

Abstract
Background
The gut microbiome is essential for physiological host responses and development of immune functions. The impact of gut microbiota on blood pressure and systemic vascular function, processes that are determined by immune cell function, is unknown.
Methods and Results
Unchallenged germ‐free mice (GF) had a dampened systemic T helper cell type 1 skewing compared to conventionally raised (CONV‐R) mice. Colonization of GF mice with regular gut microbiota induced lymphoid mRNA transcription of T‐box expression in T cells and resulted in mild endothelial dysfunction. Compared to CONV‐R mice, angiotensin II (AngII; 1 mg/kg per day for 7 days) infused GF mice showed reduced reactive oxygen species formation in the vasculature, attenuated vascular mRNA expression of monocyte chemoattractant protein 1 (MCP‐1), inducible nitric oxide synthase (iNOS) and NADPH oxidase subunit Nox2, as well as a reduced upregulation of retinoic‐acid receptor‐related orphan receptor gamma t (Rorγt), the signature transcription factor for interleukin (IL)‐17 synthesis. This resulted in an attenuated vascular leukocyte adhesion, less infiltration of Ly6G+ neutrophils and Ly6C+ monocytes into the aortic vessel wall, protection from kidney inflammation, as well as endothelial dysfunction and attenuation of blood pressure increase in response to AngII. Importantly, cardiac inflammation, fibrosis and systolic dysfunction were attenuated in GF mice, indicating systemic protection from cardiovascular inflammatory stress induced by AngII.
Conclusion
Gut microbiota facilitate AngII‐induced vascular dysfunction and hypertension, at least in part, by supporting an MCP‐1/IL‐17 driven vascular immune cell infiltration and inflammation.
Keywords: angiotensin II, arterial hypertension, gut microbiota, inflammation, monocytes, vascular dysfunction
Subject Categories: Inflammation, Vascular Biology, High Blood Pressure, Pathophysiology
Introduction
The absence of microbiota in germ‐free (GF) mice, that are born and raised in sterile flexible film isolators, demonstrated that the presence of commensal bacteria is essential for normal immune development.1, 2 The immune profile in GF mice is characterized by a default T helper cell type 2 (Th2) bias, a significant reduction in proinflammatory interleukin (IL)‐17‐producing CD4+ T cells3 as well as T regulatory cells4 and less IL‐12 formation5 compared to mice with an intact commensal gut bacterial profile. GF mice are characterized by reduced white adipose tissue inflammation and presence of gut microbiota increases macrophage content of that tissue with a polarization toward the M1 phenotype.6 Furthermore, mucosal immunity and gut microbiota have been shown to activate myelomonocytic cells, drive their development toward an inflammatory phenotype,6, 7 promote their major histocompatibility class II expression8 and to be essential for a proper systemic inflammatory response after lipopolysaccharide challenge9 or ischemia/reperfusion injury.10 Regarding the potential of the spleen to act as a reservoir for inflammatory monocytes,11 it is noteworthy that the spleen of GF mice forms significantly less macrophage colony‐stimulating factor–dependent colonies than the spleen of conventionally raised (CONV‐R) mice.8
Inflammatory monocytes and macrophages have been identified as powerful mediators of inflammation and oxidative stress in arterial hypertension and are crucial prerequisites in the pathogenesis of high blood pressure and vascular dysfunction.12, 13, 14, 15, 16, 17 Ablation of lysozyme‐M‐positive cells led to reduced angiotensin II (AngII)‐induced accumulation of myelomonocytic cells in the vessel wall and attenuated the blood pressure increase in response to AngII. We have recently identified interferon gamma (IFN‐γ), a cytokine connected to Th1 immune response, as a decisive cytokine to skew monocytes toward a proinflammatory phenotype in AngII‐induced vascular dysfunction and inflammation: Mice lacking the transcription factor, T‐box, expressed in T cells (encoded by the Tbx21 gene), which directs IFN‐γ formation in immune cells, were protected from AngII‐induced vascular injury.18 Additionally, IL‐17A can act as a mediator of AngII‐induced arterial hypertension19 and of inflammation‐associated vascular dysfunction20—at least in part, by recruiting neutrophil granulocytes to the aortic vessel wall. Besides, it has been shown that the development of IL‐17‐producing CD4+ T cells (Th17 cells) in the intestine is dependent on gut microbiota.21 In a mouse model of portal vein ligation, GF mice were partially protected from portal hypertension compared to colonized controls.22
It has been reported, that AngII can induce gut dysbiosis in rats; when treated with antibiotics reducing Firmicutes species, blood pressure was attenuated.23 To date, it is unknown whether the presence of gut microbiota contributes to regulation of basal vascular tone and of AngII‐induced arterial hypertension in vivo. We hypothesized that enteric microbiota might potentially play a role in facilitating myelomonocytic cells, such as monocytes, to respond to the blood pressure hormone, AngII. To address this issue, we investigated the role of gut microbiota in the pathogenesis of AngII‐induced vascular dysfunction and hypertension using the GF mouse technology. Here, we demonstrate that GF mice are protected from AngII‐induced systemic vascular inflammation, supporting a decisive role of gut microbiota for the regulation of vascular tone in arterial hypertension.
Materials and Methods
Mice
All procedures performed on mice were approved by the Institutional Animal Care and Use Committee (Landesuntersuchungsamt Rheinland‐Pfalz, Koblenz, Germany; animal experimental approval 23‐177‐07/G 12‐1‐002), following the German Law on the Protection of Animals. All mice were housed in a barrier facility (Translational Animal Research Center, University Medical Center Mainz, Mainz, Germany) with a 12‐hour light‐dark cycle and kept in EU Type II IVC cages with 2 to 5 mice per cage under specific pathogen‐free or GF conditions with standard autoclaved lab diet (PMI, St. Louis, MO) and water ad libitum and a 22±2°C room temperature. GF status of mice was tested on a weekly basis by polymerase chain reaction (PCR) for detection of 16S rDNA and by culture‐based methods. Male 10‐ to 16‐week‐old GF mice that were born and raised in sterile, flexible film isolators versus CONV‐R mice on a Swiss Webster and C57BL/6J background were used. GF mice were colonized for 14 days with normal gut microbiota to obtain conventionally derived (CONV‐D) mice. Colonization was achieved by oral gavage of ex‐GF mice with a suspension of the cecal content of a CONV‐R mice in 5 mL of PBS.
In vivo treatment of CONV‐R versus GF mice with AngII (1 mg/kg per day for 7 days) versus sham was achieved by osmotic minipumps in the case of GF mice with a specific sterile implantation technique. In brief, Alzet miniosmotic pumps (model 1007D; Alzet, Cupertino, CA) were filled and prepared according to the manufacturer′s instruction under a sterile hood, then transported in a sterile 15ml Falcon tube filled with 0.9% NaCl solution to the GF mouse isolator, where it was sterilized by spraying with a chlorine dioxide‐based desinfectant in the isolator port. Isoflurane, together with analgetics and sedation, was also transferred into the prepared experimental isolator. Implantation of the pumps was performed with heat‐sterilized surgical instruments; for anesthesia and analgesia, mice received intraperitoneal injections of midazolam (5 mg/kg; Ratiopharm GmbH, Ulm, Germany), medetomidine (0.5 mg/kg body weight; Pfizer Deutschland GmbH, Berlin, Germany), and fentanyl (0.05 mg/kg body weight; Janssen‐Cilag GmbH, Neuss, Germany). After the surgical procedure, animals were administered atipamezole (0.05 mg/kg), flumazenil (0.01 mg/kg), and naloxone (0.024 mg/kg) subcutaneously to antagonize anesthesia.
Measurement of blood pressure was performed using the noninvasive blood pressure system for mice (tail‐cuff method; Kent Scientific CODA‐STD, Torrington, CT), as well as the assessment of vascular function (vascular relaxations studies ex vivo), was performed as described previously.12 Assessment of blood pressure in the GF mice was performed within 120 minutes after export of mice out of their sterile environment.
As experimental groups, CONV‐R mice with (n=15) and without AngII treatment (n=17) and GF mice with (n=32) and without AngII (n=27) treatment as well as CONV‐R (n=5), CONV‐D (n=5), and GF mice (n=9) on Swiss Webster background were the analyzed.
Vascular Tone Experiments
We studied the vascular responsiveness to vasconstrictors and vasodilators (increasing doses of phenylephrine, prostaglandin F2 alpha, acetylcholine, and nitroglycerin in a range form 10−9–10−4 mol/L) of isolated aortic rings of GF mice versus CONV‐R and CONV‐D mice. Isolated aortas were cut into 4 mm segments and mounted on force transducers (Kent Scientific Corporation; Powerlab; ADInstruments, Spechbach, Germany) in organ chambers filled with Krebs‐Henseleit solution (37°C, pH 7.35, containing 98.93 mmol/L of NaCl, 4.69 mmol/L of KCl, 2.49 mmol/L of CaCl2, 1.2 mmol/L of MgSO4, 0.613 mmol/L of K2HPO4, 25 mmol/L of NaHCO3, and 11.1 mmol/L of d‐glucose) bubbled with carbogen gas (95% O2/5% CO2) and containing 10 μmol/L of indomethacin to prevent endogenous synthesis of prostaglandins. To test for vasorelaxation in response to acetylcholine (ACh) and nitroglycerin (glycerol trinitrate; GTN), aortic segments were stretched gradually over 30 minutes to reach a resting tension of 1.0 g. Following preconstriction with prostglandin F2α (3.3 μmol/L) and phenylephrine (cumulative concentration‐constriction‐curves, 10−8–10−5.5 mol/L) to reach 50% to 80% of maximal tone induced by KCl, cumulative concentration‐relaxation curves were recorded in response to increasing concentrations of ACh and GTN (10−9–10−4.5 mol/L).
Detection of Reactive Oxygen Species Formation Using L012‐Enhanced Chemiluminescence and Fluorescence Oxidative Microtopography
We analyzed oxidative burst of white blood cells in whole blood by L012 enhanced chemiluminescence (CL) and oxidative stress in the vasculature by dihydroxyethidium staining of aortic cryosections. First, 200 IU of heparine was injected into the beating heart of the mouse and venous blood then drawn from the right ventricle. Heparinized blood was kept at room temperature and CL was measured immediately. L012 enhanced chemiluminescence (ECL) signals were counted in 10μL samples with 100 μmol/L of L012 in the presence of PdBU (10 μmol/L) at intervals of 1 minute using a Lumat LB9507 from Berthold Technologies (Bad Wildbad, Germany). CL was expressed as counts per minute after incubation for 10 minutes. Thoracic cryosections of aortas were stained with the superoxide‐sensitive dye, dihydroethidium (DHE; 1 μmol/L) for fluorescence oxidative microtopography12: Aortas were rinsed and cleaned and then cut into 4‐mm sections to then be incubated in Krebs–Henseleit solution containing 0.1 mg/mL of aprotinin, 0.2 mg/mL of pepstatin, and 0.5 mg/mL of leupeptin for 10 minutes at 37°C and then embedded in Tissue‐Tek and frozen in liquid nitrogen. We cut aortic cryosection of 8 μm, stained with DHE, and incubated them for 30 minutes at 37°C. Green autofluorescence from aortic lamina and red ethidium fluorescence inside the reactive oxygen species (ROS)‐producing cells was detected by fluorescence light microscopy (Zeiss Axiovert 40 CFL microscope, Zeiss lenses and Axiocam MRm camera; Zeiss, Oberkochen, Germany) and analyzed with the AxioVision data acquisition software (Zeiss).
mRNA Expression Analysis
Total RNA was isolated from aortic tissue or splenic CD4+ lymphocytes (stimulated on anti‐CD3‐coated plates in RPMI containing 5% FCS and 1% penicilin/streptomycin and anti‐CD28 [1:500] overnight) using guanidinium thiocyanate after controlled crushing with TissueLyser (Qiagen, Hilden, Germany).
Real‐time quantitative reverse‐transcriptase PCR (qRT‐PCR) was performed using one‐step qRT‐PCR: 0.05 μg/μL of total RNA was used for analysis with the QuantiTect Probe RT‐PCR kit (Qiagen). A Taq‐Man Gene Expression assay for the denoted primers was used as probe‐and‐primer set (Applied Biosystems, Foster City, CA).
Analysis was performed for mRNA expression of the corresponding transcription factors, (T‐bet; Mm0045090‐m1), forkhead box protein p3 (Foxp3 marks regulatory T‐cell development; Mm01351178_g1), GATA binding protein 3 (GATA‐3; marks Th2 cells; Mm00484683_m1), (retinoic‐acid receptor‐related orphan receptor gamma t (RORγt) directs IL‐17 formation; Mm01261022_m1) as well as inducible nitric oxide synthase (iNOS; Mm00440485_m1), NADPH oxidase 2 (Nox2; Mm00514478_m1), chemokine (C‐C motif) ligand 2 (Ccl‐2; Mm00441242_m1), angiotensin type 1 receptor (Agtr1a; Mm01957722_s1), and vascular cell adhesion molecule 1 (VCAM‐1; (Mm00449197_m1). The relative expression levels of the respective samples to TATA‐box binding protein as endogenous control (housekeeping gene) were calculated with the delta‐delta threshold cycle method.24
Cytokine Formation
The polarization pattern of the systemic immune profile was assessed by analyzing spleen‐derived lymphocytes. We measured cytokine formation of these cells after antiCD3 and antiCD28 stimulation eliciting a T‐cell response by ELISA and by the bead‐based Bio‐Plex assay (96‐well plate, Luminex 200; Life Technologies, Carlsbad, CA) (IFN‐γ, IL‐4, and IL‐10) according to a published protocol.18
Fluorescence‐Activated Cell Sorting Analysis of Immune Cells
The immunological phenotype of mice was examined by flow cytometric analysis of B220+ and TCRß+ lymphoid cells (representing B cells and T cells), as well as CD11b+, Gr‐1+ (Ly6G+, Ly6C+), and F4/80+ myeloid cells (representing neutrophils and monocytes/macrophages) in the whole blood, spleen, aorta, and kidney. Blood erythrocytes were hemolyzed by BD (BD Biosciences, San Jose, CA) fluorescence‐activated cell sorting (FACS) lysing solution. For the analysis of cells in the mouse aorta, the total aorta was digested by using collagenase II (1 mg/mL) and DNase I (50 μg/mL) or Liberase (1 mg/mL) for 30 minutes at 37°C. For flow cytometric analysis of kidney cells, the kidney was flushed blood free and digested with collagenase D (2 mg/mL) and DNase (100 μg/mL) for 30 minutes at 37°C and further homogenized with gentleMACS C tubes (Miltenyi Biotenc, Cambridge, MA). To block nonspecific Fc‐receptor‐mediated binding sites, cells were preincubated with unlabeled antibody against CD16/CD32 for 10 minutes (Fc‐block). We also phenotyped vascular immune cells in GF versus CONV‐R mice.
Intravital Fluorescence Microscopy
Mice were anesthetized with midazolam (5 mg/kg; Ratiopharm GmbH), medetomidine (0.5 mg/kg body weight; Pfizer Deutschland GmbH), and fentanyl (0.05 mg/kg body weight; Janssen‐Cilag GmbH) given as an intraperitoneal injection. The right and the left common carotid artery of each mouse were dissected free. First, 100 μL of acridine orange (0.5 mg/mL; Sigma‐Aldrich, St. Louis, MO) was injected by a jugular vein catheter (0.28 mm ID, 0.61 mm OD; Smiths Medical Deutschland GmbH, Grasbrunn, Germany) to stain circulating leukocytes in vivo. A high‐speed wide‐field Olympus BX51WI fluorescence microscope using a long‐distance condenser and a 10× (NA 0.3) water immersion objective with a monochromator (MT 20E; Olympus Deutschland GmbH, Hamburg, Germany) and a charge‐coupled device camera (ORCA‐R2; Hamamatsu Photonics, Hamamatsu City, Japan) was used for microscopical analysis. Image acquisition and analysis was performed with Realtime Imaging System eXcellence RT (Olympus Deutschland GmbH) software. Leukocytes were quantified in 4 fields of view (150×100 μm) per carotid artery and could be graduated in adhering and rolling leukocytes.25
Histology and Immunohistology
Cardiac fibrosis was determined on paraffin‐embedded cross‐sections through the left ventricle using Masson's trichrome (MTC) stain. Macrophages were visualized using monoclonal antibodies against mouse Mac2 (clone M3/38; BIOZOL Diagnostic, Munich, Germany). Neutrophils were detected using polyclonal antibodies against mouse neutrophil elastase (ELANE; abcam, Cambridge, MA). Results were quantified by automatically determining the positive area (blue signal for MTC, red‐brown signal for Mac2) using image analysis software (Image ProPlus, version 7.0; Media Cybernetics, Inc., Bethesda, MD) or by manually counting the number of positive cells (ELANE). In each case, 2 or more 200× microscope fields were evaluated and results averaged per mouse.
Cardiomyocyte membranes were visualized on acetone‐fixed cardiac cryosections using FITC‐labeled wheat germ agglutinin (WGA; Molecular Probes, Eugene, OR) followed by determination of the single cardiomyocyte cross‐sectional area. Per cross‐section, 100 randomly selected cardiomyocytes were evaluated and results averaged.
Cardiac Ultrasound Measurements
Anesthesia of mice was induced in a chamber (2–4% isoflurane mixed with 0.2 L/min 100% O2) and maintained with a face mask (1–2% isoflurane with 0.2 L/min 100% O2). Animals were kept on a heated table mounted on a rail system (VisualSonics, Toronto, Ontario, Canada). Ultrasound was performed with the Vevo 770 System and a 40 MHz mouse transducer (VisualSonics). Heart rate was determined and body temperature was monitored using a rectal probe and maintained at 37°C. Left ventricular wall thickness, intraventricular septum thickness, left ventricular end‐diastolic, end‐systolic volumes, and left ventricular shortening fraction were determined. Assessment of cardiac function in GF mice was performed within 120 minutes after bringing mice out of their sterile environment.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism software (version 5; GraphPad Software, Inc., La Jolla, CA). Data were analyzed for normal distribution with the Kolmogorow–Smirnow test.
When normal distribution was given, the 2‐tailed unpaired Student's t‐test, 1‐way ANOVA test with Bonferroni post‐hoc test or 2‐way‐ANOVA were applied. If there was no normal distribution, Wilcoxon–Mann–Whitney or the Kruskal–Wallis test with Dunn's multiple comparison or comparison of selected columns were used as appropriate and indicated in the figure legends.
Data are presented as box plots with median and interquartile range; P values of <0.001, <0.01, and <0.05 were considered statistically significant and marked by 3, 2, and 1 asterisks, respectively.
Results
AngII‐Induced Vascular Inflammation Is Attenuated in GF Mice
To address the impact of gut microbiota on vascular tone in unchallenged animals, we studied GF and CONV‐R mice on a Swiss Webster background. Vascular constriction in response to phenylephrine as well as endothelium‐dependent (ACh) and endothelium‐independent (GTN) vascular relaxation was not statistically different between GF and CONV‐R Swiss Webster mice (Figure S1A through S1C). Interestingly, T‐box 21 (Tbx21) mRNA expression in isolated splenocytes was reduced in GF, compared to CONV‐R, mice. GF mice that were recolonized with gut microbiota to derive the status of CONV‐R mice (CONV‐D mice) had splenocytic Tbx21 mRNA expression levels comparable to CONV‐R mice (Figure S1D). Accordingly, IFN‐γ, the signature cytokine of a type 1 immune response, was significantly reduced in supernatants of antiCD3‐ and antiCD28‐stimulated splenocytes isolated from GF mice compared with CONV‐R counterparts and partially recovered in CONV‐D. The same tendency was observed for IL‐4 and IL‐10 (Figure S1E). The proinflammatory role of IFN‐γ negatively impacts on vascular relaxation. Consequently, we observed an augmented vascular constriction and a mild endothelial dysfunction in CONV‐D, compared to CONV‐R and GF mice (Figure S1A and S1B). These results indicate a microbiota‐induced augmentation of vascular type 1 immune response and a role of the microbiota for endothelial function.
Based on the concept that a type 1 immune response could favor AngII‐induced vascular injury,18, 26, 27 we further investigated the impact of microbiota on vascular dysfunction. For this purpose, we used C57BL/6 mice because this mouse strain is known to be the most suitable inbred strain to study arterial disease attributed to default Th1 cell skewing, as observed, for instance, in a more‐rapid development of atherosclerosis.28, 29 AngII‐infused (1 mg/kg per day for 7 days) GF C57BL/6 mice were protected from ROS formation in the vessel wall as well as in whole blood compared to CONV‐R mice (Figure 1A and 1B). AngII‐infused CONV‐R mice had increased aortic mRNA expression of the retinoic‐acid receptor‐related orphan receptor gamma t (Rorγt) directing IL‐17A formation and of the Th2 cell transcription factor, GATA‐3, which were not changed in the GF mouse model (Figure 1C). Vascular mRNA levels of the phagocyte type NADPH oxidase, nox2, and of inducible nitric oxide synthase (inos)—both indicating invasion of inflammatory cells as well as the generation of ROS/reactive nitrogen species—were reduced in AngII‐infused GF mice compared to CONV‐R mice (Figure 1D). These results suggest that conductance vessels of GF mice are less prone to vascular inflammation and production of vascular ROS upon AngII challenge and that this could be attributed to changed T‐cell skewing.
Figure 1.
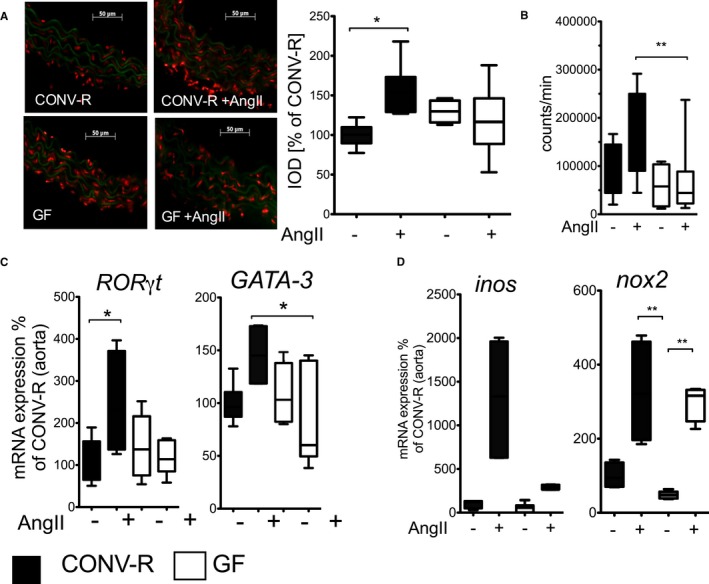
Germ‐free (GF) mice are protected from AngII‐induced vascular oxidative stress and inflammatory gene expression. CONV‐R versus GF mice on C57BL/6 background±AngII (1 mg/kg per day) were studied after 7 days of in vivo treatment. A, Aortic superoxide formation. Left: Representative dihydoethidium photomicrotopographs of aortic cryosections; superoxide formation appears in red. Right: Quantification, n=3 to 6 mice per group, Kruskal–Wallis test with Dunn′s multiple comparison. B, Respiratory burst in whole blood in the presence of the phorbolester, PDBu (100 nmol/L), measured by L012 (100 μmol/L) ECL. 1‐way ANOVA with Bonferroni post‐hoc test, n=5 to 13 mice per group. C, Aortic Ror‐γt and GATA‐3 mRNA expression. Kruskal–Wallis test with Dunn′s multiple comparison of selected columns (Rorγt: n=3 to 10 mice per group, CONV‐R vs CONV‐R+AngII and GF vs GF+AngII were compared. GATA‐3: n=3 to 8 mice per group, CONV‐R+AngII and GF+AngII were compared). D, Aortic iNOS and Nox2 mRNA expression. Kruskal–Wallis test with Dunn′s multiple comparison, iNOS: n=4 to 12 mice per group, Nox2: n=4 to 12 mice per group. AngII indicates angiotensin II; CONV‐R, conventionally raised; ECL, enhanced chemiluminescence; GATA‐3, GATA binding protein 3; iNOS, inducible nitric oxide synthase; Nox2, NADPH oxidase 2; Rorγt, retinoic acid‐related orphan nuclear receptor gamma t.
AngII‐Induced Aortic Recruitment of Myelomonocytic Cells Is Reduced in Germfree Mice
The interplay and mutual activation of pro‐inflammatory T cells with myelomonocytic cells is of great importance in the development of arterial hypertension.30, 31, 32 Aortic mRNA expression of Ccl‐2 encoding for monocyte chemoattractant protein 1 (MCP‐1), the most relevant chemokine for monocyte attraction in hypertension,15 increased in response to AngII and was attenuated in GF mice. In contrast, neither aortic mRNA expression levels of Agtr nor of VCAM‐1, both important for the direct action of AngII on the vasculature and for leukocyte adhesion, were influenced by the absence of microbiota (Figure 2A and Figure S2A). We observed attenuated rolling and adhesion of leukocytes when analyzing the carotid arteries in AngII‐treated CONV‐R and GF mice by intravital videomicroscopy (IVM; Figure 2B and Figure S2B). These results indicate a role for Ccl‐2 in leukocyte recruitment to the arterial vessel wall and for vascular dysfunction in colonized mice. In line with the reduced recruitment of myelomonocytic cells in GF mice, we detected a significant accumulation of CD45+ inflammatory cells in aortas of AngII‐treated CONV‐R mice that was blunted in GF mice (Figure 2C). More specifically, flow cytometric analysis of isolated aortic tissue revealed an increase of CD11b+ myelomonocytic cells and of CD11b+Ly6G+ neutrophils as well as inflammatory CD11b+Ly6G−Ly6Chi monocytes (Figure 2D) in response to AngII, which was attenuated in the absence of gut microbiota. These findings suggest that MCP‐1‐dependent chemotaxis of myelomonocytic cells is mitigated in GF mice, leading to attenuated vascular inflammation.
Figure 2.
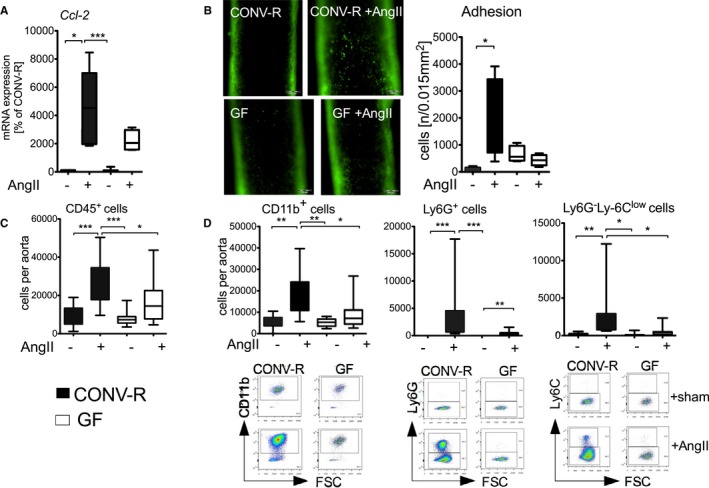
Absence of gut microbiota attenuates AngII‐induced vascular infiltration of myelomonocytic cells into the aortic vessel wall. CONV‐R versus GF mice on C57BL/6 background±AngII (1 mg/kg per day) were studied after 7 days of in vivo treatment. A, Aortic Ccl‐2 mRNA expression. Kruskal–Wallis test with Dunn′s multiple comparison test, n=4 to 12 mice per group. B, Intravital videomicroscopy imaging of rolling leukocyte in carotid arteries of AngII‐infused GF and CONV‐R mice. Left panel, representative pictures; right panel, quantification. Kruskal–Wallis test with Dunn′s multiple comparison test, n=3 to 5 mice per group. C, Flow cytometric analysis of total CD45.2+ cells per aorta. Kruskal–Wallis test with Dunn′s multiple comparison test, n=10 to 11 mice per group. D, Flow cytometric analysis of CD11b+ cells as well as CD11b+Ly6G+ and CD11b+Ly6G−Ly6C+ cells per aorta. Pregating on living CD45+ cells. Total cell numbers are shown in bar graphs. Kruskal–Wallis test with Dunn′s multiple comparison test, n=10 to 12 mice per group. Below, representative fluorescence‐activated cell sorting plots are given. AngII indicates angiotensin II; Ccl‐2, chemokine (C‐C motif) ligand 2; CONV‐R, conventionally raised; GF, germ‐free.
AngII‐Induced Vascular Dysfunction, Arterial Hypertension, and Endorgan Damage Are Blunted in the Absence of Gut Microbiota
In contrast to CONV‐R mice, GF mice were protected from AngII‐induced vascular endothelial and smooth muscle dysfunction (assessed by concentration‐relaxation curves of isolated aortic rings in response to ACh and GTN, respectively; Figure 3A and 3B). Importantly, the blunted immune phenotype of GF mice was not only linked to improved vascular function, but also prevented AngII‐induced blood pressure increase in GF, compared to CONV‐R, mice (Figure 3C). In parallel, levels of infiltrating CD45.2+ and CD11b+ cells were reduced in kidneys of AngII‐treated GF mice in comparison to AngII‐treated CONV‐R mice (Figure 3D and 3E).
Figure 3.
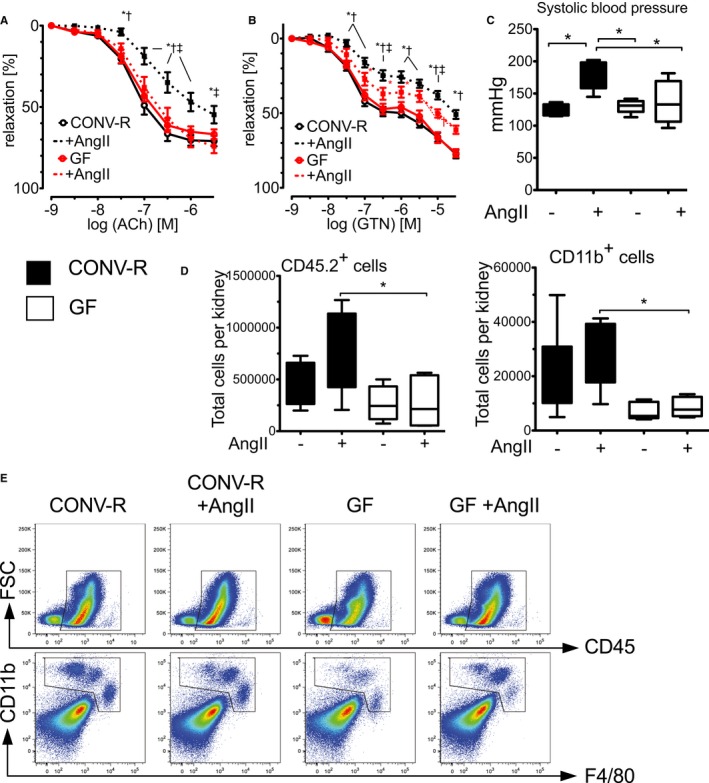
Germ‐free (GF) mice are protected from AngII‐induced vascular dysfunction and blood pressure increase. CONV‐R versus GF mice on C57BL/6 background±AngII (1 mg/kg per day) were studied after 7 days of in vivo treatment. A and B, Cumulative concentration relaxation curves of isolated aortic rings in response to endothelium‐dependent (ACh) and endothelium‐independent vasodilatators (GTN). Two‐way ANOVA, both n=6 to 15 mice per group. *P<0.05 versus CONV‐R; † P<0.05 versus GF; ‡ P<0.05 versus GF+AngII. C, Blood pressure of CONV‐R and GF mice with and without AngII challenge, measured by tail‐cuff method. Kruskal–Wallis test with Dunn′s multiple comparison, n=4 to 5 mice per group. D, Flow cytometric analysis of kidneys of CONV‐R control mice and GF mice with and without AngII treatment. CD45+ as well as CD11b+ cells (pregated on living CD45+ cells) per kidney in CONV‐R control mice and GF mice±AngII treatment are shown in bar graphs. Statistical analysis was performed with 1‐way ANOVA with Bonferroni post‐hoc test, n=5 to 8 mice per group. Below, representative fluorescence‐activated cell sorting plots are given. ACh indicates acetylcholine; AngII, angiotensin II; CONV‐R, conventionally raised; GTN, glyceryl trinitrate.
Clinical outcomes of patients with arterial hypertension are largely driven by end‐organ damage, such as hypertensive heart or kidney disease. AngII infusion increased cardiac fibrosis and caused an accumulation of MAC‐2+ as well as ELANE+ myelomonocytic cells in the heart of CONV‐R mice, which was attenuated in GF controls (Figure 4A). Cardiomyocytes were hypertrophic in AngII‐infused CONV‐R, but not in GF mice (Figure 4B). Accordingly, cardiac AngII‐induced hypertrophy and ‐contractility was reduced in GF, compared to CONV‐R, mice, indicating that hypertension‐induced end‐organ damage was attenuated by the absence of gut microbiota (Figure 5).
Figure 4.
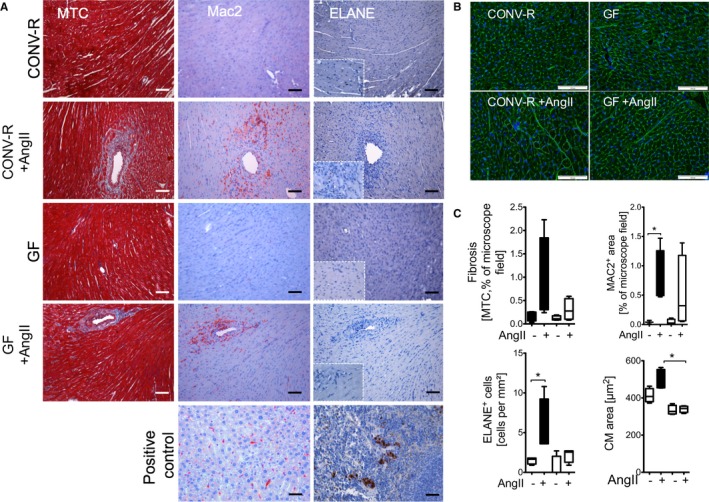
AngII‐triggered cardiac fibrosis, as well as accumulation of myelomonocytic cells, is attenuated in GF mice. CONV‐R versus GF mice±AngII (1 mg/kg per day) were studied after 7 days of in vivo treatment. A, Cardiac sections were stained with Masson's trichrome (MTC) staining to assess fibrosis and analyzed by immunohistochemistry staining for MAC‐2+ as well as ELANE + cells to assess macrophage as well as neutrophil accumulation, respectively. B, Representative images of cardiomyocyte cross‐sectional areas of cardiac sections. FITC‐labeled wheat germ agglutinine probed cardiac membranes appear in green. C, Quantification. MAC‐2, ELANE, and MTC: Kruskal–Wallis test with Dunn′s multiple comparison of selected columns (CONV‐R vs CONV‐R AngII, GF vs GF AngII), n=3 to 4 mice per group. Cardiomyocyte (CM) area: Kruskal–Wallis test with Dunn′s multiple comparison of selected columns (GF vs GF AngII, CONV‐R AngII vs GF AngII), n=3 to 4 mice per group. AngII indicates angiotensin II; CONV‐R, conventionally raised; GF, germ‐free.
Figure 5.
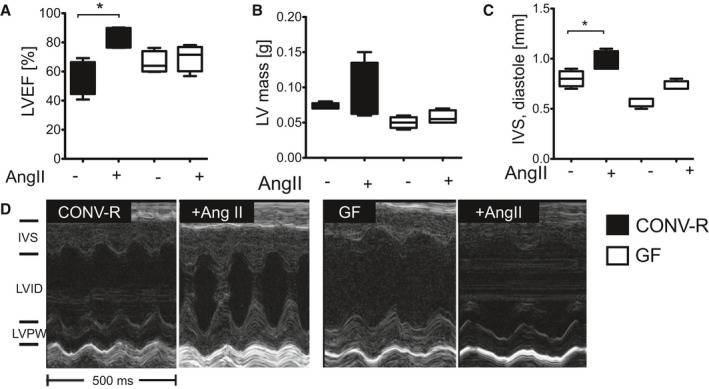
End‐organ damage in AngII‐induced hypertension is attenuated in GF mice. CONV‐R versus GF mice±AngII (1 mg/kg per day) were studied after 7 days of in vivo treatment. A through C, Left ventricular (LV) ejection fraction (LVEF; %), LV mass (g), as well as intraventricular septum (IVS) thickness at diastole (mm) were measured and delineated by high‐frequency ultrasound. Kruskal–Wallis test with Dunn′s multiple comparison. n=4 mice per group; *P<0.05. D, Representative ultrasound pictures including IVS, left‐ventricular internal diameter (LVID), as well as the left‐ventricular posterior wall (LVPW) at diastole (over 500 ms). AngII indicates angiotensin II; CONV‐R, conventionally raised; GF, germ‐free.
Discussion
We demonstrate here that the absence of gut microbiota protects mice from AngII‐induced arterial hypertension, vascular dysfunction, and hypertension‐induced end‐organ damage. This protection appears to be mediated by inhibition of accumulation of inflammatory myelomonocytic cells in the vasculature. It was discovered recently that a T‐bet gradient determines development of innate lymphoid cells (ILCs) characterized by expression of RORγt in gut lymphoid tissue. These ILCs are capable of forming IFN‐γ (controlled by T‐bet) and IL‐17 (controlled by RORγt) and share features of natural killer cells and cytotoxic T cells.33 GF mice are characterized by altered IL‐12 formation, T‐bet/IFN‐γ signaling,33 and RORγt/IL‐17 signaling.3, 21 In accord with that, mice that had been depleted of their enteric microbiota were partially protected from experimental encephalomyelitis attributed to an attenuated proinflammatory response.34 Vice versa, GF mice develop an exaggerated Th2 immune response in an allergic airway inflammation model with less pulmonary infiltration of CD11b+ cells and less alveolar macrophages as compared to CONV‐R mice.35
Importantly, all of the cytokine signaling outlined above has been shown to be causally involved in AngII‐induced vascular dysfunction and arterial hypertension.18, 19, 36 Our data indicate that this signaling is mechanistically involved in AngII‐induced vascular dysfunction and is attenuated in the absence of gut microbiota.
Blood pressure was not different between GF and CONV‐R mice under baseline conditions. This is compatible with observations made in mice where the gut microbiota has been decimated by antibiotic treatment.37, 38 However, the absence of gut microbiota conferred protection from blood pressure increase in response to AngII in vivo. The observation that both the IL‐17 and the IFN‐γ pathways, triggering mobilization, activation, and vascular infiltration of Ly6G+ and Ly6Chi myelomonocytic cells, were attenuated in AngII‐infused GF mice, at least in part explains the critical role of the gut microbiota for promoting systemic inflammatory responses. The dampened type 1 immune response phenotype in GF mice resulted in blunted vascular oxidative stress as well as reduced vascular nox2 and inos expression, which is mainly driven by inflammatory cells accumulating in the vasculature.12, 39
In addition, aortic mRNA expression of Ccl‐2 encoding for MCP‐1, the ligand of the chemokine receptor, CCR‐2, which is most important for myelomonocytic cell recruitment in response to AngII,15, 16 was reduced in GF, compared to CONV‐R, mice. In our study, vascular Agtr1 levels were not affected by the absence of gut microbiota, and infiltrating myelomocytic cells chemoattracted by MCP‐1 as well as by IL‐17A are known to be required to mediate blood pressure increase in response to AngII.12, 19 Interestingly, lack of T‐bet in the myeloid department will result in reduced IL‐12 formation as well as in attenuation of the MCP‐1/CCR‐2 axis in the vasculature, with implications for AngII‐induced hypertension.18 Conversely, we have recently shown that monocytes with increased CCR‐2 expression adhere more to the vascular endothelium.25 It is therefore very likely that systemic attenuation of T‐bet/IFN‐γ33 and RORγt/IL‐173, 21 as well as MCP‐1/CCR‐2 signaling in GF mice could contribute to attenuation of AngII‐induced vascular injury.
There are preliminary data that GF ApoE−/− mice are protected from high‐fat‐diet–induced atherosclerosis, the classical animal model of a chronic inflammatory vascular disease.40 ApoE−/− mice eradicated by broad‐spectrum antibiotics and fed a choline‐rich diet were partially protected from vascular lesion formation and aortic infiltration of F4/80+ macrophages.41 It has been demonstrated that depletion of gut microbiota attenuates cardiac damage in experimental myocardial infarction.42 We show here that AngII‐induced expansion of CD11b+ myelomonocytic cells in kidneys was reduced in GF mice. This is important, given that the kidney is both a site for blood pressure regulation as well as an organ damaged by MCP‐1‐,43 IL‐17A‐, and IFN‐γ‐driven inflammation44 implicated in hypertension. Likewise, GF mice were protected from AngII‐induced cardiac inflammation and remodeling. Additionally, cardiac function, which is impaired by AngII‐driven and IFN‐γ‐dependent immune cell infiltration,26, 45 was partially preserved in GF mice. This further corroborates the concept that microbiota facilitate the systemic inflammatory alterations induced by AngII and thereby may promote development of high blood pressure in vivo. Formation of commensal microbiota, an ecosystem that is acquired directly after birth, could therefore represent an environmental factor promoting AngII‐induced high blood pressure and supporting development of AngII‐induced cardiac hypertrophy. In conclusion, targeting the molecular pathways of systemic inflammation that are triggered by the gut microbiome might add to the therapeutic options to treat hypertension in the future.
Sources of Funding
This work was supported by a grant from the German Foundation for Heart Research (F/34/14) and the German Research Foundation (DFG WE 4361/4‐1) to Wenzel and an intramural project grant from the Naturwissenschaftlich‐Medizinisches Forschungszentrum (NMFZ) to Reinhardt and Wenzel. Karbach and Reinhardt received funding by the German Research Foundation (DFG KA 4035/1‐1, RE 3450/3‐1 and 3450/5‐1) related to this work. Karbach, Brandão, Kossmann, Knorr, Brandt, Reinhardt, Schäfer, Münzel, and Wenzel received funding by the Federal Ministry of Education and Research (BMBF 01EO1003 and 01EO1503) related to this study.
Disclosures
None.
Supporting information
Figure S1. Vascular function in mice in the presence and absence of gut microbiota. A through C, Force evoked by aortic rings of GF, conventionally raised (CONV‐R), and conventionally derived (CONV‐D) mice after incubation with phenylephrine (vascular constriction, A) and vascular relaxation curves of aortas of GF, CONV‐R, and CONV‐D mice after incubation with acetylcholine (Ach; endothelium‐dependent vasorelaxation, B), glyceryl trinitrate (GTN), and nitric oxide (NO; endothelium‐independent vasorelaxation, C) are shown. In contrast to vascular constriction and endothelial function, smooth‐muscle–dependent relaxation was unaffected by recolonization (Figure 1C). Cumulative curves were analyzed by 2‐way‐ANOVA and the maximal aortic evoked force was analyzed by 1‐way ANOVA with Bonferroni post‐hoc test, n=8 to 15 aortic rings per group. *P<0.05 versus GF; # P<0.05 versus CONV‐R. D, T‐box 21 (Tbx21) mRNA expression in CD4+ splenocytes of GF, CONV‐R, and CONV‐D mice given as percentage of CONV‐R mice (left graph) and of GF mice (right graph). Student t test, n=5 to 8 per group (left) and Mann–Whitney test, n=16 per group (right). E, ELISA of the serum for interferon (IFN)‐γ, interleukin (IL)‐4, and IL‐10 detection, Kruskal–Wallis test with Dunn´s multiple comparison, n=4 to 6 mice per group.
Figure S2. A, Aortic vascular cell adhesion molecule (VCAM)‐1 and angiotensin type 1 receptor (Agtr1) mRNA expression. Kruskal–Wallis test with Dunn´s multiple comparison test, n=4 to 12 mice per group. B, Leukocyte rolling assessed by intravital videomicroscopy imaging of the carotid arteries. n=3 to 4 mice per group.
Acknowledgments
We thank Kathy Perius and Klaus‐Peter Derreth for excellent technical support. We are grateful to Fredrik Bäckhed, Wallenberg Laboratory, University of Gothenburg, Sweden for scientific discussion and support. We would like to express our gratitude to Carina Arvidsson and Anna Hallén, Wallenberg Laboratory, for providing germ‐free mouse colonies.
(J Am Heart Assoc. 2016;5:e003698 doi: 10.1161/JAHA.116.003698)
References
- 1. Abt MC, Artis D. The intestinal microbiota in health and disease: the influence of microbial products on immune cell homeostasis. Curr Opin Gastroenterol. 2009;25:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. [DOI] [PubMed] [Google Scholar]
- 3. Niess JH, Leithauser F, Adler G, Reimann J. Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J Immunol. 2008;180:559–568. [DOI] [PubMed] [Google Scholar]
- 4. Gaboriau‐Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, Eberl G, Snel J, Kelly D, Cerf‐Bensussan N. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. [DOI] [PubMed] [Google Scholar]
- 5. Hrncir T, Stepankova R, Kozakova H, Hudcovic T, Tlaskalova‐Hogenova H. Gut microbiota and lipopolysaccharide content of the diet influence development of regulatory T cells: studies in germ‐free mice. BMC Immunol. 2008;9:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caesar R, Reigstad CS, Backhed HK, Reinhardt C, Ketonen M, Lunden GO, Cani PD, Backhed F. Gut‐derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut. 2012;61:1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tlaskalova‐Hogenova H, Tuckova L, Stepankova R, Hudcovic T, Palova‐Jelinkova L, Kozakova H, Rossmann P, Sanchez D, Cinova J, Hrncir T, Kverka M, Frolova L, Uhlig H, Powrie F, Bland P. Involvement of innate immunity in the development of inflammatory and autoimmune diseases. Ann N Y Acad Sci. 2005;1051:787–798. [DOI] [PubMed] [Google Scholar]
- 8. Nicaise P, Gleizes A, Sandre C, Forestier F, Kergot R, Quero AM, Labarre C. Influence of intestinal microflora on murine bone marrow and spleen macrophage precursors. Scand J Immunol. 1998;48:585–591. [DOI] [PubMed] [Google Scholar]
- 9. Amaral FA, Sachs D, Costa VV, Fagundes CT, Cisalpino D, Cunha TM, Ferreira SH, Cunha FQ, Silva TA, Nicoli JR, Vieira LQ, Souza DG, Teixeira MM. Commensal microbiota is fundamental for the development of inflammatory pain. Proc Natl Acad Sci USA. 2008;105:2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Souza DG, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, Teixeira MM. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol. 2004;173:4137–4146. [DOI] [PubMed] [Google Scholar]
- 11. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez‐Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. [DOI] [PubMed] [Google Scholar]
- 13. Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II‐infused macrophage colony‐stimulating factor‐deficient mice: evidence for a role in inflammation in angiotensin‐induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:2106–2113. [DOI] [PubMed] [Google Scholar]
- 15. Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, Egashira K. Critical role of monocyte chemoattractant protein‐1 receptor CCR2 on monocytes in hypertension‐induced vascular inflammation and remodeling. Circ Res. 2004;94:1203–1210. [DOI] [PubMed] [Google Scholar]
- 16. Bush E, Maeda N, Kuziel WA, Dawson TC, Wilcox JN, DeLeon H, Taylor WR. CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II‐induced hypertension. Hypertension. 2000;36:360–363. [DOI] [PubMed] [Google Scholar]
- 17. Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal. 2014;20:102–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, Knorr M, Hu H, Kroller‐Schon S, Schonfelder T, Grabbe S, Oelze M, Daiber A, Munzel T, Becker C, Wenzel P. Angiotensin II‐induced vascular dysfunction depends on interferon‐gamma‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol. 2013;33:1313–1319. [DOI] [PubMed] [Google Scholar]
- 19. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, Koukes L, Yogev N, Nikolaev A, Reissig S, Ullmann A, Knorr M, Waldner M, Neurath MF, Li H, Wu Z, Brochhausen C, Scheller J, Rose‐John S, Piotrowski C, Bechmann I, Radsak M, Wild P, Daiber A, von Stebut E, Wenzel P, Waisman A, Munzel T. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis‐like skin disease. Arterioscler Thromb Vasc Biol. 2014;34:2658–2668. [DOI] [PubMed] [Google Scholar]
- 21. Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota‐induced IL‐1beta, but not IL‐6, is critical for the development of steady‐state Th17 cells in the intestine. J Exp Med. 2012;209:251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moghadamrad S, McCoy KD, Geuking MB, Sagesser H, Kirundi J, Macpherson AJ, De Gottardi A. Attenuated portal hypertension in germ‐free mice: function of bacterial flora on the development of mesenteric lymphatic and blood vessels. Hepatology. 2015;61:1685–1695. [DOI] [PubMed] [Google Scholar]
- 23. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 25. Wenzel P, Rossmann H, Muller C, Kossmann S, Oelze M, Schulz A, Arnold N, Simsek C, Lagrange J, Klemz R, Schonfelder T, Brandt M, Karbach SH, Knorr M, Finger S, Neukirch C, Hauser F, Beutel ME, Kroller‐Schon S, Schulz E, Schnabel RB, Lackner K, Wild PS, Zeller T, Daiber A, Blankenberg S, Munzel T. Heme oxygenase‐1 suppresses a pro‐inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J. 2015;36:3437–3446. [DOI] [PubMed] [Google Scholar]
- 26. Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Muller DN. Interferon‐gamma signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension. 2012;60:1430–1436. [DOI] [PubMed] [Google Scholar]
- 27. Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, Fujita T. Imbalance of T‐cell subsets in angiotensin II‐infused hypertensive rats with kidney injury. Hypertension. 2003;42:31–38. [DOI] [PubMed] [Google Scholar]
- 28. Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172:1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laurat E, Poirier B, Tupin E, Caligiuri G, Hansson GK, Bariety J, Nicoletti A. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E‐knockout mice. Circulation. 2001;104:197–202. [DOI] [PubMed] [Google Scholar]
- 30. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, Iwakura Y, Blinder Y, Rahman A, Quyyumi AA, Harrison DG. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A. A T‐bet gradient controls the fate and function of CCR6‐RORgammat+ innate lymphoid cells. Nature. 2013;494:261–265. [DOI] [PubMed] [Google Scholar]
- 34. Ochoa‐Reparaz J, Mielcarz DW, Ditrio LE, Burroughs AR, Foureau DM, Haque‐Begum S, Kasper LH. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:6041–6050. [DOI] [PubMed] [Google Scholar]
- 35. Herbst T, Sichelstiel A, Schar C, Yadava K, Burki K, Cahenzli J, McCoy K, Marsland BJ, Harris NL. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med. 2011;184:198–205. [DOI] [PubMed] [Google Scholar]
- 36. Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II‐induced hypertension and vascular injury. Hypertension. 2011;57:469–476. [DOI] [PubMed] [Google Scholar]
- 37. Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti‐Peterdi J, Caplan MJ. Olfactory receptor responding to gut microbiota‐derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morton J, Coles B, Wright K, Gallimore A, Morrow JD, Terry ES, Anning PB, Morgan BP, Dioszeghy V, Kuhn H, Chaitidis P, Hobbs AJ, Jones SA, O'Donnell VB. Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and IFNgamma‐dependent iNOS expression in the vasculature of healthy mice. Blood. 2008;111:5187–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, Brahler M, Knorr M, Brandt M, Karbach SH, Becker C, Oelze M, Bauersachs J, Widder J, Munzel T, Daiber A, Wenzel P. Inflammatory monocytes determine endothelial nitric‐oxide synthase uncoupling and nitro‐oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Caesar R, Fak F, Backhed F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med. 2010;268:320–328. [DOI] [PubMed] [Google Scholar]
- 41. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lam V, Su J, Koprowski S, Hsu A, Tweddell JS, Rafiee P, Gross GJ, Salzman NH, Baker JE. Intestinal microbiota determine severity of myocardial infarction in rats. FASEB J. 2012;26:1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007;292:F330–F339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end‐organ inflammation. J Clin Invest. 2015;125:1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation. 2009;119:2904–2912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Vascular function in mice in the presence and absence of gut microbiota. A through C, Force evoked by aortic rings of GF, conventionally raised (CONV‐R), and conventionally derived (CONV‐D) mice after incubation with phenylephrine (vascular constriction, A) and vascular relaxation curves of aortas of GF, CONV‐R, and CONV‐D mice after incubation with acetylcholine (Ach; endothelium‐dependent vasorelaxation, B), glyceryl trinitrate (GTN), and nitric oxide (NO; endothelium‐independent vasorelaxation, C) are shown. In contrast to vascular constriction and endothelial function, smooth‐muscle–dependent relaxation was unaffected by recolonization (Figure 1C). Cumulative curves were analyzed by 2‐way‐ANOVA and the maximal aortic evoked force was analyzed by 1‐way ANOVA with Bonferroni post‐hoc test, n=8 to 15 aortic rings per group. *P<0.05 versus GF; # P<0.05 versus CONV‐R. D, T‐box 21 (Tbx21) mRNA expression in CD4+ splenocytes of GF, CONV‐R, and CONV‐D mice given as percentage of CONV‐R mice (left graph) and of GF mice (right graph). Student t test, n=5 to 8 per group (left) and Mann–Whitney test, n=16 per group (right). E, ELISA of the serum for interferon (IFN)‐γ, interleukin (IL)‐4, and IL‐10 detection, Kruskal–Wallis test with Dunn´s multiple comparison, n=4 to 6 mice per group.
Figure S2. A, Aortic vascular cell adhesion molecule (VCAM)‐1 and angiotensin type 1 receptor (Agtr1) mRNA expression. Kruskal–Wallis test with Dunn´s multiple comparison test, n=4 to 12 mice per group. B, Leukocyte rolling assessed by intravital videomicroscopy imaging of the carotid arteries. n=3 to 4 mice per group.