

Abstract
Background
Activated T cells and dendritic cells (DCs) are colocalized in atherosclerotic plaques in association with plaque rupture. Oxidized low‐density lipoprotein (oxLDL) promotes immune activation and inflammation. We studied the effects of statins (atorvastatin and simvastatin) on human DC maturation and T‐cell activation.
Methods and Results
Human peripheral blood monocytes were differentiated to DCs and stimulated with oxLDL. T cells were isolated from carotid endarterectomy specimens from patients undergoing carotid endarterectomy or from healthy individuals. Naïve T cells were cocultured with pretreated DCs. The effects of statin were studied. OxLDL induced DC maturation and T‐cell activation. OxLDL induced atherogenic heat shock proteins (HSP) 60 and 90 and decreased potentially atheroprotective heat shock protein 27, effects restored by atorvastatin. T cells exposed to oxLDL‐treated DCs produced interferon‐γ and interleukin (IL)‐17. Atorvastatin and simvastatin suppressed the DC maturation showing lower expression of CD80, CD83, and CD86, and limited their production of tumor necrosis factor‐α, IL‐1β and IL‐6, and increased transforming growth factor‐β and IL‐10 secretion. Statin‐treated DCs inhibited Th1 and/or Th17 polarization by downregulation of transcriptional factors T‐bet and RORγt expression, and induced T regulatory cells with IL‐10 production. OxLDL‐induced miRNA let7c and phosphorylation of Akt and ERK were repressed by statins. Let‐7c had a pivotal role in mediating effect of oxLDL. Experiments on T cells derived from carotid atherosclerotic plaques or healthy individuals showed similar results.
Conclusions
Statins repress human DC maturation induced by oxLDL, limit T‐cell activation, and repress an atherogenic heat shock protein profile and promote induction of T regulatory cells. MicroRNA let‐7c is integral to the effects.
Keywords: atherosclerosis, dendritic cells, immune system, microRNA, oxidized low‐density lipoprotein, statin, T cells
Subject Categories: Inflammation, Lipids and Cholesterol, Vascular Biology, Translational Studies
Introduction
During recent years it has become clear that atherosclerosis, the main cause of cardiovascular disease (CVD), is an inflammatory process in the artery wall, where activated immune‐competent cells including T cells, dendritic cells (DCs), monocytes/macrophages and others, which actively produce factors as cytokines, are abundant.1, 2
A proatherogenic role of T‐cell subsets is indicated by studies using mouse models, other experimental models, and also human ex vivo studies. For example, transfer of CD4+ T cells to immunodeficient atherosclerosis‐prone mice accelerates atherosclerosis progression and abrogation of transforming growth factor‐beta (TGF‐β) signaling in T cells alters atherosclerotic plaques.3, 4 Also, studies in humans support a pathologic role for pro‐inflammatory T cells.5 DCs most likely play an important role in atherosclerosis at different stages as specialized antigen‐presenting cells.6, 7, 8 Furthermore, DCs and T cells colocalize in plaques, which suggests participation in local immune reactions.9
Another essential feature of atherosclerotic lesions is the presence of lipids, especially low‐density lipoproteins (LDL), where oxidation and/or modification of LDL by enzymes as phospholipases (OxLDL) appears to be a prerequisite for uptake in lesions.2 Furthermore, oxLDL in contrast to LDL activates DC and T cells into a pro‐inflammatory phenotype and oxLDL‐induced T‐cell activation could be of importance for plaque rupture, where heat shock protein 60 (HSP60) (and potentially HSP90) may play a central role also as antigens.10
The enzyme 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase, which catalyses the conversion of 3‐hydroxy‐3‐methylglutaryl coenzyme A to mevalonic acid, is essential for cholesterol synthesis. A common denominator for different statins is inhibition of this enzyme.11 Statins for prevention of atherosclerosis complications have been much studied and are widely used.12, 13, 14 Clinical studies such as the Jupiter study, which targeted patients at CVD risk with increased C‐reactive protein levels, suggested that statin effects could also be related to inflammation,15 a notion also supported by much experimental evidence.16 Pleiotropic effects of statins have been much discussed, including anti‐inflammatory, and even immune modulatory.17 Still, to the best of our knowledge, there is no information about the role of statins in specific immune reactions of direct potential importance in plaque rupture and thus CVD. LDL lowering per se may be a significant factor in reducing risk in acute coronary syndromes.18 One mechanism could be that lower LDL also could imply lower levels of immune stimulatory modified LDL, which causes activation of DC and T cells from atherosclerotic plaques.10
We here report that statins, by a novel mechanism involving the microRNA (miRNA) let‐7c, repress human DC maturation induced by oxLDL, limit T‐cell activation, and promote an anti‐inflammatory cell response and induction of T regulatory cells. The implications of these findings are discussed.
Materials and Methods
Differentiation and Treatment of Monocyte‐Derived DCs
Peripheral blood mononuclear cells were isolated using the standard Ficoll‐Paque PLUS (GE Healthcare, Sweden). For monocyte isolation by adherence, 50×106 peripheral blood mononuclear cells were seeded per Petri dish (10‐cm diameter) in 10 mL RPMI/10% Fetal Calf serum, and allowed to adhere in a 5% CO2 incubator at 37°C for 2 hours. Nonadherent cells were removed and the adherent cells were carefully washed twice with culture medium. Recovered monocytes were >90% pure as assessed by CD14 labeling. Monocytes were differentiated to immature DCs during 7 days with 50 ng/mL human recombinant granulocyte‐macrophage colony‐stimulating factor and 25 ng/mL human recombinant interleukin (IL)‐4 in RPMI 1640 supplemented with 2 mmol/L glutamine, 10 mmol/L HEPES, and 1% penicillin/streptavidin. For statin treatment, an aliquot of immature DCs at day 5 were treated with 2 μmol/L atorvastatinor 5 μmol/L simvastatin (Sigma‐Aldrich, Germany) for 24 hours, then DCs were incubated with 2 μmol/L atorvastatinor 5 μmol/L simvastatin, in the presence or absence of 10 μg/mL oxLDL (Copper‐oxidized LDL from Source BioScience, UK) and/or 100 μmol/L mevalonate (Mev) (Sigma‐Aldrich, Germany) for another 24 hours. Cells were harvested and analyzed. Cell viability was >90%.
Plaque T‐Cell Isolation
Carotid endarterectomy specimens (atherosclerotic plaques) were from patients undergoing carotid endarterectomy at the Department of Surgery, Karolinska University Hospital, Stockholm, Sweden. All patients had clinical symptoms of transitory ischemic attacks or minor stroke. All specimens contained advanced atherosclerotic lesions type IV.19 The plaque specimens were kept in RPMI‐medium and were transferred immediately to the laboratory for separation of cells. The study was approved by the local ethics committee of Karolinska Hospital, participating subjects gave informed content, institutional review board approval was obtained, and the study was conducted in accordance with the Helsinki Declaration.
The experimental procedure has been described in a recent publication.10 Plaque was rinsed intensively with phosphate‐buffered saline and medium RPMI‐1640 to reduce the risk of contamination. Mononuclear cell populations were isolated according to Grivel et al with modification.20 Briefly, the plaque was dissected into multiple blocks, and were digested with the enzyme mixture containing collagenase IV (1.25 mg/mL; Life Technologies Europe BV, Sweden), Liberase DL (25 μg/mL; Roche Applied Science, Sweden), and DNase I (0.2 mg/mL; Roche Applied Science) for 1.5 hours at 37°C. The released cells and tissue ghosts were strained through a 100‐μm Celltrics filter (Millipore AB, Sweden). The supernatant was collected for mononuclear cell isolation using the standard Ficoll‐Paque PLUS (GE Healthcare, Sweden). T cells were further purified by Dynabeads® Untouched™ Human T Cells (Life Technologies Europe BV,).
T Cell–DC Cocultures
Peripheral blood mononuclear cells were isolated by density gradient centrifugation on Ficoll‐Hypaque. CD4+ T cells were purified from peripheral blood mononuclear cells by negative selection using the untouched CD4+ T cell Isolation kit (Miltenyi Biotech, Sweden), according to the manufacturer's instructions.
T cell–DC cocultures were conducted in 96‐well flat‐bottom culture plates. DCs were generated as above, collected at day 7, extensively washed, and resuspended in RPMI/10% Fetal Calf serum. Two methods were employed to analyze the T‐cell proliferation: (1), carboxyfluorescein succinimidyl ester (CFSE) staining. 2×105 T cells prelabeled by CFSE (5 μmol/L) were then cocultured with autologous DCs in 200 μL of complete culture medium at 10:1 T:DC cells ratio. After 4 days, culture supernatant was frozen for cytokine analysis and cells were harvested to test the CFSE expression by flow cytometry. Cells were stained with anti‐CD4 Phycoerythrin‐conjugated (BD Biosciences, Sweden) for 20 minutes at 4°C. Results were expressed as percentage of proliferating CFSElow/CD4+ cells. (2), Bromodeoxyuridine incorporation assay: 2×105 T cells were cocultured with autologous DCs for 5 days in 200 μL of complete culture medium at 10:1 T:DC cells ratio. Bromodeoxyuridine (10 μmol/L) (Roche Diagnostics Scandinavia AB, Sweden) was present in the last 16 hours. The cells were fixed and processed according to the manual provided. Proliferative response was evaluated as optical density value.
T‐Cell Differentiation and Suppression
CD4+ T cells were depleted of CD45RO+ cells using anti‐CD45RO‐coupled magnetic beads and LD negative selection columns (Miltenyi Biotech, Sweden). In the purified cells the proportion of CD4+CD45RO−CD45RA+ was consistently >90%. 1×106 CD4+CD45RO− T cells were cultured with 1×105 autologous DCs in 1 mL of X‐vivo 15 medium (Biowhittaker, Sweden). After 6 days, recombinant human IL‐2 (20 U/mL) was added, and cells were expanded for an additional 7 days. Thirteen days after initiation of the culture, T cells were collected, washed, and analyzed for their functions. T cells stimulated with oxLDL‐treated DC referred to as T (DC [oxLDL]), and T cells stimulated with (atorvastatin [AVA]+oxLDL)‐treated DC referred to as T (DC [AVA+oxLDL]).
To test for the capacity of these T cells to suppress proliferation and/or cytokine production, autologous CD4+CD45RO− T cells prelabeled by CFSE (5 μmol/L) were then stimulated with mature DCs (10:1; 105T:104DC), in the absence or presence of T (DC), T (DC [oxLDL]), or T (DC [AVA+oxLDL]) cells (1:1 ratio) in a final volume of 200 μL of complete medium in 96‐well round‐bottom plates. Cells were cultured for 5 days and CFSE expression was tested by flow cytometry. Results were expressed as percentage of proliferating CFSElow/CD4+ cells.
Intracellular Staining of Cytokines
Intracellular cytokines were detected by flow cytometry. Briefly, T cells (1×106/mL) were stimulated with immobilized anti‐CD3 (1 μg/mL; OKT3, eBioscience) and tetradecanoylphorbol acetate (10 ng/mL; Sigma) for 6 hours in complete medium. Prior to the culture, the plates were centrifuged for 5 minutes at 800g. Three hours after activation, monensin (10 μg/mL; Sigma) was added. T cells were then collected, washed in phosphate‐buffered saline, and fixed with 2% formaldehyde. After fixation, T cells were permeabilized in phosphate‐buffered saline supplemented with 2% Fetal Calf serum and 0.5% saponin (Sigma). Permeabilized T cells were incubated with anti‐IL‐17, anti‐IL‐4, anti‐interferon‐γ, anti‐IL‐10, or anti‐FoxP3 monoclonal antibodies. All monoclonal antibodies were obtained from BD PharMingen. After washing, cells were analyzed using LSRII Fortessa™ cell analyzer (BD Biosciences, CA), and data were analyzed with Flowjo software. Quadrant markers were set accordingly to isotype‐matched controls.
Phospho‐Flow Cytometry
Dendritic cells were fixed with 500 μL BD Cytofix (BD Biosciences) at 37°C, 15 minutes, permeabilized with ice‐cold Perm Buffer III (BD Biosciences) for 30 minutes on ice. Cells were stained with Alexa Fluor® 647 or Alexa Fluor® 488 conjugated Mouse anti‐Akt (pS473), Alexa Fluor® 488 or PerCP‐Cy™5.5 conjugated Mouse anti‐MEK1 (pS298). Isotype controls were used to establish positive expression limits. After 60 minutes incubation, cells were analyzed on an LSRII Fortessa™ cell analyzer.
Enzyme‐Linked Immunosorbent Assay
The amounts of tumor necrosis factor‐alpha (TNF‐α), IL‐10, IL‐1β, TGF‐β, and IL‐6 in cell cultures were determined by enzyme‐linked immunosorbent assay. Culture supernatants of DCs were collected at day 7 after a 24‐hour treatment as indicated, and level of TNF‐α, IL‐10, IL‐1β, TGF‐β, and IL‐6 was measured according to the manufacturer's instructions (R&D Systems Europe Ltd, UK).
Quantitative Real‐Time Reverse Transcription Polymerase Chain Reaction
Total RNA was isolated and cDNA was reversely transcribed using Superscript II (Gibco BRL) according to manufacturer's instructions. Real‐time quantitative polymerase chain reaction was performed for selected genes. We tested genes TBX21 (Hs00203436_m1), GATA3 (Hs00231122_m1), RORC (Hs01076122_m1), FOXP3 (Hs01085834_m1), HSP60 (Hs01102298_g1), HSP90 (Hs00743767_sH), (Hs00356629_g1), and PRDM1 (Hs00153357_m1; Applied Biosystems, CA). The expression level of the housekeeping gene GAPDH (Hs02758991_g1) was used to normalize for differences in input cDNA. Real‐time quantitative polymerase chain reaction was carried out using TaqMan gene expression presynthesized reagents and master mix (Applied Biosystems, CA) in Applied Biosystems 7500 Real‐Time PCR System. The expression ratio was calculated using the ΔΔCT method.
Micro RNA
To detect miRNAs from cells, total RNA was isolated using Trizol Reagent (Invitrogen). MiRNA‐specific cDNA was generated with TaqMan MicroRNA RT kit (Applied Biosystems, CA). Primer sets for let‐7c (000379), miR‐155 (000479), miR‐146 (002163), miR‐17 (002308), and miR‐23b (000400) were from Applied Biosystems, CA. The polymerase chain reaction was directly monitored by the Applied Biosystems 7500 Real‐Time PCR System. U6 RNA was used as an endogenous control.
miRNA/Small Interference RNA Transfection and Functional Study
DCs were transfected with the miRNA inhibitors, let‐7c (MH12580), mimic control (MC12580) using Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) in a 20‐nmol/L concentration. oxLDL was added 6 hours after transfection, and cells were further incubated for 24 hours followed with functional study and RNA/protein analysis.
Statistical Analysis
Statistical significance was determined with 1‐way ANOVA. The statistical significance is based on Bonferroni corrected values. P‐value is presented for individual experiments (*P<0.05, **P<0.01). The sample sizes are for each condition (eg, untreated, oxLDL, AVA+oxLDL, etc) and not for the total number of data points.
Results
oxLDL‐Induced Maturation of DCs Is Limited by Statin
The effect of different statins, atorvastatin and simvastatin, on the maturation of DCs was studied. Mevalonate was included in the treatment to test whether the effect of statin on DC maturation was mediated through the specific inhibition of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase. The expression of costimulatory molecules CD86, CD40, CD83, and HLA‐DR were upregulated in oxLDL‐treated DCs (Figures 1A and 2A). By treatment with atorvastatin or simvastatin, their upregulation induced by oxLDL was significantly repressed. Mevalonate abrogated the effect of statins on oxLDL‐induced DC maturation.
Figure 1.
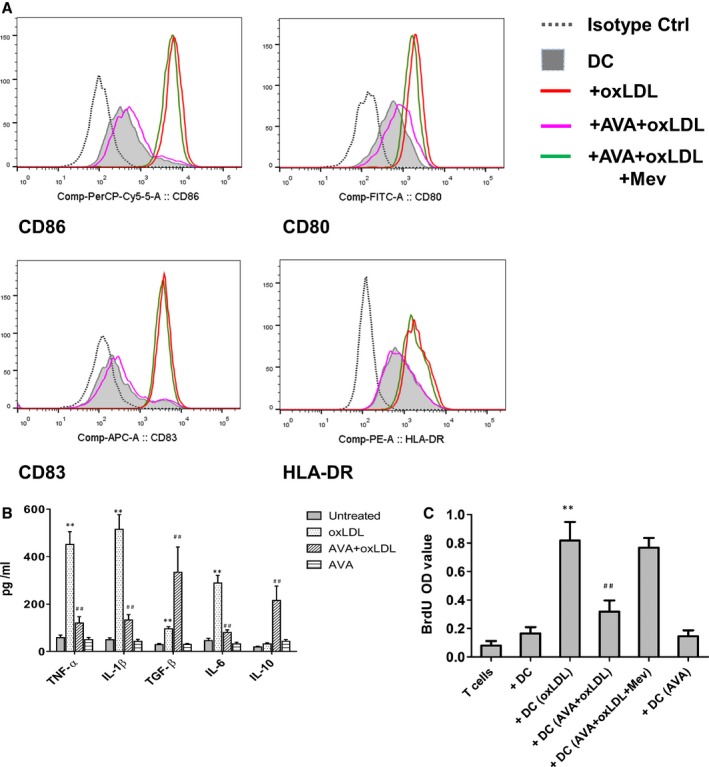
Dendritic cell (DC) maturation, proinflammatory cytokine production, and the subsequent T‐cell proliferation induced by oxidized low‐density lipoprotein (OxLDL) were suppressed by statins. DCs at day 6 were treated with 10 μg/mL oxLDLfor 24 hours. For atorvastatin treatment, an aliquot of DCs on day 5 were treated with 2 μmol/L atorvastatin (AVA) for 24 hours, then stimulated with 2 μmol/L atorvastatin in the presence or absence of 10 μg/mL oxLDL and/or 100 μmol/L mevalonate (Mev) for another 24 hours. A, The expression of CD80, CD86, CD83, and HLA‐DR on day 7 DCs was analyzed by flow cytometry. A representative of 10 independent experiments is shown. B, Cytokine production tested by ELISA. DC supernatants were collected at day 7. Results represent the mean concentration±SEM of 8 independent experiments. **P<0.01, DC+OxLDL vs DC; ## P<0.01, DC+AVA+oxLDL vs DC+OxLDL. C, T‐cell proliferation. After the indicated treatment, DCs were washed and cocultured in triplicate for 5 days with autologous T cells. BrdU (bromodeoxyuridine), 10 μmol/L, was present in the last 16 hours. Proliferative response was evaluated as OD value. Results show mean±SD (n=6). **P<0.01, DC+OxLDL vs DC; ## P<0.01, DC+AVA+oxLDL vs DC+OxLDL. IL indicates interleukin; TGF, transforming growth factor; OD, optical density; TNF, tumor necrosis factor.
Figure 2.
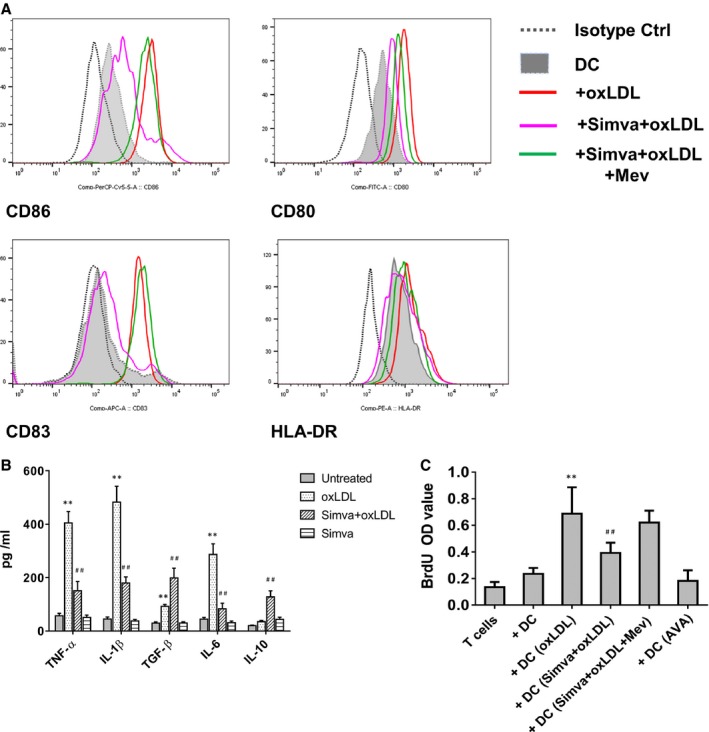
Simvastatin suppressed oxidized low‐density lipoprotein (oxLDL)‐induced dendritic cell (DC) maturation, cytokine production, and the subsequent T‐cell proliferation. DCs at day 6 were treated with 10 μg/mL oxLDLfor 24 hours. An aliquot of DCs on day 5 were treated with 5 μmol/L simvastatin (Simva) for 24 hours, then stimulated with 5 μmol/L simvastatin in the presence or absence of 10 μg/mL oxLDL and/or 100 μmol/L mevalonate (Mev) for further 24 hours. A, The expression of CD80, CD86, CD83, and HLA‐DR on DCs were detected by flow cytometry. A representative of 4 independent experiments is shown. B, Cytokine production was tested by ELISA. Supernatants of DC were collected at day 7. Results represent the mean concentration±SD of 5 independent experiments. **P<0.01, DC+oxLDL vs DC; ## P<0.01, DC+Simva+oxLDL vs DC+oxLDL. C, T‐cell proliferation. After the indicated treatment, DCs were washed and cocultured in triplicate for 5 days with autologous T cells. BrdU (bromodeoxyuridine), 10 μmol/L, was present in the last 16 hours. Proliferative response was evaluated as OD value. Results show mean±SD (n=4). **P<0.01, DC+oxLDL vs DC; ## P<0.01, DC+Simva+oxLDL vs DC+oxLDL. AVA indicates atorvastatin; OD, optical density.
The expression of costimulatory molecules CD86, CD40, CD83, and HLA‐DR was upregulated in oxLDL‐treated DCs (Figures 1A and 2A). By treatment with atorvastatin or simvastatin, their upregulation induced by oxLDL was significantly repressed. Mevalonate abrogated the effect of statins on oxLDL‐induced DC maturation.
To characterize the impact of oxLDL on DC function, we analyzed the production of cytokines from DCs (Figure 1B). oxLDL induced a significantly increased secretion of TNF‐α (452±53 pg/mL versus 59±11 pg/mL, n=6, P<0.01), IL‐1β (515±61 pg/mL versus 49±9 pg/mL, n=6, P<0.01), IL‐6 (289±32 pg/mL versus 46±10 pg/mL, n=6, P<0.01), and TGF‐β (96±9 pg/mL versus 29±5 pg/mL, n=6, P<0.01). A low level of IL‐10 could be detected in supernatants of oxLDL‐treated DCs and immature DCs (32±5 pg/mL and 19±3 pg/mL, respectively). In contrast to oxLDL‐treated‐only DC cultures, atorvastatin inhibited the production of TNF‐α, IL‐1β, and IL‐6 but increased substantially secretion of IL‐10 (215±61 pg/mL) and TGF‐β (335±106 pg/mL) in the DC cultures (Figure 1B). Simvastatin showed a similar effect (Figure 2B).
T‐Cell Activation Stimulated by oxLDL‐Treated DC Is Suppressed by Statins
Mature DCs are of major importance for stimulation of naïve T cells, in order to test the effect of statin on T‐cell proliferation. DCs were thus treated with atorvastatin and/or oxLDL, and applied in DC–T‐cell coculture experiments. We demonstrate that oxLDL‐treated DCs stimulate T‐cell proliferation (Figure 1C). With treatment by atorvastatin, T‐cell proliferation induced by oxLDL‐treated DCs was significantly reduced (Figure 1C). Simvastatin treatment showed an effect similar to atorvastatin (Figure 2C).
Th1 and Th17 Polarization Induced by oxLDL‐Treated DCs Were Inhibited, While T Regulatory Cells Were Induced by Atorvastatin
Differentiation of naïve CD4+ T cells into different T‐cell subsets is driven by the lineage‐specifying transcription factors. Th1, Th2, Th17, and Treg subsets are driven by T‐bet/TBX21, GATA3, RORγt/RORC, and FOXP3, respectively. TBX21, GATA3, RORC, and FOXP3 gene expression was evaluated by real‐time polymerase chain reaction (Figure 3A). oxLDL‐treated DCs strongly induced TBX21 and RORC expression in naïve T‐cell populations. However, on pretreatment with statin, expression of TBX21 and RORC was inhibited while FoxP3 was strikingly induced.
Figure 3.
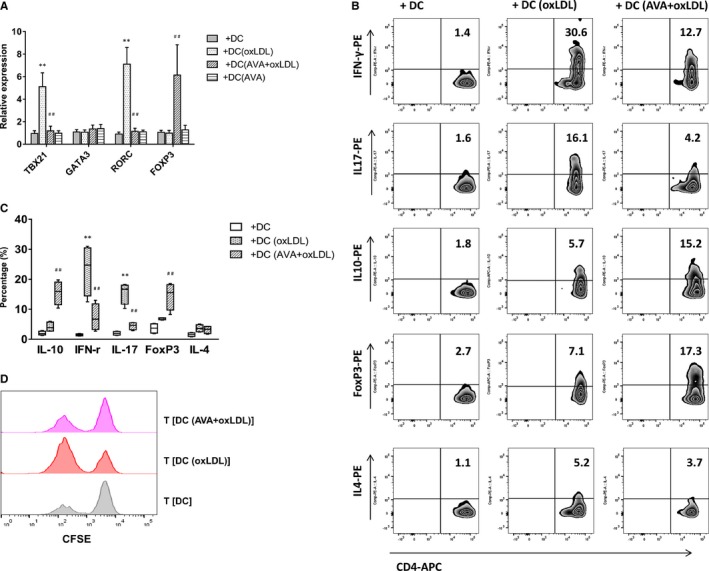
Th1 and Th17 polarization induced by oxidized low‐density lipoprotein (oxLDL)‐treated dendritic cells (DCs) were inhibited, while T regulatory cells were induced by atorvastatin. A, T‐cell polarization‐related transcription factors. Quantitative PCR data represent mean±SD of 6 independent experiments. B, Intracellular staining of cytokine production in T cells. Naïve CD4+ T cells were cultured with oxLDL‐ or atorvastatin (AVA)+oxLDL–treated autologous DCs for 13 days. After stimulation, T cells were activated with anti‐CD3 monoclonal antibody and intracellular staining of cytokine production. Percentages of interferon‐γ (IFN‐γ)‐, interleukin (IL)‐17‐, IL‐10‐producing cells, and FoxP3‐positive cells in T (DC), T (DC [oxLDL]), or T (DC [AVA+oxLDL]) cells are presented. Figure depicts a representative experiment of 5. C, Frequency of T‐cell subsets is summarized. Results show mean±SD (n=5). D, T (DC [AVA+oxLDL]) cells suppressed primary T‐cell proliferation. T‐cell proliferation was measured by CFSE staining. Naïve CD4+ T cells were stimulated with mature DCs alone or in the presence of T (DC), T (DC [oxLDL]), or T (DC [AVA+oxLDL]) cells at a 1:1 ratio. Results show a representative experiment of 4. **P<0.01, T (DC [oxLDL]) vs T (DC); ## P<0.01, T (DC [AVA+oxLDL]) vs T (DC [oxLDL]). CFSE indicates carboxyfluorescein succinimidyl ester; PCR, polymerase chain reaction.
A considerable proportion of T cells stimulated with oxLDL‐treated DCs produced interferon‐γ (23.3±8.5% versus 1.5±0.3%, n=5, P<0.01), whereas no significant induction of IL‐4‐producing cells could be detected. Interestingly, a small population of IL‐17‐producing T cells was also induced (15.5±3.6% versus 2.0±0.6%, n=5, P<0.01) (Figure 3B and 3C). This indicated that oxLDL might tune T cells to initiate both Th1 and Th17, but not Th2 polarization. A low level of IL‐10‐producing T cells was detected in cultures stimulated with oxLDL‐treated DCs or immature DCs (5.1±2.0% and 2.0±0.6%, respectively, n=5). In contrast to oxLDL‐only‐treated DC cultures, T cells obtained after stimulation with DC (AVA+oxLDL) contained a low proportion of interferon‐γ‐ and IL‐4‐producing cells (10.8±3.6% and 4.2±1.1%, respectively, n=5, P<0.01), but increased a significant proportion of IL‐10‐producing and FoxP3‐positive T cells (16.8±3.2% and 17.6±3.8%, respectively, n=5) (Figure 3B and 3C).
T (DC [AVA+oxLDL]) cells were tested for their ability to suppress autologous CD4+ T cells activated with mature DCs. Proliferation of naive CD4+ T cells stimulated with mature DCs was significantly reduced by addition of T (DC [AVA+oxLDL]) cells at a 1:1 ratio (Figure 3D). Thus, T cells generated with DC (AVA+oxLDL) suppress primary T‐cell responses.
miRNA let‐7c and PI3k/ERK Signaling Pathways Were Involved in the Effect of oxLDL and Statins
miRNAs that related to DC maturation and tolerogenicity were tested. Expression of miR155 and miR146 was induced by oxLDL (Figure 4A). Pretreatment with statin limited this induction but induced miR‐17 and miR‐23b that related to tolerogenicity. Interestingly, let‐7c was the most prominently induced by oxLDL but it was abolished by statin pretreatment.
Figure 4.
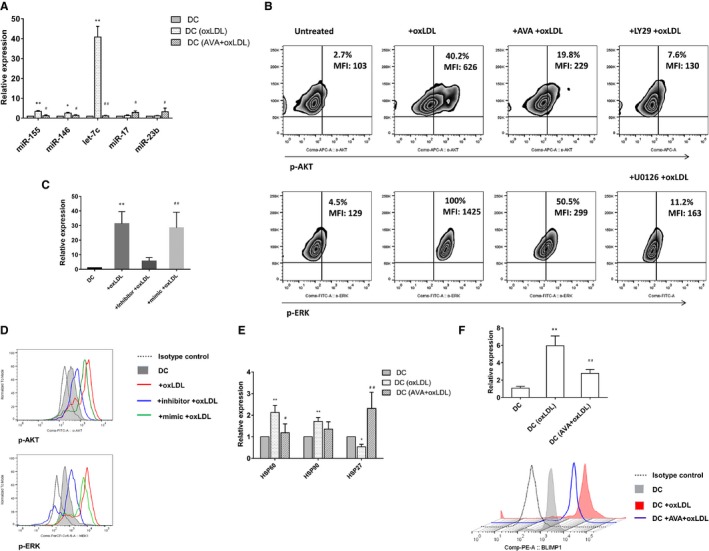
MicroRNA (miRNA) let‐7c and PI3k/ERK signaling pathways were involved in the effect of oxidized low‐density lipoprotein (oxLDL) and statins. A, Expression of miRNAs in dendritic cells (DCs) was tested by quantitative reverse transcription polymerase chain reaction (qRT‐PCR). The value was normalized as fold change to that of nontreated DC samples. Results represent the mean±SEM of 7 experiments. *P<0.05, **P<0.01, DC+oxLDL vs DC; # P<0.05, ## P<0.01, DC+ atorvastatin (AVA)+oxLDL vs DC+oxLDL. B, Phosphorylation of Akt and ERK induced in oxLDL‐treated DCs were confined by AVA. OxLDL‐induced phosphorylation of Akt and ERK were tested by phospho‐flow using specific antibody anti‐Akt (pS473) and anti‐MEK1 (pS298). Results show 1 representative experiment of 5. LY294002 (PI3k inhibitor) and U0126 (MEK inhibitor) are used as control. C, Inhibition of let‐7c in DCs was tested by qRT‐PCR. DCs at day 6 were treated with 10 μg/mL oxLDL for 24 hours. For inhibition of let‐7c, inhibitor of miRNA let‐7c, mimic control was transfected with Lipofectamine® RNAiMAX (Invitrogen) in a 20 nmol/L concentration at day 6. oxLDL was added 6 hours after transfection, and cells were further incubated for 24 hours. The value was normalized as fold change to that of nontreated DC samples. Results represent the mean±SD of 4 experiments. **P<0.01, DC+oxLDL vs DC; ## P<0.01, DC+inhibitor+oxLDL vs DC+mimic+oxLDL. D, Downregulation of let‐7c restricted the phosphorylation of Akt and ERK in the oxLDL‐treated DC. One of 4 experiments is shown. E, Expression of heat shock proteins (HSPs) in DCs was tested by qRT‐PCR. Results represent the mean±SD of 4 experiments. *P<0.05, **P<0.01, DC+oxLDL vs DC; # P<0.05, ## P<0.01, DC+AVA+oxLDL vs DC+oxLDL. F, Expression of B lymphocyte–induced maturation protein‐1 (BLIMP1) in DCs was tested by qRT‐PCR showing the mean±SD of 4 experiments, and flow cytometry showing a representative of 4 experiments. **P<0.01, DC+oxLDL vs DC; ## P<0.01, DC+AVA+oxLDL vs DC+oxLDL. MFI indicates median fluorescence intensity.
Phosphorylation of Akt and extracellular signal‐regulated kinase (ERK) mitogen‐activated protein kinase (MAPK) were tested by phospho‐flow using specific antibody anti‐Akt (pS473) and anti‐MEK1 (pS298). Both were induced in oxLDL‐treated DCs and the effect was confined by atorvastatin (Figure 4B). Specific inhibitors of PI3k and MEK, LY294002 and U0126, respectively, were treated as positive control. Expression of let‐7c was strikingly downregulated by specific inhibitor, and miRNA mimics were administered as control (Figure 4C). Furthermore, downregulation of let‐7c restricted the phosphorylation of Akt and ERK in the oxLDL‐treated DC (Figure 4D).
In our recent article, we demonstrated that HSP is essential for oxLDL‐induced DC‐ and T‐cell activation, and we suggest that T cells recognize oxLDL‐induced HSP as antigens and thus that HSP (60) is a major immunological antigen related to oxLDL.10 Expression of HSP60 and HSP90 was upregulated but that of HSP27 was downregulated in oxLDL‐treated DCs. Atorvastatin treatment suppressed the expression of HSP60, whereas it significantly upregulated the HSP27 expression (2.31±0.75% versus 0.54±0.23%, n=4, P<0.01; Figure 4E).
Recently the transcriptional repressor B lymphocyte–induced maturation protein‐1 (BLIMP1) was implicated in DC activation with let‐7c.21 We therefore investigated the role of BLIMP1 in our oxLDL‐related systems. BLIMP1 expression in DCs was induced by oxLDL treatment; however, in the presence of statin the induction of BLIMP1 was dampened (Figure 4F).
miRNA let‐7c Was Essential to the oxLDL‐Mediated Effect on DC Maturation, Cytokine Production, and the Subsequent T‐Cell Proliferation
In comparison with the mimic control, inhibition of let‐7c decreased considerably the expression of CD86 and CD83 on DCs (Figure 5A). The production of TNF‐α, IL‐1β, and IL‐6 was limited but IL‐10 secretion was increased in the DC cultures (Figure 5B). In the subsequent T‐cell proliferation assay, inhibition of let‐7c significantly reduced T‐cell proliferation compared with that in mock‐treated cultures (0.38±0.14 versus 0.84±0.13, n=6, P<0.01; Figure 5C).
Figure 5.
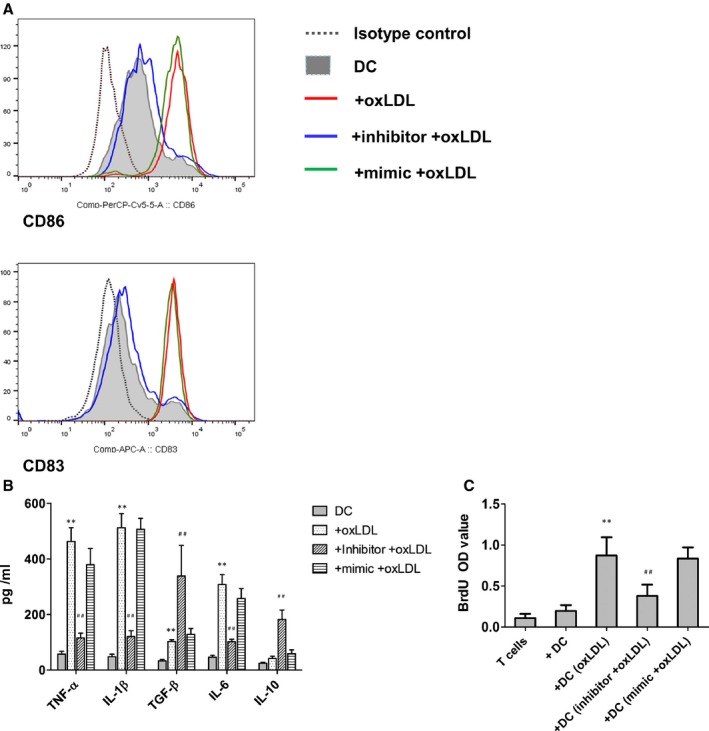
MicroRNA let‐7c mediated the effect of oxidized low‐density lipoprotein (oxLDL) on dendritic cell (DC) maturation, cytokine production, and T‐cell proliferation. A, let‐7c was involved in the DC maturation. The expression of CD86 and CD83 were analyzed by flow cytometry. Results show 1 representative of 6 independent experiments. B, Cytokine production in DC cultures after let‐7c downregulation. DC supernatants were collected at day 7, and tested by ELISA. Results represent the mean concentration±SEM of 6 independent experiments. **P<0.01, DC+OxLDL vs DC; ## P<0.01, DC+inhibitor+oxLDL vs DC+OxLDL. IL indicates interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor. C, T‐cell proliferation. After the indicated treatment, DCs were washed and cocultured with autologous T cells in triplicates for 5 days. Bromodeoxyuridine (BrdU) 10 μmol/L was present in the last 16 hours. Proliferative response was evaluated as OD value. Results show mean±SD of 6 independent experiments. **P<0.01, DC+oxLDL vs DC; ## P<0.01, DC+inhibitor+oxLDL vs DC+mimic+oxLDL. OD indicates optical density.
Statin Suppressed the oxLDL‐Induced DC Maturation, Proinflammatory Cytokine Production, and Subsequent T‐Cell Proliferation Derived From Patients
DCs were generated from peripheral blood monocytes of patients who underwent carotid endarterectomy with higher basal expression of CD86 and CD83 on DCs, and they were further enhanced by oxLDL stimulation. Pretreatment with statin repressed the induction (Figure 6A). We analyzed the cytokine production from DCs. Patient DCs had a higher basal level of TNF‐α, IL‐6, and IL‐10. oxLDL induced production of TNF‐α, IL‐1β, IL‐6, and TGF‐β. However, atorvastatin inhibited this induction but promoted secretion of IL‐10 and TGF‐β (335±106 pg/mL) (Figure 6B).
Figure 6.
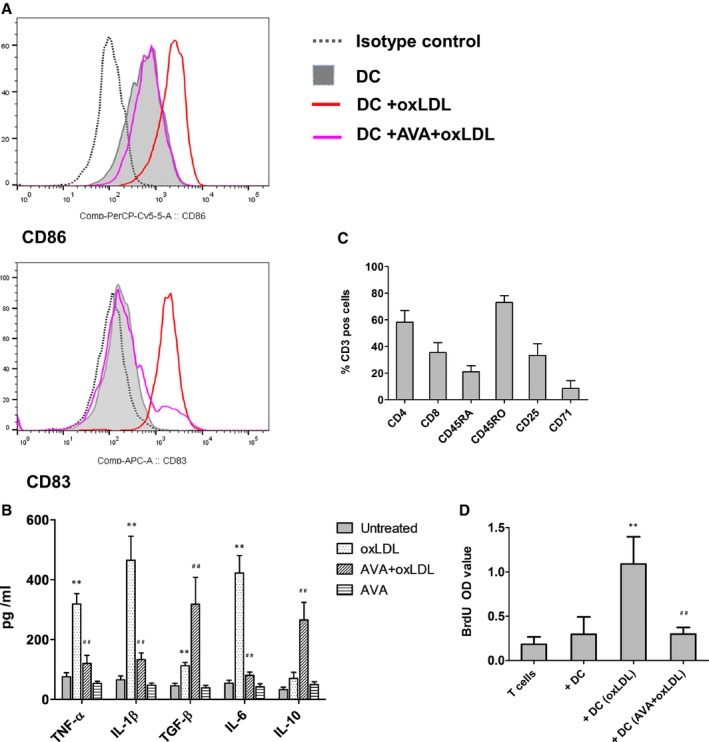
Dendritic cell (DC) maturation, proinflammatory cytokine production, and the subsequent T‐cell proliferation from plaque patients induced by oxidized low‐density lipoprotein (oxLDL) were suppressed by statins. DCs were generated from peripheral blood monocytes of patients who underwent carotid endarterectomy. The same protocol was used as that for DCs of normal donors. Briefly, DCs at day 6 were treated with 10 μg/mL oxLDL for 24 hours. An aliquot of DCs at day 5 were treated with 2 μmol/L atorvastatin for 24 hours, and continued with 2 μmol/L atorvastatin in the presence or absence of 10 μg/mL oxLDL for an additional 24 hours. A, The expression of CD80, CD86, CD83 and HLA‐DR was tested on day 7 DCs derived from patient blood. Figure depicts the expression of CD86 and CD83 tested by flow cytometry. A representative of 6 independent experiments is shown. B, Cytokine production in DC cultures derived from patient blood. Culture supernatants were collected after 24 hours treatment. Cytokine level was determined by ELISA. Results represent the mean±SD from 6 independent experiments. IL indicates interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor. C, Percentage of CD4, CD8, CD45RO, CD45RA, CD25, and CD69/71 positive cells in plaque T cells are presented. Results represent the mean±SD generated from 5 plaques. Plaque mononuclear cells were isolated from single cell populations using the standard Ficoll‐Paque PLUS, and followed by T‐cell purification using Dynabeads® Untouched™ Human T Cells. D, Plaque T‐cell proliferation induced by oxLDL‐treated DC was suppressed by atorvastatin (AVA). After the indicated treatment for 24 hours, DCs derived from patient peripheral blood monocyte were washed and cocultured with autologous plaque T cells in triplicates for 5 days. Bromodeoxyuridine (BrdU) was present in the last 16 hours. **P<0.01, DC+oxLDL vs DC and DC+AVA+oxLDL. ## P<0.01, DC+ (AVA)+oxLDL vs DC+oxLDL.
Furthermore, we studied the human plaque T‐cell response to autologous oxLDL‐treated DCs. T cells were isolated from carotid atherosclerotic plaques, and phenotype and activation status of T cells were analyzed by flow cytometry (Figure 6C). CD4 T cells were major populations and constituted 58±9% of T cells. Naive (CD45RA positive) and memory (CD45RO positive) T cells were 21±5% and 73±11%, respectively. We tested the expression of activation markers CD25, as well as CD71. There were 30±11% and 9±6% of T cells expressing CD25 and CD71, respectively. We demonstrated that oxLDL‐treated DCs, which were generated from patient peripheral blood monocytes, stimulated autologous plaque T‐cell proliferation. The induced plaque T‐cell proliferation was abrogated by atorvastatin treatment (Figure 6D).
Inhibition of let‐7c in DCs Abrogated the Plaque T‐Cell Proliferation
We tested the expression of let‐7c in DCs that derived from patient peripheral blood monocytes. Basal level of let‐7c in patient DCs had higher expression. The expression was induced by oxLDL and pretreatment with statin limited this induction (Figure 7A). We demonstrated that oxLDL‐treated DCs, which were generated from patient peripheral blood monocytes, stimulated autologous plaque T‐cell proliferation. The induced plaque T‐cell proliferation was abrogated by atorvastatin treatment. Downregulation of let‐7c by specific inhibitor in the oxLDL‐treated DCs abrogated T‐cell proliferation (Figure 7B).
Figure 7.
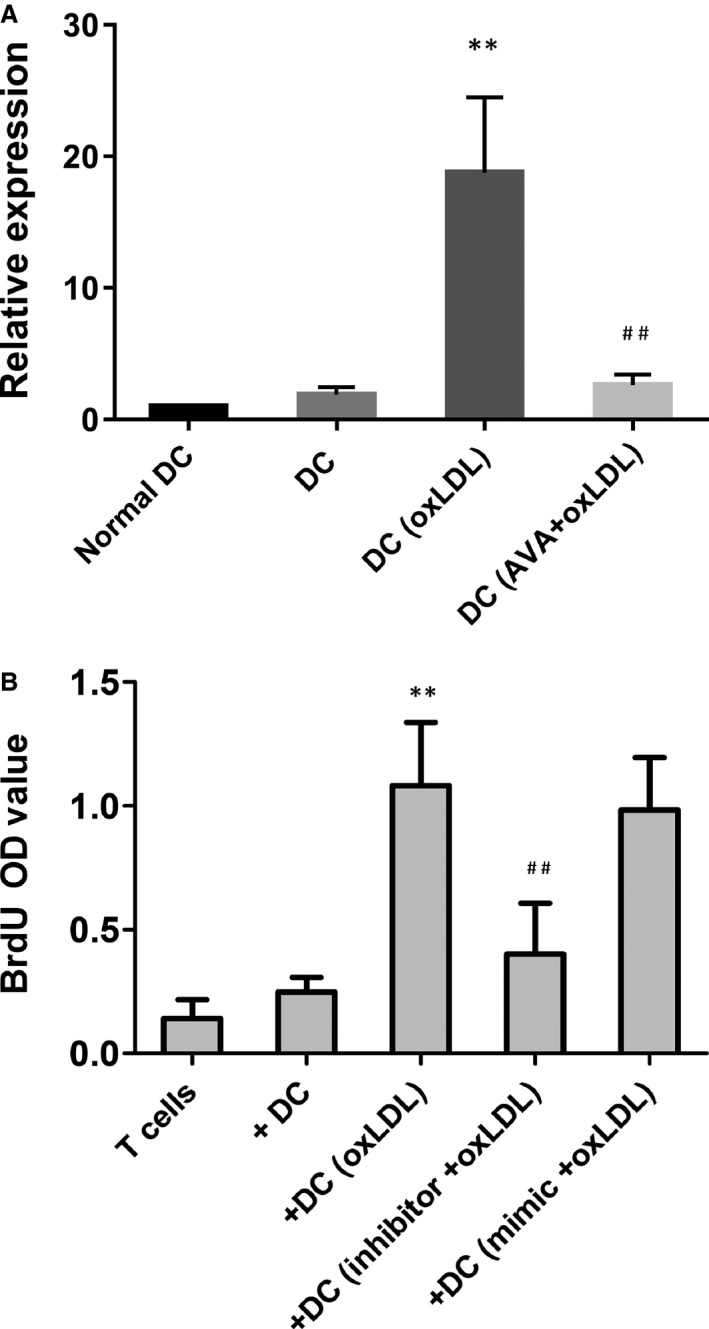
Downregulation of let‐7c in dendritic cells (DCs) abrogated the plaque T‐cell proliferation. A, Expression of let‐7c in patient DCs was tested by quantitative reverse transcription polymerase chain reaction, with normal dendritic cell (DC) samples as controls. The value was normalized as fold change to that of nontreated normal DC samples. Results represent the mean±SD of 4 experiments. **P<0.01, DC+ oxidized low‐density lipoprotein (oxLDL) vs DC; ## P<0.01, DC+ atorvastatin (AVA)+oxLDL vs DC+oxLDL. B, Downregulation of let‐7c abrogated the plaque T‐cell proliferation. After downregulation of let‐7c, oxLDL‐treated DCs were washed and cocultured in triplicates for 5 days with autologous plaque T cells. Bromodeoxyuridine (BrdU) was present in the last 16 hours and proliferative response was evaluated as OD value. **P<0.01 compared to that of mock‐treated samples. OD indicates optical density.
Discussion
We here report that OxLDL induces T‐cell activation as determined by proliferation, through DCs from the same donor, and from both blood and atherosclerotic plaques. Furthermore, OxLDL induced DC activation with expression of CD86, and other markers including CD83, CD40, HLA‐DR, CCR2, and CX3CR1. These findings confirm and extend previous reports.10, 22, 23
These effects of oxLDL on DC‐mediated proliferation of T cells were abolished by atorvastatin and simvastatin. There are now several other statins in use, since the discovery of the first one, which is a product of yeast as penicillin. From an evolutionary point of view this could represent a defense against bacteria, in the case of statins possibly inhibition of build‐up of bacterial cell walls.24 Even though our findings indicate common effects of atorvastatin and simvastatin, we cannot exclude the possibility that other statins differ in some respect in relation to T‐cell activation as described herein, but we focus on atorvastatin, being one of the most commonly used statins.
Modification including oxidized LDL is most likely a major antigen and pro‐inflammatory compound in the plaques.2, 10, 25 Our finding that oxLDL‐induced DC‐mediated activation of T cells from vulnerable atherosclerotic plaques is inhibited by statins could thus represent a novel specific immunomodulatory mechanism, unrelated to LDL‐lowering. The benefits of statins in patients with CVD could be at least partly caused by mechanisms described herein. This is not least the case in coronary artery disease, where inflammation and immune activation is common and plaques typically are vulnerable. Even though the situation in stroke is more complex, emerging evidence points to a role in ameliorating inflammation and stabilizing plaque in carotid arteries (the origin of T cells from plaque tested herein).26 Such an effect is in line with recent imaging studies, where statins decrease inflammation in atherosclerotic plaques.27, 28
Statins, including atorvastatin and simvastatin, exert their blood lipid–lowering effect by inhibition of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase, which is a pivotal step in the production of mevalonate, which is a precursor of cholesterol. Mevalonate reverts the effects on statin‐induced inhibition of OxLDL‐induced DC‐mediated activation of T cells, thus indicating specificity of the effects described herein.
HSP60 was originally reported by Wick and coworkers to be atherogenic, and immunization with HSP60 promotes atherosclerosis in animal models. An interesting possibility is that stress to the arterial wall, for example, at sites exposed to stress as in hypertension, the induced HSP could promote atherosclerosis through cross‐reactivity with bacterial HSPs.29 We reported that oxLDL induces HSP60 in monocytes30 and that HSP60 could be of major importance in oxLDL‐induced DC‐mediated T‐cell activation.10 Also, HSP90 may be proatherogenic,31, though it appears to be less studied than HSP60. Furthermore, HSP90 also plays a role in oxLDL‐induced activation of T cells from atherosclerotic plaques.10 HSP27 may, on the other hand, have atheroprotective properties.32 We here report that atorvastatin restores the proatherogenic HSP profile induced by OxLDL, adding yet another immunological mechanism by which statins could modulate immune reactions pivotal to atherosclerosis and CVD.
In line with our previous report,10 DC exposed to oxLDL promoted a pro‐inflammatory T‐cell profile with development of Th1 and Th17 cells. The role of IL‐17 (from Th17 cells) at different stages of atherosclerosis development has been discussed.33, 34, 35 It appears that in human atherosclerosis, the Th17 phenotype36 and, in our previous studies, a Th1 type of cytokine profile is dominant in advanced atherosclerotic plaques,1 a finding that has been reported in recent studies in vulnerable human plaques.37 Of note, human plaque rupture (leading to CVD) is known to be difficult to mimic in mouse models.38
We also report that when T cells are activated by OxLDL through DC and then cocultured with naive T cells in the presence of matured DC, both types of T cells were activated to proliferate, indicating a synergy effect, which could be mediated by pro‐inflammatory cytokines, in addition to specific T‐cell activation.39
Furthermore, oxLDL‐induced DC‐mediated T‐cell polarization, in the form of Th1 cytokines, was reversed by atorvastatin, instead promoting T regulatory cells and thus a different type of T‐cell activation. Regulatory T cells are of major importance in regulation of T‐cell immune responses. They suppress pro‐inflammatory T‐cell effector functions, and an imbalance with decreased proportion of T regulatory cells may promote atherosclerosis (and also autoimmune diseases such as systemic lupus erythematosus). T regulatory cells may also have other beneficial effects in the context of atherosclerosis and its complications including inhibition of foam cell formation, and induction of anti‐inflammatory macrophages. Therefore, increasing T regulatory cells has been discussed as a potential therapy in atherosclerosis and CVD.40
Interestingly, immune modulatory properties of statins are also reported to include inhibition of Th17‐related T‐cell reactions and induction of T regulatory cells. Experimental evidence supports a potential role in autoimmune disease, for this reason. However, even though there are now many studies in different autoimmune diseases where statins have been used, evidence of an effect on these conditions by statins is insufficient.41 Previous findings also indicate that statins can inhibit MHC class II interaction with antigen.42
Another central finding herein is that we report for the first time both that oxLDL specifically induces the miRNA Let‐7c in DC and that this novel effect is abolished by atorvastatin. Furthermore, atorvastatin constrained the dysregulated upregulation of pAkt and pERK in DCs induced by oxLDL. ERK and the PI3K‐Akt signaling pathway were downstream of the effect of atorvastatin, since treatment with an ERK inhibitor or a PI3K inhibitor during DC maturation could directly induce CD4+CD25+ Foxp3+ T reg. Downregulation of let‐7c in DCs abrogated the plaque T‐cell proliferation, thus indicating that Let‐7c is pivotal for oxLDL‐induced DC‐mediated T‐cell activation through DC and for the effects of atorvastatin.
Our findings are in line with a recent study using a mouse model where let7c is involved in the activation of DC. Furthermore, it was demonstrated that increased BLIPM1 expression promotes DC maturation and increases let7c expression.21 In line with these findings, we here report that BLIMP1 expression is induced by oxLDL but in the presence of statin the induction was dampened. The role of BLIMP1 in relation to let7‐c thus deserves further study.
Let‐7 was one of the first miRNAs that was initially discovered in the nematode Caenorhabditis elegans. The let‐7 family has several roles, all of which are not fully known.
The expression of the let‐7 family is required for developmental timing and tumor suppressor function, but must be suppressed for the self‐renewal of stem cells. This implies that a careful control of the expression of this miRNA is essential. Let‐7 expression is especially high during embryogenesis and brain development.43 Let‐7 miRNAs are evolutionarily conserved and present in many animal species, but not in plants, and could play an important role as a regulator of gene expression. Still, much of Let‐7 functions are not known in humans.43 It is thus not clear what physiological significance the specific oxLDL induction could have for pathophysiology in plaques and for atherosclerosis, CVD, and other conditions in general. Still it is interesting to note that in our previous report, oxLDL was reported to induce differentiation of a monocytic tumor cell line in addition to monocytes from healthy donors.44
However, this finding could have implications for statins in general. Let‐7c is downregulated and is characterized by low expression (and poorer survival) in many tumors.45 The possibility that downregulation of Let‐7c by statins could pose a risk should be considered.
Whether statin use is associated with or even causatively influences the risk of cancer has been much debated, and is beyond the topic of the present study. However, there appears to be no clear general evidence of an increased risk of cancer among statin‐treated individuals, though it cannot be excluded that subgroups of cancer are influenced in different ways by statins. Inflammation per se could be a risk factor in some forms of cancer.46, 47, 48
Even though statins are still debated, most experts agree that they are beneficial for secondary prevention after coronary artery disease. They are also widely used among patients with known risk factors, as supported by clinical evidence. The beneficial effects for women and the elderly is more debated. In general, there are also critical voices in relation to statin treatment. However, this whole discussion is beyond the scope of the present study.49
Statin's inhibition of the mevalonate pathway and its isoprenoid formation are believed to be the underlying cause of much of the pleiotropic effects described for statins, since prenylation of protein is an important step in intracellular signaling.16 In the present study, it is not clear if this is the underlying effect, though it is downstream of mevalonate formation.
Taken together, our data indicate that OxLDL activates human T cells through DCs and that this effect was abolished by a statin, atorvastatin, and also by simvastatin, though the focus was on the former. A specific mechanism where Let‐7c plays a role was described. Our findings could provide novel explanations for the effects of statins on CVD.
Sources of Funding
This work was supported by the Swedish Heart Lung Foundation, the Swedish Research Council, the Stockholm County (ALF), the King Gustav V 80th Birthday Fund, Swedish Association against Rheumatism, Vinnova, AFA, and Torsten Söderberg′s Foundation. This work was also supported by the 6th Framework Program of the European Union (grant LSHM‐CT‐2006‐037227 CVDIMMUNE) with Frostegård as coordinator. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosures
None.
(J Am Heart Assoc. 2016;5:e003976 doi: 10.1161/JAHA.116.003976)
Mr Zhang is currently located at the Department of Pathophysiology, Basic College of Medicine, Jilin University, Changchun, China.
Dr Yan is currently located at the Department of Urology, Qilu hospital, Shandong University, Jinan, China.
Contributor Information
Johan Frostegård, Email: johan.frostegard@ki.se.
Anquan Liu, Email: anquan.liu@ki.se.
References
- 1. Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro‐inflammatory (Th1) and macrophage‐stimulating cytokines. Atherosclerosis. 1999;145:33–43. [DOI] [PubMed] [Google Scholar]
- 2. Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013;11:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. 2000;102:2919–2922. [DOI] [PubMed] [Google Scholar]
- 4. Gojova A, Brun V, Esposito B, Cottrez F, Gourdy P, Ardouin P, Tedgui A, Mallat Z, Groux H. Specific abrogation of transforming growth factor‐beta signaling in T cells alters atherosclerotic lesion size and composition in mice. Blood. 2003;102:4052–4058. [DOI] [PubMed] [Google Scholar]
- 5. Kolbus D, Ljungcrantz I, Andersson L, Hedblad B, Fredrikson GN, Bjorkbacka H, Nilsson J. Association between CD8+ T‐cell subsets and cardiovascular disease. J Intern Med. 2013;274:41–51. [DOI] [PubMed] [Google Scholar]
- 6. Millonig G, Niederegger H, Rabl W, Hochleitner BW, Hoefer D, Romani N, Wick G. Network of vascular‐associated dendritic cells in intima of healthy young individuals. Arterioscler Thromb Vasc Biol. 2001;21:503–508. [DOI] [PubMed] [Google Scholar]
- 7. Yilmaz A, Lochno M, Traeg F, Cicha I, Reiss C, Stumpf C, Raaz D, Anger T, Amann K, Probst T, Ludwig J, Daniel WG, Garlichs CD. Emergence of dendritic cells in rupture‐prone regions of vulnerable carotid plaques. Atherosclerosis. 2004;176:101–110. [DOI] [PubMed] [Google Scholar]
- 8. Liu P, Yu YR, Spencer JA, Johnson AE, Vallanat CT, Fong AM, Patterson C, Patel DD. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler Thromb Vasc Biol. 2008;28:243–250. [DOI] [PubMed] [Google Scholar]
- 9. Bobryshev YV, Watanabe T. Ultrastructural evidence for association of vascular dendritic cells with T‐lymphocytes and with B‐cells in human atherosclerosis. J Submicrosc Cytol Pathol. 1997;29:209–221. [PubMed] [Google Scholar]
- 10. Liu A, Ming JY, Fiskesund R, Ninio E, Karabina SA, Bergmark C, Frostegard AG, Frostegard J. Induction of dendritic cell‐mediated T‐cell activation by modified but not native low‐density lipoprotein in humans and inhibition by annexin a5: involvement of heat shock proteins. Arterioscler Thromb Vasc Biol. 2015;35:197–205. [DOI] [PubMed] [Google Scholar]
- 11. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG‐CoA reductase. Science. 2001;292:1160–1164. [DOI] [PubMed] [Google Scholar]
- 12. Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–1307. [DOI] [PubMed] [Google Scholar]
- 13. Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, Langendorfer A, Stein EA, Kruyer W, Gotto AM Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. [DOI] [PubMed] [Google Scholar]
- 14. Kang S, Wu Y, Li X. Effects of statin therapy on the progression of carotid atherosclerosis: a systematic review and meta‐analysis. Atherosclerosis. 2004;177:433–442. [DOI] [PubMed] [Google Scholar]
- 15. Ridker PM. Moving beyond JUPITER: will inhibiting inflammation reduce vascular event rates? Curr Atheroscler Rep. 2013;15:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tousoulis D, Psarros C, Demosthenous M, Patel R, Antoniades C, Stefanadis C. Innate and adaptive inflammation as a therapeutic target in vascular disease: the emerging role of statins. J Am Coll Cardiol. 2014;63:2491–2502. [DOI] [PubMed] [Google Scholar]
- 17. Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM; Investigators I‐I . Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. [DOI] [PubMed] [Google Scholar]
- 19. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. [DOI] [PubMed] [Google Scholar]
- 20. Grivel J‐C, Ivanova O, Pinegina N, Blank PS, Shpektor A, Margolis LB, Vasilieva E. Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011;31:2929–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim SJ, Gregersen PK, Diamond B. Regulation of dendritic cell activation by microRNA let‐7c and BLIMP1. J Clin Invest. 2013;123:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perrin‐Cocon L, Coutant F, Agaugue S, Deforges S, Andre P, Lotteau V. Oxidized low‐density lipoprotein promotes mature dendritic cell transition from differentiating monocyte. J Immunol. 2001;167:3785–3791. [DOI] [PubMed] [Google Scholar]
- 23. Atout R, Karabina SA, Dollet S, Carreras M, Payre C, Andre P, Lambeau G, Lotteau V, Ninio E, Perrin‐Cocon L. Human group X secreted phospholipase A2 induces dendritic cell maturation through lipoprotein‐dependent and ‐independent mechanisms. Atherosclerosis. 2012;222:367–374. [DOI] [PubMed] [Google Scholar]
- 24. Tobert JA. Lovastatin and beyond: the history of the HMG‐CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. [DOI] [PubMed] [Google Scholar]
- 25. Frostegard J, Wu R, Giscombe R, Holm G, Lefvert AK, Nilsson J. Induction of T‐cell activation by oxidized low density lipoprotein. Arterioscler Thromb. 1992;12:461–467. [DOI] [PubMed] [Google Scholar]
- 26. Artom N, Montecucco F, Dallegri F, Pende A. Carotid atherosclerotic plaque stenosis: the stabilizing role of statins. Eur J Clin Invest. 2014;44:1122–1134. [DOI] [PubMed] [Google Scholar]
- 27. Tahara N, Kai H, Ishibashi M, Nakaura H, Kaida H, Baba K, Hayabuchi N, Imaizumi T. Simvastatin attenuates plaque inflammation: evaluation by fluorodeoxyglucose positron emission tomography. J Am Coll Cardiol. 2006;48:1825–1831. [DOI] [PubMed] [Google Scholar]
- 28. Tawakol A, Fayad ZA, Mogg R, Alon A, Klimas MT, Dansky H, Subramanian SS, Abdelbaky A, Rudd JH, Farkouh ME, Nunes IO, Beals CR, Shankar SS. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multicenter fluorodeoxyglucose‐positron emission tomography/computed tomography feasibility study. J Am Coll Cardiol. 2013;62:909–917. [DOI] [PubMed] [Google Scholar]
- 29. Xu Q, Dietrich H, Steiner HJ, Gown AM, Schoel B, Mikuz G, Kaufmann SH, Wick G. Induction of arteriosclerosis in normocholesterolemic rabbits by immunization with heat shock protein 65. Arterioscler Thromb. 1992;12:789–799. [DOI] [PubMed] [Google Scholar]
- 30. Frostegard J, Kjellman B, Gidlund M, Andersson B, Jindal S, Kiessling R. Induction of heat shock protein in monocytic cells by oxidized low density lipoprotein. Atherosclerosis. 1996;121:93–103. [DOI] [PubMed] [Google Scholar]
- 31. Madrigal‐Matute J, Lopez‐Franco O, Blanco‐Colio LM, Munoz‐Garcia B, Ramos‐Mozo P, Ortega L, Egido J, Martin‐Ventura JL. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res. 2010;86:330–337. [DOI] [PubMed] [Google Scholar]
- 32. Martin‐Ventura JL, Nicolas V, Houard X, Blanco‐Colio LM, Leclercq A, Egido J, Vranckx R, Michel JB, Meilhac O. Biological significance of decreased HSP27 in human atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:1337–1343. [DOI] [PubMed] [Google Scholar]
- 33. Lim H, Kim YU, Sun H, Lee JH, Reynolds JM, Hanabuchi S, Wu H, Teng BB, Chung Y. Proatherogenic conditions promote autoimmune T helper 17 cell responses in vivo. Immunity. 2014;40:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, Wang Y, Bockler D, Katus HA, Dengler TJ. Inhibition of IL‐17A attenuates atherosclerotic lesion development in apoE‐deficient mice. J Immunol. 2009;183:8167–8175. [DOI] [PubMed] [Google Scholar]
- 35. Gistera A, Robertson AK, Andersson J, Ketelhuth DF, Ovchinnikova O, Nilsson SK, Lundberg AM, Li MO, Flavell RA, Hansson GK. Transforming growth factor‐beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin‐17‐dependent pathway. Sci Transl Med. 2013;5:196ra100. [DOI] [PubMed] [Google Scholar]
- 36. Liu Z, Lu F, Pan H, Zhao Y, Wang S, Sun S, Li J, Hu X, Wang L. Correlation of peripheral Th17 cells and Th17‐associated cytokines to the severity of carotid artery plaque and its clinical implication. Atherosclerosis. 2012;221:232–241. [DOI] [PubMed] [Google Scholar]
- 37. Erbel C, Dengler TJ, Wangler S, Lasitschka F, Bea F, Wambsganss N, Hakimi M, Bockler D, Katus HA, Gleissner CA. Expression of IL‐17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol. 2011;106:125–134. [DOI] [PubMed] [Google Scholar]
- 38. Campbell IC, Weiss D, Suever JD, Virmani R, Veneziani A, Vito RP, Oshinski JN, Taylor WR. Biomechanical modeling and morphology analysis indicates plaque rupture due to mechanical failure unlikely in atherosclerosis‐prone mice. Am J Physiol Heart Circ Physiol. 2012;304:H473–H486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fei GZ, Huang YH, Swedenborg J, Frostegard J. Oxidised LDL modulates immune‐activation by an IL‐12 dependent mechanism. Atherosclerosis. 2003;169:77–85. [DOI] [PubMed] [Google Scholar]
- 40. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. 2015;35:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ulivieri C, Baldari CT. Statins: from cholesterol‐lowering drugs to novel immunomodulators for the treatment of Th17‐mediated autoimmune diseases. Pharmacol Res. 2014;88:41–52. [DOI] [PubMed] [Google Scholar]
- 42. Ghittoni R, Napolitani G, Benati D, Ulivieri C, Patrussi L, Laghi Pasini F, Lanzavecchia A, Baldari CT. Simvastatin inhibits the MHC class II pathway of antigen presentation by impairing Ras superfamily GTPases. Eur J Immunol. 2006;36:2885–2893. [DOI] [PubMed] [Google Scholar]
- 43. Lee H, Han S, Kwon CS, Lee D. Biogenesis and regulation of the let‐7 miRNAs and their functional implications. Protein Cell. 2016;7:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frostegard J, Nilsson J, Haegerstrand A, Hamsten A, Wigzell H, Gidlund M. Oxidized low density lipoprotein induces differentiation and adhesion of human monocytes and the monocytic cell line U937. Proc Natl Acad Sci USA. 1990;87:904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nair VS, Maeda LS, Ioannidis JP. Clinical outcome prediction by microRNAs in human cancer: a systematic review. J Natl Cancer Inst. 2012;104:528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tan P, Wei S, Tang Z, Gao L, Zhang C, Nie P, Yang L, Wei Q. LDL‐lowering therapy and the risk of prostate cancer: a meta‐analysis of 6 randomized controlled trials and 36 observational studies. Sci Rep. 2016;6:24521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boudreau DM, Yu O, Johnson J. Statin use and cancer risk: a comprehensive review. Expert Opin Drug Saf. 2010;9:603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhong S, Zhang X, Chen L, Ma T, Tang J, Zhao J. Statin use and mortality in cancer patients: systematic review and meta‐analysis of observational studies. Cancer Treat Rev. 2015;41:554–567. [DOI] [PubMed] [Google Scholar]
- 49. Stone NJ, Turin A, Spitz JA, Valle CW, Kazmi S. Statin therapy across the lifespan: evidence in major age groups. Expert Rev Cardiovasc Ther. 2016;14:341–366. [DOI] [PubMed] [Google Scholar]