

Abstract
Background
Sepsis patients with cardiac dysfunction have significantly higher mortality. Although several pathways are associated with myocardial damage in sepsis, the precise cause(s) remains unclear and treatment options are limited. This study was designed to develop a new model to investigate the early events of cardiac damage during sepsis progression.
Methods and Results
Francisella tularensis subspecies novicida (Ft.n) is a Gram‐negative intracellular pathogen causing severe sepsis syndrome in mice. BALB/c mice (N=12) were sham treated or infected with Ft.n through the intranasal route. Serial electrocardiograms were recorded at multiple time points until 96 hours. Hearts were then harvested for histology and gene expression studies. Similar to septic patients, we illustrate both cardiac electrical and structural phenotypes in our murine Ft.n infection model, including prominent R' wave formation, prolonged QRS intervals, and significant left ventricular dysfunction. Notably, in infected animals, we detected numerous microlesions in the myocardium, previously observed following nosocomial Streptococcus infection and in sepsis patients. We show that Ft.n‐mediated microlesions are attributed to cardiomyocyte apoptosis, increased immune cell infiltration, and expression of inflammatory mediators (tumor necrosis factor, interleukin [IL]‐1β, IL‐8, and superoxide dismutase 2). Finally, we identify increased expression of microRNA‐155 and rapid degradation of heat shock factor 1 following cardiac Ft.n infection as a primary cause of myocardial inflammation and apoptosis.
Conclusions
We have developed and characterized an Ft.n infection model to understand the pathogenesis of cardiac dysregulation in sepsis. Our findings illustrate novel in vivo phenotypes underlying cardiac dysfunction during Ft.n infection with significant translational impact on our understanding of sepsis pathophysiology.
Keywords: cardiac damage, electrocardiogram, immunology, microRNA, myocardial inflammation, myocyte apoptosis and necrosis, pathogenesis, sepsis
Subject Categories: Basic Science Research, Animal Models of Human Disease, Inflammation, Pulmonary Biology, Contractile function
Introduction
Severe sepsis and septic shock are the primary causes of death in intensive care units (ICUs), accounting for approximately 2 million infections and 300 000 deaths annually.1 Sepsis is a complex disease, defined as life‐threatening organ dysfunction caused by a dysregulated host response to infection.2 The initial hyperinflammatory response (increased production of tumor necrosis factor [TNF], interleukin [IL]‐1β, IL‐6, and chemokines) during onset of sepsis is followed by an anti‐inflammatory immunosuppressive phenotype (IL‐10, IL‐1 receptor antagonist, soluble receptors of TNF and IL‐1, and glucocorticoids)3 that favors the growth of microbial pathogens. Additionally, the switching time from inflammatory to anti‐inflammatory condition varies in patients, which leads to inappropriate treatment strategies.4 Secondary nosocomial infections (Pseudomonas spp. and Staphylococcus spp.) in sepsis patients are common and a persistent problem in health care today, specifically in ICUs.5, 6 Although there are standard treatment options for controlling bacterial growth and inflammation during early onset of sepsis, these patients frequently develop secondary complications (between 14 and 50 days) that lead to higher mortality.5, 6
The heart is essential for supplying oxygen and nutrients throughout the body, and cardiac dysfunction is an important component of multiorgan failure induced by severe sepsis.7, 8, 9 Critically ill patients present with greater risk for cardiac complications during sepsis, with ≈90% of cardiac events, such as infarction, arrhythmia, and heart failure, occurring within the first 7 days and >50% occurring within 24 hours.10, 11 More than 60% of sepsis patients admitted to the ICU show a clinical picture of cardiac dysfunction that has been recognized as a serious manifestation of severe sepsis, with a mortality rate ranging from 70 to 90%, in contrast with 20% mortality for patients without cardiac involvement.12, 13 Development of arrhythmia and heart failure following myocardial infarction and ischemia is closely linked with profound changes in cardiac structure, electrical activity, and, ultimately, pump function.14, 15, 16 Clinical assessment for cardiac dysfunction during sepsis relies mainly on electrocardiograms (ECGs)17, 18 and serum troponin levels.19 However, it is unclear whether these diagnostic modalities from critically ill patients are attributed to the administration of various drugs or damage created by bacterial infection that influences treatment options for severe sepsis.
In order to advance the treatment approaches for cardiac dysfunction during sepsis, it is necessary to develop a sepsis model that closely mimics human sepsis and provides sufficient time to understand the cardiac deregulation events during the initial stages of sepsis progression. We used Francisella tularensis subspecies novicida (Ft.n), an opportunistic intercellular pathogen, as a surrogate to study sepsis‐associated cardiac dysfunction, myocyte function, and molecular phenotypes.
Here, we present novel observations with regard to a significant change in cardiac electrophysiology, defects in conduction (R' wave formation), and prolonged QRS intervals in Ft.n‐infected mice, similar to those reported in damaged hearts of culture‐positive sepsis patients.18 Notably, we found evidence of Ft.n invasion into the cardiac tissue and describe the molecular basis for microlesion formation in the myocardium. We also demonstrate that Ft.n‐mediated microlesions are attributed to cardiomyocyte apoptosis caused by Ft.n infection. Our data show the increased infiltration of leukocytes, increased expression of inflammatory mediators, and modified expression in myocytes of 2 molecular signatures of cardiac function, microRNA (miR)‐155 and heat shock factor 1 (HSF‐1). This Ft.n infection model provides a unique opportunity to study the early events of cardiac dysfunction in a highly relevant model of sepsis that arises from a pulmonary source.10, 11, 14, 20
Methods
Animals
For all control and infection experiments, BALB/c wild‐type (WT) mice were used (aged 8 weeks). All animal studies were conducted in accord with the American Physiological Society Guiding Principles for Research Involving Animals and Human Beings and approved by The Ohio State University Institutional Animal Care and Use Committee (protocol approval number 2014 A00000054). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Subsurface Electrocardiography
Subsurface ECG recordings were obtained from control and Ft.n (50 colony forming units [CFUs]/animal)‐infected mice at baseline and at regular time intervals postinfection (0, 24, 48, 72, and 96 hours postinfection [HPI]). Anesthesia was induced using 2% isoflurane in oxygen at a flow rate of 1.0 L/min. Mice were then placed in the prone position on a heated pad to maintain body temperature. For the duration of the experiment, anesthesia was maintained at 1% isoflurane at 1.0 L/min. Subcutaneous electrodes were placed in the lead II configuration, and ECGs were recorded for 20 minutes on a Powerlab 4/30 (ADInstruments, Houston, TX).21 ECG traces were analyzed using LabChart 7 Pro (AD Instruments).
Echocardiography
To assess cardiac function in vivo, two‐dimensional echocardiography (Vevo 2100; VisualSonics Inc., Toronto, Ontario, Canada) was performed at baseline and at 96 HPI. Mice were anesthetized in an induction chamber at 2% isoflurane in oxygen at a flow rate of 1.0 L/min. Mice were then placed in the supine position on a heated stage, and hair was removed from the chest using depilatory lotion. Anesthesia was maintained at 1.5% isoflurane for the duration of the experiment. Heart rate was monitored throughout to ensure proper anesthetic dosage. Using a MS‐400 transducer, proper anatomical orientation was determined by imaging of the long axis of the heart. Once proper orientation was achieved, the transducer was turned 90 degrees to visualize the short axis of the left ventricle. M‐mode images were recorded at the level of the papillary muscles. Images were analyzed to assess ejection fraction, chamber diameters, and left ventricular wall thicknesses.
Bacterial Strains
Ft.n U112 was obtained from the American Type Culture Collection (Manassas, VA), and Francisella tularensis Schu‐4 (Ft.Schu4) was generously provided by Dr Rick Lyons (Colorado State University, Fort Collins, CO). Bacteria were cultured on Chocolate II plates (Becton Dickinson, Sparks, MD). For some experiments, Schu‐4 was passaged through THP‐1 cells. All work with the type A Schu‐4 strain was carried out in The Ohio State University BSL3 Select Agent facility in accord with national and local approved BSL3 facility and safety plans.
Reagents and Antibodies
Dulbecco's PBS with and without Ca2+ and Mg2+, RPMI 1640 medium with l‐glutamine (RPMI) and Iscove's Modified Dulbecco's Medium (IMDM) were purchased from GIBCO‐Invitrogen (Invitrogen Life Technologies, Carlsbad, CA). Mouse monoclonal F. tularensis antibody (Ab) was purchased from Abcam (Cambridge, MA), nuclear stain 4',6‐diamidino‐2‐phenylindole (DAPI; Molecular Probes, Carlsbad), antiphalloidin CruzFlour‐594 and β‐actin were purchased from Santa Cruz Biotechnology (Dallas, TX), and HSF‐1 antibody from Cell Signaling Technology (Danvers, MA). Mouse monoclonal anti‐Ft.n lipopolysaccharide (LPS) primary antibody Fn8.2‐producing cell lines were purchased from Immuno‐Precise Antibodies Ltd. (Victoria, British Columbia, Canada). Anti‐CD45 antibody was from BD Bioscience (San Jose, CA).
Isolation and Culture of Bone‐Marrow–Derived Macrophages
Bone‐marrow–derived macrophages (BMDMs) were isolated from femurs of WT BALB/c as previously described.22 Briefly, femurs were harvested and bone marrow was flushed out using 29‐gauge needles and cultured in Iscove's media supplemented with 50% L‐cell–conditioned media, 20% FBS, and penicillin/streptomycin cocktail for 5 days. Differentiated macrophages were harvested and used for our experiment.
Macrophage Infection, Lysis, and Immunoblotting
BMDMs were prepared by culturing bone marrow cells in IMDM containing 20% FBS and 50% L‐cell–conditioned medium.22 After 5‐day differentiation, cells were harvested and plated in six‐well tissue culture plates for infection. Ft. novicida and Schu‐4 were prepared as described above. BMDM monolayers were washed with warm RPMI 1640, replenished with IMDM, and prechilled at 4°C for 10 minutes, bacteria were added to BMDMs, and infections were synchronized by centrifuging at 4°C, 250g, for 10 minutes. BMDMs were then returned to 37°C 5% CO2 and incubated for the specified times. Cells were washed with PBS and lysed with TN‐1 lysis buffer (50 mmol/L of Tris [pH 8.0], 10 mmol/L of EDTA, 10 mmol/L of Na4P2O7, 10 mmol/L of NaF, 1% Triton X‐100, 125 mmol/L of NaCl, 10 mmol/L of Na3VO4, and 10 μg/mL each of aprotinin and leupeptin), incubated at 4°C for 5 minutes, and then centrifuged at 16 000g at 4°C for 10 minutes to pellet the cell debris. Protein concentrations of the cleared cell lysates were measured using the Pierce BCA‐protein assay kit (Thermo Scientific, Rockford, IL). Samples were subjected to separation by SDS‐PAGE and analyzed by western blot with the different Abs of interest.
Cytokine Assays
Blood from infected (96‐hours post infection) and uninfected mice was collected immediately after euthanasia and incubated at 4°C for 1 hour. The blood samples were centrifuged at 5000×g for 10 minutes and the cleared supernatants were collected and stored at −80°C. Cytokine (TNF and IL‐1β) levels in the serum (96 hours) were analyzed by ELISA (R&D Systems, Minneapolis, MN). All assays were performed at least 3 times and the values obtained were plotted as mean±SD.
Cardiac Troponin Assay
Serum cardiac‐specific troponin‐I (cTnI) was determined by using the high‐sensitivity mouse cardiac troponin‐I ELISA kit from Life Diagnostics, Inc (West Chester, PA). Briefly, serum was isolated from sham‐ and Ft.n‐infected mice at different time points (24, 48, 72, and 96 HPI), as described earlier, and used for the assay as directed. All assays were performed with at least 3 animals, and the values obtained were plotted as mean±SD. Significance was P<0.005.
Isolation of Cardiomyocytes
Infected (96 HPI) and uninfected BALB/c mice were anesthetized with tribromoethanol (0.025 g/mL, intraperitoneally) and hearts were excised. Using blunt dissection, the aorta is isolated from the surrounding tissue. A cut was made inferior to the aortic arch and a steel cannula was inserted. The heart was perfused with Hank's solution (Life Technologies) to clear the cardiac vasculature (retrograde perfusion). Hearts were then rapidly switched to a Langendorff apparatus and modified Tyrode solution was perfused through the heart. Modified Tyrode containing Protease (Sigma P‐5147; Sigma‐Aldrich, St. Louis, MO) and Collagenase Type II (Worthington 4176; Worthington Biochemical Corp, Lakewood, NJ) were then perfused to digest the heart and isolate single cells. Isolated cells were filtered through a cell strainer and washed twice to enrich for live cardiomyocytes.23
RNA Extraction From Heart Tissue and Quantitative Reverse‐Transcription Polymerase Chain Reaction
BALB/C mice were left uninfected or infected with Ft.n (50 CFUs) through the intranasal route; at 96 HPI, mice were sacrificed, the heart tissue was cut into small pieces and washed with PBS to remove the blood. Heart tissue was homogenized in TRIzol and total RNA was extracted, treated with DNAse 1, and purified with RNAeasy column (Qiagen, Hilden, Germany). RNA (100 ng) was reverse transcribed to cDNA by reverse‐transcriptase (RT) enzyme (SuperScript III; Invitrogen), and quantitative RT polymerase chain reaction (qRT‐PCR) was performed using a mouse IL‐1β, TNF, IL‐8, and superoxide dismutase 2 (SOD2) TaqMan gene expression kit (Applied Biosystems, Foster City, CA). IL‐1β, TNF, IL‐8, and SOD2 amplification was normalized to β‐actin as a housekeeping gene for relative gene expression. Triplicate samples were analyzed in duplicate wells in each experiment (n=3).
miRNA Extraction From Heart and Cardiomyocytes, and qRT‐PCR
BALB/C mice were left uninfected or infected with Ft.n (50 CFUs) through the intranasal route; at 96 HPI, mice were sacrificed, heart tissue was cut into small pieces and washed with PBS to remove the blood. Heart tissue was homogenized in TRIzol, and total RNA was extracted. Cardiomyocytes from Ft.n‐infected mice (96 HPI) or uninfected mice were isolated as described above. miR‐155 cDNA was generated from total RNA by the Taqman MicroRNA Reverse Transcription kit (Applied Biosystems). Expression of miR‐155 was determined by qRT‐PCR using Taqman MicroRNA Assays with sno412 as housekeeping control (Applied Biosystems). Negative controls in all qRT‐PCR included no reverse transcriptase and no template (cDNA) groups in the reactions. Triplicate samples were analyzed in duplicate wells in each experiment.
Pathological Methods and Immunohistochemistry
Hearts from infected (96 HPI) and uninfected BALB/c mice were isolated and washed in cold PBS before being fixed in 10% formalin overnight at 4°C. Hearts were then switched to 20% sucrose. Hearts were then embedded in paraffin and sectioned for analysis. Hematoxylin and Eosin (H&E) staining was used for visualization of gross cardiac structure. Masson's trichrome staining was used to identify fibrotic replacement of cardiac tissue. To examine bacterial dissemination in the heart, heart sections were incubated with anti‐Ft.n LPS antibody (Fn 8.2) for 1 hour in a humidified chamber and followed by antimouse Alexa Fluor 488 secondary Ab. Nuclei were stained with DAPI. Leukocyte staining with CD45 was performed with formalin‐fixed, paraffin‐embedded sections. Briefly, slides underwent deparaffinization and an antigen retrieval step before they were incubated with anti CD45 antibody for 1 hour, washed, and further incubated with biotinylated anti‐Rat antibody for 30 minutes.
TUNEL Assay
Paraffin‐embedded heart sections from sham‐ and Ft.n‐infected hearts were deparaffinized with xylene followed by different concentrations of ethanol. Apoptotic cells were stained by the Click‐it plus terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay for in situ apoptosis detection and Alexa Fluor 488 dye from Molecular Probes Life Technology (Thermo Scientific). Nuclei were stained with Hoechst 33342 solution and examined under confocal microscopy. Ten random images were taken from each heart (n=3), and mean fluorescent intensities (MFIs) were calculated by using ImageJ software (NIH, Bethesda, MD). Average MFIs were calculated by multiplying the intensities by area of each image.
Ft.n infection and survival studies
Survival studies were performed with a group of 5 BALB/c mice per treatment (sham, Ft.n infection alone, Ft.n infection+PBS, and Ft.n infection+gentamicin in PBS). After 4 days of infection, 1 group of mice received PBS (200 μL of PBS) or gentamycin in PBS (10 mg/kg, 200 μL) through the intraperitoneal route. The cardiac function was monitored by echocardiography on day 4 of Ft.n infection and on day 9 of gentamycin treatment. The survival of mice was recorded.
Statistical Analysis
A paired 2‐tailed Student t test was used to analyze the differences between 2 groups in figures showing 1 representative figure of 3independent experiments unless specified in figure legends. An unpaired 1‐tailed Student t test was used to analyze differences between 2 groups in figures showing cumulative data from 3 independent experiments. Significance was P<0.05, P<0.005, and P<0.0005.
Results
Ft.n Infection Causes Cardiac Dysfunction
We selected Ft.n to study the relationship between sepsis and myocardial phenotypes. This bacterial infection reveals several significant advantages over other acute sepsis infection models: (1) Similar to humans, Ft.n‐infected animals survive over 6 days following a biologically relevant low‐dose infection22, 24, 25, 26; (2) Ft.n infection causes severe sepsis, and the signs of cardiac arrhythmia are comparable to those of culture‐positive sepsis patients18; (3) disease outcomes are similar to the hospital‐acquired Streptococcus pneumoniae 20; and (4) Ft.n infection is a highly reproducible and tractable animal model for understanding underlying mechanisms of cardiac dysfunction.
Although sepsis causes multiorgan failure (including the heart) that leads to increased mortality, the influence of bacterial sepsis on cardiac damage is not well studied. We examined the changes in cardiac troponin release in the serum of infected and uninfected animals. BALB/c mice were infected with Ft.n (25 CFUs) or received PBS through intranasal delivery. Ninety‐six HPI, we determined the bacterial load in lungs by CFU assay and measured cTnI levels in the serum. We observed more than 8 log bacteria in lungs (Figure 1A), an indicator of severe pneumonia. The bacterial titer in the lung was increased along with an increase in troponin‐I release in serum, a clinical marker of cardiac injury (Figure 1B). Gross pathology is shown in Figure 1C for sham mice and one‐dimensional Ft.n‐infected mice. Yellow arrows indicate pulmonary abscess caused by Ft.n in mice. Together, these data demonstrate that Ft.n infection causes severe pneumonia and cardiac damage, the latter indicated by increased serum cTnI levels.
Figure 1.
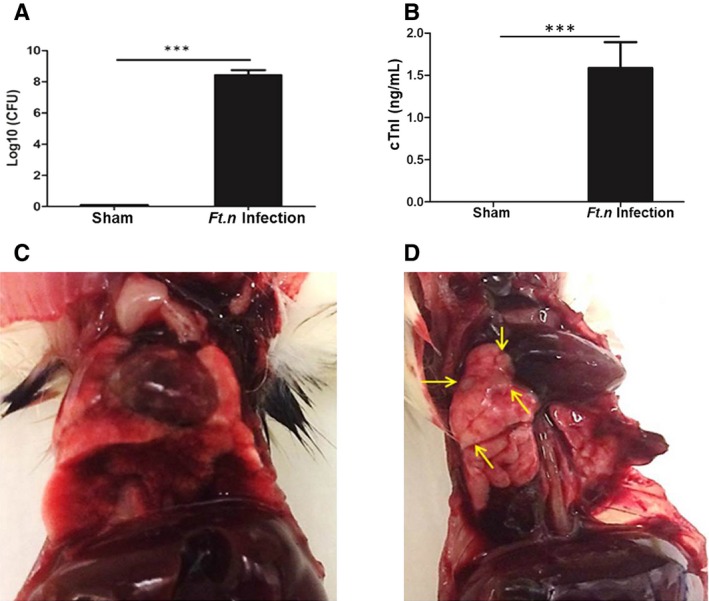
Ft.n bacterial load correlates with cardiac damage. Wild‐type BALB/C mice were infected with Ft.n (25 CFUs) through the intranasal route, and after 96 hours of incubation, mice were euthanized and lungs and blood were retrieved for further analysis. (A) Bacterial burden was determined by CFU assay by serial dilution and plating of the whole‐lung homogenates on chocolate blood agar plates (2 independent experiments; N=6). (B) Serum cardiac troponin‐I (cTnI) levels were determined by an ELISA assay kit (***P<0.0005; 2 independent experiments; N=6). Images shown in (C) and (D) are lungs and hearts of sham‐treated and Ft.n‐infected mice. Yellow arrows indicate bacterial colonization (lesions) in lungs of infected animals, a representative image from 3 independent experiments (N=12). CFU(s) indicates colony forming unit(s); ELISA, enzyme‐linked immunosorbent assay.
Ft.n Infection Alters Cardiac Electrical Phenotypes
Given that Ft.n infection causes cardiac damage as indicated by increased cTnI (Figure 1B), we next examined whether cardiac injury was associated with altered cardiac electrical phenotypes. We performed limb‐lead ECG analysis in sham‐ and Ft.n‐infected animals (24, 48, 72, and 96 HPI). Interestingly, all infected animals (N=12) showed an abnormal ECG pattern, specifically the presence of prominent R' wave formation, indicative of bundle branch block (BBB). These ECG abnormalities were not found in mice treated with vehicle (Figure 2A and 2B). Interestingly, we observed R' wave formation as early as 48 HPI with an R' frequency of 48±5.6%. Amplitude of R' wave formation gradually increased after 48 hours of infection, and R' wave frequency achieved 100% of recorded ECGs at 72 and 96 HPI (Figure 2C), whereas in uninfected mice ECGs were normal (Figure 2A). We observed an increased level of cardiac troponin in the serum of Ft.n‐infected mice over time in concert with cardiac dysfunction (Figure 2D). Serum cardiac troponin levels peaked at 72 HPI, indicating that Ft.n infection caused acute cardiac damage similar to that observed in human sepsis patients. Notably, the bacterial load in lung positively correlated with ECG abnormalities that we observed in Ft.n‐infected mice (Figure 2E). Furthermore, we determined the heart rate (HR), QRS intervals, and PR intervals from recorded ECG. Ft.n‐infected animals showed significantly elevated HR compared to sham‐treated mice (Figure 2F), a phenotype observed in culture‐positive human sepsis patients.18 Additionally, we measured PR and QRS intervals (Figure 2G and 2H) in both infected and uninfected animals and observed significantly increased QRS duration in Ft.n‐infected animals, indicative of BBB.18 Together, these data provide strong evidence that Ft.n infection causes severe cardiac damage, producing significant cardiac conduction defects (BBB, increased QRS interval).
Figure 2.
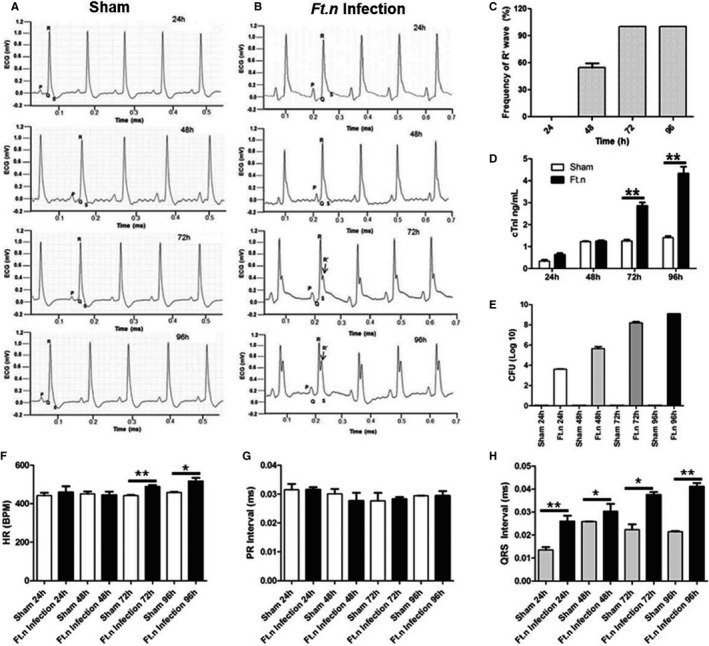
Ft.n infection alters cardiac electrophysiology. Subsurface electrocardiogram (ECG) recordings were obtained from control and Ft.n‐infected mice at baseline and at regular time intervals postinfection (24, 48, 72, and 96 hours postinfection). (A) Representative ECG data from sham‐treated mice (N=4) and (B) are Ft.n‐infected mice (N=4) at different time points (3 independent experiments). After Ft.n infection, note the gradual appearance of an R' wave (black arrow). (C) R' wave frequency was determined in 3 independent experiments (total number of mice=12). From 20 minutes of recorded ECG data, we calculated frequency of R' wave formation at different time points. (D) Serum cardiac troponin‐I levels were determined in sham‐ and Ft.n‐treated mice at different time points. (E) Bacterial burden in lungs was determined by CFU assay using lung homogenates from 4 mice in each group. From 20 minutes of recorded ECG data, we calculated average heart rate (F), PR intervals (G), and QRS intervals (H). Graphs are cumulative data from 4 mice per group and representative of 2 independent experiment (*P<0.05; **P<0.005; ***P<0.0005). BPM indicates beats per minute; CFU, colony forming unit; CTnI indicates cardiac troponin‐I; HR, heart rate.
Cardiac Dysfunction in Mice Following Ft.n Infection
To further characterize the cardiac damage produced during Ft.n infection, we elected to perform cardiac ultrasound of mice 96 HPI. Consistent with our earlier findings (Figure 2A and 2B), we observed atypical R' wave formation (Figure 3A and 3B) in Ft.n‐infected mice by ECGs gathered during the ultrasound procedure. Interestingly, in contrast to systolic dysfunction witnessed in CLP and LPS‐induced murine sepsis models,27, 28, 29 Ft.n infection significantly increased ejection fraction (Figure 3C) and fractional shortening (Figure 3D). Further analysis revealed that Ft.n infection significantly reduced the left ventricular internal dimension during diastole (LVID, d; Figure 3E) and systole (LVID, s; Figure 3F). As a result of decreased chamber dimensions, stroke volume (as defined by stroke volume (SV)=end diastolic volume (EDV) – end systolic volume [ESV]) was significantly diminished (Figure 3G). This decrease in SV subsequently led to significant decreases in cardiac output in Ft.n‐infected mice (Figure 3H). We further found that posterior ventricular wall thickness (LVPW, d and LVPW, s) was significantly increased upon infection (Figure 3I and 3J). Interestingly, the heart weight‐to‐tibia length ratio was elevated in Ft.n.‐infected mice compared to control animals (8.93±0.74 and 7.71±0.13 mg/mm, respectively); however, the difference was not statistically significant (P=0.151).
Figure 3.
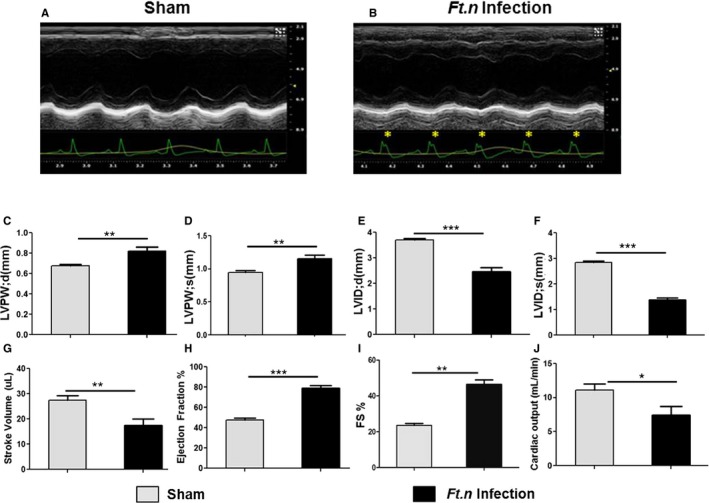
Cardiac pump dysregulation following Ft.n infection. To assess cardiac function in vivo, two‐dimensional echocardiography was performed at baseline and at 96 hours postinfection. (A) Representative cardiac patch of uninfected mice (N=9) and (B) a cardiac patch at 96 hours post‐Ft.n infection (N=9). Note the presence of a prominent R' wave present in Ft.n‐infected mice (yellow asterisks). Posterior ventricular wall thickness (LVPW; d and LVPW; s) during (C) diastole and systole (D), left ventricular internal diameter during (E) diastole (LVID; d) and (F) systole (LVID; s), (G) stoke volume, (H) ejection fraction, (I) % fractional shortening (FS), and (J) cardiac output were determined from sham‐treated and Ft.n‐infected mice. N=3 (3 mice/group; *P<0.05; **P<0.005; ***P<0.0005).
Because of the significant impairment in cardiac function after Ft.n infection, we next conducted survival analysis in these animals. Animals were infected at day 0, as previously described, and followed for 12 days total. By 6 days postinfection, 100% (5/5) of Ft.n‐infected animals had suffered mortality. In contrast, 100% of sham‐treated animals survived until day 12 of the protocol. An alternative explanation for the aforementioned cardiac phenomena is that they arise secondary attributed to insufficient blood volume (ie, hypovolemia) following infection. In an effort to curtail this effect, we elected to conduct fluid resuscitation experiments. Briefly, Ft.n‐infected animals were infected at day 0 and were followed to day 4 (96 hours) as previously described. At this point, mice began receiving fluid resuscitation (PBS, 10 mL/kg, intraperitoneally) planned for the remaining 8 days. However, PBS fluid treatment alone did not significantly delay mortality in this cohort (5 of 5 mice died by day 7). In an effort to extend survivability in Ft.n‐infected animals, we supplemented our fluid replacement regimen with the antibiotic, gentamycin (10 mg/kg intraperitoneally, volume equal to 10 mL/kg). Unlike fluid replacement alone, PBS+gentamycin significantly prolonged the life span of Ft.n‐infected animals. Because of lower death in this study arm, we next conducted echocardiographic analysis. As expected, Ft.n‐infected mice displayed the stereotypic increase in ejection fraction compared to uninfected controls. Interestingly, after receiving 5 days of resuscitative treatment, echocardiographic parameters (ejection fraction [EF], LVID; d, LVID; s, wall thicknesses) were still significantly different compared to uninfected controls at the same time point (Figure 4A through 4F). Together, these results suggest a unique scenario of cardiac functional impairment following Ft.n infection in mice. Furthermore, although overall survivability is improved, these cardiac functional phenotypes persist in the presence of resuscitative treatment (Figure 4G).
Figure 4.
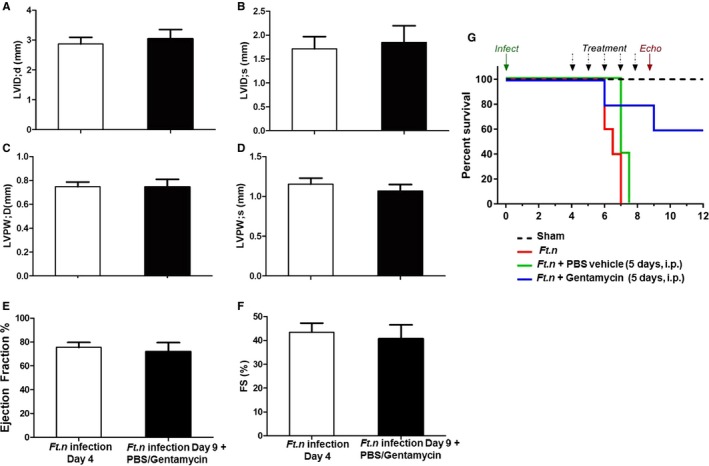
Fluid replacement does not rescue cardiac pump dysregulation during Ft.n infection. BALB/c mice were infected with Ft.n (25 CFUs) through the intranasal route, and after 96 hours of incubation, we supplemented infected mice with fluid replacement at 10% total blood volume (200 μL of PBS, intraperitoneally, or gentamycin [10 mg/kg] in PBS) for 5 days. On days 4 and 9, mice were anesthetized in an induction chamber at 2% isoflurane in oxygen at a flow rate of 1.0 L/min. To assess cardiac function in vivo, two‐dimensional echocardiography (Vevo 2100; VisualSonics Inc., Toronto, Ontario, Canada) was performed at baseline and at day 4 postinfection. Left ventricular internal diameter during (A) diastole (LVID; d) and (B) systole (LVID; s), posterior ventricular wall thickness (LVPW; d and LVPW; s) during (C) diastole and systole (D), ejection fraction (E), and % fractional shortening (FS; F) were determined from with and without fluid resuscitation (PBS, 10 mL/kg intraperitoneally) of Ft.n‐infected mice. N=5 mice/group. (G) Survival studies were performed with a group of 5 mice per treatement (sham, Ft.n infection alone, Ft.n infection+PBS, and Ft.n infection+gentamicin in PBS). After 96 hours of infection, 1 group of mice received PBS (200 μL of PBS, intraperitoneally) or gentamycin in PBS (10 mg/kg intraperitoneally, volume equal to sham treatment [200 μL]); survival of mice was monitored.
Evidence of Ft.n Dissemination into the Heart Tissue
Ft.n disseminates into various organs,22, 30, 31 prompting us to examine whether Ft.n directly disseminated into heart tissue. When heart sections of infected BALB/c mice were analyzed with anti‐Ft.n LPS antibody, we observed the presence of Ft.n in the posterior and septal regions as well as in the pericardium (Figures 5A through 5C). To further confirm the internalization of Ft.n by cardiomyocytes and fibroblasts, we infected isolated primary purified myocytes with green fluorescent protein (GFP)‐expressing Ft.n (multiplicity of infection [MOI], 10:1). After 60 minutes, cells were washed, incubated with DAPI, and analyzed by confocal microscopy. Interestingly, we found that Ft.n was internalized more efficiently by cardiomyocytes than fibroblasts (6.96±0.58‐fold less bacteria; Figure 5D and 5E). The z‐stack analysis of infected mice heart showed that bacteria invaded into heart tissue (Figure 5F). These results strongly indicate that the cardiac damage is partly attributed to bacterial invasion and activation of immune cells in cardiac tissue.
Figure 5.
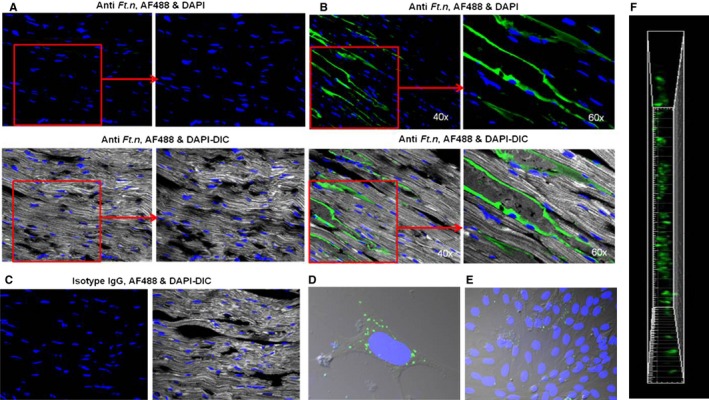
Ft.n disseminates into heart tissue. Cardiac sections from control and Ft.n‐infected mice were stained with anti‐Ft.n LPS Ab or isotype control Ab followed by FITC 488 secondary Ab. Nuclei were stained with DAPI and examined for the presence of Ft.n by confocal microscopy. (A) Representative images from 3 sham‐treated mice. (B) Representative images from Ft.n‐infected mice. (C) Isotype controls for anti‐Ft.n antibody. (D) Neonatal cardiomycytes and (E) fibroblasts were isolated from BALB/c mice and plated on coverslips. Cardiomycytes were incubated with GFP‐Ft.n (MOI, 20:1) for 60 minutes, washed 3 times with PBS, and fixed with 4% PFA. Nuclei were stained with DAPI and examined by confocal microscopy. Representative images from 2 independent experiments. (F) Snap shot of vertical view of heart section from the Z‐stack of heart section from Ft.n‐infected mice. Ab indicates antibody; DAPI, 4',6‐diamidino‐2‐phenylindole; DIC, differential interference contrast; GFP, green fluorescent protein; IgG, immunoglobulin G; LPS, lipolysaccharide; MOI, multiplicity of infection.
Presence of Cardiac Microlesions in Ft.n‐Infected Mice
Hearts from Ft.n‐infected animals had more damage compared with uninfected animals (Figure 6A and 6B). In order to assess cardiac damage during bacterial infection, we stained heart sections with Masson's trichrome and H&E. We observed the presence of distinct microlesions, randomly dispersed throughout the intraventricular septum and myocardium of the left and right ventricular free wall of Ft.n‐infected mice (Figure 6D and 6F). In contrast, cardiac sections from sham‐treated mice did not present with visible microlesions (Figure 6C and 6E). H&E staining clearly showed that no infiltrating immune cells were present within lesions and surrounding tissue. These lesions were not present in the pericardium, indicating that they were likely formed because of inflammation and apoptosis by direct bacterial colonization of the myocardium. Additionally, Masson's trichrome staining revealed significant cardiac fibrosis during Ft.n infection. As shown in Figure S1, Ft.n infection enhanced cardiac interstitial and perivascular fibrosis compared to sham‐treated mice.
Figure 6.
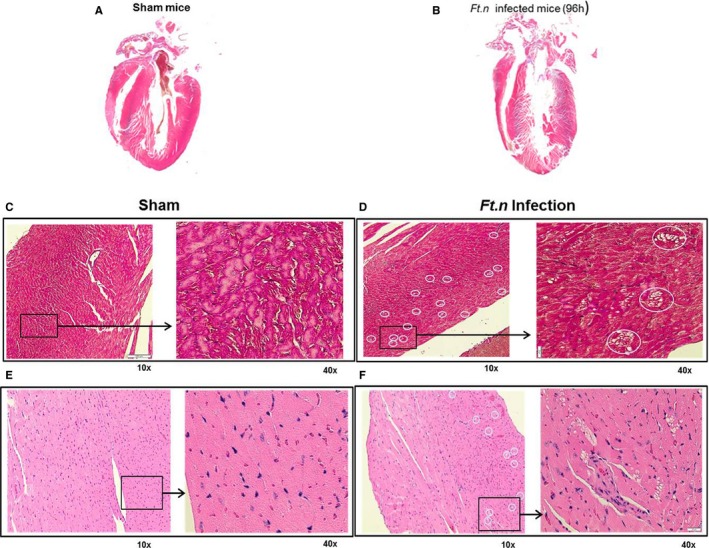
Presence of cardiac microlesions in Ft.n‐infected mice. Cross‐sections of hearts obtained from BALB/c mice after 96 hours of Ft.n challenge. (A) Four‐chamber view of hearts isolated from uninfected mice and (B) Ft.n‐infected mice. Overall size of the heart was reduced and showed significant tissue damage. Representative of 3 infected and uninfected hearts. The 4‐chamber view sections from uninfected (C) and Ft.n‐infected (D) mice were stained with Masson's trichrome to identify fibrotic replacement of cardiac tissue. Hematoxylin and eosin staining of uninfected (E) and Ft.n‐infected (F) heart sections and white circles show the regions of lesions by 10× magnification, and the area in the square was enlarged to 40× magnification. Cardiac microlesions were randomly dispersed throughout the septa and myocardium (N=6). White circled areas indicate lesion areas in the section.
Ft.n Infection Induces Cardiac Inflammation
Given that Ft.n infection caused microlesions in cardiac tissue, we next examined immune cell infiltration in hearts of Ft.n‐infected and sham mice. Examination of CD45‐stained sections revealed a substantial infiltration of hematopoietic immune cells in the superior aspect, the left ventricle free wall of the heart, and around the great vessels (Figure 7A, lower panel). The number of CD45‐positive cells were less in heart sections from sham‐treated animals (Figure 7A, top panel). As shown in Figure 7B, the MFIs (MFI/μm2) of CD45 staining is significantly increased in Ft.n‐infected animals compared to sham‐treated mice. To determine whether the microlesions were formed because of inflammation of cardiac tissue, we examined the hearts for CD45‐positive cells in the septal region (Figure 7C and 7D). Surprisingly, we observed that the lesions themselves were not positive for CD45 staining, except for small numbers of positive cells juxtaposed with the lesions (Figure 7D). Also, we observed an increased expression of IL‐1β, TNF, and IL‐8 genes in cardiac tissue of Ft.n‐infected mice (Figure S2A through S2C). Concomitantly, we observed increased IL‐1β and TNF in serum of infected mice (Figure S2E and S2F). Additionally, we observed increased expression of SOD2 in infected cardiac tissue (Figure S2D).Together, these data suggest that the infected heart is inflamed by immune cell infiltration, but the lesions formed are attributed to bacterial dissemination and not attributed to the infiltration and accumulation of inflammatory cells.
Figure 7.
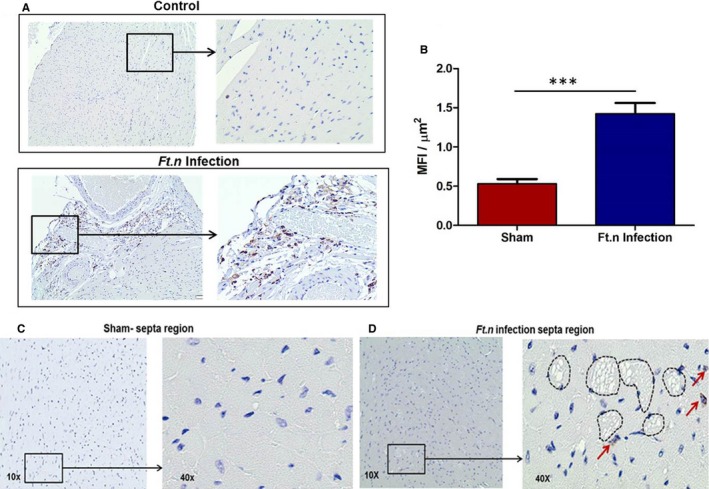
Ft.n sepsis induces inflammation that is not the cause of the microlesons. Four‐chamber view sections from uninfected (A, upper panel) and Ft.n‐infected (A, lower pannel) mice were stained with CD45. CD45 staining of uninfected and infected heart sections by 10× magnification, and the area in the square was enlarged to 40× magnification. (B) Mean fluorescent intensities (MFI) of CD45 staining per μm2 indicates infiltration of immune cells in the heart. The images clearly show infiltration of immune cells in the posterior region of heart tissue. Photomicrographs of sham‐treated (C) and Ft.n‐infected (D) septa region are shown with 10× and 40× magnifications. Microlesions of septa region of Ft.n‐infected mice do not show any leukocyte infiltration. Images shown are representative of 6 animals per group.
Increased Cardiac Apoptosis Following Ft.n Infection
We next assessed whether Ft.n infection causes apoptosis in the heart. Microlesions and cardiac scar are often attributed to induction of apoptosis in cardiomyocytes.32 As expected, we observed increased numbers of apoptotic cells in hearts from Ft.n‐infected mice. Furthermore, we detected abundant apoptotic cells in the posterior region of the heart (Figure 8A). The septal region also showed evidence of cardiomyocyte apoptosis (Figure 8B). We observed that most of the lesions were positive for TUNEL staining (Figure 8C; lower panel shows a single microlesion), and, overall, the MFIs (MFI; Figure 8D) for TUNEL staining was significantly increased in Ft.n‐infected cardiac tissue, suggesting that these lesions are irreversible and may associate with defects in electrical conduction. We also observed that most of the CD45‐positive regions were positive for TUNEL, indicating a correlation of inflammation with apoptotic cell death in cardiac tissue. Therefore, our data indicate that Ft.n infection causes increased apoptosis in the heart. To further confirm Ft.n‐mediated cardiomyocyte apoptosis, we infected neonatal myocytes with GFP‐expressing Ft.n, and after 6 hours of infection, cells were analyzed for actin cytoskeleton staining. Consistent with our in vivo data, Ft.n infection altered cardiac myofilaments and cytoskeletal structures within 6 hours of infection (Figure S3). These data strongly support that Ft.n infection causes significant apoptosis, leading to irreversible myocardial damage, which would be expected to lead to increased mortality during severe bacterial sepsis. Together, these data are consistent with Ft.n infection causing microlesions through induction of myocyte apoptosis.
Figure 8.
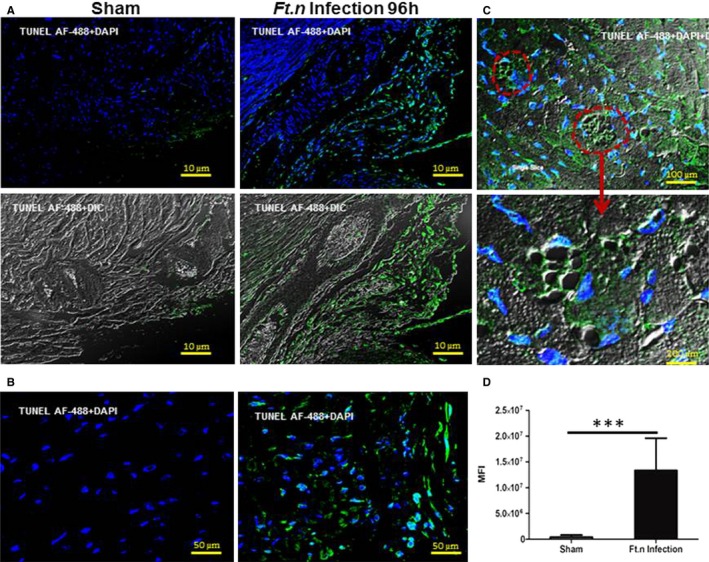
Apoptosis of heart tissue during Ft.n infection. Heart sections from sham‐ and Ft.n‐infected mice were deparaffinized by xylene, and apoptotic cells were stained by the Click‐it plus TUNEL assay kit with Alexa Fluor 488 dye. (A) Representative image from the posterior region of heart sections of sham‐treated mice (right panel) and Ft.n‐infected mice (left panel). (B) Representative image of the septa region of Ft.n‐infected mice. (C) Representative image from the microlesion in septa region of Ft.n‐infected heart sections. Lower panel shows an enlarged image of microlesions in an Ft.n‐infected heart section. Dotted circle shows a lesion caused by Ft.n infection. (D) Graph shows cumulative data of mean florescent intensities (MFI) of apoptotic cells (AF488; N=3; 10 images/mouse; ***P<0.0005). AF488 indicates Alexa Fluor 488; DAPI, 4',6‐diamidino‐2‐phenylindole; DIC, differential interference contrast; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
miR‐155 Expression Is Increased in the Heart Following Ft.n Infection
To elucidate the molecular mechanism for cardiac inflammation and altered electrophysiology, we studied 2 known inflammatory mediators, miR‐155 and HSF‐1. miR‐155 expression is enhanced in lungs, spleen, and liver of Ft.n‐infected mice33 associated with robust immune cell infiltration and inflammation in hearts of infected animals. Furthermore, miR‐155 regulates inflammation by targeting the tyrosine phosphatase, SH2‐containing inositol phosphatase 1.34 miR‐155 expression was significantly increased in whole‐heart tissue of Ft.n‐infected mice (Figure 9A), whereas in isolated cardiomyocytes, miR‐155 expression was not significantly increased (Figure 9B). Together, these data indicate that Ft.n infection upregulates expression of miR‐155 in the heart and, potentially, in immune cells that have infiltrated cardiac tissue.
Figure 9.
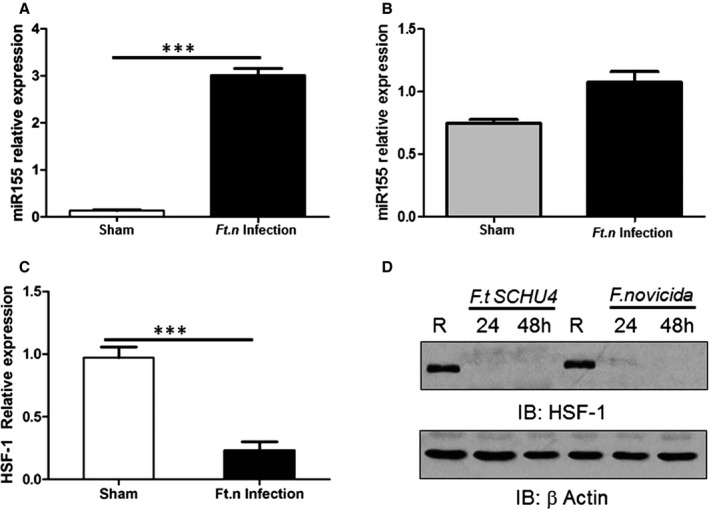
Ft.n infection enhances microRNA (miR)‐155 expression in the heart. Total RNA from hearts of Ft.n‐infected (25 CFUs) or uninfected BALB/c mice (3 per group) was extracted after 96 hours. Animals were sacrificed, hearts harvested immediately, cut into pieces to remove blood, washed 3×, and homogenized in RNA later. (A) miR‐155 expression was measured using the TaqMan MicroRNA assay kit and subsequent quantitative reverse‐transcriptase polymerase chain reaction (qRT‐PCR). N=2. (B) Myocytes from Ft.n‐infected or uninfected mice were isolated and lysed in TRIzol reagent, and total RNA was extracted. miR‐155 expression was determined by using the Taqman MicroRNA assay kit and subsequent qRT‐PCR. (C) Total RNA was extracted from infected and uninfected heart tissue to measure heat shock factor 1 (HSF‐1) expression by qRT‐PCR. N=3. (D) Bone‐marrow–derived macrophages were infected with Ft.n (MOI, 10:1) or left uninfected (R), and cells were lysed 24 and 48 HPI. Protein‐matched cell lysates were used to examine HSF‐1 levels by western blot and reprobed with anti‐β‐actin antibody as a loading control. Representative graph from 3 experiments (N=3; ***P<0.0005). CFU indicates colony forming unit; HPI, hours postinfection; IB, immunoblot; MOI, multiplicity of infection.
Ft.n Infection Decreases HSF‐1 Expression in Heart
HSF‐1 regulates expression of various stress response proteins,35 and its transcriptional activity is an essential element for protection against various pathogens by regulating expression of various inflammatory genes and preserving cardiomyocyte viability and function.36, 37, 38, 39 Therefore, we next determined expression of HSF‐1 in Ft.n‐infected animals. Ft.n infection decreases expression of HSF‐1 in the heart within 24 hours compared to sham‐treated mice. Results in Figure 9C indicate that HSF‐1 mRNA levels are significantly decreased by Ft.n infection in cardiac tissue, suggesting the dysregulation of expression of this stress response protein in the heart. To further examine whether the decreased expression of HSF‐1 occurred in immune cells, we infected BMDMs with Ft.n and determined the expression of HSF‐1 by immunoblot. We observed that Ft.n infection leads to rapidly decreased expression of HSF‐1 in macrophages (Figure 9D). These data suggest that Ft.n infection promotes inflammation and apoptosis, in part, by targeting HSF‐1 expression during infection.
Discussion
The results presented here provide evidence in support of the hypothesis that the alteration in cardiac electrophysiology during bacterial sepsis culminates in multiple organ failure and increased mortality. Sepsis‐mediated cardiac damage in patients is frequently characterized by several ECG changes. In this study, we have developed an instructive mouse model of sepsis by introducing low‐dose Ft.n (≈20 CFUs) through the respiratory infection route to study the early pathophysiological and immunological changes in the heart during development of sepsis. Within 48 hours after Ft.n infection, we observed abnormalities in the ECG waveform, specifically the development of a notable R' wave, lengthening of the QRS interval, and increased heart rate (Figure 2A and 2B). The magnitude of ECG changes was further increased at 96 hours, showing a severe defect in electrical conduction as evidenced by an increase in QRS duration and clear R wave formation. Similarly, Iuliano et al40 demonstrated that prolongation of the QRS interval is an indicator of increased mortality and sudden death in patients with heart failure. Also the increased QRS duration observed in a surface echocardiogram is associated with left ventricular dysfunction and causes the conduction defect.41, 42 We postulate that cardiac damage is initiated during the early stage of infection, as early as 48 HPI, as indicated by an abnormal ECG with mainly R' wave and prolonged QRS interval. Consistent with our sepsis model, Rich et al18 reported prolonged QRS and development of a BBB in patients with septic shock, providing further support that our model mimics human sepsis with respect to heart function.
Bacterial infections are often associated with abscess formation, bacterial persistence, microlesions, and release of inflammatory mediators.20, 22, 43 The data presented in this article provide evidence for bacterial invasion and colonization into the myocardium (Figure 5), consistent with bacterial colonization in cardiac tissue during the early stages of sepsis development. This may be the main cause for the changes in electrical and structural changes in the heart given that pathological examination of hearts from Ft.n‐infected animals showed multiple microlesions in the intraventricular septum and left ventricular free wall. These cardiac microlesions can be linked to cardiac dysfunction, physical interruption of electrical conduction pathways, and cardiomyocyte death. Brown et al20 observed similar microlesions in the heart of Streptococcus pneumonia–infected mice and sepsis patients, suggesting that this is a characteristic phenomenon of cardiac damage during bacterial sepsis. Notably, Ft.n infection in mice causes cardiac interstitial and perivascular fibrosis (Figure S1), a general pathological condition associated with chronic inflammation found in the cecal ligation and puncture (CLP)‐induced sepsis model.44 Importantly, histological analysis of microlesions in the Ft.n‐infected heart tissue reveals the absence of immune cell infiltration. In contrast, we observed robust infiltration of immune cells in the posterior region and myocardium of the heart (Figure 7), suggesting that the Ft.n infection causes cardiac inflammation and damage.
Apoptosis is a programmed cell death process and deregulation in this process is linked with many human diseases, including heart failure and ischemia‐reperfusion injury.45 Our TUNEL assay reveals that the microlesions are composed of apoptotic cells without immune cells (Figure 8). Furthermore, our in vitro studies with isolated neonatal myocytes show a rapid degradation of myocyte cytoskeletal structure following Ft.n treatment. Consistent with these results, we found an increased level of cTnI in the serum of Ft.n‐infected mice, indicative of cardiomyocyte damage. Herein, we postulate that bacterial sepsis induces apoptosis in cardiac tissue and causes irreversible damage, which may be further amplified by secondary infections, thereby increasing the risk for mortality in these patients.
Our novel murine model of sepsis also presents with a distinct cardiac functional phenotype. In contrast to the systolic dysfunction witnessed in CLP‐ and LPS‐induced murine models of sepsis, Ft.n infection produces a marked increase in left ventricular EF (>75%). These mice also present with decreased stroke volume, leading to depressed cardiac output.46, 47 The decreased stroke volume in these severely septic mice is attributed to the reduced left ventricular internal dimension during both systole and diastole of the heart (Figure 3B). Furthermore, our strategy of fluid and antibiotic resuscitation did not significantly rescue cardiac functional parameters in Ft.n‐infected animals (Figure 4). Given our data, it is intriguing to speculate that Ft.n infection was causing diastolic dysfunction in mice. Indeed, several reports have linked sepsis to diastolic dysfunction, such as in patients with S. pneumoniae septicemia48 and severe sepsis patients with increased mortality.49 These results suggest that these cardiac phenomena arise because of the infectious presence of Ft.n. Alternatively, we cannot discount the hypothesis that these dysfunctional phenotypes are caused, in part, by blood fluid volume changes postinfection (ie, hypovolemia). However, our current echocardiographic data are insufficient to exclude either hypothesis and must be noted as a major limitation of this study. Our data further demonstrate increased LVPW diastolic and LVPW systolic thickness in Ft.n‐infected mice. Although insignificant (P=0.151), a trend for increased heart weight/tibia length ratio was also witnessed in Ft.n‐infected animals. These results, coupled with the histological, biochemical, and electrocardiogram data, indicate significant cardiac dysfunction following Ft.n infection.
An increased production of inflammatory mediators and myocardial inflammation are generally thought to be associated with cardiac death, severe arrhythmia, and heart failure.50 Our findings of electrophysiology changes are correlated with increased serum cytokine expression and protein levels of TNF, IL‐1β, IL‐8, and SOD2 in the heart tissue. The increased expression of TNF and IL‐1β in heart tissue suggests that cardiomyocyte death is attributed to the TNF‐mediated death pathway51 and IL‐1β‐mediated apoptosis.52 miRNAs play a significant role in the regulation of inflammatory signaling cascades, and, notably, we observed increased expression of miR‐155 in heart tissue, suggesting a mechanism for increased inflammation during Ft.n infection. Additionally, cardiomyocyte apoptosis has been shown to be regulated by heat shock protein (HSP) 70, whose expression is regulated by the transcription factor, HSF‐1.37 Notably, we show, for the first time, that HSF‐1 is downregulated during Ft.n infection in heart tissue. Our finding suggests that HSF‐1 is rapidly degraded in heart tissue during Ft.n infection, which suggests that cardiomyocytes are undergoing the stress that leads to increased apoptosis. Importantly, overexpression of HSP‐27 protects cardiac dysfunction mediated by ischemia through stabilization of cardiac troponin I and T.38
To our knowledge, this is the first study demonstrating cardiac damage, abnormal electrical conductivity, and dysfunction in a bacterial sepsis model that mimics several aspects of human sepsis. We observed a pronounced conduction defect, R' wave formation, prolonged QRS, and cardiac dysfunction in hearts of Ft.n‐infected mice. The underlying mechanisms include bacterial colonization in the heart, increased expression of inflammatory genes, and immune cell infiltration into cardiac tissue. Finally, we observed increased apoptosis in cardiac tissue, which may be attributed to increased miR‐155 expression and rapid degradation of myofilaments and HSF‐1 in the heart. These findings uncover a new sepsis model that enables the study of early events in cardiac dysfunction, unlike current sepsis models, and will allow us to uncover new treatment strategies for treatment of sepsis.
Disclosures
None.
Supporting information
Figure S1. Ft.n infection causes cardiac fibrosis. Cross‐sections of hearts obtained from BALB/c mice after 96 hours of Ft.n challenge. The 4‐chamber view sections from uninfected (A) and Ft.n‐infected (B and C) mice were stained with Masson's trichrome to identify fibrosis in cardiac tissue. Image shown in (B) is interstitial fibrosis, and (C) is perivascular fibrosis from Ft.n‐infected mice hearts. The image shown in right panel is 60× magnification and representative of 6 animals per group.
Figure S2. Ft.n infection increases expression of inflammatory mediators in the heart as well as serum cytokine levels. BALB/c mice were infected with Ft.n (25 CFUs) through the intranasal route, and after 96 hours incubation, mice were sacrificed to harvest the heart, which was cut into small pieces, washed with PBS, and immediately homogenized with Trizol reagent. Total RNA was extracted and purified by using RNAeasy column and converted into cDNA. Quantitative real‐time RT‐PCR was used to determine (A) IL‐1β, (B) TNF, (C) IL‐8, and (D) SOD2 mRNA levels. Data ware normalized to the β‐actin gene, and relative gene expression was determined. Blood samples from Ft.n‐infected or sham‐treated mice were kept at 4°C for 1 hour and centrifuged at 1500g for 10 minutes at 4°C. Serum samples were used to determine levels of inflammatory cytokines by ELISA. Graph shown in (E) IL‐1β and (F) TNF is a representative graph from 3 experiments (N=3; **P<0.005; ***P<0.0005).
Figure S3. Ft.n infection induces cardiomyocyte apoptosis. Neonatal myocytes were isolated from BALB/C mice and infected with GFP‐Ft.n (MOI, 10:1), and after 6 hours of incubation, cells were washed, fixed, permeabilized, and stained with Alexa Fluor 594 (AF594)‐conjugated phalloidin antibody. Myocytes were examined under confocal microscopy, and the image shown is representative of 10 infected myocytes.
Acknowledgments
The authors thank The Ohio State University Campus Microscopy and Imaging Facility for training with confocal imaging and the Comparative Pathology and Mouse Phenotyping Shared Resource (CPMPSR), College of Veterinary Medicine, for heart sectioning and immunohistochemistry analysis. The authors thank the Small Animal Imaging facility at the Davis Heart and Lung Research Institute and, finally, Dr Mohamood Khan for initial ECG recordings.
(J Am Heart Assoc. 2016;5:e003820 doi: 10.1161/JAHA.116.003820)
References
- 1. Munford RS. Ch. 271: Severe Sepsis and Septic Shock. New York: McGraw‐Hill; 2011:2223–2231. [Google Scholar]
- 2. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, Van der Poll T, Vincent JL, Angus DC. The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA. 2016;315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83–96. [DOI] [PubMed] [Google Scholar]
- 4. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. [DOI] [PubMed] [Google Scholar]
- 5. Young LS, Stevens P, Kaijser B. Gram‐negative pathogens in septicaemic infections. Scand J Infect Dis Suppl. 1982;31:78–94. [PubMed] [Google Scholar]
- 6. Brun‐Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, Mercier JC, Offenstadt G, Regnier B. Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA. 1995;274:968–974. [PubMed] [Google Scholar]
- 7. Court O, Kumar A, Parrillo JE, Kumar A. Clinical review: myocardial depression in sepsis and septic shock. Crit Care. 2002;6:500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rudiger A, Singer M. Mechanisms of sepsis‐induced cardiac dysfunction. Crit Care Med. 2007;35:1599–1608. [DOI] [PubMed] [Google Scholar]
- 9. Zanotti‐Cavazzoni SL, Hollenberg SM. Cardiac dysfunction in severe sepsis and septic shock. Curr Opin Crit Care. 2009;15:392–397. [DOI] [PubMed] [Google Scholar]
- 10. Corrales‐Medina VF, Serpa J, Rueda AM, Giordano TP, Bozkurt B, Madjid M, Tweardy D, Musher DM. Acute bacterial pneumonia is associated with the occurrence of acute coronary syndromes. Medicine (Baltimore). 2009;88:154–159. [DOI] [PubMed] [Google Scholar]
- 11. Corrales‐Medina VF, Musher DM, Wells GA, Chirinos JA, Chen L, Fine MJ. Cardiac complications in patients with community‐acquired pneumonia: incidence, timing, risk factors, and association with short‐term mortality. Circulation. 2012;125:773–781. [DOI] [PubMed] [Google Scholar]
- 12. Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242. [DOI] [PubMed] [Google Scholar]
- 13. Vieillard‐Baron A, Caille V, Charron C, Belliard G, Page B, Jardin F. Actual incidence of global left ventricular hypokinesia in adult septic shock. Crit Care Med. 2008;36:1701–1706. [DOI] [PubMed] [Google Scholar]
- 14. Corrales‐Medina VF, Suh KN, Rose G, Chirinos JA, Doucette S, Cameron DW, Fergusson DA. Cardiac complications in patients with community‐acquired pneumonia: a systematic review and meta‐analysis of observational studies. PLoS Med. 2011;8:e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Griffin AT, Wiemken TL, Arnold FW. Risk factors for cardiovascular events in hospitalized patients with community‐acquired pneumonia. Int J Infect Dis. 2013;17:e1125–e1129. [DOI] [PubMed] [Google Scholar]
- 16. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35:569–582. [DOI] [PubMed] [Google Scholar]
- 17. Mehta S, Granton J, Lapinsky SE, Newton G, Bandayrel K, Little A, Siau C, Cook DJ, Ayers D, Singer J, Lee TC, Walley KR, Storms M, Cooper J, Holmes CL, Hebert P, Gordon AC, Presneill J, Russell JA. Agreement in electrocardiogram interpretation in patients with septic shock. Crit Care Med. 2011;39:2080–2086. [DOI] [PubMed] [Google Scholar]
- 18. Rich MM, McGarvey ML, Teener JW, Frame LH. ECG changes during septic shock. Cardiology. 2002;97:187–196. [DOI] [PubMed] [Google Scholar]
- 19. John J, Woodward DB, Wang Y, Yan SB, Fisher D, Kinasewitz GT, Heiselman D. Troponin‐I as a prognosticator of mortality in severe sepsis patients. J Crit Care. 2010;25:270–275. [DOI] [PubMed] [Google Scholar]
- 20. Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, Faverio P, Restrepo MI, Halade GV, Mortensen EM, Lindsey ML, Hanes M, Happel KI, Nelson S, Bagby GJ, Lorent JA, Cardinal P, Granados R, Esteban A, LeSaux CJ, Tuomanen EI, Orihuela CJ. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA, Wright PJ, Unudurthi SD, Snyder J, Bennett V, Hund TJ, Mohler PJ. Ankyrin‐G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014;115:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajaram MV, Ganesan LP, Parsa KV, Butchar JP, Gunn JS, Tridandapani S. Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol. 2006;177:6317–6324. [DOI] [PubMed] [Google Scholar]
- 23. Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin‐dependent protein kinase II‐based regulation of voltage‐gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharma J, Li Q, Mishra BB, Pena C, Teale JM. Lethal pulmonary infection with Francisella novicida is associated with severe sepsis. J Leukoc Biol. 2009;86:491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ojeda SS, Wang ZJ, Mares CA, Chang TA, Li Q, Morris EG, Jerabek PA, Teale JM. Rapid dissemination of Francisella tularensis and the effect of route of infection. BMC Microbiol. 2008;8:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage‐associated molecular patterns. Infect Immun. 2008;76:3001–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL, Li C. Attenuation of Cardiac dysfunction in polymicrobial sepsis by MicroRNA‐146a is mediated via targeting of IRAK1 and TRAF6 expression. J. Immunol. 2015;195:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gobbetti T, Coldewey SM, Chen J, McArthur S, Le FP, Cenac N, Flower RJ, Thiemermann C, Perretti M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc Natl Acad Sci USA. 2014;111:18685–18690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knuefermann P, Nemoto S, Misra A, Nozaki N, Defreitas G, Goyert SM, Carabello BA, Mann DL, Vallejo JG. CD14‐deficient mice are protected against lipopolysaccharide‐induced cardiac inflammation and left ventricular dysfunction. Circulation. 2004;106:2608–2615. [DOI] [PubMed] [Google Scholar]
- 30. Parsa KV, Ganesan LP, Rajaram MV, Gavrilin MA, Balagopal A, Mohapatra NP, Wewers MD, Schlesinger LS, Gunn JS, Tridandapani S. Macrophage pro‐inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2006;2:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajaram MV, Butchar JP, Parsa KV, Cremer TJ, Amer A, Schlesinger LS, Tridandapani S. Akt and SHIP modulate Francisella escape from the phagosome and induction of the Fas‐mediated death pathway. PLoS ONE. 2009;4:e7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neviere R, Fauvel H, Chopin C, Formstecher P, Marchetti P. Caspase inhibition prevents cardiac dysfunction and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care Med. 2001;163:218–225. [DOI] [PubMed] [Google Scholar]
- 33. Cremer TJ, Ravneberg DH, Clay CD, Piper‐Hunter MG, Marsh CB, Elton TS, Gunn JS, Amer A, Kanneganti TD, Schlesinger LS, Butchar JP, Tridandapani S. MiR‐155 induction by F. novicida but not the virulent F. tularensis results in SHIP down‐regulation and enhanced pro‐inflammatory cytokine response. PLoS ONE. 2012;4:e8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR‐155. Proc Natl Acad Sci USA. 2009;106:7113–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vihervaara A, Sistonen L. HSF1 at a glance. J Cell Sci. 2014;127:261–266. [DOI] [PubMed] [Google Scholar]
- 36. Murapa P, Ward MR, Gandhapudi SK, Woodward JG, D'Orazio SE. Heat shock factor 1 protects mice from rapid death during Listeria monocytogenes infection by regulating expression of tumor necrosis factor alpha during fever. Infect Immun. 2011;79:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jayakumar J, Suzuki K, Sammut IA, Smolenski RT, Khan M, Latif N, Abunasra H, Murtuza B, Amrani M, Yacoub MH. Heat shock protein 70 gene transfection protects mitochondrial and ventricular function against ischemia‐reperfusion injury. Circulation. 2001;104:I303–I307. [DOI] [PubMed] [Google Scholar]
- 38. Lu XY, Chen L, Cai XL, Yang HT. Overexpression of heat shock protein 27 protects against ischaemia/reperfusion‐induced cardiac dysfunction via stabilization of troponin I and T. Cardiovasc Res. 2008;79:500–508. [DOI] [PubMed] [Google Scholar]
- 39. Zhao Y, Wang W, Qian L. Hsp70 may protect cardiomyocytes from stress‐induced injury by inhibiting Fas‐mediated apoptosis. Cell Stress Chaperones. 2007;12:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iuliano S, Fisher SG, Karasik PE, Fletcher RD, Singh SN. QRS duration and mortality in patients with congestive heart failure. Am Heart J. 2002;143:1085–1091. [DOI] [PubMed] [Google Scholar]
- 41. Nada A, Gintant GA, Kleiman R, Gutstein DE, Gottfridsson C, Michelson EL, Strnadova C, Killeen M, Geiger MJ, Fiszman ML, Koplowitz LP, Carlson GF, Rodriguez I, Sager PT. The evaluation and management of drug effects on cardiac conduction (PR and QRS intervals) in clinical development. Am Heart J. 2013;165:489–500. [DOI] [PubMed] [Google Scholar]
- 42. Murkofsky RL, Dangas G, Diamond JA, Mehta D, Schaffer A, Ambrose JA. A prolonged QRS duration on surface electrocardiogram is a specific indicator of left ventricular dysfunction [see comment]. J Am Coll Cardiol. 1998;32:476–482. [DOI] [PubMed] [Google Scholar]
- 43. Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009;23:3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tomita K, Takashina M, Mizuno N, Sakata K, Hattori K, Imura J, Ohashi W, Hattori Y. Cardiac fibroblasts: contributory role in septic cardiac dysfunction. J Surg Res. 2015;193:874–887. [DOI] [PubMed] [Google Scholar]
- 45. Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55:801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Celes MR, Prado CM, Rossi MA. Sepsis: going to the heart of the matter. Pathobiology. 2013;80:70–86. [DOI] [PubMed] [Google Scholar]
- 47. Wang Q, Yokoo H, Takashina M, Sakata K, Ohashi W, Abedelzaher LA, Imaizumi T, Sakamoto T, Hattori K, Matsuda N, Hattori Y. Anti‐inflammatory profile of levosimendan in cecal ligation‐induced septic mice and in lipopolysaccharide‐stimulated macrophages. Crit Care Med. 2015;43:e508–e520. [DOI] [PubMed] [Google Scholar]
- 48. De SN, Hoton D, Castanares‐Zapatero D, Hantson P. Acute ventricular wall thickening: sepsis, thrombotic microangiopathy, or myocarditis? Case Rep Cardiol 2015;2015:275825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown SM, Pittman JE, Hirshberg EL, Jones JP, Lanspa MJ, Kuttler KG, Litwin SE, Grissom CK. Diastolic dysfunction and mortality in early severe sepsis and septic shock: a prospective, observational echocardiography study. Crit Ultrasound J. 2018;4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grun S, Schumm J, Greulich S, Wagner A, Schneider S, Bruder O, Kispert EM, Hill S, Ong P, Klingel K, Kandolf R, Sechtem U, Mahrholdt H. Long‐term follow‐up of biopsy‐proven viral myocarditis: predictors of mortality and incomplete recovery. J Am Coll Cardiol. 2012;59:1604–1615. [DOI] [PubMed] [Google Scholar]
- 51. Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. 2007;117:2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen Y, Qin J, Bu P. Pathways involved in interleukin‐1beta‐mediated murine cardiomyocyte apoptosis. Tex Heart Inst J. 2015;42:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ft.n infection causes cardiac fibrosis. Cross‐sections of hearts obtained from BALB/c mice after 96 hours of Ft.n challenge. The 4‐chamber view sections from uninfected (A) and Ft.n‐infected (B and C) mice were stained with Masson's trichrome to identify fibrosis in cardiac tissue. Image shown in (B) is interstitial fibrosis, and (C) is perivascular fibrosis from Ft.n‐infected mice hearts. The image shown in right panel is 60× magnification and representative of 6 animals per group.
Figure S2. Ft.n infection increases expression of inflammatory mediators in the heart as well as serum cytokine levels. BALB/c mice were infected with Ft.n (25 CFUs) through the intranasal route, and after 96 hours incubation, mice were sacrificed to harvest the heart, which was cut into small pieces, washed with PBS, and immediately homogenized with Trizol reagent. Total RNA was extracted and purified by using RNAeasy column and converted into cDNA. Quantitative real‐time RT‐PCR was used to determine (A) IL‐1β, (B) TNF, (C) IL‐8, and (D) SOD2 mRNA levels. Data ware normalized to the β‐actin gene, and relative gene expression was determined. Blood samples from Ft.n‐infected or sham‐treated mice were kept at 4°C for 1 hour and centrifuged at 1500g for 10 minutes at 4°C. Serum samples were used to determine levels of inflammatory cytokines by ELISA. Graph shown in (E) IL‐1β and (F) TNF is a representative graph from 3 experiments (N=3; **P<0.005; ***P<0.0005).
Figure S3. Ft.n infection induces cardiomyocyte apoptosis. Neonatal myocytes were isolated from BALB/C mice and infected with GFP‐Ft.n (MOI, 10:1), and after 6 hours of incubation, cells were washed, fixed, permeabilized, and stained with Alexa Fluor 594 (AF594)‐conjugated phalloidin antibody. Myocytes were examined under confocal microscopy, and the image shown is representative of 10 infected myocytes.