

Summary
Glutamate is a major excitatory neurotransmitter present in the central nervous system. The glutamate/cystine antiporter system xc– connects the antioxidant defense with neurotransmission and behaviour. Overactivation of ionotropic glutamate receptors induces neuronal death, a pathway called excitotoxicity. Glutamate-induced oxidative stress is a major contributor to neurodegenerative diseases including cerebral ischemia, Alzheimer’s and Huntington’s disease. Curcuma has a wide spectrum of biological activities regarding neuroprotection and neurocognition. By reducing the oxidative damage, curcumin attenuates a spinal cord ischemia-reperfusion injury, seizures and hippocampal neuronal loss. The rat pheochromocytoma (PC12) cell line exhibits many characteristics useful for the study of the neuroprotection and neurocognition. This investigation was carried out to determine whether the neuroprotective effects of curcumin can be observed via the glutamate-PC12 cell model. Results indicate that glutamate (20 mM) upregulated glutathione peroxidase 1, glutathione disulphide, Ca2+ influx, nitric oxide production, cytochrome c release, Bax/Bcl-2 ratio, caspase-3 activity, lactate dehydrogenase release, reactive oxygen species, H2O2, and malondialdehyde; and downregulated glutathione, glutathione reductase, superoxide dismutase and catalase, resulting in enhanced cell apoptosis. Curcumin alleviates all these adverse effects. Conclusively, curcumin can effectively protect PC12 cells against the glutamate-induced oxidative toxicity. Its mode of action involves two pathways: the glutathione-dependent nitric oxide-reactive oxygen species pathway and the mitochondria-dependent nitric oxide-reactive oxygen species pathway.
Key words: curcumin, caspase, apoptotic pathways, glutamate cytotoxicity, PC12 cell line, glutathione, nitric oxide, reactive oxidative substances
Introduction
Glutamate is a major excitatory neurotransmitter present in the central nervous system (1). The glutamate/cystine antiporter system xc– transports cystine into cells in exchange for the important neurotransmitter glutamate at a ratio of 1:1 (2). Glutamate exported by system xc– is largely responsible for the extracellular glutamate concentration in the brain, whereas the imported cystine is required for the synthesis of the major endogenous antioxidant, glutathione (2).
Excess extracellular glutamate level could induce brain lesions and other pathological changes in several organs associated with endocrine function (3). Overactivation of ionotropic glutamate receptor induces neuronal death, a pathway called excitotoxicity coined by Olney (3).
The existing data reveal that oxidative stress is a causal factor in the neuropathology of several adult neurodegenerative disorders (4). Apart from that, high level of extracellular glutamate could induce oxidative stress, contributing to neurodegenerative diseases by stimulating the generation of reactive oxygen species (ROS), mitochondrial hyperpolarization and lipid peroxidation in neuronal cells (5). Physiopathologically, glutamate-induced excitotoxicity involves a combination of ferroptosis, necrosis and the mitochondria-associated apoptosis-inducing factor (AIF)-dependent apoptosis (5). Etiologically, glutamate-induced excitotoxicity has been implicated in the pathogenesis of many central nervous system (CNS) diseases, including cerebral ischemia, Alzheimer’s disease (AD), Huntington’s disease (HD), epilepsy, and amyotrophic lateral sclerosis (6).
Curcuma (Curcuma longa Zinziberaceae), a well known herbal spice widely distributed in India, Indonesia, Malaysia and Southwest China, is popularly formulated in curry (Fig. 1a), whereas curcumin (diferuloylmethane; Fig. 1b) (7) is frequently found in European diet. Phytotherapeutically, curcumin exhibits a wide spectrum of biological and pharmacological activities regarding neuroprotection and neurocognition.
Fig. 1.

Curcuma longa Linn. and the structure of curcumin: a) the morphology of Curcuma longa Linn. and the flowering plant (upper panel), its rhizoma (middle panel) and cross section (lower panel) (courtesy of National Institute of Health, Taiwan); b) chemical structure of curcumin (7)
Preclinically, curcumin alone or curcumin in combination with quercetin has been shown effective for treating spinal cord injury by reducing the oxidative damage (8–10). Curcumin attenuated the spinal cord ischemia-reperfusion injury in rabbits by reducing the oxidative damage (10). It inhibited hippocampal neuronal loss (11) and bipolar disorder (12). In conjunction with stem cell therapy, curcumin synergistically improved recovery from acute traumatic spinal cord injury (SCI) (13), underlying the potential regenerative action of curcumin if appropriately mediated by the neural stem cells. Beneficial protection of axons from degeneration by suppressing the local neuroinflammation is also attributted to curcumin (14). Curcumin rescued α-synuclein-induced cell death (15). By targeting histone deacetylase (HDAC), curcumin promisingly prevented apoptosis and improved motor deficits in Parkinson’s disease (PD) in rat model (16, 17).
Curcumin modulated the growth of several tumour cells through regulation of multiple cell signalling pathways like the cell proliferation pathways (cyclin D1 and c-myc), cell survival pathways (Bcl-2, Bcl-xL, cFLIP, XIAP and c-IAP1), caspase activation pathways (caspase-8, -3, and -9), tumour suppressor pathways (p53 and p21), death receptor pathways (DR4 and DR5), mitochondrial pathways, and protein kinase pathways (JNK, Akt and AMPK) (18).
More relevantly, curcumin has been shown to effectively inhibit the glutamate release from rat prefrontocortical nerve terminals by reducing the voltage-dependent Ca2+ entry and controlling the synaptic vesicle recruitment and exocytosis through the suppression of MAPK/ERK activation and the synapsin I phosphorylation (19). It is worth mentioning that despite the lower bioavailability, the therapeutic efficacy of curcumin against various human diseases like cancer, cardiovascular diseases, arthritis, neurological diseases, Crohn’s disease (20) and diabetic neuropathic pain (21) has also been documented.
There still lacks data on biological and pharmacological effects of curcumin; therefore, we have conducted an experiment with the PC12 cell model to establish whether curcumin can exhibit the potential therapeutic activity to protect the neurons from glutamate-induced injury. The major relevant mechanism of action regarding the curcumin therapy in the glutamate-induced PC12 cell damage model was pertinently examined.
Materials and Methods
Chemicals
Curcumin (grade A, 95% purity) was purchased from Hangzhou Greensky Biological Tech Co., Ltd (Suzhou, PR China). Aluminium trichloride hexahydrate (AlCl3·6H2O), 2,2’-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS), aprotinin, ammonium persulphate, butylated hydroxytoluene (BHT), bovine serum albumin (BSA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulphoxide (DMSO), l-glutamic acid, peroxidase, poly-l-lysine hydrobromide, phenylmethylsulfonyl fluoride (PMSF), sulphanilamide, sodium dodecyl sulphate, 1,1,3,3-tetramethoxypropane (TEP), 2-thiobarbituric acid (TBA), trichloroacetic acid (TCA), Trypan Blue solution, Tween 20 and oxidized glutathione were all provided by Sigma-Aldrich Co. (St. Louis, MO, USA).
Preparation of reagents
Preparation of glutamate solution
The modified method of Kawakami et al. (22) was adopted to dissolve glutamic acid. Briefly, the required amount of glutamic acid was first dissolved in incomplete medium. After the pH was adjusted to 7.0, the solution was stirred to facilitate the dissolution and filtered through a 0.2-µm Micropore filter (Bangalore, Karnataka, India). The fresh filtrate was used for further analyses.
Preparation of stock curcumin solution
Curcumin (2.8 mg) was accurately weighed, dissolved in 1.5 mL of DMSO, and stored in the brown coloured Eppendorf vial (Sigma-Aldrich Co.) maintained at –20 °C for use. For experiment, the stock solution was diluted with the medium to obtain different concentrations of test solutions.
Preparation of complete medium
To each 500 mL of RPMI 1640 cell culture medium, 10% horse serum, 5% heat-inactivated fetal bovine serum (FBS, previously inactivated at 56 °C for 30 min to eliminate the complement), 1% antibiotics (100 IU of penicillin and 100 mg of streptomycin) and 1% glutamine were added. After thorough mixing, the medium was stored at 4 °C in an ice box. For experiment, the medium was rewarmed to 37 °C before use.
Preparation of incomplete medium
To each 500 mL of RPMI 1640, 1% antibiotics (100 IU of penicillin and 100 mg of streptomycin), and 1% glutamine were added, mixed well and stored at 4 °C in an ice box. The medium was rewarmed to 37 °C immediately before use.
Cultivation of PC12 cell line
Rat adrenal pheochromocytoma cell line (PC12 cell line) was obtained from the Bioresource Collection and Research Center of Food Industry Research and Development Institute (Hsinchu, Taiwan). The cultivation was carried out according to the method previously described by Chang et al. (23, 24).
Briefly, the PC12 cell line was incubated in the complete RPMI 1640 medium and incubated at 37 °C under 5% CO2 atmosphere until 80% confluent. To cultivate the subculture passage, 15 mL of the cell culture were transferred into the centrifuge tube and centrifuged at 1250×g for 5 min. The supernatant was removed. The sediment pellets were rinsed twice with phosphate-buffered saline (PBS) and centrifuged at 1250×g for 5 min each time. The final supernatant was removed. A volume of 3 to 4 mL of complete culture medium was added to the rinsed cell pellets. Unless otherwise stated, all culture plates were previously coated with 0.1 mg/mL of polylysine and left to stand for 24 h before the seeding of the cells.
Cell viability affected by the glutamate-induced cytotoxicity
To each well, PC12 cells were seeded at a density of 104 cell/well and incubated at 37 °C under 5% CO2 atmosphere until completely adhered. The medium was changed to incomplete culture media that contained 0.0, 1.0, 5.0, 10.0, 15.0 and 20.0 mM of glutamate. The culture was inspected at 24 and 48 h. At each set point, MTT (0.5 g/L) was added (note: the reaction vessel should be kept in the dark to avoid the direct sunlight). Cultivation was continued at 37 °C under 5% CO2 atmosphere for 2 to 4 h to facilitate the reaction. The supernatant containing non-reacted MTT was removed. To each well, DMSO (200 µL) was added to dissolve the purple crystalline formazan. The absorbance was read with ELISA Reader (ClarioStar, BMG Labtech Japan Ltd., Saitama, Japan) at 570 nm. Cell viability was calculated according to Eq. 1:
where As is the absorbance of the sample, and Ac is the absorbance of the control.
Cytotoxicity of curcumin to the PC12 cells
The protocol used for this experiment was conducted similarly as mentioned above. Instead of glutamate, 0.0, 1.0, 5.0, 10.0 and 50.0 µM of curcumin were used. The cell viability was calculated according to Eq. 1.
Protective effect of curcumin against the glutamate- -induced cytotoxicity
The PC12 cells were seeded at a density of 104 cells/well and incubated for 24 h until adhesion. The medium was changed to the incomplete media containing curcumin at 0.0, 1.0, 5.0, 10.0 and 50.0 µM. To each plate, 20 mM of glutamate were added and the incubation was continued for 48 h. The MTT assay was conducted as mentioned above. The inspection point was set at 24 and 48 h to inspect the viability in the presence and absence of curcumin, and the cell viability was calculated according to Eq. 1.
Determination of the glutathione content
To 2·106 PC12 cells separately harvested from the three experiments mentioned above, 400 µL of ice-cooled PBS were added and homogenized on ice. The homogenate was centrifuged at 12 000×g for 30 min at 4 °C. The obtained supernatant was transferred into a 1.5-mL microcentrifuge tube, to which 100 µL of TCA (5%, containing 1 mM of EDTA) were added and mixed well. The solution was cooled on ice for 20 min and centrifuged at 12 000×g for 30 min at 4 °C. The supernatant was separated and the glutathione (GSH) content was assayed with the Glutathione Assay Kit (Cayman Chemical Co., Ann Arbor, MI, USA) according to the manufacturer’s instructions. The level of GSH was expressed in nmol per mg of protein.
Assay of the activity of glutathione peroxidase
Methods described by Mantha et al. (25) were followed for the assay of glutathione peroxidase (GPX). One unit of GPX is defined as the amount of enzyme that causes the oxidation of 1 nmole of NADPH to NADP+ per min at 25 °C and pH=8.0. The activity of GPX was expressed as mU per mg of protein (25).
Assay of the activity of glutathione reductase
Procedure described by Mohandas et al. (26) was followed to determine the activity of glutathione reductase (GR). The molar absorption coefficient of NADPH of 6.22·103 M–1cm–1 measured at 340 nm was used for the calculation of GR activity.
Determination of the calcium influx
The protocol used to determine the intracellular calcium ion level was carried out as previously described by Chang et al. (24). The fluorescence was excited at the wavelength of 488 nm and the intensity of emission was taken at 532 nm. The untreated sample was taken as the control. On the other hand, 100.0 mg of authentic calcium oxide (Wako Pure, Osaka, Japan) were accurately weighed and dissolved in 10 mL of mixed solvent (HCl/acetic acid=1:4). The standard solution was used to establish the calibration curve from which the intracellular calcium ion concentration was obtained. The experiments were repeated with triplicate samples.
Determination of the total intracellular reactive oxygen species
The method for the determination of total intracellular reactive oxygen species (ROS) was carried out as previously described by Chang et al. (24). The chemifluorescence was excited at the wavelength of 488 nm and the emission was taken at 532 nm. The untreated sample was taken as the control (27). The determination was repeated with triplicate samples and the total ROS were calculated.
Determination of the cytochrome c release
The assay for determination of cytochrome c was conducted using the Mitochondria Isolation Kit for Tissue Cultured Cells (Amsbio LLC, Lake Forest, CA, USA) according to the instructions given by the manufacturer.
Isolation of mitochondria
To 2·106 PC12 cells harvested from the three experiments as mentioned above, 1 mL of ice-cooled PBS was added, agitated with a vibrator for 1 min, and centrifuged at 600×g for 10 min. Rinsing was repeated twice and the supernatant was discarded. Mitochondrial Isolation Buffer (MIB 1×, 2 mL; Sigma-Aldrich Co.) was added to the cell pellets and homogenized on ice. The homogenate was transferred to a centrifuge tube and centrifuged at 600×g for 10 min at 4 °C. The supernatant was separated, transferred to a microcentrifuge tube, recentrifuged at 12 000×g for 15 min at 4 °C, and then discarded. The precipitate was resuspended in 0.5 mL of MIB (1×) and homogenized. The homogenate was transferred to a new microcentrifuge tube and centrifuged at 600×g for 10 min at 4 °C. The supernatant was separated, centrifuged at 12 000×g for 15 min at 4 °C, and then discarded. The precipitate was suspended in 50–100 µL of Mitochondrial Storage Buffer (Sigma-Aldrich Co.) and stored on ice for use (as mitochondrial pellet).
Lysis of mitochondria
To obtain the mitochondrial enzyme proteins, the mitochondrial pellet was resuspended in 100 µL of lysis buffer K105-25 (BioVision Inc., Milpitas, CA, USA) with protease inhibitors. The proteins conjugated with mitochondria were thus obtained, and the amount of conjugates required for experiment ranged between 0.5 and 1.0 mg/mL. The enzyme sample was stored at –80 °C for use.
Assay of the enzyme activity
To 850 µL (1×) of enzyme assay buffer, 100 µL of mitochondrial protein (the enzyme sample) and 50 µL of ferrocytochrome c substrate were added. The initial absorbance was read at 550 nm five seconds after the beginning and then the readings were repeated every 10 s.
To calculate the activity of cytochrome c oxidase in the samples, the linearity developed at the largest slopes was used, from which the maximum rate was obtained from Eq. 2:
where α is the activity (unit per mL), Vs is the volume of sample (mL), ∆A is As–Ac, X is fold of dilution, As is the change of absorbance of the sample per min, Ac is the change of absorbance of the control per min (usually within 0.001–0.003), 21.84 is the difference of molar absorption coefficient (∆εm) at 550 nm between ferrocytochrome c and ferricytochrome c. The relative activity was expressed in percentage in relation to the control.
Determination of the release of lactate dehydrogenase
To 2·106 PC12 cells harvested respectively from the three experiments as mentioned above, 1 mL of PBS was added, homogenized on ice and centrifuged at 14 000×g for 10 min at 4 °C. The supernatant was transferred into a microcentrifuge tube and frozen to –80 °C. The residual cell debris was rinsed twice with fresh PBS, each time using 2 mL of fresh PBS. The rinses were centrifuged at 14 000×g for 10 min at 4 °C. The supernatant and the rinses were combined, frozen to –80 °C and immediately delivered to the Biomedical Center (Taichung, Taiwan) for assay of the lactate dehydrogenase activity, which was expressed in mU per mL.
Assay of the activity of superoxide dismutase
The determination method for superoxide dismutase (SOD) activity was conducted according to the manufacturer’s instructions (Cayman Chemical Co.). The absorbance was read at 450 nm. The activity of SOD was expressed in U per mg of protein.
Assay of hydrogen peroxide content
Method instructed by Cell Biolabs Inc. (San Diego, CA, USA) was followed to carry out the assay of hydrogen peroxide content. The absorbance of the final solution was read at 620 nm against the blank. The authentic hydrogen peroxide (Cell Biolabs Inc.) was used to establish the calibration curve, from which the content of H2O2 was calculated and expressed in nmol per mg of protein.
Determination of the thiobarbituric acid reactive substances
The assay for thiobarbituric acid reactive substances (TBARs) was carried out according to the protocol previously reported by Chang et al. (23). Alternatively, the product formed from malondialdehyde (MDA) reacting with thiobarbituric acid exhibits a molar absorption coefficient ε532=1.56·105 M−1cm−1 at 532 nm (28), from which the amount of MDA in the sample can also be calculated. The level of MDA was expressed in µmol per µg of protein.
Determination of nitric oxide
The determination of nitric oxide (NO) was carried out as previously described (24). The absorbance was measured at 552 nm with an ELISA microreader (ClarioStar, BMG Labtech Japan Ltd.). Similar experiments were repeated in triplicate. The obtained data were statistically analyzed. The level of NO was expressed in µmol per mg of protein.
Western blotting
The Western blotting for anti-β-actin, anti-Bcl-2, anti-Bax, cytochrome c, and anti-cleaved caspase-3 was carried out as previously described by Chang et al. (24).
TUNEL assay
The terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labelling (TUNEL) assay was carried out as previously described by Chang et al. (24).
Statistical analysis
ANOVA was done using statistical analysis system SPSS v. 10.0 (SPSS Inc., Chicago, IL, USA) software to analyze the variances. Duncan’s multiple range tests were used to test the significance of differences between paired means. Data were presented as mean±standard deviation. Significance of the difference was determined by a confidence level of p<0.05.
Results and Discussion
Effect of glutamate, curcumin and combined therapy on the cell viability
Brain tissue contains an unusually high concentration of glutamate, approx. 5–15 mmol/kg (29). Most of this glutamate is found in neurons. Its concentration in the cytoplasm of glutamatergic neurons is approx. 5–10 mM (29–33).
The estimated content of intracellular glutamate in brain reported by Tossman and Ungerstedt (34) is 10 mM, while that of extracellular glutamate concentration reported by Bouvier et al. (35) is about 0.6 mM. Excess glutamate at concentrations 1–20 mM inhibited cell viability in a dose- and time-responsive manner (Fig. 2a). Cao et al. (36) indicated an EC50 of 5 mM for glutamate-induced injury of PC12 cells. By interpolation, the EC50 of glutamate was approx. 17 mM (Fig. 2a). Hence, glutamate level at 20 mM was adopted in our experiment. Literature data report ambient glutamate concentrations of 1–4 µM in vivo (37–39). If glutamate was present tonically at low micromolar concentrations, many receptors, especially the high-affinity N-methyl-d-aspartate (NMDA) receptors (or NMDARs), would be activated or desensitized, resulting in substantially altered neuronal excitability. A recent study of Herman and Jahr (40) indicated that the baseline concentration of glutamate is much lower, near 25 nM, in hippocampal slice, suggesting different sensitivity of different cell lines to glutamate that may depend on the cell type, source of cells, the function of cells and the environment under which the cells are cultured.
Fig. 2.
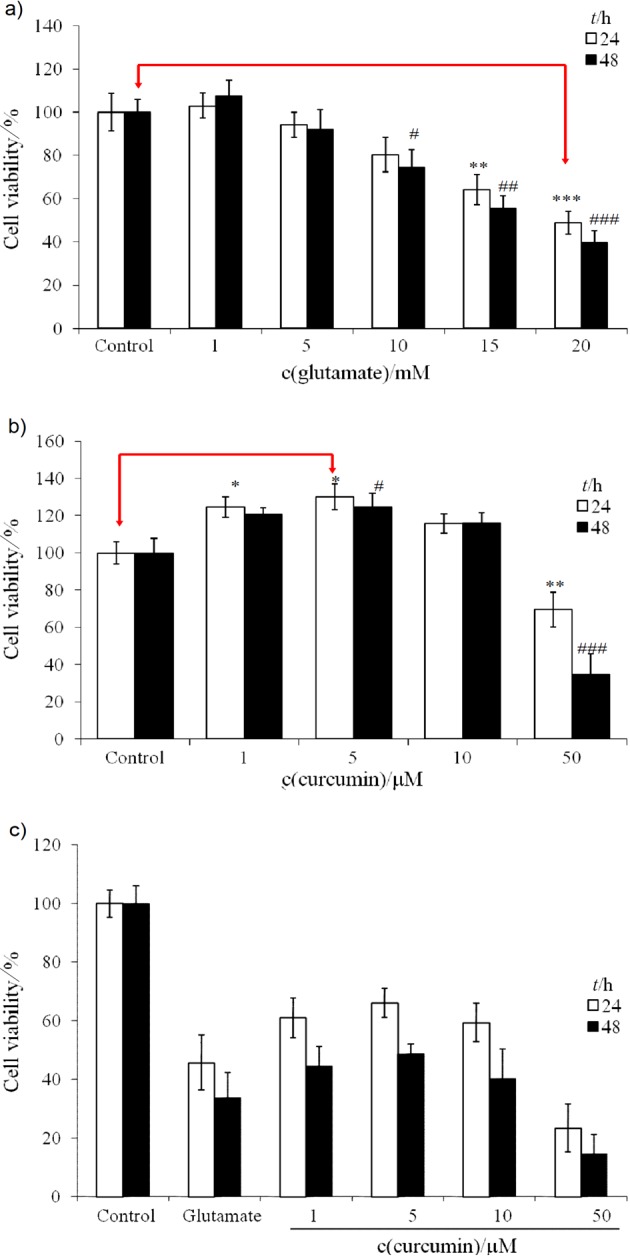
Dose- and time-responsive cell viability (in % of control) of PC12 cell line affected by different treatments: a) glutamate (1–20 mM) alone, b) curcumin (1–50 µM) alone, and c) combined therapy with glutamate 20 mM and curcumin 1–50 µM. * and # indicate significant difference from the control after incubation for 24 and 48 h, respectively. Singlet, doublet, and triplet symbols indicate statistical difference at confidence level of p<0.05, p<0.01, and p<0.001, respectively (N=6)
At lower doses, curcumin seemed to be a cell-proliferating agent (Fig. 2b). Higher dose of curcumin, e.g. ≥50 µM, inhibited cell viability (Fig. 2b). The inhibition reached 30 and 63% at 24 and 48 h, respectively. By interpolation, the EC50 of curcumin for PC12 cells was approximated to be 50 µM (Fig. 2b). Curcumin in the cotherapy (with 20 mM of glutamate) dose- and time-dependently protected the viability of PC12 cells up to 5 µM. The effect declined starting with 20 mM of glutamate and 10 µM of curcumin, drastically at doses ≥20 µM (Fig. 2c). In reality, the results indicated in Fig. 2 were not contradictory to each other. We selected the concentration of glutamate of 20 mM to insult the PC12 cells. Although curcumin alone at 10 µM was proved to be cell-proliferating (Fig. 2b), yet it counteracted the glutamate (20 mM)-induced injury. As a consequence, part of its efficiency at 10 µM was lost and the cell viability slightly declined with cotherapy of 10 µM of curcumin and 20 mM of glutamate (Fig. 2c). Apparently, curcumin exerted its effects by increasing cell survival in toxic environment, and not by activating the cell proliferation.
Mendonça et al. (41) demonstrated that curcumin at doses of 1.36–348 µM significantly reduced the total frequency of micronuclei induced by cisplatin. This fact implicated that a low dose of curcumin could be effective to alleviate the glutamate oxidative toxicity. In contrast, higher doses of curcumin were required for treatment of cisplatin genotoxicity (41). Literature suggests that determining the cytotoxic and genotoxic/antigenotoxic effects of such a frequently used antioxidant in a neuronal model is important to assess possible hazards when combined with other chemical agents, including chemotherapy drugs used in cancer therapy (41).
Curcumin alleviated the glutamate-suppressed cell viability in a dose-dependent manner up to 5 µM (Fig. 2c). On the other hand, higher concentration of curcumin was cytotoxic (14). At a dose ≥10 µM, a substantially suppressive effect occurred (Fig. 2c), suggesting that the biological interactions vary with the molar ratio of glutamate to curcumin, which was also consistent with the results of Mendonça et al. (41). The bioefficiency of curcumin against glutamate-induced injury was approx. 4000 (20 mM glutamate/5 µM curcumin), which means a single curcumin molecule was able to protect against the adverse effect elicited by 4000 glutamate molecules. As can be seen in Fig. 3, the cell viability was greatly reduced by glutamate- -induced insult (20 mM) (middle panel), while curcumin at 5 µM was shown to substantially protect the cell viability (lower panel) when compared with the control (upper panel).
Fig. 3.

Viability of PC12 cells before and after the exposure to glutamate (20 mM) alone or combined with curcumin (5 µM). Glutamate stimulated the production of intracellular ROS and reduced the cell viability (middle panel), which was effectively alleviated by curcumin (5 µM) (lower panel). The intracellular ROS were assayed with 2’,7’-dichlorofluorescine diacetate (H2DCFDA) fluorescence imaging
Effect of glutamate and curcumin on glutathione peroxidase and related oxidative defense system
Glutamate activated glutathione peroxidase (GPX) and at the same time suppressed the activity of glutathione reductase (GR), resulting in increased glutathione disulphide (GSSG) and reduced glutathione (GSH) (Table 1).
Table 1. Biochemical parameters affected by glutamate and the combined therapy with curcumin.
| Parameters | Control | Glutamate 20 mM |
Glutamate 20 mM +curcumin 5 µM |
|---|---|---|---|
| GPX/(mU/mg) | 11.2±0.4 | (28±3)* | 11.5±0.6 |
| GR/(mU/mg) | 9.4±0.6 | (2.3±0.4)* | 7.8±0.5 |
| b(GSH)/(nmol/mg) | 8.5±0.6 | (2.6±0.5)* | 8.3±0.7 |
| b(GSSG)/(nmol/mg) | 0.8±0.1 | (1.7±0.2)* | 0.8±0.1 |
| c(Ca2+-influx)/(nmol/L) | 118±17 | (351±21)** | (112±16)* |
| Cyt c release | 100±5 | (189±11)* | 102±8 |
| LDH/(mU/mL) | 313±1 | (614±1)** | (451±2)* |
| SOD/(U/mg) | 55±6 | (23±5)** | 52±6 |
| w(ROS)/% | 100±6 | (138±9)* | 109±4 |
| b(H2O2)/(nmol/mg) | 8.0±0.8 | (19±1)** | 7.8±0.4 |
| Catalase/(U/mg) | 18.7±0.4 | (10.3±0.5)** | 18.3±0.6 |
| b(TBARs (MDA))/(µmol/µg) | 8.0±0.6 | (13±1)* | 8.3±0.7 |
| b(NO)/(µmol/mg) | 26±2 | (35±3)** | (18±2)* |
Data are expressed as mean values±standard error of the mean (S.E.M.) of six determinations (N=6). Asterisks in the same row indicate statistically different results compared with the control (*p<0.05 and **p<0.001) GPX=glutathione peroxidase, GR=glutathione reductase, GSH=glutathione, GSSG=oxidized glutathione, Cyt c=cytochrome c, LDH=lactate dehydrogense, SOD=superoxide dismutase, ROS=reactive oxygen species, TBARs=thiobarbituric acid reactive species, MDA=malondialdehyde, NO=nitric oxide
The change of GPX and GSSG was 2.52- and 2.11-fold. Simultaneously, the levels of GR and GSH were suppressed to 2.3 mU per mg of protein and 2.6 nmol per mg of protein, respectively, compared to the respective control values of (9.4±0.6) mU per mg of protein and 8.5 nmol per mg of protein (Table 1). The levels of other antioxidant-related parameters were as follows (control vs. glutamate-induced injury): SOD 54.6 vs. 22.9 U per mg of protein, ROS 100 vs. 138%, H2O2 8.0 vs. 19.4 nmol per mg of protein, catalase 18.7 vs. 10.3 U per mg of protein, and MDA 8.0 vs. 12.8 µmol per mg of protein (Table 1). Treatment with curcumin apparently ameliorated most of these defects (Table 1). Current literature data indicate that a decrease in GSH levels, the main redox regulator, can be observed in neurodegenerative diseases as well as in schizophrenia (42). The major transcription factor that regulates GSH metabolism is the ubiquitously expressed protein NF-E2-related factor 2 (Nrf2) (43, 44). Several key enzymes of GSH metabolism are transcriptionally regulated by Nrf2, including the catalytic and regulatory subunits of glutamate-cysteine ligase (GCL), GSH synthase, GPX2, glutathione-S-transferases (GST), and GR (43). Hence, Nrf2 is thought to represent a key transcriptional regulator of GSH metabolism. As mentioned, glutamate induces cell death via two distinct pathways: the glutamate excitotoxicity and the oxidative glutamate toxicity (3). Glutamate oxidative toxicity triggers the expression of the cysteine-glutamate antiporter system and eventually leads to GSH depletion and cell death (4, 5). The oxidative stress- -inducible cystine/glutamate exchange system, system xc−, transports one molecule of cystine, the oxidized form of cysteine, into the cells and thereby releases one molecule of glutamate into the extracellular space (45). The enhanced release of glutamate in exchange with cystine may trigger neurodegeneration due to glutamate-induced cytotoxic processes (45). Alternatively, the extrinsically added glutamate retards the outward transport of intracellular glutamate, thereby the release of cystine molecules into the intracellular space is inhibited, leading to reduced GSH production (Table 1). Conversely, increased levels of extracellular cysteine provide a reducing microenvironment required for proper cell signalling and communication, e.g. as already shown for the mechanism of T cell activation (45). It is noteworthy that the cystine/glutamate exchanger (xCT) can provide neuroprotection by enhancing glutathione export from meningeal cells and even non-neuronal cells such as astrocytes. Furthermore, xCT is critical for cell proliferation during development in vitro and possibly in vivo (46). Other reports also indicated that curcumin increased GSH levels in astrocytes and neurons by increasing the activity of the rate-limiting synthesizing enzyme, GCL (42). The modifier subunit of GCL, GCLM, is essential for the upregulation of GCL activity induced by curcumin (42).
Effect of glutamate and curcumin on the Ca2+ influx, the release of cytochrome c and LDH, and nitric oxide production
Glutamate induced severe Ca2+ influx, cytochrome c release, lactate dehydrogenase release, and substantial nitric oxide production. The intracellular Ca2+ ion concentration increased abruptly to (350.5±21) vs. control (118±17) nmol per mg of protein (Table 1), yielding a change of 2.98-fold. Regarding other parameters, the changes were shown to be (control vs. the glutamate-induced, respectively): cytochrome c ((100±5) vs. (189±11) %), LDH release ((313±1) vs. (614±1) mU/mL)), and NO ((26±2) vs. (35±3) µmol per mg of protein). Curcumin effectively alleviated most of these changes except the LDH release and the total ROS (Table 1).
Oxidative glutamate toxicity may activate a cell death pathway involving mitochondrial dysfunction and the generation of oxygen radicals, which subsequently stimulates the intracellular calcium transport (3).
Curcumin suppressed Ca2+ influx and reduced NO production (Table 1). Doroshenko and Doroshenko (47) and Stridh et al. (48) suggested that the influx of extracellular Ca2+ may pass through the L-type Ca2+ channels mediated by oxidative stress which results from the depletion of GSH following the inhibition of glutathione reductase, similar to our results (Table 1). Astroglia treated with 30 µM of curcumin increased the cellular content of GSH in parallel to elevated basal and stimulated Ca2+ efflux (48). Curcumin remodelled the coupling of antioxidant enzyme system of SOD, hydrogen peroxide, catalase, and glutamate peroxidase disturbed by glutamate (Table 1). When glutathione is depleted and GPX is unable to remove H2O2, the accumulated H2O2 evokes cell apoptosis (49). We also showed that the increased level of MDA caused by glutamate-induced injury was effectively ameliorated by curcumin (Table 1). Prakobwong et al. (50) demonstrated that curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signalling pathways. The data shown in Table 1 also reveal such a tendency.
In addition, Bal-Price et al. (51) reported that neuronal NO is involved in the signalling pathway of the NMDA receptor in the central nervous system (CNS). Low concentrations of NO increase cell proliferation via cGMP, while high concentrations of NO block proliferation via inhibition of both glycolysis and respiration, causing energy depletion (51). Mitochondria are involved in both types of NO-induced cell death: necrosis by respiratory inhibition and apoptosis by opening the permeability transition pore (52). Yuyama et al. (53) and Pytlowany et al. (54) demonstrated that NO at (0.01–0.10) mol/m3 caused a significant increase of apoptosis-inducing factor (AIF) protein level in mitochondria, chromatin fragmentation and condensation.
Conversely, higher level of NO (0.5 mol/L) together with cyclooxygenases and lipoxygenases synergistically induced massive cell death (54). Moreover, curcumin significantly downregulated the ratio of Bax/Bcl-2 (Fig. 4a) and the cleaved caspase-3 protein (Fig. 4b) increased by glutamate. Results by other researchers (55) report that substantial decrease in the ratio of Bax/Bcl-2 in reality implicates an increased ability of Bcl-2(+) cells to maintain mitochondrial pyridine nucleotides in a reduced redox state under conditions of oxidative stress induced by Ca2+, similar to our results (Table 1).
Fig. 4.
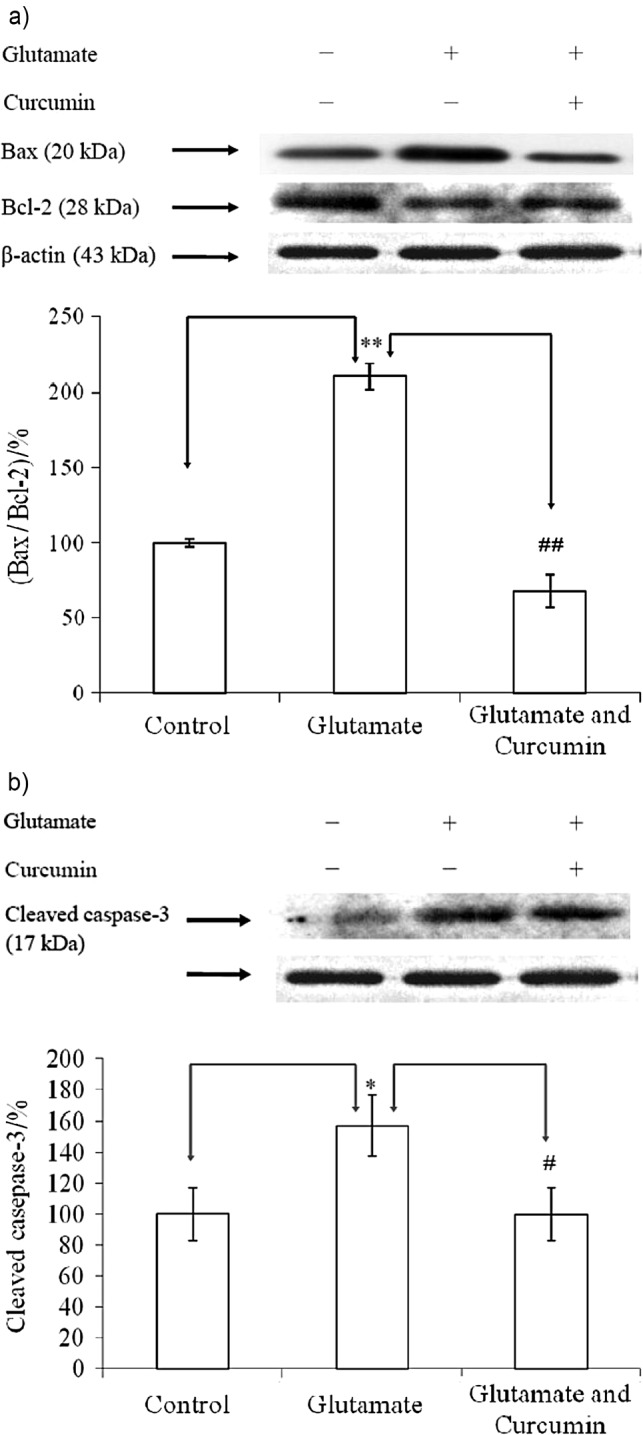
Expressions of signal proteins in PC12 cells affected by different treatments: a) proapoptotic and anti-proapoptotic protein ratio, Bax/Bcl-2; and b) cleaved caspase-3 protein. Data were statistically treated with Student’s t-test and expressed as mean±standard deviation (N=6). * indicates significant difference from the control, while # indicates significant difference between the glutamate (20 mM) and the glutamate (20 mM) and curcumin (5 µM) treatment after incubation for 24 h. Singlet and doublet symbols indicate statistical difference from each other at confidence level of p<0.05 and p<0.01, respectively (N=6)
Overall, the oxidative glutamate toxicity can be a component of the excitotoxicity cascade (56). Observed increase in calcium ions and subsequent reactions, including NO production, are characteristic of excitotoxicity (56). Along with ionotropic and metabotropic glutamate receptors, the cystine/glutamate antiporter xc– may play a critical role in CNS pathology (56). High levels of extracellular glutamate inhibit the import of cystine, resulting in the depletion of glutathione and a form of cell injury called oxidative glutamate toxicity (56).
Effect of glutamate and curcumin on the expression of caspase-3
The caspase-3 activity was upregulated up to 1.6-fold when insulted by glutamate, which was completely alleviated by curcumin (Figs. 4b and 5). Modified TUNEL assay consolidated such beneficial outcomes (Fig. 5).
Fig. 5.

The terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labelling (TUNEL) assay showing curcumin-rescued apoptosis of PC12 cell line induced by glutamate. The doses of curcumin and glutamate were 5 µM and 20 mM, respectively. Data were obtained from six different samples. DAPI=2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride, usually used for staining of the nuclei. Arrows represent the DAPI-stained nuclei damaged in the presence of gluta- mate
Cai et al. (57) suggested caspase-3 to be a key executioner caspase involved in neuronal apoptosis, which is closely associated with the progression of Alzheimer’s disease and other neurological diseases. The activity of caspase-3 is controlled by the mitochondria- and death receptor-dependent pathways (58). Caspase activation can be triggered by the release of cytochrome c from the mitochondria in response to several apoptotic stimuli including neurotoxins (59). Active caspase-9 causes cleavage and activation of downstream caspases, e.g. caspase-3 (59).
Alternatively, cell death induced by physiologically low concentrations of NO can be mediated by ROS production in mitochondria, most likely resulting from the inhibition of cytochrome c oxidase, with ROS acting as an initiator of caspase-independent cell death. NO mediated by ROS can deplete GSH (53).
More recently, curcumin in conjunction with stem cell therapy synergistically improved recovery from severe acute traumatic spinal cord injury (SCI) (60), underlying, in some instances, the neuroregenerative action of curcumin to be mediated through the neural stem cells. Fig. 6 shows the summarized therapeutic bioactivity of curcumin.
Fig. 6.

Summarized biological effects of curcumin in rescuing the PC12 cell apoptosis induced by glutamate: curcumin protective activity against glutamate-induced injury through two pathways: the GSH-dependent NO-ROS pathway and the mitochondria-dependent NO-ROS pathway, which actually involves the enhancing of GPX cycle (reaction 1 and 2) to restore the reduced glutathione, increasing the antioxidant defensive capability, and protecting the mitochondrial damages by inhibiting calcium ion influx (reaction 3) and subsequent cyt c release (reaction 4), the NO production (reaction 5), downregulating the Ca2+-induced complex I inhibition (reaction 6), the ratio Bax/Bcl-2 (reaction 10) and cleaved caspase-3 (reaction 11) to avoid apoptosis. In parallel with this, curcumin alleviated the NO- -ROS-caspase-independent pathway (reaction 5→14→15→16) and oxidative lipid peroxidation (reaction 20→21→12→(13)→22→23→24) to protect PC12 cells from apoptosis. CAT=catalase, Cur= curcumin, Cyt c=cytochrome c, l-glu=l-glutamate, GSH=glutathione, GPX=glutathione peroxidase, GR=glutathione reductase, GSSG=oxidized glutathione, MDA=malondialdehyde, NOS=nitric oxide synthase, NO=nitric oxide, PT pore=permeability transition pore, ROS=reactive oxygen species, SOD=superoxide dismutase
It is worth mentioning that even though curcumin may reveal promising therapeutic properties against a diversity of neurodiseases (20), the drawback of its application is its low bioavailability. To improve this, numerous approaches have been undertaken which involve the use of: (i) adjuvant-like piperine that interferes with glucuronidation, (ii) liposomal curcumin, (iii) curcumin nanoparticles (20), (iv) curcumin phospholipid complex, and (v) structural analogues of curcumin (e.g. EF-24) (20). The last has the advantage of rapid absorption by intestine and a longer half-life in plasma (20).
Thus, to enhance the bioavailability of curcumin in treating human neurodiseases, such a promising natural product, either curcumin or curcumin analogue, is highly recommended.
Conclusions
High level of glutamate tends to upregulate the serum level of GPX, GSSG, caspase-3 activity, stimulate the NO production, Ca2+ influx, cytochrome c release and LDH release, and elevate the Bax/Bcl-2 ratio, the formation of ROS, H2O2 and MDA. Conversely, high level of glutamate downregulates glutathione reductase, GSH, SOD and catalase, resulting in enhanced cell apoptosis. Curcumin can alleviate all these adverse effects. Taken together, curcumin can effectively protect PC12 cells against the oxidative glutamate toxicity, and apparently its mode of action involves two pathways: the GSH-dependent NO- -ROS pathway and the mitochondria-dependent NO-ROS pathway.
Acknowledgements
This work was supported by the funding of Kuang- -Tien General Hospital, Taichung, Taiwan (HK-KTOH- -102-03), partial funding from the National Science Council NSC 102-2313-B-018-001–MY3 and NSC 101-2320-B-039-040, and Taipei Medical University (TMU101-AE1-B11).
References
- 1.Doble A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol Ther. 1999;81:163–221. 10.1016/S0163-7258(98)00042-4 [DOI] [PubMed] [Google Scholar]
- 2.Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A. Mechanisms of oxidative glutamate toxicity: The glutamate/cystine antiporter system xc- as neuroprotective drug target. CNS Neurol Disord Drug Targets. 2010;9:373–82. 10.2174/187152710791292567 [DOI] [PubMed] [Google Scholar]
- 3.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–21. 10.1126/science.164.3880.719 [DOI] [PubMed] [Google Scholar]
- 4.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. 10.1126/science.7901908 [DOI] [PubMed] [Google Scholar]
- 5.Kang Y, Tiziani S, Park G, Kaul M, Paternostro G. Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun. 2014;5:3672. 10.1038/ncomms4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–55. 10.1016/j.neuint.2007.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiserodt R, Hartman TG, Ho CT, Rosen RT. Characterization of powdered turmeric by liquid chromatography-mass spectrometry and gas chromatography-mass spectrometry. J Chromatogr A. 1996;740:51–63. 10.1016/0021-9673(96)00103-3 [DOI] [Google Scholar]
- 8.Kim KT, Kim MJ, Cho DC, Park SH, Hwang JH, Sung JK, et al. The neuroprotective effect of treatment with curcumin in acute spinal cord injury: Laboratory investigation. Neurol Med Chir (Tokyo). 2014;54:387–94. 10.2176/nmc.oa.2013-0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar P, Choonara YE, Modi G, Naidoo D, Pillay V. Cur(Que)min: A neuroactive permutation of curcumin and quercetin for treating spinal cord injury. Med Hypotheses. 2014;82:437–41. 10.1016/j.mehy.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 10.Kurt G, Yildirim Z, Cemil B, Celtikci E, Kaplanoglu GT. Effects of curcumin on acute spinal cord ischemia-reperfusion injury in rabbits: Laboratry investigation. J Neurosurg Spine. 2014;20:464–70. 10.3171/2013.12.SPINE1312 [DOI] [PubMed] [Google Scholar]
- 11.Kiasalari Z, Roghani M, Khalili M, Rahmati B, Baluchnejadmojarad T. Antiepileptogenic effect of curcumin on kainate-induced model of temporal lobe epilepsy. Pharm Biol. 2013;51:1572–8. 10.3109/13880209.2013.803128 [DOI] [PubMed] [Google Scholar]
- 12.Gazal M, Valente MR, Acosta BA, Kaufmann FN, Braganhol E, Lencina CL, et al. Neuroprotective and antioxidant effects of curcumin in a ketamine-induced model of mania in rats. Eur J Pharmacol. 2014;724:132–9. 10.1016/j.ejphar.2013.12.028 [DOI] [PubMed] [Google Scholar]
- 13.Ormond DR, Shannon C, Oppenheim J, Zeman R, Das K, Murali R, et al. Stem cell therapy and curcumin synergistically enhance recovery from spinal cord injury. PLoS One. 2014;9:e88916. 10.1371/journal.pone.0088916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tegenge MA, Rajbhandari L, Shrestha S, Mithal A, Hosmane S, Venkatesan A. Curcumin protects axons from degeneration in the setting of local neuroinflammation. Exp Neurol. 2014;253:102–10. 10.1016/j.expneurol.2013.12.016 [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Yu Y, Li X, Ross CA, Smith WW. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol Res. 2011;63:439–44. 10.1016/j.phrs.2011.01.004 [DOI] [PubMed] [Google Scholar]
- 16.Chiu S, Terpstra KJ, Bureau Y, Hou J, Raheb H, Cernvosky Z, et al. Liposomal-formulated curcumin [Lipocurc™] targeting HDAC (histone deacetylase) prevents apoptosis and improves motor deficits in Park 7 (DJ-1)-knockout rat model of Parkinson's disease: Implications for epigenetics-based nanotechnology-driven drug platform. J Complement Integr Med. 2013;10:75–88. 10.1515/jcim-2013-0020 [DOI] [PubMed] [Google Scholar]
- 17.Qualls Z, Brown D, Ramlochansingh C, Hurley LL, Tizabi Y. Protective effects of curcumin against rotenone and salsolinol-induced toxicity: Implications for Parkinson's disease. Neurotox Res. 2014;25:81–9. 10.1007/s12640-013-9433-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: How many ways can curry kill tumor cells selectively? AAPS J. 2009;11:495–510. 10.1208/s12248-009-9128-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin TY, Lu CW, Huang SK, Wang SJ. Curcumin inhibits glutamate release from rat prefrontal nerve endings by affecting vesicle mobilization. Int J Mol Sci. 2012;13:9097–109. 10.3390/ijms13079097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: Problems and promises. Mol Pharm. 2007;4:807–18. 10.1021/mp700113r [DOI] [PubMed] [Google Scholar]
- 21.Banafshe HR, Hamidi GA, Noureddini M, Mirhashemi SM, Mokhtari R, Shoferpour M. Effect of curcumin on diabetic peripheral neuropathic pain: Possible involvement of opioid system. Eur J Pharmacol. 2014;723:202–6. 10.1016/j.ejphar.2013.11.033 [DOI] [PubMed] [Google Scholar]
- 22.Kawakami Z, Kanno H, Ikarashi Y, Kase Y. Yokukansan, a Kampo medicine, protects against glutamate cytotoxicity due to oxidative stress in PC12 cells. J Ethnopharmacol. 2011;134:74–81. 10.1016/j.jep.2010.11.063 [DOI] [PubMed] [Google Scholar]
- 23.Chang CH, Chen CY, Chiou JY, Peng RY, Peng CH. Astaxanthine secured apoptotic death of PC12 cells induced by β-amyloid peptide 25–35: Its molecular action targets. J Med Food. 2010;13:548–566. 10.1089/jmf.2009.1291 [DOI] [PubMed] [Google Scholar]
- 24.Chang CH, Wang HE, Liaw PY, Peng CC, Peng RY. Antrodia cinnamomea exhibits a potent neuroprotective effect in the PC12 cell-Aβ25–35 model – Pharmacologically through adenosine receptors and mitochondrial pathway. Planta Med. 2012;78:1813–23. 10.1055/s-0032-1315397 [DOI] [PubMed] [Google Scholar]
- 25.Mantha SV, Prasad M, Kalra J, Prasad K. Antioxidant enzymes in hypercholesterolemia and effects of vitamin E in rabbits. Atherosclerosis. 1993;101:135–44. 10.1016/0021-9150(93)90110-G [DOI] [PubMed] [Google Scholar]
- 26.Mohandas J, Marshall JJ, Duggin GG, Horvath JS, Tiller D. Differential distribution of glutathione and glutathione related enzymes in rabbit kidneys: Possible implication in analgesic neuropathy. Cancer Res. 1984;44:5086–91.6149017 [Google Scholar]
- 27.Kerry NL, Abbey M. Red wine and fractionated phenolic compounds prepared from red wine inhibit low density lipoprotein oxidation in vitro. Atherosclerosis. 1997;135:93–102. 10.1016/S0021-9150(97)00156-1 [DOI] [PubMed] [Google Scholar]
- 28.Hsieh CL, Lin YC, Ko WS, Peng CH, Huang CN, Peng RY. Inhibitory effect of some selected nutraceutic herbs on LDL glycation induced by glucose and glyoxal. J Ethnopharmacol. 2005;102:357–63. 10.1016/j.jep.2005.06.044 [DOI] [PubMed] [Google Scholar]
- 29.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- 30.Ottersen OP, Storm-Mathisen J, Bramham C, Torp R, Laake J, Gundersen V. A quantitative electron microscopic immunocytochemical study of the distribution and synaptic handling of glutamate in rat hippocampus. Prog Brain Res. 1990;83:99–114. 10.1016/S0079-6123(08)61244-3 [DOI] [PubMed] [Google Scholar]
- 31.Ottersen OP, Zhang N, Walberg F. Metabolic compartmentation of glutamate and glutamine: Morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience. 1992;46:519–34. 10.1016/0306-4522(92)90141-N [DOI] [PubMed] [Google Scholar]
- 32.Osen KK, Storm-Mathisen J, Ottersen OP, Dihle B. Glutamate is concentrated in and released from parallel fiber terminals in the dorsal cochlear nucleus: A quantitative immunocytochemical analysis in guinea pig. J Comp Neurol. 1995;357:482–500. 10.1002/cne.903570311 [DOI] [PubMed] [Google Scholar]
- 33.Bramham CR, Torp R, Zhang N, Storm-Mathisen J, Ottersen OP. Distribution of glutamate-like immunoreactivity in excitatory hippocampal pathways: A semiquantitative electron microscopic study in rats. Neuroscience. 1990;39:405–17. 10.1016/0306-4522(90)90277-B [DOI] [PubMed] [Google Scholar]
- 34.Tossman U, Ungerstedt U. Microdialysis in the study of extracellular levels of amino acids in the rat brain. Acta Physiol Scand. 1986;128:9–14. 10.1111/j.1748-1716.1986.tb07943.x [DOI] [PubMed] [Google Scholar]
- 35.Bouvier M, Szatkowski M, Amato A, Attwell D. The glial cell glutamate uptake carrier countertransports pH-changing anions. Nature. 1992;360:471–4. 10.1038/360471a0 [DOI] [PubMed] [Google Scholar]
- 36.Cao LL, Du GH, Wang MW. The effect of salidroside on cell damage induced by glutamate and intracellular free calcium in PC12 cells. J Asian Nat Prod Res. 2006;8:159–65. 10.1080/1028602042000325645 [DOI] [PubMed] [Google Scholar]
- 37.Lerma J, Herranz AS, Herreras O, Abraira V, Del Rio RM. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–55. 10.1016/0006-8993(86)91230-8 [DOI] [PubMed] [Google Scholar]
- 38.Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. Neuroscience. 2002;22:9134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nyitrai G, Kékesi KA, Juhász G. Extracellular level of GABA and Glu: in vivo microdialysis-HPLC measurements. Curr Top Med Chem. 2006;6:935–40. 10.2174/156802606777323674 [DOI] [PubMed] [Google Scholar]
- 40.Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. Neuroscience. 2007;27:9736–41. 10.1523/JNEUROSCI.3009-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendonça LM, Dos Santos GC, Antonucci GA, Dos Santos AC, Antonio Cardozo DS, De Lourdes Pires BM, et al. Evaluation of the cytotoxicity and genotoxicity of curcumin in PC12 cells, Mut. Res. Mutat Res. 2009;675:29–34. 10.1016/j.mrgentox.2009.02.003 [DOI] [PubMed] [Google Scholar]
- 42.Lavoie S, Chen Y, Dalton TP, Gysin R, Cuénod M, Steullet P, et al. Curcumin, quercetin, and tBHQ modulate glutathione levels in astrocytes and neurons: importance of the glutamate cysteine ligase modifier subunit. J Neurochem. 2009;108:1410–22. 10.1111/j.1471-4159.2009.05908.x [DOI] [PubMed] [Google Scholar]
- 43.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. 10.1146/annurev.pharmtox.46.120604.141046 [DOI] [PubMed] [Google Scholar]
- 44.Zhang DD. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev. 2006;38:769–89. 10.1080/03602530600971974 [DOI] [PubMed] [Google Scholar]
- 45.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system xc−: Cystine supplier and beyond. Amino Acids. 2012;42:231–46. 10.1007/s00726-011-0867-5 [DOI] [PubMed] [Google Scholar]
- 46.Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–23. 10.1523/JNEUROSCI.3178-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doroshenko N, Doroshenko P. The glutathione reductase inhibitor carmustine induces an influx of Ca2+ in PC12 cells. Eur J Pharmacol. 2004;497:17–24. 10.1016/j.ejphar.2004.06.043 [DOI] [PubMed] [Google Scholar]
- 48.Stridh MH, Correa F, Nodin C, Weber SG, Blomstrand F, Nilsson M, et al. Enhanced glutathione efflux from astrocytes in culture by low extracellular Ca2+ and curcumin. Neurochem Res. 2010;35:1231–8. 10.1007/s11064-010-0179-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim-Han JS, Sun AY. Protection of PC12 cells glutathione peroxidase in L-DOPA induced cytotoxicity. Free Radic Biol Med. 1998;25:512–8. 10.1016/S0891-5849(98)00076-8 [DOI] [PubMed] [Google Scholar]
- 50.Prakobwong S, Gupta SC, Kim JH, Sung B, Pinlaor P, Hiraku Y, et al. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis. 2011;32:1372–80. 10.1093/carcin/bgr032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bal-Price A, Gartlon J, Brown GC. Nitric oxide stimulates PC12 cell proliferation via cGMP and inhibits at higher concentrations mainly via energy depletion. Nitric Oxide. 2006;14:238–46. 10.1016/j.niox.2005.10.002 [DOI] [PubMed] [Google Scholar]
- 52.Bal-Price A, Brown GC. Nitric oxide-induced necrosis and apoptosis in PC12 cells mediated by mitochondria. J Neurochem. 2000;75:1455–64. 10.1046/j.1471-4159.2000.0751455.x [DOI] [PubMed] [Google Scholar]
- 53.Yuyama K, Yamamoto H, Nishizaki I, Kato T, Sora I, Yamamoto T. Caspase-independent cell death by low concentrations of nitric oxide in PC12 cells: involvement of cytochrome c oxidase inhibition and the production of reactive oxygen species in mitochondria. J Neurosci Res. 2003;73:351–63. 10.1002/jnr.10669 [DOI] [PubMed] [Google Scholar]
- 54.Pytlowany M, Strosznajder JB, Jeśko H, Cakała M, Strosznajder RP. Molecular mechanism of PC12 cell death evoked by sodium nitroprusside, a nitric oxide donor. Acta Biochim Pol. 2008;55:339–47. [PubMed] [Google Scholar]
- 55.Kowaltowski AJ, Vercesi AE, Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–10. 10.1038/sj.cdd.4400722 [DOI] [PubMed] [Google Scholar]
- 56.Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai L, Wang H, Li Q, Qian Y, Yao W. Salidroside inhibits H2O2-induced apoptosis in PC12 cells by preventing cytochrome c release and inactivating of caspase cascade. Acta Biochim Biophys Sin (Shanghai). 2008;40:796–802. 10.1093/abbs/40.9.796 [DOI] [PubMed] [Google Scholar]
- 58.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethalityin CPP32-deficient mice. Nature. 1996;384:368–72. 10.1038/384368a0 [DOI] [PubMed] [Google Scholar]
- 59.Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–10. 10.1016/j.bbrc.2004.12.066 [DOI] [PubMed] [Google Scholar]
- 60.Ormond DR, Shannon C, Oppenheim J, Zeman R, Das K, Murali R, et al. Stem cell therapy and curcumin synergistically enhance recovery from spinal cord injury. PLoS One. 2014;9:e88916. 10.1371/journal.pone.0088916 [DOI] [PMC free article] [PubMed] [Google Scholar]