genome‐wide association studies of the PR interval in African Americans. PLoS Genet. 2011;7:e1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kolder IC, Tanck MW, Bezzina CR. Common genetic variation modulating cardiac ECG parameters and susceptibility to sudden cardiac death. J Mol Cell Cardiol. 2012;52:620–629. [DOI] [PubMed] [Google Scholar]
- 35. Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, Kupershmidt S, Chopra N, Magnuson MA, Tanabe N, Knollmann BC, George AL Jr, Roden DM. Striking in vivo phenotype of a disease‐associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baro I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. [DOI] [PubMed] [Google Scholar]
- 37. Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J. 2012;76:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation. 2002;105:707–713. [DOI] [PubMed] [Google Scholar]
- 39. Berne P, Brugada J. Brugada syndrome 2012. Circ J. 2012;76:1563–1571. [DOI] [PubMed] [Google Scholar]
- 40. Ishikawa T, Sato A, Marcou CA, Tester DJ, Ackerman MJ, Crotti L, Schwartz PJ, On YK, Park JE, Nakamura K, Hiraoka M, Nakazawa K, Sakurada H, Arimura T, Makita N, Kimura A. A novel disease gene for Brugada syndrome: sarcolemmal membrane‐associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ Arrhythm Electrophysiol. 2012;5:1098–1107. [DOI] [PubMed] [Google Scholar]
- 41. Liu H, Chatel S, Simard C, Syam N, Salle L, Probst V, Morel J, Millat G, Lopez M, Abriel H, Schott JJ, Guinamard R, Bouvagnet P. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One. 2013;8:e54131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerrone M, Lin X, Zhang M, Agullo‐Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin‐2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watanabe H, Minamino T. Genetics of Brugada syndrome. J Hum Genet. 2016;61:57–60. [DOI] [PubMed] [Google Scholar]
- 44. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Schulze‐Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt‐Hansen J, Olesen MS, Kaab S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bezieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A‐SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alings M, Wilde A. “Brugada” syndrome: clinical data and suggested pathophysiological mechanism. Circulation. 1999;99:666–673. [DOI] [PubMed] [Google Scholar]
- 46. Aizawa Y, Takatsuki S, Kimura T, Nishiyama N, Fukumoto K, Tanimoto Y, Tanimoto K, Miyoshi S, Suzuki M, Yokoyama Y, Chinushi M, Watanabe I, Ogawa S, Antzelevitch C, Fukuda K. Ventricular fibrillation associated with complete right bundle branch block. Heart Rhythm. 2013;10:1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lloyd‐Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, D'Agostino RB, Massaro JM, Beiser A, Wolf PA, Benjamin EJ. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110:1042–1046. [DOI] [PubMed] [Google Scholar]
- 48. Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, Hedley PL, Christiansen M, Olesen SP, Haunso S, Schmitt N, Jespersen T, Svendsen JH. High prevalence of long QT syndrome‐associated SCN5A variants in patients with early‐onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–459. [DOI] [PubMed] [Google Scholar]
- 49. Ellinor PT, Nam EG, Shea MA, Milan DJ, Ruskin JN, MacRae CA. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008;5:99–105. [DOI] [PubMed] [Google Scholar]
- 50. Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, Arking DE, Muller‐Nurasyid M, Krijthe BP, Lubitz SA, Bis JC, Chung MK, Dorr M, Ozaki K, Roberts JD, Smith JG, Pfeufer A, Sinner MF, Lohman K, Ding J, Smith NL, Smith JD, Rienstra M, Rice KM, Van Wagoner DR, Magnani JW, Wakili R, Clauss S, Rotter JI, Steinbeck G, Launer LJ, Davies RW, Borkovich M, Harris TB, Lin H, Volker U, Volzke H, Milan DJ, Hofman A, Boerwinkle E, Chen LY, Soliman EZ, Voight BF, Li G, Chakravarti A, Kubo M, Tedrow UB, Rose LM, Ridker PM, Conen D, Tsunoda T, Furukawa T, Sotoodehnia N, Xu S, Kamatani N, Levy D, Nakamura Y, Parvez B, Mahida S, Furie KL, Rosand J, Muhammad R, Psaty BM, Meitinger T, Perz S, Wichmann HE, Witteman JC, Kao WH, Kathiresan S, Roden DM, Uitterlinden AG, Rivadeneira F, McKnight B, Sjogren M, Newman AB, Liu Y, Gollob MH, Melander O, Tanaka T, Stricker BH, Felix SB, Alonso A, Darbar D, Barnard J, Chasman DI, Heckbert SR, Benjamin EJ, Gudnason V, Kaab S. Meta‐analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cerrone M, Cummings S, Alansari T, Priori SG. A clinical approach to inherited arrhythmias. Circ Cardiovasc Genet. 2012;5:581–590. [DOI] [PubMed] [Google Scholar]
- 52. The Encode Project Consortium , Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]