

Abstract
Allosteric HIV‐1 integrase (IN) inhibitors (ALLINIs) bind at the dimer interface of the IN catalytic core domain (CCD), and potently inhibit HIV‐1 by promoting aberrant, higher‐order IN multimerization. Little is known about the structural organization of the inhibitor‐induced IN multimers and important questions regarding how ALLINIs promote aberrant IN multimerization remain to be answered. On the basis of physical chemistry principles and from our analysis of experimental information, we propose that inhibitor‐induced multimerization is mediated by ALLINIs directly promoting inter‐subunit interactions between the CCD dimer and a C‐terminal domain (CTD) of another IN dimer. Guided by this hypothesis, we have built atomic models of inter‐subunit interfaces in IN multimers by incorporating information from hydrogen‐deuterium exchange (HDX) measurements to drive protein‐protein docking. We have also developed a novel free energy simulation method to estimate the effects of ALLINI binding on the association of the CCD and CTD. Using this structural and thermodynamic modeling approach, we show that multimer inter‐subunit interface models can account for several experimental observations about ALLINI‐induced multimerization, including large differences in the potencies of various ALLINIs, the mechanisms of resistance mutations, and the crucial role of solvent exposed R‐groups in the high potency of certain ALLINIs. Our study predicts that CTD residues Tyr226, Trp235 and Lys266 are involved in the aberrant multimer interfaces. The key finding of the study is that it suggests the possibility of ALLINIs facilitating inter‐subunit interactions between an external CTD and the CCD‐CCD dimer interface.
Keywords: HIV‐1 integrase, allosteric HIV‐1 integrase inhibitor, protein‐protein docking, protein‐ligand binding
Introduction
HIV‐1 Integrase (IN) is an important drug target for developing anti‐AIDS therapy. IN not only catalyzes the insertion of viral DNA into host chromosomes, but also plays an important role in virus particle maturation.1 While three catalytic site inhibitors targeting the strand transfer step of viral DNA integration have become available in recent years,2 new inhibitors targeting alternative IN sites or functions are being actively sought in order to counter the drug resistance that threatens to reduce the efficacy of the existing drugs.3
Allosteric HIV‐1 IN inhibitors (ALLINIs) have attracted significant interest in recent years.4, 5, 6, 7 ALLINIs target the LEDGF/p75 (lens epithelium‐derived growth factor/transcriptional co‐activator p75) binding pocket, located at the dimer interface of the IN catalytic core domain [CCD, see Fig. 1(A)]. LEDGF/p75 tethers the IN‐DNA complex to active genes during viral DNA integration.8 ALLINIs have a multi‐mode, complex mechanism of action9: Although the binding of ALLINIs can disrupt LEDGF/p75‐IN binding, recent studies have established that ALLINIs inhibit HIV‐1 replication primarily by promoting the formation of aberrant, higher‐order IN multimers, which impairs both the catalytic activity of IN and virus core maturation.1, 10, 11, 12, 13
Figure 1.
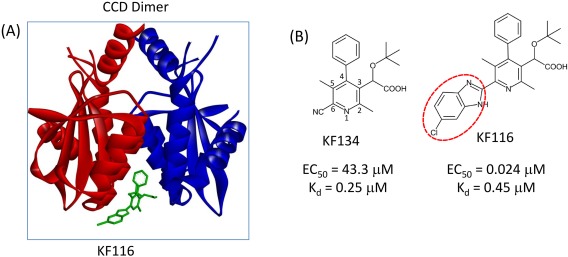
(A) ALLINI KF116 (green stick) bound at the HIV‐1 IN CCD dimer interface. Subunit 1 and subunit 2 are shown in red and blue ribbons, respectively. (B) Pyridine‐based ALLINIs KF134 and KF116.
Little is known about the structure of the aberrant higher‐order multimers induced via ALLINI binding. IN consists of three independently folded domains, the N‐terminal domain (NTD), CCD, and C‐terminal domain (CTD), connected by flexible linkers. While structures of one or two domain constructs exist, experimental structures of full‐length HIV‐1 IN are not available. Currently, several important questions regarding the mechanisms of ALLINI‐induced multimerization remain unanswered. The main goal is to understand at an atomic level how small molecule inhibitor binding to the CCD‐CCD dimer promotes higher‐order multimerization of full‐length IN. In particular, we are interested in elucidating additional ALLINI‐IN and IN‐IN interactions required for aberrant protein multimerization. Such structural information will provide much needed details for the mode of action of ALLINIs and will facilitate the development of improved inhibitors.
Understanding the mechanisms of specific drug resistance mutations is also an important issue to address. While some IN mutations such as H171T are known to operate by weakening ALLINI binding to the CCD dimer,14 others cannot be explained on the basis of binding affinity: the A128T mutation blocks aberrant IN multimerization for several ALLINIs15, 16 without significantly affecting cognate affinity for the CCD dimer.15 Knowledge of the protein‐protein interactions in the multimer can shed light on the mechanisms of these resistance mutations.
Based on recent experimental observations that specific regions of IN are involved in drug‐induced multimerization,12, 17 we propose the following possible mechanism to explain higher‐order IN multimerization: the ALLINI at the CCD dimer interface stabilizes inter‐subunit interactions between the CCD dimer and a CTD of another IN dimer, leading to the formation of higher order IN multimers. Based on this hypothesis, we build structural models of multimer inter‐subunit interfaces by incorporating information from hydrogen‐deuterium exchange (HDX)‐mass spectrometry (MS) experiments17 into a protein‐protein docking algorithm.18, 19 We also develop a novel thermodynamic free energy cycle to estimate the effect of inhibitor binding on modulating protein‐protein association. Our multimer models can account for several key experimental observations regarding inhibitor‐induced multimerization, including large differences in the potency of ALLINI compounds, the mechanism of resistance mutation A128T, and the crucial role of specific chemical R‐groups in the unusually high potency of certain ALLINIs. Our modeling study also generated experimentally testable predictions on hot spot residues critically involved in the aberrant multimer interfaces and insights on how to further improve the potency of ALLINI compounds.
Results and Discussion
Recent studies have provided clues for the molecular basis of IN multimerization. Importantly, HDX‐MS studies revealed that regions showing increased protection upon multimerization are located near the ALLINI binding pocket at the CCD dimer interface, together with downstream regions in the CTD.17 The involvement of both the CCD and CTD in higher‐order multimerization was also indicated by a study that analyzed various biophysical properties of IN truncation constructs.12 Another important observation is that many ALLINIs that contain a substituent group extended from the core of the molecule, such as compound 32 (Boehringer Ingelheim), 73 (Gilead), 80 (Bristol‐Myers Squibb) (Supporting Information Fig. S1), and KF116 20 (Fig. 1, red dashed oval), exhibit the highest potencies among ALLINIs.6, 20 To understand how such extended functional groups interact with the CCD, we have docked ligand 80 into the LEDGF/p75 pocket at the CCD dimer interface; we found that the extended R‐group is located outside the binding pocket and is largely solvent exposed (Supporting Information Fig. S2). Using free energy simulations we have estimated that the solvent exposed R‐group contributes little to the binding affinity with the CCD (Supporting Information Fig. S3). Therefore the role of the extended R‐groups in the exceptional potencies apparently cannot be attributed to enhancement of CCD binding affinity.
To further explore the functional significance of R‐groups, we have synthesized KF134 and compared its activity with its KF116 counterpart [Fig. 1(B)]. Both KF134 and KF116 contain pyridine cores but unlike KF116, KF134 lacks the 5‐chlorobenzimidazole group. Figure 1 shows that KF116 and KF134 bind the CCD dimer with similar affinities as seen from the small difference in their K d for CCD binding. However, KF11620 is markedly (≥ 1800‐fold) more potent than KF134 (Supporting Information Fig. S4). These results indicate the functional significance of the R‐group, which in the context of full length IN could establish additional interactions extending beyond the CCD‐CCD dimer.
Based on these observations, we have formed the following hypotheses: aberrant IN multimerization is mediated directly by the ALLINI bound at the CCD dimer interface, which stabilizes inter‐subunit interactions between the CCD dimer and the CTD of another IN dimer through direct interactions, and by displacing water molecules from the inter‐subunit interface in the multimer (dewetting). Water molecules confined in protein‐protein interfaces can be thermodynamically unstable, due to the loss of entropy and the inability to form extensive hydrogen bonds.21, 22 The displacement of such waters to the bulk solvent lowers the free energy of the system and stabilizes the protein‐protein interface. Figure 2(A) shows a schematic diagram that illustrates these effects of ALLINI‐binding on multimerization.
Figure 2.
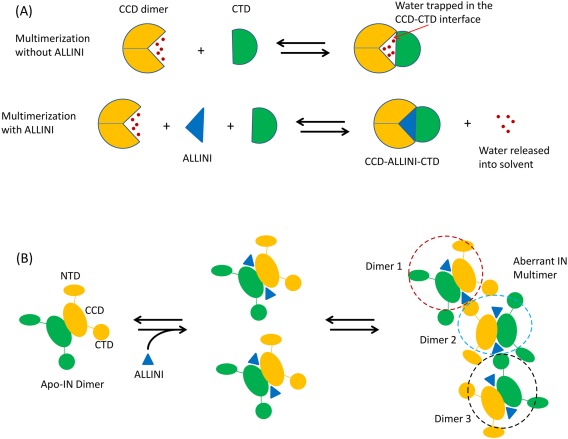
(A) Schematic diagram illustrating how ALLINI binding stabilizes the inter‐subunit interaction by expelling waters from the LEDGF/p75 binding pocket and increasing the effective protein‐protein interface. (B) Cartoon diagram showing a CTD of one IN dimer binding to the CCD‐CCD interface of another dimer in a chain reaction leading to higher order oligomerization, stabilized by the bound ALLINI at the CCD‐dimer interface.
Since IN dimerizes with picomolar dissociation constant,23 in our model, a higher order multimer is assembled from the dimer as the basic building block. Figure 2(B) illustrates how multimerization can be initiated from dimers: an inter‐subunit interface is formed between a CTD of one dimer and a CCD‐CCD interface of another dimer, stabilized by an ALLINI at the LEDGF/p75 pocket.
A key element in our hypothesis is that the neighborhood of the LEDGF/p75 binding pocket directly participates in the inter‐subunit contacts in the multimer. This view is consistent with a recent report showing that LEDGF/p75 binding is able to inhibit ALLINI‐induced multimerization.24 Presumably the binding of the comparatively large LEDGF/p75 protein makes this region of the CCD dimer unavailable for inter‐subunit interactions required for multimerization.
Our proposed mechanism for multimerization provides a physical basis for the exceptionally high potency of ALLINIs carrying an extended, solvent exposed substituent group (Fig. 1, Supporting Information Fig. S1, red ovals): compared with ALLINI KF134, the 5‐chlorobenzimidazole group extended from position 6 of the pyridine core in KF116 increases the non‐polar solvent accessible surface area by ≈ 100 Å2 which enhances the effective inter‐subunit interaction area with the external CTD upon multimerization (Supporting Information Fig. S5). Protein‐protein binding affinity is often dominated by a small number of key interface residues (hot spots).25, 26, 27 We suggest that here the extended substituent group attached to the core of KF116 is likely to mimic the role of amino acid hot spot residue(s) that serve to enhance protein‐protein interactions in the IN multimers.
We next used the HADDOCK protein‐protein program18, 19 to build structural models of the inter‐subunit interfaces between the CTD and CCD dimer. Figure 3(A) shows the top ranked structure of the CTD docked to the CCD dimer in the presence of inhibitor ALLINI‐2. As a control, we constructed the inter‐subunit interface formed in the absence of the ALLINI by docking the CTD to the apo‐CCD dimer [Fig. 3(B)]. The inter‐subunit interface structure docked with ALLINI‐2 has a significantly more favorable docking energy score compared to that from the inter‐subunit structures built without the ALLINI (−108.0 kcal/mol vs. −98.0 kcal/mol). This result is consistent with the central role of ALLINI binding in promoting IN multimerization. In the docked structure without ALLINI, there is a large volume in the center of the CCD dimer‐CTD interface filled with solvent; the same space is occupied by the inhibitor in the structure built with ALLINI‐2.
Figure 3.
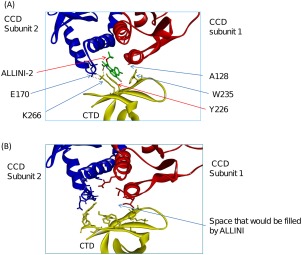
Models of the inter‐subunit interface of IN multimers constructed using: (A) CTD and CCD‐ALLINI‐2; and (B) CTD and apo CCD. ALLINI‐2 is shown in green sticks in (A).
It is noted that in the inter‐subunit interface model containing ALLINI‐2 [Fig. 3(A)], Ala128 is buried in a nonpolar cavity formed by the side chains of Trp235 of the CTD and the chlorobenzene moiety of ALLINI‐2 in the CCD‐ALLINI‐CTD interface. This could explain why mutations such as A128T15 and A128N16 confer resistance to ALLINI‐2 and similar inhibitors: both substitutions introduce a polar side chain buried in the nonpolar pocket of the protein‐protein interface, which is generally thermodynamically unstable as it is associated with a high desolvation penalty. Our model therefore suggests that the resistance mutation A128T acts by decreasing the hydrophobicity around the CCD‐dimer interface region that forms part of the aberrant multimer inter‐subunit interface.
Another feature found in the inter‐subunit interface model containing ALLINI‐2 is that the amino group of the side chain of Lys266 of the CTD forms a bifurcated intermolecular salt bridge with Glu170 of the CCD and the carboxylate ion of ALLINI‐2 [Fig. 3(A)]. Experimentally, it was found that mutations K266A/K264A deter ALLINI‐2 induced multimerization.17 The intermolecular salt bridge involving Lys266 seen in our model is consistent with this observation.
In our model, both Tyr226 and Trp235 on the CTD make extensive intermolecular interactions with the CCD‐ALLINI‐2. We therefore predict that amino acid mutations W235A/Y226A could weaken the multimerization caused by ALLINI‐2.
We have performed protein‐protein docking to generate models of multimer inter‐subunit interfaces involving other ALLINIs (Fig. 4). It can be seen that the CCD‐ALLINI‐CTD protein‐protein docking energies correlate with experimental EC50 values, which is in general agreement with the central experimental observation that ALLINI potency is determined by the ability of the drug to promote aberrant multimerization. In contrast, ALLINI binding affinities to the CCD‐CCD dimer do not always correlate with their potencies. This is particularly clear when comparing two pyridine‐based compounds KF134 and KF116. While these two compounds bind the CCD‐CCD dimer with comparable affinities, there is a very large difference in their EC50 values (Supporting Information Fig. S420). Such markedly different potencies between KF134 and KF116 can be explained based on the CCD‐ALLINI‐CTD docking energies. Note that KF116 and KF134 differ only in the substituent at the 6‐position of the pyridine core. According to the docked CCD‐KF116‐CTD structure, the chlorobenzimidazole substituent is embedded in a cavity in the CCD‐CTD inter‐subunit interface (Supporting Information Fig. S6), which explains the critical role of this expanded R‐group in stabilizing inter‐subunit interactions in multimerization.
Figure 4.
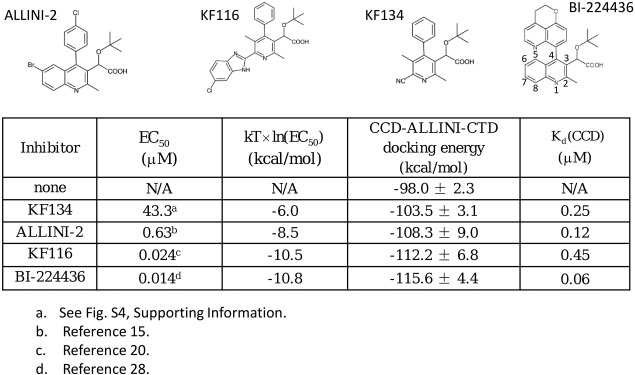
Comparisons of experimental antiviral activities of EC50 and calculated multimerization docking energies and the CCD binding affinities for several ALLINIs. The values of K d are converted from calculated values of binding free energy obtained from double decoupling calculations (Materials and Methods).
The results in Figure 4 also provide physical explanations for the high potency of BI‐224436, once a clinical candidate16, 28; the inter‐subunit interface model containing this ALLINI shows that the bulky tricyclic arene substituent at the C4 position of the quinoline core is almost completely buried in the CCD‐ALLINI‐CTD inter‐subunit interface (Supporting Information Fig. S7), which contributes significantly to inter‐subunit interaction through expelling unfavorable waters from the binding interface.
The docking scoring function used in protein‐protein docking generally does not include effects such as the protein strain free energy. Here, we also use a thermodynamic free energy cycle (“Materials and Methods” section), which in principle can better capture some physical aspects of protein‐protein binding, to examine in more detail whether docked models of the inter‐subunit interface can explain the trend in ALLINI potency. Using this free energy cycle, we have estimated that , the free energy contributions of compound binding to the stability of the CCD‐ALLINI‐CTD inter‐subunit interface, are −2.7 ± 1.1 kcal/mol and −5.7 ± 0.9 kcal/mol for ALLINI‐2 and KF116, respectively. These numbers support the idea that inhibitor binding stabilizes the CCD‐CTD inter‐subunit interaction. The values predict that KF116 is ∼ 330 times more effective than ALLINI‐2 in promoting multimerization. Experimentally KF116 is found to be ∼ 26 times more potent than ALLINI‐2.15, 20 The free energy calculations are therefore in qualitative agreement with experiments. We also examined the explicit solvent molecular dynamics trajectories generated from these free energy simulations to estimate the number of waters expelled from the CCD‐ALLINI‐CTD protein‐protein interface: on average, between 5 and 8 trapped water molecules were released to the bulk solvent by ALLINI‐2.
In this study, we propose that ALLINI binding at the CCD dimer cleft provides a crucial anchor point for inter‐subunit interactions during multimerization and that the multimer inter‐subunit interface is formed between CCD‐ALLINI and CTD. In multimerization caused by different ALLINIs, other IN segments such as the NTD and/or linker regions between different domains may also form inter‐subunit interactions with the CCD‐ALLINI surface.
It has been suggested that the CTD‐CTD crystal contacts observed in the crystal structure of the CCD‐CTD 2‐domain construct29 might form the inter‐subunit interface in ALLINI‐induced aberrant IN multimers.12 We note that in this crystal structure, the average distances between the CTD and ALLINI binding pocket on the CCD dimer are greater than 65 Å.29 Thus, long range allosteric conformational changes would be required to account for the essential role of ALLINI binding in promoting multimerization based on this crystal contact. In this study, we chose to investigate a simpler mechanism that involves direct interactions between the ALLINI‐CCD and the CTD.
Based on our models of KF116 and BI‐224436 induced inter‐subunit interfaces (Supporting Information Figs. S6 and S7), we suggest that a bulky aryl substituent at the C4 position of the pyridine core, combined with an expanded and not too polar substitution in the direction of the C6 position of the pyridine, is likely to yield a highly potent inhibitor of HIV‐1 IN.
Conclusion
We have proposed and examined the concept that ALLINI binding at the CCD dimer interface stabilizes inter‐subunit CCD‐CTD interactions which lead to aberrant multimerization. We have shown that the atomic models constructed based on this idea are able to provide physical explanations for a number of key experimental results concerning the multimerization, including the exceptional potency in both quinoline and pyridine based ALLINIs. The work suggests that the neighborhood of the ALLINI binding site on the CCD plays a central role in nucleating and stabilizing protein‐protein interactions that drive aberrant IN multimerization and hence the potent antiviral activities of this novel drug class. The study shed lights on the mechanism of action of the allosteric inhibitors of HIV‐1 IN and provides insights into the design of more potent compounds.
Materials and Methods
The details of the synthesis of KF134, experimental determination of antiviral activities, protein‐protein docking, and the new thermodynamic free energy cycle are described in the Supporting Information.
Supporting information
Supporting Information
Acknowledgments
The calculations were run on XSEDE allocation resources (to RML) and a shared computing cluster at Temple University supported by NIH S10 OD020095. We thank Dr. Emilio Gallicchio for stimulating discussions on the novel thermodynamic free energy cycle used in this work.
References
- 1. Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras‐Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A (2013) Allosteric integrase inhibitor potency is determined through the inhibition of HIV‐1 particle maturation. Proc Natl Acad Sci USA 110:8690–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Engelman A, Cherepanov P (2014) Retroviral integrase structure and DNA recombination mechanism. Microbiol Spect 2:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masuda T (2011) Non‐enzymatic functions of retroviral integrase: the next target for novel anti‐HIV drug development. Front Microbiol 2:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z (2010) Rational design of small‐molecule inhibitors of the LEDGF/p75‐integrase interaction and HIV replication. Nat Chem Biol 6:442–448. [DOI] [PubMed] [Google Scholar]
- 5. Engelman A, Kessl JJ, Kvaratskhelia M (2013) Allosteric inhibition of HIV‐1 integrase activity. Curr Opin Chem Biol 17:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Demeulemeester J, Chaltin P, Marchand A, De Maeyer M, Debyser Z, Christ F (2014) LEDGINs, non‐catalytic site inhibitors of HIV‐1 integrase: a patent review (2006 – 2014). Expert Opin Ther Pat 24:609–632. [DOI] [PubMed] [Google Scholar]
- 7. Feng L, Larue RC, Slaughter A, Kessl JJ, Kvaratskhelia M, HIV‐1 integrase multimerization as a therapeutic target In: Torbett BE, Goodsell DS, Richman DD, Eds. (2015) The future of HIV‐1 therapeutics. Vol. 389 Switzerland: Springer International Publishing; pp 93–119. Available from http://link.springer.com/10.1007/82_2015_439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Craigie R, Bushman FD (2012) HIV DNA integration. Cold Spring Harb Perspect Med 2:a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kessl JJ, Jena N, Koh Y, Taskent‐Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Le Grice SFJ, Engelman A, Fuchs JR, Kvaratskhelia M (2012) Multimode, cooperative mechanism of action of allosteric HIV‐1 integrase inhibitors. J Biol Chem 287:16801–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desimmie B, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z (2013) LEDGINs inhibit late stage HIV‐1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balakrishnan M, Yant SR, Tsai L, O'Sullivan C, Bam RA, Tsai A, Niedziela‐Majka A, Stray KM, Sakowicz R, Cihlar T (2013) Non‐catalytic site HIV‐1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS One 8:e74163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, Van Duyne GD, Bushman FD (2014) Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains. J Biol Chem 289:20477–20488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, Steven AC (2015) Distribution and redistribution of HIV‐1 nucleocapsid protein in immature, mature, and integrase‐inhibited virions: a role for integrase in maturation. J Virol 89:9765–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Slaughter A, Jurado KA, Deng N, Feng L, Kessl JJ, Shkriabai N, Larue RC, Fadel HJ, Patel PA, Jena N, Fuchs JR, Poeschla E, Levy RM, Engelman A, Kvaratskhelia M (2014) The mechanism of H171T resistance reveals the importance of Ndelta‐protonated His171 for the binding of allosteric inhibitor BI‐D to HIV‐1 integrase. Retrovirology 11:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, Engelman A, Fuchs JR, Kvaratskhelia M (2013) The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV‐1 integrase inhibitors. J Biol Chem 288:15813–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fenwick C, Amad M, Bailey MD, Bethell R, Bos M, Bonneau P, Cordingley M, Coulombe R, Duan J, Edwards P, Fader LD, Faucher A‐M, Garneau M, Jakalian A, Kawai S, Lamorte L, LaPlante S, Luo L, Mason S, Poupart M‐A, Rioux N, Schroeder P, Simoneau B, Tremblay S, Tsantrizos Y, Witvrouw M, Yoakim C (2014) Preclinical profile of BI 224436, a novel HIV‐1 non‐catalytic‐site integrase inhibitor. Antimicrob Agents Chemother 58:3233–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shkriabai N, Dharmarajan V, Slaughter A, Kessl JJ, Larue RC, Feng L, Fuchs JR, Griffin PR, Kvaratskhelia M (2014) A critical role of the C‐terminal segment for allosteric inhibitor‐induced aberrant multimerization of HIV‐1 integrase. J Biol Chem 289:26430–26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dominguez C, Boelens R, Bonvin AMJJ (2003) HADDOCK: a protein−protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125:1731–1737. [DOI] [PubMed] [Google Scholar]
- 19. van Zundert GCP, Rodrigues JPGLM, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, Bonvin AMJJ (2016) The HADDOCK2.2 web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol 428:720–725. [DOI] [PubMed] [Google Scholar]
- 20. Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, Bushman FD, Fuchs JR, Kvaratskhelia M (2014) A new class of multimerization selective inhibitors of HIV‐1 integrase. PLoS Pathog 10:e1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem 49:6177–6196. [DOI] [PubMed] [Google Scholar]
- 22. Young T, Abel R, Kim B, Berne BJ, Friesner RA (2007) Motifs for molecular recognition exploiting hydrophobic enclosure in protein‐ligand binding. Proc Natl Acad Sci USA 104:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsiang M, Jones GS, Hung M, Mukund S, Han B, Liu X, Babaoglu K, Lansdon E, Chen X, Todd J, Cai T, Pagratis N, Sakowicz R, Geleziunas R (2009) Affinities between the binding partners of the HIV‐1 integrase dimer‐lens epithelium‐derived growth factor (IN Dimer‐LEDGF) complex. J Biol Chem 284:33580–33599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng L, Dharmarajan V, Serrao E, Hoyte A, Larue RC, Slaughter A, Sharma A, Plumb MR, Kessl JJ, Fuchs JR, Bushman FD, Engelman AN, Griffin PR, Kvaratskhelia M (2016) The competitive interplay between allosteric HIV‐1 integrase inhibitor BI/D and LEDGF/p75 during the early stage of HIV‐1 replication adversely affects inhibitor potency. ACS Chem Biol 11:1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280:1–9. [DOI] [PubMed] [Google Scholar]
- 26. Moreira IS, Fernandes PA, Ramos MJ (2007) Hot spots‐a review of the protein‐protein interface determinant amino‐acid residues. Proteins 68:803–812. [DOI] [PubMed] [Google Scholar]
- 27. Deng N‐J, Cieplak P (2009) Insights into affinity and specificity in the complexes of α‐lytic protease and its inhibitor proteins: binding free energy from molecular dynamics simulation. Phys Chem Chem Phys 11:4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, LaPlanate S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bos M, Bonneau P, Poupart M‐A, Faucher A‐M, Simoneau B, Fenwick C, Yoakim C, Tsantrizos Y (2014) Discovery of BI 224436, a noncatalytic site integrase inhibitor (NCINI) of HIV‐1. ACS Med Chem Lett 5:422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen JC‐H, Krucinski J, Miercke LJW, Finer‐Moore JS, Tang AH, Leavitt AD, Stroud RM (2000) Crystal structure of the HIV‐1 integrase catalytic core and C‐terminal domains: a model for viral DNA binding. Proc Natl Acad Sci USA 97:8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information