

Abstract
De novo protein design offers templates for engineering tailor‐made protein functions and orthogonal protein interaction networks for synthetic biology research. Various computational methods have been developed to introduce functional sites in known protein structures. De novo designed protein scaffolds provide further opportunities for functional protein design. Here we demonstrate the rational design of novel tumor necrosis factor alpha (TNFα) binding proteins using a home‐made grafting program AutoMatch. We grafted three key residues from a virus 2L protein to a de novo designed small protein, DS119, with consideration of backbone flexibility. The designed proteins bind to TNFα with micromolar affinities. We further optimized the interface residues with RosettaDesign and significantly improved the binding capacity of one protein Tbab1‐4. These designed proteins inhibit the activity of TNFα in cellular luciferase assays. Our work illustrates the potential application of the de novo designed protein DS119 in protein engineering, biomedical research, and protein sequence‐structure‐function studies.
Keywords: protein design, protein‐protein interaction, TNF, inhibitor, de novo
INTRODUCTION
Rational design of protein‐protein interactions is a powerful tool for creating novel protein drugs, diagnostic probes or modulating the cellular signaling pathways.1, 2, 3, 4 The small modular scaffolds such as SH3 and PDZ domains play central roles in mediating protein‐protein interactions, and thus the applications of these domains in protein design have been extensively studied.5 To create novel protein binding interfaces and increase the structural diversity of protein engineering toolkit, de novo designed protein scaffolds can be incorporated into various computational design methods.6 This procedure offers a possibility of optimizing the overall shape of binding scaffolds while matching them to different targets.7, 8, 9 By far a diverse range of protein folds has been designed from scratch, including Top7 (an α/β protein), four helix bundles and DS119 (a βαβ motif).10, 11, 12 Top7 has been engineered to display conformation‐specific HIV‐1 epitopes;13 four helix bundles and DS119 have been designed to incorporate metal‐binding sites,14, 15 which indicates their versatility for protein engineering purpose. Importantly, de novo designed scaffolds usually exhibit high thermal stability, making them suitable for anchoring functional residues.7, 11
De novo designed protein scaffolds can be viewed as “prototype structures” in the protein evolution process.16, 17 They provide a valuable platform for testing our knowledge about protein‐protein interactions if we can design binding partners using these novel folds. Previous studies have illustrated descriptive models for protein interfaces, including the “hot spot” residues dominating the binding event and the “o‐ring” of polar interactions surrounding the central interacting residues.9, 18, 19 These principles have been proved useful in protein design. For example, Liu et al. grafted “hot spot” resides from human erythropoietin to the rat PLCδ1‐PH domain and generated a strong EPOR binding protein.20 Karanicolas et al. combined computational design and directed evolution to create a protein binding pair (Prb and Pdar) with nanomolar binding affinity based on the “o‐ring” model.9 In the current study, we aim to test this knowledge on the de novo designed protein, DS119, using computational methods. We chose DS119 because it has high stability and a singular βαβ motif which does not exist in natural proteins.11 Specifically, we want to first graft key interaction residues from a natural protein complex onto DS119 and look for hints of binding capacity. Then we will optimize the surrounding residues to enhance the interactions. With these procedures we can simultaneously test current models on a “prototype structure” and develop a comprehensive method for rational design of protein interactions.
We chose tumor necrosis factor alpha (TNFα) as our design target. TNFα is a proinflammatory cytokine that interacts with membrane receptors (TNFR1 and TNFR2) to initiate the downstream NF‐κB, JNK or caspase3 pathways.21, 22 The primary role of TNFα is the regulation of immune reactions. Its malfunction contributes to autoimmune disorders such as asthma and rheumatoid arthritis.23, 24 Hence, TNFα binding proteins and inhibitors are potential drugs for the treatment of autoimmune diseases.25 We engineered DS119 to bind TNFα using key residue grafting combined with interface optimization methods. We applied a range of biophysical characterization and found several candidates with micromolar binding affinity. We also verified the activity of designed proteins in cell‐based assays, demonstrating the potential usage of these proteins as TNFα inhibitors.
RESULTS
General flow of the computational design methods
We first searched for all natural binding partners of TNFα with known complex structures and found the poxvirus 2L protein with the highest binding affinity (KD = 43 pM).26 2L also shows high binding specificity to TNFα as it does not bind to TNFβ or other TNF family members in contrast to the membrane receptor TNFRs. TNFα is a homotrimer and each 2L binds at the interface of two monomers [Fig. 1(A)]. The 2L binding site is composed of a shallow groove between two adjacent chains of TNFα monomers and this groove is also crucial for TNFα binding to TNFRs. We then analyzed the binding interface between TNFα and 2L within the complex structural model (PDB ID: 3IT8, 2.8 Å resolution) to identify key interacting residues. Because there is no alanine scan data available, we applied a simple rule of buried area to quickly estimate which 2L residues contribute the most to the binding.27 We calculated the buried surface area of all residues in 2L and identified E99, Y160, and M161 as the ones that contact tightly with TNFα and have the largest change in solvent accessibility [Fig. 1(A), Supporting Information Fig. S1]. We chose these three residues as the hot spot residues for the initial grafting step.
Figure 1.
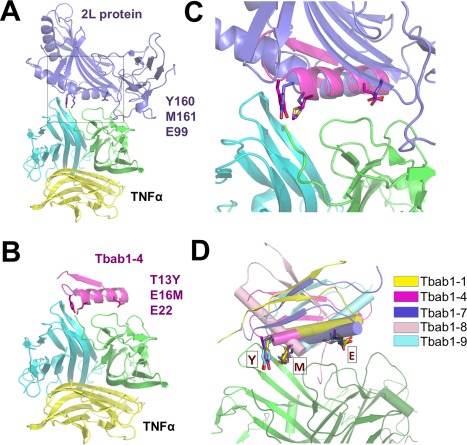
Key residue grafting generated Tbab1 design models that resemble the binding mode between TNFα and 2L protein. (A) Structural model of TNFα and 2L protein complex (PDB ID: 3IT8). (B) Design model of TNFα and Tbab1‐4 complex. (C) Comparison of key residue conformations between 2L protein and Tbab1‐4. (D) Comparison of key residue conformations among Tbab1 proteins.
Second, we used the native conformation of each key residue (E99, Y160, and M161) in 2L and the solution structure of DS119 (PDB ID: 2KI0) as inputs for our grafting method. We have previously developed an efficient program, AutoMatch, for transplanting functional residues onto any protein scaffolds.28 With the constraints to keep the grafted residues in native conformations, AutoMatch searched for the best combination of mutagenesis sites to accommodate E, Y and M on DS119. We considered the backbone flexibility with a backrub motion algorithm in AutoMatch. Furthermore, the intrinsic backbone flexibility of DS119 was evaluated in our calculation by incorporating the 20 NMR structural models of DS119 as input scaffolds.11 When the searching finished, AutoMatch automatically ranked its solutions according to their binding energy score and active site conformation score. In this case we obtained 73 solutions and the top‐ranking ones are illustrated in Supporting Information Table SI. We named the designed proteins as Tbab1 (TNFα binding proteins with βαβ motif) and used numbers to indicate their rankings (Tbab1‐1 is the model with the highest score in the first round of design and so forth).
We superimposed the key residues from top‐ranking designs to the template structure (TNFα‐2L complex) and found their conformations matched well [Fig. 1(C)]. The root‐mean‐square deviation (RMSD) between these residues are in the range of 0.91 Å to 1.20 Å. Based on the location of key residues on DS119, the designed models were classified into three types: (1) E, Y and M locate at the N‐cap region; (2) at the head of the helix; (3) at the tail of the helix (Supporting Information Fig. S2). We chose the representative ones from each type for experimental validation.
In a further design step of binding interface optimization, we applied RosettaDesign to mutate the residues that are within 6.0 Å to both TNFα and the key residues in Tbab1 proteins. RosettaDesign iteratively optimizes the conformations of main chain and side chains using Monte Carlo sampling and searches for mutants that have lower free energies.29 The resulting sequences are named as Tbab2 (Table 1).
Table 1.
Sequences of Tbab2 proteins in comparison to their Tbab1 partners
| Tbab1‐1 | GSG QVRTIWV GG TPEELEKLKEEAKMY NIRVTFWGD |
| Tbab2‐1 | GSN TVRTIWV GG TPDELEELKAEAKMY NIRVTFWGD |
| Tbab1‐4 | GSG QVRTIWV GG YPEMLKKLKEEAKKA NIRVTFWGD |
| Tbab2‐4 | GSG SVRTIWV GG YEEMLKHLKERAKKA NIRVTFWGD |
| Tbab1‐9 | GSG QVRTIWV GY MPEELEKLKEEAKKA NIRVTFWGD |
| Tbab2‐9 | GSG QVRTIYV GY MPEELEKLKEEAKKA NIRVTFWGM |
The mutation sites suggested by RosettaDesign are colored in blue and the mutation sites suggested by AutoMatch are colored in red. The residues identified by the 6.0Å criteria are indicated with the underline.
Characterization of binding affinities and structures for Tbab1 proteins
We first explored whether the grafting procedure can generate TNFα‐binding affinities among the Tbab1 proteins. In the top ranking list we excluded the ones with mutations at the hydrophobic core. We also kept G11, K21 and R30 intact in the chosen models because they are important for keeping DS119 stable and monomeric, as suggested by protein folding experiments.30 We purified the resulting nine proteins (Supporting Information Table SI) as GST‐fusion proteins and then removed the GST tag. The mass spectra indicate all the proteins have high purities (>99%, Supporting Information Fig. S8).
We then applied surface plasmon resonance (SPR) to estimate their binding capacity as well as the interaction rates (k on and k off). TNFα was immobilized on a dextran coated flow cell through lysine ligation and Tbab1 proteins were loaded onto the surface for the detection of interactions. We found five out of nine Tbab1 proteins exhibited binding capacities to TNFα (KD = 0.85 μM–62 μM) with dose response curves (Table 2, Supporting Information Fig. S3). As a control, DS119 exhibited no affinity to the immobilized TNFα. The rate constants further indicate that Tbab1‐1 and Tbab1‐4 have “fast bind, fast dissociate” kinetics in comparison to the others (Supporting Information Table SII).
Table 2.
Biophysical characterizations of Tbab proteins
| % Secondary Structures | Aggregation State |
Binding Affinity (ITC KD/μM) |
Binding Affinity (SPR KD/μM) |
|
|---|---|---|---|---|
| Tbab1‐1 | 90 | 0.89 | n. a. | 62.7 ± 6.5 |
| Tbab1‐2 | 0 | n. a. | n. a. | n. a. |
| Tbab1‐3 | 100 | 1.15 | n. a. | n. a. |
| Tbab1‐4 | 84 | 1.02 | 21.2 ± 1.1 | 3.2 ± 0.9 |
| Tbab1‐5 | 97 | 2.15 | n. a. | n. a. |
| Tbab1‐6 | 100 | 2.06 | n. a. | n. a. |
| Tbab1‐7 | 55 | 1.14, 0.91 | 39.2 ± 2.8 | 0.85 ± 0.08 |
| Tbab1‐8 | 69 | 0.94 | 25.8 ± 3.5 | 0.96 ± 0.06 |
| Tbab1‐9 | 49 | 1.14, 0.91 | 32.9 ± 3.8 | 3.8 ± 0.6 |
| Tbab2‐4 | 54 | 1.01 | 7.2 ± 0.8 | 0.36 ± 0.05 |
The secondary structural content was measured with CD using the CD signal of DS119 as the reference (100%). The aggregation state was determined by analytical size exclusion chromatography. The binding affinity was determined by SPR. n. a., not available
We verified the binding affinities of the five Tbab proteins with isothermal titration calorimetry (ITC, Fig. 3, Supporting Information Fig. S4). The ITC results suggested that four proteins feature weak binding affinity to TNFα (Table 2, KD =21.2–39.2 μM). Furthermore, for Tbab1‐7 and Tbab1‐9 the stoichiometry of the Tbab‐TNFα interaction was not 1:1, probably due to their aggregation states (Supporting Information Table SIII).
Figure 3.
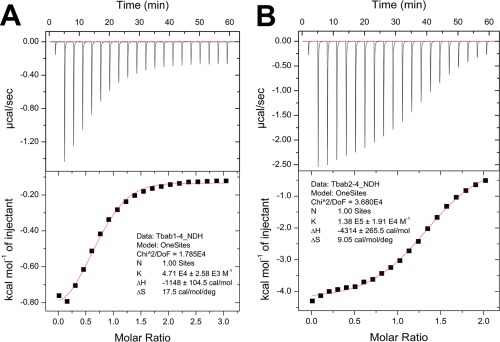
Binding affinities of Tbab1‐4 and Tbab2‐4 to TNFα measured by ITC. ITC titration curves are fitted to a one site binding model and the resulting KD for Tbab1‐4 (A) and Tbab2‐4 (B) are 21.2 μM and 7.2 μM, respectively.
We performed circular dichroism (CD) and size exclusion chromatography to determine whether Tbab1 proteins retained the monomeric βαβ structure. Eight out of nine proteins have good secondary structures and their far‐UV CD spectra are similar to that of DS119 [Fig. 2(A), Supporting Information Fig. S5]. Using the CD signal of DS119 at 222 nm as a reference, we also estimated the secondary structure content of each designed protein (Table 2). Interestingly, the ones with the highest structural contents (Tbab1‐1, 3, 4, 5, and 6) have either fast dissociation kinetics or no binding affinity. In contrast, Tbab1‐7, 8, and 9, which feature higher binding affinity to TNFα, all possess relatively lower structural content, indicating their backbone have higher flexibility. We further demonstrated through size exclusion chromatography that Tbab1‐1, 4 and 8 are monomers in solution, while Tbab1‐7 and Tbab1‐9 exhibit fast exchange of monomeric and dimeric states (Fig. 2B, Supporting Information Fig. S6, Table III).
Figure 2.
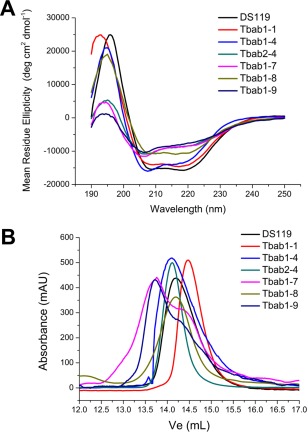
Tbab proteins with binding affinities to TNFα have good secondary structure and are mostly monomeric. (A) CD spectra of Tbab proteins indicate they have similar βαβ structural component as DS119. (B) Size exclusion chromatography of Tbab proteins indicates they are monomers except for Tbab1‐7 and Tbab1‐9.
Improvement of binding affinity through interface optimization
With further mutations around the key residues, we aim to increase the binding capacities of Tbab1 proteins through the formation of additional polar interactions. We selected Tbab1‐1, Tbab1‐4, and Tbab1‐9 for further design based on their binding capacity and stoichiometry, secondary structure content, and aggregation states. The calculations on Tbab1‐1, Tbab1‐4, and Tbab1‐9 returned reasonable results with a significant decrease of the binding free energies (Table 1). We have successfully purified Tbab2‐4 and Tbab2‐9, corresponding to the optimization of Tbab1‐4 and Tbab1‐9, respectively. CD spectra suggested Tbab2‐9 is unstructured (Supporting Information Fig. S5). Hence, we only tested Tbab2‐4 for its binding affinity and stability.
We found Tbab2‐4 had significantly elevated binding capacity in comparison with Tbab1‐4 (Table 2). The KD measured by SPR is 0.36 μM, which is ten times smaller than that of Tbab1‐4 (3.2 μM). SPR also suggested Tbab2‐4 features slow dissociation kinetics when it interacts with TNFα ( Supporting Information Fig. S3) and the corresponding k off decreased seven times compared to that of Tbab1‐4 (Supporting Information Table SII). To further validate this improvement, we applied ITC to determine the KD between TNFα and the two proteins. By fitting the titration curves to a one‐site binding model (one Tbab protein binds to one TNFα monomer), we obtained the corresponding KD, which shows the binding affinity of Tbab2‐4 has increased three times [Fig. 3, KD (Tbab1‐4) = 21.2 μM, KD (Tbab2‐4) = 7.2 μM]. The ITC measurements are in concert with the SPR results and demonstrate that the interface redesign improves the binding affinity.
We further confirmed that Tbab2‐4 has similar secondary structure to DS110, but with decreased thermodynamic stability in comparison to Tbab1‐4 (Fig. 2). Thermal denaturation experiments indicated the melting temperatures of Tbab1‐4 and Tbab2‐4 are 85°C and 55°C, respectively (Supporting Information Fig. S7). Thus the additional mutations on Tbab2‐4 led to a decrease of 30°C in the melting temperature, which implies the limitation of a small protein scaffold like DS119 (34 residues). Overall, Tbab2‐4 exhibits cooperative folding and thermal stability comparable to natural proteins.
Cellular activity of Tbab1‐4 and Tbab2‐4
As TNFα binding proteins, Tbab1‐4 and Tbab2‐4 can potentially block the interactions between TNFα and its receptors, leading to the inhibition of TNFα signaling pathway.31 To validate their bioactivity we performed a luciferase assay with ΗΕΚ293Τ cells transected with NF‐κB and other signaling elements.31 The stimulation of NF‐κB pathway with TNFα leads to the expression of luciferase, which can be detected through luciferin luminance reaction. When we incubated TNFα with the designed proteins, we found the luminescence signal decreased in a dose dependent manner (Fig. 4). By fitting the signal‐concentration curve with the Hill model, we estimated the half maximal inhibitory concentration (IC50) values for Tbab1‐4 and Tbab2‐4 as 12.5 μM and 8.9 μM, respectively. As a control, DS119 did not show inhibition effect. We also observed a high Hill coefficient (n = 5‐6) in both cases, indicating a cooperative inhibition interaction. We think it may be due to the known phenomena of TNFα receptors clustering on the cell membrane, so that the inhibition of downstream signals may require the blocking of several TNFα molecules simultaneously.22, 32
Figure 4.
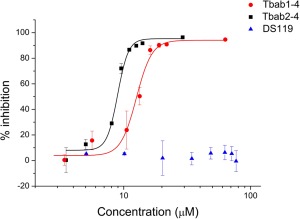
Luriferase assay indicates the ex vivo activity of Tbab1‐4 and Tbab2‐4 as TNFα inhibitors. Tbab1‐4, Tbab2‐4 and DS119 were incubated with TNFα before they were added to ΗΕΚ293Τ cells. TNFα stimulates NFκB and other signaling elements leading to the expression of luciferase. We measured the level of TNFα as the readout of luciferin luminance signals. The data were reported as means ± standard errors from three independent experiments.
DISCUSSION
The two elements for a successful hot spot grafting are a suitable scaffold and the key residues.20, 33 In the current study, the scaffold was fixed as the de novo designed protein DS119 and the key residues were identified through buried surface area calculations. In cases where the alanine scan data is absent, this method is feasible for various binding interfaces. In our case this analysis suggested reasonable residues [Fig. 1(A)]: (1) E99 on a β‐strand forms strong salt‐bridge with TNFα R31 and R32 and may be critical for both binding affinity and specificity. (2) Y160 deeply stretched into the surface pocket of TNFα and forms hydrogen bonds with R82. (3) M161 has hydrophobic interaction with TNFα Y87 and V91 (26). The binding affinities of five designed Tbab1 proteins further justified the usage of E, Y and M as key residues.
Recent studies have demonstrated significant improvement in the accuracy of grafting methods. Functional motifs including linear epitopes and discontinuous motifs have been transplanted to heterologous proteins and strong binders have been obtained as novel antigens or artificial antibodies.34, 35, 36 Thus, key residue grafting has remained an efficient strategy to create binding capacity and it is now combined with directed evolution or computational optimization to further advance protein engineering.37 This combinatorial strategy has been successfully applied in designing binders to influenza hemagglutinin and matrix metalloproteinases, as well as in our study.36, 37 We learnt from Tbab1 designs that incorporating backbone flexibility may be crucial for key residue grafting. Among the eight Tbab1 proteins (excluding Tbab1‐2 which is unstructured), the ones with rigid structures (Tbab1‐1, 3, 4, 5, 6) showed little binding affinity or fast dissociation kinetics, as suggested by SPR screen (Supporting Information Fig. S3). In contrast, the CD spectra suggested Tbab1‐7, 8 and 9, which possess higher affinities to TNFα, have more dynamic structures or more flexible regions. We posit that in these three proteins the key residues can sample bigger conformational space and accordingly adjust their conformations when they approach TNFα. This difference suggests we should capture this feature and incorporate more backbone motions in AutoMatch calculations in addition to the backrub motion.28 It may also explain why RosettaDesign can significantly improve the binding affinity for Tbab2‐4 through finer sampling of the backbone conformations.38, 39, 40
The mutations suggested by RosettaDesign can either add to the interactions with TNFα (an “o‐ring” surrounding the hot spots) or stabilize the conformation of key residues (the second layer interactions). In the case of Tbab2‐4, H19 may directly interact with N34 in TNFα; E14 and R23 can stabilize the conformation of Y13 and E22, respectively (Fig. 5). Due to the small size of DS119, we cannot allow more mutations while keeping the protein stable (for Tbab2‐4 about 30% of non‐hydrophobic residues have been mutated in comparison to DS119). If the sizes of protein scaffolds or the number of mutations are not limited in the design process, we posit that RosettaDesign optimization have potential of drastically improving the binding affinity or specificity.
Figure 5.
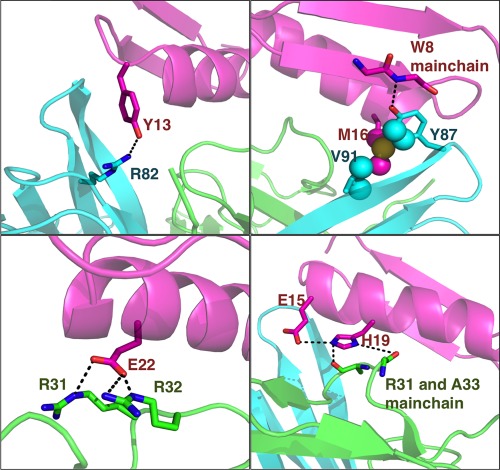
Mutations in Tbab2‐4 suggested by RosettaDesign contact with TNFα residues in the design model (magenta: Tbab2‐4, green and cyan: TNFα).
The differences between the ITC‐ and SPR‐measured binding affinities suggest that the binding capacities for Tbab1‐7, Tbab1‐8 and Tbab2‐4 are significantly overestimated in the SPR measurement. Furthermore, the SPR binding curves of Tbab1‐7, Tbab1‐8 and Tbab2‐4 feature both non‐specific and specific binding signatures. We think there are two possible reasons for this phenomenon. First, the Tbab proteins may interact non‐specifically with dextran coating of the CM5 chip. Tbab1‐7, Tbab1‐8 and Tbab2‐4 possess high isoelectronic points (8.4, 9.3 and 9.8, respectively), indicating they are highly negatively charged (relatively high charge/volume ratios) in the solution (pH 7.4). They may interact with the weakly acidic dextran coating the surface of the chips. In contrast, most of the Tbab1 proteins that exhibit little or no binding capacity on SPR have low isoelectronic points (4.8–6.3, Supporting Information Table SIII). Second, the CD spectra indicate that for Tbab1‐7, Tbab1‐8 and Tbab2‐4 have relatively low secondary structure contents. We speculate that certain hydrophobic residues in these proteins have high possibility to be flexible or partially exposed, which may lead to non‐specific interactions. We further posit that in our circumstance, the SPR binding assay is best suited for picking out the “Yes/No” binding candidates in the initial screen, while the ITC is used to estimate the binding capacities. Based on the SPR results, we did not select the proteins exhibiting significant non‐specifically binding (Tbab1‐7 and Tbab1‐8) in the RosettaDesign procedure.
The advantage of using de novo designed scaffolds in protein engineering is these scaffolds have not “co‐evolved” with other cellular proteins and hence we can develop orthogonal interaction pairs from them, which are useful for synthetic biology studies.1, 41 In our case DS119 neither interacts with TNFα nor has background bioactivity. Thus, engineering the protein‐protein interactions from scratch is beneficial for achieving a high specificity in cellular environment. Rational design of proteins with increased affinity or novel binding specificities can provide valuable reagents for biomedical research.39 Our methods should be useful for a wide range of applications.
MATERIALS AND METHODS
Key residue grafting with AutoMatch
The inputs of AutoMatch calculations include scaffold protein structure, target protein structure and the information of key residues and key atoms. We first defined the key atoms of functional residues to be grafted (E, Y, and R). AutoMatch looks for the solutions that have a small RMSD of these key atoms in comparison to the native conformation. Backbone flexibility is partly achieved using backrub motion algorithm. A grid‐based binding energy scoring method was used to screening and sorting solutions.
The 20 NMR conformations of DS119, which embody the backbone flexibility in solution, were employed as the scaffold for accepting the graft of the three hot‐spot residues. The three N terminal residues (G1, S2, and G3) and C terminal residue D36 are very flexible and may not play critical role for interaction, therefore we think key residues should not be on these sites and set these residues to be un‐mutable. The three TNFα chains in structural model 3IT8 were employed as the target protein structure, and key residues E99, Y160, M161 of chain D in PDB 3IT8 were used as the hot spots template.
Key atoms of each of the three key residues were determined using the automatic determination method in AutoMatch (side‐chain atoms within 4.2 Å of target protein). Nearly all the side‐chain atoms of these three residues were within 4.2 Å of TNF‐α except M161 Cγ and E99 Cβ. Max amplitude of backbone backrub was set to be 24° and the maximum accepted RMSD of the active atoms between the designed and the template is set to be 1.2 Å. A grid‐based binding energy scoring method was used to screen and sort solutions. The threshold for solution acceptance was that the binding energy score should be lower than −16.0. Other parameters for AutoMatch running were set to default value.
The solutions using the 20 scaffold conformations were merged together by deleting redundancy. The binding energy score (negative value) and the active‐site conformation score (positive value) of each solution were combined together as:
The solution with lowest (most negative) hybrid score in a group of redundant solutions was retained. After the merging process, 73 solutions were obtained in the design process.
Interface optimization using RosettaDesign
Interface residues on Tbab1 (6.0Å within TNF‐α) were further designed using the software Rosetta (version 2.3.0). During this interface optimization process, the important residues for folding and preventing aggregation were kept intact. To preserve the stability of the proteins, Mutations on G11 were not permitted; W9 and W34 only allowed be mutated to F or Y to keep the aromatic stacking; I8, V10, L17, L20, I29, V31, and F33 could only be mutated to hydrophobic residues; R6, K21, E23, and R30 could only be mutated to hydrophilic residues.
For each Tbab protein, we ran ten rounds of RosettaDesign. RosettaDesign iteratively optimized the conformations of main chains and side chains using Monte Carlo sampling. During each sampling step, only one residue was allowed to change and the resulting mutations with lower free energy were saved. In the final output, different mutations are combined and ranked based on the scoring function in RosettaDesign. We selected the sequence with the least mutation sites or kept the mutations shared among different designs.
Protein expression and purification
The genes encoding Tbab proteins were synthesized and cloned into the BamHI and XhoI sites of pGEX4T‐1 vectors (Invitrogen, Gaithersburg, MD) as reported before.14 Point mutations were generated with site‐directed mutagenesis kits (Saibaisheng, Beijing, China) according to the manufacturer's instructions. All constructs were confirmed by DNA sequencing.
All proteins were expressed and purified as previously described.14 Briefly, they were expressed in Escherichia coli as a GST‐fusion protein (GST: glutathione S‐transferase) and purified on a GST‐affinity column (GE Healthcare, Piscataway, NJ) and then by reversed‐phase HPLC. Lyophilized samples were obtained and their molecular weights were confirmed by high‐resolution mass spectrometry (Supporting Information Fig. S8).
Circular dichroism and size exclusion chromatography
CD spectra were measured on a MOS 450 AF/CD device (Bio‐Logic, Claix, France) at room temperature. Thermal denaturation curves were obtained with a Peltier accessory from 1°C to 97°C at the speed of 1°C/min. In both experiments, the protein concentrations were kept at 0.2 mg/mL in 50 mM PB buffer (Na2HPO4/NaH2PO4 pH 7.3).
In size exclusion experiments, protein samples were dissolved in 50 mM PB (pH 7.3) buffers with 100 mM NaCl and loaded onto a SuperdexTM Peptide 10/300 GL column (GE Healthcare, Piscataway, NJ). The UV absorbance at 280 nm was monitored.
Surface plasmon resonance
Binding interactions between Tbab proteins and TNFα were measured on a BIAcore 3000 biosensor system (BIAcore, GE). The protein samples were dissolved in PBS‐EP buffer (10 mM NaH2PO4/Na2HPO4, 150 mM NaCl, 3.7 mM EDTA, 0.005% surfactant P20, pH 7.4). TNFα was immobilized on a CM‐5 sensor chip via an amine coupling reaction to a response of 2000 resonance units (RU). A reference flow cell was activated and blocked in the absence of protein.
The analyte, Tbab proteins, was injected over the chip at various concentrations (Supporting Information Fig. S3). The observed dissociation constant, KD, was calculated by using the average RU under steady state conditions. Data were fitted globally by using the Biacore BIAevaluation 4.1 software.
Isothermal titration calorimetry
An iTC200 Microcalorimeter (MicroCal, USA) was used to measure the binding affinities (KD) between Tbab proteins and TNFα: 30 μM TNFα proteins in PBS buffer (Na2HPO4/NaH2PO4, 100 mM NaCl, pH 7.3) was loaded into the cell, and 300 μM Tbab protein dissolved in the same buffer was titrated by syringe. The titration was performed at 25°C with nineteen 2 μL injections. The spacing between each injection was 150 s. Data were collected every 5 s and then analyzed using MicroCal Origin software by fitting to the single‐site binding model. Correction for ligand dilution and other nonspecific interactions was carried out by performing a control experiment, using the same Tbab protein solutions, and the heat effect was subtracted in the original data.
Luciferase activity assay
The celluar assay were carried out as described previously.31 HEK293T cells were grown to 70% confluency in 96‐well plates at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS; Gibco), and treated with EntransterTM‐H (Engreen) transfection reagent (0.1 μL) and purified plasmids [0.25 μg pGL4.74 (hRluc/TK)] and 0.25 μg pGL4.32 (luc2P/NF‐kB‐RE/Hygro plasmid) in 50 μL DMEM/10% FBS per well. After 24 h, 50 μL preincubated mixture of TNFα and Tbab proteins was added to stimulate the cells for 12 h and the luciferase assays were carried out using the Dual‐Glo Luciferase Assay System (Promega) with a BioTek synergy 4 Multi‐Mode Microplate Reader. The final concentration of TNFα in each well was 10 ng/mL. Equal amounts of TNFα in the absence of Tbab proteins were added to the cells as a negative control. The data were reported as means ± errors from three independent experiments. In each individual experiment, results were calculated from three parallel wells.
Supporting information
Supporting Information
References
- 1. Kapp GT, Liu S, Stein A, Wong DT, Remenyi A, Yeh BJ, Fraser JS, Taunton J, Lim WA, Kortemme T (2012) Control of protein signaling using a computationally designed GTPase/GEF orthogonal pair. Proc Natl Acad Sci USA 109:5277–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Procko E, Berguig GY, Shen BW, Song YF, Frayo S, Convertine AJ, Margineantu D, Booth G, Correia BE, Cheng YH, Schief WR, Hockenbery DM, Press OW, Stoddard BL, Stayton PS, Baker D (2014) A computationally designed inhibitor of an Epstein‐Barr viral Bcl‐2 protein induces apoptosis in infected cells. Cell 157:1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker D (2014) Centenary Award and Sir Frederick Gowland Hopkins Memorial Lecture. Protein folding, structure prediction and design. Biochem Soc Trans 42:225–229. [DOI] [PubMed] [Google Scholar]
- 4. Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal V, Connell MJ, Stevens E, Schroeter A, Chen M, Macpherson S, Serra AM, Adachi Y, Holmes MA, Li Y, Klevit RE, Graham BS, Wyatt RT, Baker D, Strong RK, Crowe JE, Jr , Johnson PR, Schief WR (2014) Proof of principle for epitope‐focused vaccine design. Nature 507:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300:445–452. [DOI] [PubMed] [Google Scholar]
- 6. Der BS, Kuhlman B (2013) Strategies to control the binding mode of de novo designed protein interactions. Curr Opin Struct Biol 23:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koga N, Tatsumi‐Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D (2012) Principles for designing ideal protein structures. Nature 491:222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang PS, Love JJ, Mayo SL (2007) A de novo designed protein protein interface. Protein Sci 16:2770–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karanicolas J, Corn JE, Chen I, Joachimiak LA, Dym O, Peck SH, Albeck S, Unger T, Hu W, Liu G, Delbecq S, Montelione GT, Spiegel CP, Liu DR, Baker D (2011) A de novo protein binding pair by computational design and directed evolution. Mol Cell 42:250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D (2003) Design of a novel globular protein fold with atomic‐level accuracy. Science 302:1364–1368. [DOI] [PubMed] [Google Scholar]
- 11. Liang H, Chen H, Fan K, Wei P, Guo X, Jin C, Zeng C, Tang C, Lai L (2009) De novo design of a beta alpha beta motif. Angew Chem Int Ed Engl 48:3301–3303. [DOI] [PubMed] [Google Scholar]
- 12. Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF (2006) Computational de novo design and characterization of a four‐helix bundle protein that selectively binds a nonbiological cofactor (vol 127, pg 1346, 2005). J Am Chem Soc 128:663–663. [DOI] [PubMed] [Google Scholar]
- 13. Viana IFT, Soares TA, Lima LFO, Marques ETA, Krieger MA, Dhalia R, Lins RD (2013) De novo design of immunoreactive conformation‐specific HIV‐1 epitopes based on Top7 scaffold. RSC Adv 3:11790–11800. [Google Scholar]
- 14. Zhu C, Zhang C, Liang H, Lai L (2011) Engineering a zinc binding site into the de novo designed protein DS119 with a betaalphabeta structure. Protein Cell 2:1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF (2002) Computational de novo design, and characterization of an A(2)B(2) diiron protein. J Mol Biol 321:923–938. [DOI] [PubMed] [Google Scholar]
- 16. Samish I, MacDermaid CM, Perez‐Aguilar JM, Saven JG (2011) Theoretical and computational protein design. Ann Rev Phys Chem 62:129–149. [DOI] [PubMed] [Google Scholar]
- 17. Beasley JR, Hecht MH (1997) Protein design: the choice of de novo sequences. J Biol Chem 272:2031–2034. [DOI] [PubMed] [Google Scholar]
- 18. Desrosiers DC, Peng ZY (2005) A binding free energy hot spot in the ankyrin repeat protein GABPbeta mediated protein‐protein interaction. J Mol Biol 354:375–384. [DOI] [PubMed] [Google Scholar]
- 19. del Sol A, O'Meara P (2005) Small‐world network approach to identify key residues in protein‐protein interaction. Proteins 58:672–682. [DOI] [PubMed] [Google Scholar]
- 20. Liu S, Liu SY, Zhu XL, Liang HH, Cao AN, Chang ZJ, Lai LH (2007) Nonnatural protein‐protein interaction‐pair design by key residues grafting. Proc Natl Acad Sci USA 104:5330–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dempsey PW, Doyle SE, He JQ, Cheng G (2003) The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 14:193–209. [DOI] [PubMed] [Google Scholar]
- 22. Mukai Y, Shibata H, Nakamura T, Yoshioka Y, Abe Y, Nomura T, Taniai M, Ohta T, Ikemizu S, Nakagawa S, Tsunoda S‐i, Kamada H, Yamagata Y, Tsutsumi Y (2009) Structure‐function relationship of tumor necrosis factor (TNF) and its receptor interaction based on 3D structural analysis of a fully active TNFR1‐selective TNF mutant. J Mol Biol 385:1221–1229. [DOI] [PubMed] [Google Scholar]
- 23. Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 104:487–501. [DOI] [PubMed] [Google Scholar]
- 24. Bradley JR (2008) TNF‐mediated inflammatory disease. J Pathol 214:149–160. [DOI] [PubMed] [Google Scholar]
- 25. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL (2003) Anti‐TNF‐alpha therapies: The next generation. Nat Rev Drug Discov 2:736–746. [DOI] [PubMed] [Google Scholar]
- 26. Yang Z, West AP, Jr , Bjorkman PJ (2009) Crystal structure of TNF alpha complexed with a poxvirus MHC‐related TNF binding protein. Nat Struct Mol Biol 16:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alsop JD, Mitchell JC (2015) Interolog interfaces in protein‐protein docking. Proteins 83:1940–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang C, Lai L (2012) AutoMatch: target‐binding protein design and enzyme design by automatic pinpointing potential active sites in available protein scaffolds. Proteins 80:1078–1094. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y, Kuhlman B (2006) RosettaDesign server for protein design. Nucleic Acids Res 34:W235–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu C, Dai Z, Liang H, Zhang T, Gai F, Lai L (2013) Slow and bimolecular folding of a de novo designed monomeric protein DS119. Biophys J 105:2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang C, Shen Q, Tang B, Lai L (2013) Computational design of helical peptides targeting TNFalpha. Angew Chem Int Ed Engl 52:11059–11062. [DOI] [PubMed] [Google Scholar]
- 32. Mukai Y, Nakamura T, Yoshikawa M, Yoshioka Y, Tsunoda S, Nakagawa S, Yamagata Y, Tsutsumi Y (2010) Solution of the structure of the TNF‐TNFR2 complex. Sci Signal 3:ra83. [DOI] [PubMed] [Google Scholar]
- 33. Jacobs TM, Kuhlman B (2013) Using anchoring motifs for the computational design of protein‐protein interactions. Biochem Soc Trans 41:1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azoitei ML, Correia BE, Ban YEA, Carrico C, Kalyuzhniy O, Chen L, Schroeter A, Huang PS, McLellan JS, Kwong PD, Baker D, Strong RK, Schief WR (2011) Computation‐guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 334:373–376. [DOI] [PubMed] [Google Scholar]
- 35. Azoitei ML, Ban YE, Julien JP, Bryson S, Schroeter A, Kalyuzhniy O, Porter JR, Adachi Y, Baker D, Pai EF, Schief WR (2012) Computational design of high‐affinity epitope scaffolds by backbone grafting of a linear epitope. J Mol Biol 415:175–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tlatli R, Nozach H, Collet G, Beau F, Vera L, Stura E, Dive V, Cuniasse P (2013) Grafting of functional motifs onto protein scaffolds identified by PDB screening–an efficient route to design optimizable protein binders. FEBS J 280:139–159. [DOI] [PubMed] [Google Scholar]
- 37. Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D (2011) Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Z, Schindler CE, Lange OF, Zacharias M (2015) Application of enhanced sampling Monte Carlo methods for high‐resolution protein‐protein docking in Rosetta. PLoS One 10:e0125941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mandell DJ, Kortemme T (2009) Computer‐aided design of functional protein interactions. Nat Chem Biol 5:797–807. [DOI] [PubMed] [Google Scholar]
- 40. Humphris EL, Kortemme T (2008) Prediction of protein‐protein interface sequence diversity using flexible backbone computational protein design. Structure 16:1777–1788. [DOI] [PubMed] [Google Scholar]
- 41. Kortemme T, Joachimiak LA, Bullock AN, Schuler AD, Stoddard BL, Baker D (2004) Computational redesign of protein‐protein interaction specificity. Nat Struct Mol Biol 11:371–379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information