

Abstract
X‐ray diffractometry dominates protein studies, as it can provide 3D structures of these diverse macromolecules or their molecular complexes with interacting partners: substrates, inhibitors, and/or cofactors. Here, we show that under cocrystallization conditions the results could reflect induced protein folds instead of the (partially) disordered original structures. The analysis of synchrotron radiation circular dichroism spectra revealed that the Im7 immunity protein stabilizes the native‐like solution structure of unfolded NColE7 nuclease mutants via complex formation. This is consistent with the fact that among the several available crystal structures with its inhibitor or substrate, all NColE7 structures are virtually the same. Our results draw attention to the possible structural consequence of protein modifications, which is often hidden by compensational effects of intermolecular interactions. The growing evidence on the importance of protein intrinsic disorder thus, demands more extensive complementary experiments in solution phase with the unligated form of the protein of interest.
Keywords: induced protein folding, SRCD spectroscopy, colicin E7 nuclease, Im7 immunity protein
Introduction
Studies on colicin nucleases revealed that an intrinsic disorder might be induced by mutations of the enzymes.1, 2, 3 In independent parallel research, published recently, it was shown that certain properties of such colicin E3 and E7 mutants can be fully restored by protein–protein interactions.2, 4 Colicin E7, the nuclease toxin of Escherichia coli, is a particular example of this kind5, 6, 7, 8 as its nuclease domain (NColE7) has been extensively studied by X‐ray crystallography. The C‐terminal HNH active centre is made up of ∼40 amino acids forming a ββα‐type Zn2+‐ion binding structure.9, 10, 11 Similar HNH motifs12, 13, 14, 15 are observed in various endonucleases, such as the homing,16 restriction,17 or nonspecific enzymes.18 While this motif in NColE7 binds to the 3′ site of the scissile phosphodiester‐group in the DNA minor groove, the central helical regions also form strong nonspecific interactions in the major groove.14, 19, 20, 21, 22 The ∼40 amino acids long N‐terminal sequence is known to have no well‐defined secondary structure and to contain an Arg, which is necessary for effective nuclease activity.23, 24, 25, 26 In addition, the N‐terminus was also found to affect the structure of the enzyme. Controlling of a conserved metal binding catalytic motif through an intramolecular allosteric activation by a distal part of the protein is an intriguing mechanism in protein research. Therefore, to get a more profound view of the role of these residues, we designed eight variants of NColE7 (Figure 1), including N‐terminal truncated mutants, missing 4, 25, 45, or 69 amino acid residues,24 as well as, point mutants substituting the (T454) threonine and/or (K458) lysine and/or (W464) tryptophan amino acid with alanines.1, 2
Figure 1.
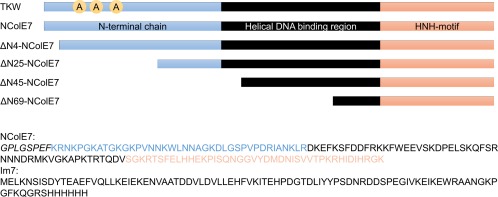
Scheme of the NColE7 variant sequences: the sequences of the purified NColE7 and Im7 proteins are also shown. The GPLGSPEF is an additional sequence encoded by the pGEX‐6P‐1 plasmid, which remained at the N‐terminus of the nucleases after enzymatic cleavage of the GST purification tag. TKW symbolizes the TKW, TK, KW, or W NColE7 point mutants, in which T454A and/or K458A and/or W464A mutations are present. For the truncated mutants X in the ΔNX notation indicates the number of deleted amino acids at their N‐termini. The Im7 protein is purified together with a hexahistidine tag at its C‐terminus.
Molecular recognition is a key issue in function of the colicin nucleases. The Im7 immunity protein is needed to prevent the cleavage of nucleic acids by NColE7 in E. coli cells during protein expression. The four antiparallel α‐helices of Im727 strongly and specifically interact with the nucleic acid binding helical regions of NColE7,15, 28 based on charge and surface‐complementarity.29, 30, 31 This as well as the interaction with the substrate DNA may affect the structure of NColE7 mutants. In the crystal structures published so far, NColE7 and its mutants are present together with either Im713, 15, 16, 29 or DNA,15, 21, 22, 27 with the exception of only two crystals: the Zn2+‐complexes of NColE7 itself20 and its ΔN4 truncated mutant.32 Independently of the interacting molecules or ions, the overall nuclease structure is retained in all crystals. Despite the detailed crystallographic information available, there is a limited knowledge on the solution structure of this protein. This inspired us to investigate the structural consequences of the interaction of mutant NColE7 proteins (shown in Figure 1) with Im7 in aqueous solution in more detail. In this study, we show how the folding of NColE7 is induced by specific protein–protein interactions using synchrotron radiation circular dichroism spectroscopy. The possible discrepancies between solution and crystallographic protein structure studies, caused by such induced structures are also discussed.
Results
The interaction of Im7 with NColE7 has minor consequences on the protein structure both in crystal and in solution phases
Comparing the crystal structures of the free and complexed NColE7 and Im7 proteins,15, 27, 28 suggests that neither of the native proteins undergo significant structural changes in their complexes [Figure 2(A)]. The superimposition of the free NColE7 structure (1M08 15) and the Im7 bound NColE7 structure (7CEI 28) revealed very few differences and the RMSD is 0.408 for 109 Cα atoms. Likewise, the free Im7 protein (1CEI 27) is very similar to the NColE7 bound Im7 protein, with an RMSD value of 0.414 for 76 Cα atoms.
Figure 2.
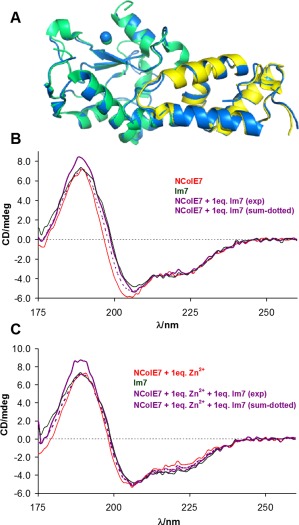
Consequences of NColE7–Im7 interaction. (A) The comparison of the structures of free and bound Im7 and NColE7 proteins visualized from their single crystal structures. The free NColE7 structure (green, 1M08 15), as well as the free Im7 structure (yellow, 1CEI 27) is superimposed to the structure of the NColE7‐Im7 complex (blue, 7CEI 28). The Zn2+‐ions are indicated by spheres. (B) The experimental SRCD spectra of NColE7 and Im7 proteins (3.2 × 10−5 M) and their equimolar mixtures (1.6 × 10−5 M, each). The mathematical sum of the component protein spectra considering the same composition as in the experiment, but assuming no interaction between the two proteins is also shown for comparison. (C) The same as (B) but the spectra were recorded in the presence of an equivalent amount of Zn2+‐ions referred to the actual NColE7 concentration.
SRCD spectra of the free NColE7 and Im7 proteins and their mixture have been recorded to verify that the above statement holds also in solution phase. NColE7 and Im7 display far‐UV SRCD spectra in correspondence with the structures being rich in α‐helices [Figure 2(B,C)], as observed in the crystalline state for both proteins. The experimentally obtained and the mathematical sum of the individual spectra are indeed very similar. The small differences between the protein spectra obtained in the presence and absence of metal ion may be related both to structural adjustment and to charge transfer transitions in the UV region arising from complexation with the Zn2+‐ion. No interaction was detected between Im7 and Zn2+ (data not shown).
The SRCD spectra of NColE7 and Im7 proteins were subjected to evaluation of the secondary structure element composition by various programs. The results are collected in Table 1, together with the calculations based on the difference spectra derived from the protein mixtures as well as from the crystal structures of the two proteins. Even the relative amounts of the secondary structure elements obtained from the crystal structure differ according to the applied algorithm, as shown, for example, for the 1M08 15 structure in the table. The alterations in the results obtained by various programs can be attributed to the different classifications of the secondary structure elements by the authors. The comparison of the results calculated with the same program, however, provides the same trends.
Table 1.
The fractions of the secondary structure elements obtained from the crystal structures with programs DSSP33 and BeStSel34 or calculated from the SRCD spectra of the reference protein databases35, 36 by BeStSel, 34 the CDPro program package, 37 and CDNN program 38
| Protein | Data | α‐Helix | β‐Sheet | Turn | Unordered | |
|---|---|---|---|---|---|---|
| NColE7 | SRCD | CDPro | 0.32 | 0.16 | 0.20 | 0.32 |
| CDNN | 0.30 | 0.26 | 0.20 | 0.24 | ||
| BeStSel | 0.20 | 0.21 | 0.16 | 0.43 | ||
| SRCD (with Zn2+) | CDPro | 0.28 | 0.24 | 0.21 | 0.27 | |
| CDNN | 0.29 | 0.26 | 0.20 | 0.25 | ||
| BeStSel | 0.16 | 0.24 | 0.16 | 0.44 | ||
| PDB id: 1M08 (with Zn2+) | DSSP | 0.38 | 0.17 | 0.15 | 0.31 | |
| BeStSel | 0.31 | 0.11 | 0.15 | 0.44 | ||
| PDB id: 1PT3 (with 8bp DNA) | 0.39 | 0.17 | 0.12 | 0.32 | ||
| SRCD (with Im7)a | CDPro | 0.35 | 0.28 | 0.13 | 0.24 | |
| CDNN | 0.31 | 0.23 | 0.19 | 0.27 | ||
| BeStSel | 0.19 | 0.25 | 0.16 | 0.40 | ||
| PDB id: 7CEI (with Im7 and Zn2+) | 0.30 | 0.15 | 0.12 | 0.43 | ||
| PDB id: 1MZ8 (with Im7 and Zn2+) | 0.36 | 0.17 | 0.20 | 0.27 | ||
| PDB id: 2ERH (with Im7b) | 0.32 | 0.17 | 0.15 | 0.36 | ||
| Im7 | SRCD | CDPro | 0.43 | 0.12 | 0.19 | 0.26 |
| CDNN | 0.44 | 0.15 | 0.18 | 0.23 | ||
| BeStSel | 0.36 | 0.15 | 0.14 | 0.34 | ||
| PDB id: 1CEI | DSSP | 0.52 | 0.02 | 0.10 | 0.36 | |
| BeStSel | 0.48 | 0.00 | 0.11 | 0.41 | ||
| SRCD (with NColE7)a | CDPro | 0.53 | 0.2 | 0.14 | 0.13 | |
| CDNN | 0.46 | 0.13 | 0.17 | 0.23 | ||
| BeStSel | 0.34 | 0.25 | 0.13 | 0.28 | ||
| PDB id: 1M08 (with NColE7) | 0.53 | 0.00 | 0.09 | 0.38 | ||
| PDB id: 7CEI (with NColE7) | 0.46 | 0.00 | 0.16 | 0.38 | ||
| PDB id: 2ERH (with NColE7b) | 0.52 | 0.02 | 0.10 | 0.36 | ||
The mathematical difference of the spectra of the mixtures and individual proteins were applied in the secondary structure determination. The result is distorted, as the fitted protein is supposed to suffer of all the conformational changes occurred in the protein complex.
Proteins with redesigned interaction interface.
Interactions of NColE7 and its N‐terminal mutants with the Im7 protein in bacteria
According to in vitro DNA cleavage assays, the nuclease activity of the mutant proteins containing single (W464A), double (K458A/W464A), or triple (T454A/K458A/W464A) amino acid changes (all including W464A mutation) drastically decreased in comparison to the wild‐type enzyme or to the TK point mutant (T454A/K458A) in which W464 was not affected. Nevertheless, all the point mutants remained cytotoxic: the coexpression of Im7 was necessary for their production, demonstrating their mutual interaction with the inhibitor.1, 2
In contrast, the N‐terminally truncated NColE7 mutants all could be overexpressed without the coexpression of Im7.24, 25 The Im7 binding of the latter proteins was therefore, studied in vitro. The genes of each NColE7 mutant were also constructed to express a GST (glutathione‐S‐transferase) fusion protein together with Im7. Then, GST pull‐down assays with the N‐terminally truncated NColE7 mutants were carried out in the presence of Im7 protein. Figure 3 shows that all mutants were able to interact with Im7, except for the shortest ΔN69‐NColE7 protein, although the latter still contains the intact Im7 binding region.
Figure 3.
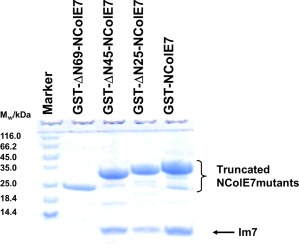
Im7 binding of truncated NColE7 mutants. The result of the GST pull‐down assay showing the interaction of Im7 with N‐terminally truncated NColE7 mutants in their GST‐tagged forms. The first lane contains the Thermo Scientific™ Unstained Protein Molecular Weight Marker. In lanes 3–5 the NColE7 proteins were shown to bind Im7 under the conditions of the GST‐affinity purification, while the shortest mutant protein in lane 2 did not.
Correlation between the Im7 and the DNA binding of NColE7 mutants
The inhibitor and the substrate bind to overlapping regions of NColE7. Therefore, the comparison of these interactions provides information on the relative binding affinity of Im7 and DNA. The DNA binding of the TKW point mutant and N‐terminally truncated NColE7 proteins have been determined previously by gel mobility shift assays.1, 2, 24, 25 The point mutant and the ΔN4‐NColE7 truncated protein showed almost the same affinity toward DNA as NColE7, but the shorter proteins gradually lost their DNA binding ability. It is intriguing that the ΔN45‐NColE7 protein could not bind DNA,24 but it bound Im7. This phenomenon was further studied using a sensitive method, such as the flow linear dichroism (FLD) spectroscopy. FLD of a double‐stranded CT‐DNA sample yields a characteristic negative signal at the absorbance maximum of the DNA bases (ca. 260 nm) since the transition moments of the base π→π* transitions are located in the plane of the bases, and these are oriented orthogonally with respect to the orientational axis of DNA double helix.39 Although the nuclease activity was completely suppressed by the addition of EDTA, a large impact of small amounts of NColE7 on DNA LD signal was observed due to DNA‐binding. In contrast, the addition of ΔN45‐NColE7 did not affect the FLD spectrum of the DNA (Figure 4), supporting the previous observations by gel electrophoresis.24
Figure 4.
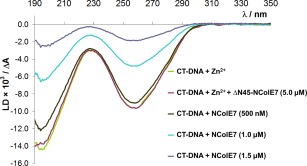
DNA binding of ΔN45‐NColE7 mutant. Flow linear dichroism spectra of a 130 μM (referred to base pairs) CT‐DNA sample containing Zn2+‐ions (50 µM) in the presence and absence of ΔN45‐NColE7 (5 µM). The spectra are compared with those of the same DNA sample incubated with increasing amounts of NColE7 (0.5–1.5 µM) in the presence of 60 µM EDTA to avoid DNA cleavage by the active enzyme.
The difference in the Im7‐ and DNA‐binding features of ΔN45‐NColE7 raised the question, whether a structural activation can occur in this severely truncated, partially disordered NColE7 variant on the binding to Im7.
Im7‐binding alters the solution structure of the intrinsically disordered NColE7 point mutants
Previously, we have shown that several N‐terminal NColE7 mutants have distinct features from NColE7. Among the T454A and/or K458A and/or W464A NColE7 mutants, those including the latter point mutation undergo severe structural changes in aqueous solutions according to their SRCD spectra. In the series of the NColE7, TK, TKW, KW, and W mutants, two distinct types of spectra were identified: the first two proteins showed the same native‐like spectral pattern, while the last three resulted in spectra characteristic for proteins with a large fraction of unordered structure.1, 2
Figure 5 shows the effect of the interaction with the Im7 protein on the spectrum of the TKW mutant. It is clear that the solution structure of the mutant (similarly to those of TW and W mutants—not shown) is recovered by binding of the Im7 protein, since the experimental SRCD spectrum of the TKW/Im7 complex is identical to that of the NColE7/Im7 complex, while the mathematical sum of the TKW and Im7 component spectra—assuming that no interaction occurs in the solution—differs significantly both in the intensity and spectral pattern.
Figure 5.
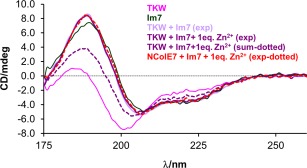
Induction of the native‐like structure of the TKW mutant in solution. Comparison of the experimental SRCD spectra of TKW mutant, NColE7, and Im7 proteins (3.2 × 10−5 M) and the equimolar mixtures of the nucleases and Im7 (1.6 × 10−5 M, each). The calculated spectral sum was constructed from the component spectra according to the initial composition of the measured system—thus, representing a spectrum of a mixture without interaction between the TKW mutant and Im7.
N‐terminally truncated variants of NColE7 and their interaction with Im7
The four N‐terminal amino acids (446‐KRNK‐449) are required for catalytic activity of NColE7.25, 26 However, they do not affect the overall fold of the protein in the crystal structure.32 The truncation including only these amino acids does not have significant effect on the DNA and Zn2+‐binding properties, as well.25 Conversely, the deletion of longer amino acid strings from the N‐terminus in ΔN25, ΔN45, and ΔN69‐NColE7 mutants gradually abolishes the ability for Zn2+ and DNA binding, and even the Im7 binding capability is lost in the latter protein (see Figure 3).
The shape and intensity of the mean residue molar SRCD spectra of the ΔN25, ΔN45‐NColE7 truncated mutants significatly differ from that of the native NColE7 [Figure 6(A)]. The decrease of the spectral intensity suggests a decrease of the helical content, as it is supported by the calculated secondary structure distributions (Table 2). This is, however, against expectations considering that sequences of mainly unordered structure were deleted in the truncated mutants, while the amino acid residues comprising the central helical region remained intact. This indicates that the N‐terminal sequence of NColE7 consisting of ∼40 amino acids stabilizes the NColE7 structure, in agreement with the observations on the effect of the W464A point mutation within this region.
Figure 6.
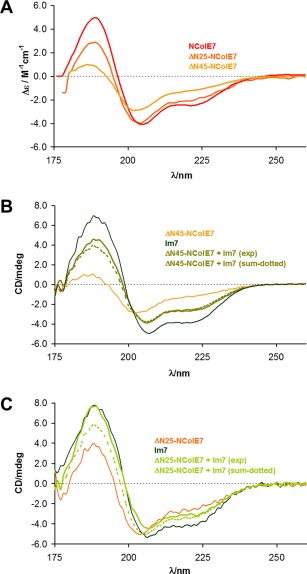
SRCD spectra of the N‐terminally truncated NColE7 mutants. (A) Comparison of the molar SRCD spectra of the ΔN25, ΔN45‐NColE7 truncated mutant proteins and that of the native NColE7. The calculation of the Δε values was based on the average amino acid residue molecular weight for each protein. (B) Experimental spectra of Im7 protein and the ΔN45‐NColE7 mutant (3.2 × 10−5 M, each) and their equimolar mixtures (1.6 × 10−5 M, each). The calculated spectra constructed from the component spectra are also shown for comparison. (C) Comparison of the experimental spectra of Im7 and the ΔN25‐NColE7 mutant proteins (3.2 × 10−5 M, each) and their equimolar mixtures (1.6 × 10−5 M, each). The calculated spectra constructed from the component spectra are also shown.
Table 2.
The fractions of the secondary structure elements calculated from the SRCD spectra of the N‐terminally truncated proteins in the presence and absence of Im7
| Protein | Data | α‐Helix | β‐Sheet | Turn | Unordered | |
|---|---|---|---|---|---|---|
| ΔN25‐NColE7 | SRCDa | CDPro | 0.23 | 0.20 | 0.20 | 0.37 |
| BeStSel | 0.13 | 0.25 | 0.16 | 0.46 | ||
| SRCD (with Im7)b | CDPro | 0.28 | 0.29 | 0.19 | 0.24 | |
| CDNN | 0.26 | 0.28 | 0.20 | 0.26 | ||
| BeStSel | 0.11 | 0.32 | 0.17 | 0.40 | ||
| PDB id: 1M08 (with Zn2+) | 0.43 | 0.17 | 0.08 | 0.32 | ||
| PDB id: 7CEI (with Im7) | 0.37 | 0.15 | 0.13 | 0.35 | ||
| ΔN45‐NColE7 | SRCDa | CDPro | 0.15 | 0.29 | 0.22 | 0.33 |
| BeStSel | 0.12 | 0.27 | 0.17 | 0.43 | ||
| SRCD (with Im7)a,b | CDPro | 0.20 | 0.20 | 0.20 | 0.31 | |
| BeStSel | 0.14 | 0.25 | 0.19 | 0.42 | ||
| PDB id: 1M08 (with Zn2+) | 0.45 | 0.15 | 0.07 | 0.33 | ||
| PDB id: 7CEI (with Im7) | 0.37 | 0.15 | 0.14 | 0.34 | ||
| ΔN69‐NColE7 | PDB id: 1M08 (with Zn2+) | 0.34 | 0.21 | 0.08 | 0.37 | |
In comparison the data derived from in silico truncated crystal structures as the appropriate fractions of NColE7, are also given.
The evaluation of the spectra of low intensity by CDNN program38 yielded unreliable data with a sum of fractions significantly higher than 1.00.
The mathematical difference of the spectra of the mixtures and individual proteins were applied in the secondary structure determination (see the footnote of Table 1).
As shown in Figure 6(B), there is only a slight difference between the experimental SRCD spectra of the ΔN45‐NColE7 mutant obtained in the presence and absence of Im7. Thus, while this mutant does bind Im7, it does not refold, suggesting that the binding to Im7 occurs through the numerous (14) positively charged side chains in the iteracting region with partially helical structure. It is likely that this interaction occurs without extending the structure stabilization effect to the rest of the protein.
At the same time, the spectra of ΔN25‐NColE7 clearly demonstrate the significant structural change caused by its interaction with Im7, as the measured spectrum of the protein–protein complex differs from the mathematical sum of the protein component spectra [Figure 6(C)].
Discussion
Solution structure of native NColE7
Circular dichroism spectroscopy is a useful tool to obtain rough information about the solution structure of proteins by evaluation the spectra concerning the secondary structure elements. Independently of the programs applied,33, 34, 35, 36, 37, 38 our calculations revealed slightly decreased helical content and overestimated amounts of turns for NColE7, or turns and β‐strands for Im7 in comparison to the crystal structures (Table 1). This may be attributed to the uncertainty of the analysis of secondary structure distribution. As there is no crystal structure available for the apo NColE7 in the Protein Data Bank (PDB), we performed solution studies to reveal whether the metal ion binding had any influence on the NColE7 SRCD spectra. No significant differences were observed compared to the apo protein—in contrary to the dramatic structural change of the zinc finger proteins with similar ββα‐metal ion binding motif on removal of the Zn2+‐ions.40, 41, 42
In case there is no structural change on protein–protein interaction, the intensities of individual CD signals are additive. This is predicted for NColE7–Im7 complex by the available crystal structures. Therefore, it is possible to analyse the difference spectra obtained by subtraction of the spectral contribution of one component from the experimental curve of the mixture. The results of such calculations revealed slight changes in the secondary structure distribution of both NColE7 and Im7 proteins (Table 1). The mutual stabilization of the more flexible protein regions can be the reason for the slight increase of the fraction of the β‐sheet and decrease in the fraction of unordered structure of either NColE7 or Im7, in contrast to the crystal data.
Mutations of NColE7 result in structural changes
Since there are numerous crystal structures on NColE7 reporting that the structure of the various mutants in complex with Im7 or DNA are all virtually the same, it was suprising to observe significant structural changes on N‐terminal modifications. An intriguing property of NColE7 is that while its active centre is situated at the C‐terminus, the N‐terminus also influences the catalytic activity, even though it is located on the surface of the protein, and is not directly involved in the formation of well defined secondary structure elements. The structural changes caused by the mutations within the N‐terminal sequence led us to the recognition of an important feature of this protein: the native‐like structure of partially disordered mutants can be induced by binding to its substrate or to its inhibitor protein. Table 3 shows the summary of the Im7 and DNA binding properties, as well as the cytotoxic and in vitro catalytic activity of the point mutant and N‐terminally truncated NColE7 proteins (Figure 1). These data demonstrate that the gradual removal of amino acids from the N‐terminus abolished the nuclease activity first in the ΔN4‐NColE7 mutant,25 then the DNA binding affinity decreased for the ΔN25‐NColE7 mutant.25 The ΔN45‐NColE7 mutant already could not interact with DNA24 as also comfirmed by the flow linear dichroism method here (Figure 4). In contrast, ΔN45‐NColE7 could still bind Im7 (Figure 3). These observations accord with the stronger interaction of NColE7 with the inhibitory protein than with the substrate DNA. To reveal the structural consequences of this interaction, maintained even in the severely truncated disordered mutant, we studied the Im7‐binding by SRCD spectroscopy in detail.
Table 3.
Effect of N‐terminal mutations on the features of the NColE7 protein
| NColE7 variant | Im7 binding | DNA binding | Cytotoxicity | In vitro DNA cleaving activity | Solution structure |
|---|---|---|---|---|---|
| NColE72, 3, 13, 15, 26 | + | + | + | +++ | |
| W464A‐mutants2, 3 | + | + | + | + | Partially disordered |
| T454A/K458A3 | + | + | + | + | NColE7‐like |
| ΔN4‐NColE725 | + | + | − | − | NColE7‐like |
| ΔN25‐NColE725 | + | + | − | − | Partially disordered |
| ΔN45‐NColE724 | + | − | − | − | Partially disordered |
| ΔN69‐NColE724 | − | − | − | − | Partially disordered |
Im7 binding induces the folding of the truncated mutants
The Zn2+‐binding site of NColE7 is preorganized2 thus, a change in the affinity for the metal ion indicates severe conformational changes in the C‐terminal HNH‐motif. The decreased metal binding affinity by the N‐terminal truncation in ΔN25, ΔN45, and ΔN69‐NColE7 mutants suggest that a part of the protein structure is altered. Indeed, the analysis of the CD spectra of both the ΔN25 and ΔN45‐NColE7 truncated mutants [Figure 6(A)] revealed an increased disorder in the structure compared to the expected one, based on the intact NColE7 structure (Tables 1 and 2).
The structure of Im7 is unaffected by the interaction with NColE7. Therefore, we supposed that any singificant changes in the CD spectra of the Im7 complexes of NColE7 mutants can be attributed to the induced folding of the nucleases. Thus, to calculate the changes in the secondary structure we subtracted the spectrum of Im7 from the spectra of its complexes with the truncated mutants and analyzed the difference spectra assigned to the latter proteins. The secondary structure distribution of the truncated proteins shown in Figure 7, revealed that the structures are at least partially—ΔN25‐NColE7 to a larger, and ΔN45‐NColE7 to a lesser extent—refolded due to the interaction with the immunity protein.
Figure 7.
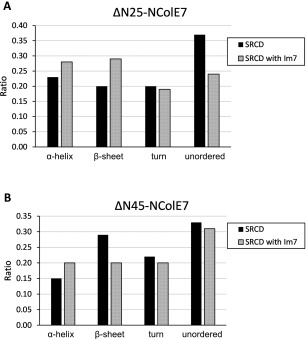
The comparison of the induced folding in the ΔNX‐NColE7–Im7 systems: change in the distribution of secondary structure elements of ΔNX‐NColE7 mutants as a consequence of the interaction with Im7 protein as calculated by the CDPro program package. The data on the NColE7 variants in complex with Im7 were obtained from the difference spectra of the complex and Im7 with the asumption that the structure of Im7 is stable and does not significantly change on complex formation. (A) ΔN25‐NColE7 mutant; (B) ΔN45‐NColE7 mutant.
Notably, the W464 residue belongs to the deleted amino acid sequences in ΔN25 and ΔN45‐NColE7. W464 (together with F499, W500, L509, F513) is a key residue in forming the hydrophobic core of the protein. These interactions are extended toward the N‐terminal chain (L465, P475, V476, P477) and also the HNH motif (V563).32 The disturbance of this hydrophobic patch results in severe structural consequences.
Structural activation as reflected by metal binding
The analysis of the SRCD spectra of the point mutants revealed a drastically decreased fraction of the ordered secondary structures on W464A mutation. Typically, the fractions of the secondary elements calculated by the CDPro program package37 comprise 16% α‐helix 22% β‐sheet, 21% turn, and 41% of unordered sequence, while BeStSel34 provides 11% α‐helix 20% β‐sheet, 18% turn, and 51% of unordered sequence for these proteins. The changes in the structure mirror the significantly decreased metal ion binding affinity of these mutants,2 which was shown to be recovered in their Im7 complexes possessing a native‐like SRCD spectral pattern (Figure 5). Although the SRCD spectra do not directly reflect the tertiary structure of the proteins, the Zn2+‐binding also became comparable to the native protein.2 This suggests that the W464 mutants have the same overall structure as the NColE7 in their Im7 complexes.
Comparison of the disordered structures of W464A mutant and N‐terminally truncated mutants—structural role of the N‐terminal loop
The fact that Im7 binding recovered the Zn2+ binding capability of the point mutants—including changes at W464 site—suggested that their interaction with Im7 is strong.2 It is well‐known that Im7 inhibits the nuclease activity of NColE7 by interacting with the central helices of the latter protein.16, 29 These regions of NColE7 otherwise provide strong DNA binding affinity.15, 21, 27 Together, these results suggest that the folding into the native‐like tertiary structure promoted by the interaction with Im7 is not strongly dependent on the W464 residue but rather on the presence of the N‐terminal chain. In this context, the structure of the W464 mutants became more unordered than that of the ΔN25‐NColE7 mutant, which is surprising as the W464 residue belongs to the deleted 25 amino acid sequence. The latter residue is thus most probably responsible for the stabilization of fine structure of the active centre through interactions within the hydrophobic pocket. This also points out that it is difficult to predict the effect of mutations in terms of their effect on protein structure or function. Performing a virtual alanine scan—changing residues 450–470 to Ala one by one—and analysing these NColE7 mutants by semiempirical quantum chemical computations, the optimized structures did not differ significantly from the wild type protein.1 Similarly, the SDM server for prediction of the effect of mutations—based on the calculated ΔΔG value of −0.90 kcal/mol for W464A NColE7 mutant—suggested only slight destabilization for W464A mutant without any functional effect,43 in contrast to the solution structure of the purified protein and its decreased catalytic activity.1 The more recent DUET server calculated ΔΔG of −2.53 kcal/mol for the above mutant.44 Detailed computational analysis of the destabilization effect of the mutations in the N‐terminal sequence of NColE7 also assigned high contribution to W464 amino acid.1
Consequences of structural activation on the possible application of NColE7 mutants in artificial nucleases
While the overall secondary structure composition of the mutant NColE7 proteins is restored by Im7, and similar changes occur in the presence of DNA,1 the catalytic activity is drastically reduced or lost compared to the native enzyme. This suggests that the main function of the N‐terminal sequence is not only to maintain the overall structure, but it directly participates in the catalytic process and in the stabilization of the fine structure of the functional active center. Such a property provides the possibility of achieving allosteric control in designed artificial nucleases.45, 46 Similar allosteric regulation has recently been suggested, for example, the c‐Src kinase, based on the interdomain interactions restricting the conformational flexibility of the N‐terminal intrinsically disordered region47 or for the Pin1 peptidyl‐prolyl isomerase.48
Significance of the induced protein folding
Recently increasing attention is being favoured to the role of intrinsically disordered regions in proteins,49, 50 including the investigation of disease related mutations or the development of potential drugs reordering a malfunctional mutated protein.51, 52, 53, 54, 55 Accordingly, studies should extend toward the activation mechanisms through rather general structure recruitment to understand the functions of these proteins. Our findings on NColE7 mutants exemplify how intermolecular interactions trigger important structural stabilization of disordered proteins in solutions leading to functional entities. As disordered proteins are difficult to crystallize on their own, the standard procedure involves the cocrystallization with substrates, inhibitors or other interacting partners.56 The results easily draw focus on the induced order in structures, overlooking or underestimating the functional significance of originally disordered regions. Therefore, it is essential to supplement these data by spectroscopic techniques (e.g., CD or NMR) sensitive to the solution structure of the individual interacting partners and to the structural changes on their interactions,1, 2, 57 as well as to the dynamic nature of the protein structure.58
A related example is the Im3 inhibitor, able to induce the native‐like structure of a disordered Colicin E3 ribonuclease mutant.4 The proof for this was primarily provided by CD and NMR spectroscopic methods,3, 4 as the X‐ray data were only available for the Im3 complexes of both the wild‐type and the mutant enzymes4, 59, 60 with essentially the same structure, or with the substrate RNA.61
Induced folding is an essential way of protein regulation.62 Such phenomenon was earlier observed for the truncated staphylococcal nuclease. This mutant has a disordered structure, that is restored by the binding of 3′,5′‐bisphospho‐2′‐deoxythymidine inhibitor.63 The ADPglucose pyrophosphorylase can also be reactivated after N‐terminal truncation.64 Furthermore, the structural disorder recognition was shown to be elementary feature of RNA and protein chaperone,65 or antibody and disordered antigen interactions.66 In the NF‐κB signaling module one of the subunits has a 30 amino acids long disordered C‐terminal unit in the DNA‐bound state, but adopts an ordered helical structure when binding to the IκBα inhibitor.67 The disordered part of the p53 tumor suppressor protein also initiated intense research, pointing to importance in phosphorylation, and cancer related mutations.53, 68, 69, 70
The above results and statements draw attention to the importance in general of investigation of proteins in solution in the absence of their inhibitors, substrate analogue compounds, or other binding partners, as intermolecular interactions often cause structural stabilization not present in the isolated protein.
Materials and Methods
Construction of the genes and the expression and purification of the proteins
The pQE70 plasmid containing the gene of NColE7 and the Im7 immunity protein was a generous gift from Prof. K.‐F. Chak, Institute of Biochemistry and Molecular Biology, National Yang Ming University, Taipei, Taiwan.5 The genes of the truncated mutants were amplified in PCR from this template as described earlier.24 Because of the cytotoxicity of the NColE7 mutants containing the T454A and/or K458A and/or W464A changes, the genes of these proteins were cloned together with the DNA fragment encoding for Im7 protein.1, 24 All the PCR fragments were inserted into a pGEX‐6P‐1 vector (GE Healthcare) providing an N‐terminal glutathione‐S‐transferase (GST) affinity fusion tag. E. coli DH10B and E. coli BL21 (DE3) cells were transformed for DNA and protein production, respectively. The detailed conditions of the protein expression and purification procedures were described elsewhere.24, 26 For the spectroscopic studies the GST fusion tag was cleaved off with Human rhinovirus C3 protease71—sold as PreScission protease by GE Healthcare. The Im7 protein was separated from the mutant proteins by adjusting the pH to 3.0 and performing HPLC separation on a Sepharose SP FF 16/10 cation exchange column. The fractions of the nuclease mutants and Im7 were concentrated by Amicon ultrafilter with 5 kDa cutoff, and the buffer was exchanged to 20 mM HEPES, pH = 7.7. The sequences of the purified mutant proteins are shown in Figure 1 in comparison with the wild‐type NColE7.
GST pull‐down assay
GST‐tagged truncated NColE7 variants, overexpressed together with the Im7 immunity protein within the bacterial cells, were obtained in PBS (pH = 7.3) buffer, and then subjected to pull‐down assay using Glutathione Sepharose 4B resin (GE Healthcare). The unbound proteins were washed away and the bound proteins were eluted from the resin by reduced glutathione solution of increasing concentration. The obtained fractions were analyzed by standard SDS gel electrophoresis using 12% polyacrylamide gel and Coomassie staining.72
SRCD spectroscopic measurements
The Synchrotron Radiation Circular Dichroism (SRCD) spectra were recorded at the CD1 beamline of the storage ring ASTRID at the Institute for Storage Ring Facilities (ISA), University of Aarhus, Denmark.73, 74 Camphor‐sulfonic acid served as a calibration material for the instrument. All spectra were recorded with 1 nm steps and a dwell time of 2 s per step, using a 100.4 μm quartz cell (SUPRASIL, Hellma GmbH, Germany), in the wavelength range of 175–260 nm. The concentration of the protein solutions was adjusted to 3.2 × 10−5 M in 10 mM HEPES, pH = 7.7. The protein mixtures contained equimolar amounts of the nuclease mutants and the Im7 protein, each in 1.6 × 10−5 M concentration. From raw spectra the corresponding baselines (water or the appropriate buffer solution) were subtracted. The experimental CD intensities are reported in mdeg units. The CD signal was converted into differential molar absorbance units given with respect to the concentration of the backbone peptide groups for the analysis of secondary structure distribution by simultaneous application of a variety of programs published in the literature.33, 34, 35, 36, 37, 38
Linear dichroism spectroscopy
Flow linear dichroism (FLD) spectra were measured on a Jasco‐815 CD spectrometer equipped for linear dichroism spectroscopy, using a microvolume Couette flow device75 with an annular gap of 0.25 mm giving a combined light path of 0.5 mm. An additional quartz lens was mounted to allow for focusing onto the sample in the Couette cell, which was positioned as close to the photomultiplier as possible in the J‐815 sample compartment. Water circulation through the metal blocks of the flow device thermostated the cell to 298 K. The sample volume was 70 µL and 3000 rpm rotation was applied. The optical bandwidth was 1 nm and the spectra were recorded in continuous mode between 190 nm and 400 nm with 50 nm/min scanning speed, 1 s integration time, 0.5 nm data pitch, and with 15 L/min nitrogen flow. Protein‐DNA binding was studied with a 130 μM (final concentration calculated for base pairs) calf thymus DNA (CT‐DNA) sample. The mixture contained 17 mM KH2PO4, 2.4 mM HEPES (the pH of both buffers was adjusted to 7.7), 50 μM Zn2+‐ions and 0–5 μM protein. 60 μM EDTA was added to the NColE7 containing samples to assure that only the DNA binding effect of the enzyme is observed. The solutions were incubated for 10 min before recording the spectra. The incubation time did not influence the results.
Acknowledgment
K.B. and R.K.B. thank to the Hungarian Scholarship Board. E.N. is currently an International Research Fellow of the Japan Society for the Promotion of Science.
50‐75‐Word Statement, Written for Broader Audience: So far, all the 11 crystal structures of WT and mutant NColE7 suggested that this protein has a stable structure not influenced by modifications. Using SRCD spectroscopy we demonstrate that a disorder does occur on certain mutations, and the native‐like structure can be induced by inhibitor binding. This case study gives insight into how the structural disorder may get hidden in experimental conditions involving a mutant protein and its interacting partners.
References
- 1. Németh E, Körtvélyesi T, Kožíšek M, Thulstrup PW, Christensen HEM, Asaka MN, Nagata K, Gyurcsik B (2014) Substrate binding activates the designed triple mutant of the colicin E7 metallonuclease. J Biol Inorg Chem 19:1295–1303. [DOI] [PubMed] [Google Scholar]
- 2. Németh E, Kožíšek M, Schilli GK, Gyurcsik B (2015) Preorganization of the catalytic Zn2+‐binding site in the HNH nuclease motif—a solution study. J Inorg Biochem 151:143–149. [DOI] [PubMed] [Google Scholar]
- 3. Walker D, Lancaster L, James R, Kleanthous C (2004) Identification of the catalytic motif of the microbial ribosome inactivating cytotoxin colicin E3. Protein Sci 13:1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papadakos G, Sharma A, Lancaster LE, Bowen R, Kaminska R, Leech AP, Walker D, Redfield C, Kleanthous C (2015) Consequences of inducing intrinsic disorder in a high‐affinity protein−protein interaction. J Am Chem Soc 137:5252–5255. [DOI] [PubMed] [Google Scholar]
- 5. Chak KF, Kuo WS, Lu FM, James R (1991) Cloning and characterization of the ColE7 plasmid. J Gen Microbiol 137:91–100. [DOI] [PubMed] [Google Scholar]
- 6. Liao CC, Hsia KC, Liu YW, Leng PH, Yuan HS, Chak KF (2001) Processing of DNase domain during translocation of colicin E7 across the membrane of Escherichia coli. Biochem Biophys Res Commun 284:556–562. [DOI] [PubMed] [Google Scholar]
- 7. Cheng YS, Shi Z, Doudeva LG, Yang WZ, Chak KF, Yuan HS (2006) High‐resolution crystal structure of a truncated ColE7 translocation domain: implications for colicin transport across membranes. J Mol Biol 356:22–31. [DOI] [PubMed] [Google Scholar]
- 8. Mora L, de Zamaroczy M (2014) In vivo processing of DNase colicins E2 and E7 is required for their import into the cytoplasm of target cells. PLoS One 9:e96549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Orlowski J, Bujnicki JM (2008) Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res 36:3552–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eastberg JH, Eklund J, Monnat R, Stoddard BL (2007) Mutability of an HNH nuclease imidazole general base and exchange of a deprotonation mechanism. Biochemistry 46:7215–7225. [DOI] [PubMed] [Google Scholar]
- 11. Mehta P, Katta K, Krishnaswamy S (2004) HNH family subclassification leads to identification of commonality in the His‐Me endonuclease superfamily. Protein Sci 13:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang H, Yuan HS (2007) The conserved asparagine in the HNH motif serves an important structural role in metal finger endonucleases. J Mol Biol 368:812–821. [DOI] [PubMed] [Google Scholar]
- 13. Ku W, Liu Y, Hsu Y, Liao C, Liang P, Yuan H, Chak K (2002) The zinc ion in the HNH motif of the endonuclease domain of colicin E7 is not required for DNA binding but is essential for DNA hydrolysis. Nucleic Acids Res 30:1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doudeva LG, Huang H, Hsia KC, Shi Z, Li CL, Shen Y, Cheng CL, Yuan HS (2006) Crystal structural analysis and metal‐dependent stability and activity studies of the ColE7 endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+ Protein Sci 15:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sui MJ, Tsai LC, Hsia KC, Doudeva LG, Ku WY, Han GW, Yuan HS (2002) Metal ions and phosphate binding in the H‐N‐H motif: crystal structures of the nuclease domain of ColE7/Im7 in complex with a phosphate ion and different divalent metal ions. Protein Sci 11:2947–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shen BW, Landthaler M, Shub DA, Stoddard BL (2004) DNA binding and cleavage by the HNH homing endonuclease I‐HmuI. J Mol Biol 342:43–56. [DOI] [PubMed] [Google Scholar]
- 17. Kriukiene E, Lubiene J, Lagunavicius A, Lubys A (2005) MnlI ‐ The member of HNH subtype of Type IIS restriction endonucleases. Biochim Biophys Acta 1751:194–204. [DOI] [PubMed] [Google Scholar]
- 18. Scholz SR, Korn C, Bujnicki JM, Gimadutdinow O, Pingoud A, Meiss G (2003) Experimental evidence for a ββα‐Me‐finger nuclease motif to represent the active site of the caspase‐activated DNase. Biochemistry 42:9288–9294. [DOI] [PubMed] [Google Scholar]
- 19. Cheng YS, Hsia KC, Doudeva LG, Chak KF, Yuan HS (2002) The crystal structure of the nuclease domain of colicin E7 suggests a mechanism for binding to double‐stranded DNA by the H‐N‐H endonucleases. J Mol Biol 324:227–236. [DOI] [PubMed] [Google Scholar]
- 20. Wang YT, Yang WJ, Li CL, Doudeva LG, Yuan HS (2007) Structural basis for sequence‐dependent DNA cleavage by nonspecific endonucleases. Nucleic Acids Res 35:584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsia KC, Chak KF, Liang PH, Cheng YS, Ku WY, Yuan HS (2004) DNA binding and degradation by the HNH protein ColE7. Structure 12:205–214. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y‐T, Wright JD, Doudeva LG, Jhang H‐C, Lim C, Yuan HS (2009) Redesign of high‐affinity nonspecific nucleases with altered sequence preference. J Am Chem Soc 131:17345–17353. [DOI] [PubMed] [Google Scholar]
- 23. Shi Z, Chak KF, Yuan HS (2005) Identification of an essential cleavage site in ColE7 required for import and killing of cells. J Biol Chem 280:24663–24668. [DOI] [PubMed] [Google Scholar]
- 24. Gyurcsik B, Czene A, Jankovics H, Jakab‐Simon IN, Slaska‐Kiss K, Kiss A, Kele Z (2013) Cloning, purification and metal binding of the HNH motif from colicin E7. Protein Express Purif 89:210–218. [DOI] [PubMed] [Google Scholar]
- 25. Czene A, Németh E, Zóka IG, Jakab‐Simon NI, Körtvélyesi T, Nagata K, Christensen HEM, Gyurcsik B (2013) The role of the N‐terminal loop in the function of the colicin E7 nuclease domain. J Biol Inorg Chem 18:309–321. [DOI] [PubMed] [Google Scholar]
- 26. Németh E, Körtvélyesi T, Thulstrup PW, Christensen HEM, Kožíšek M, Nagata K, Czene A, Gyurcsik B (2014) Fine tuning of the catalytic activity of colicin E7 nuclease domain by systematic N‐terminal mutations. Protein Sci 23:1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chak KF, Safo MK, Ku WY, Hsieh SY, Yuan HS (1996) The crystal structure of the immunity protein of colicin E7 suggests a possible colicin‐interacting surface. Proc Natl Acad Sci USA 93:6437–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS (1999) The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 7:91–102. [DOI] [PubMed] [Google Scholar]
- 29. Kolade OO, Carr SB, Kuhlmann UC, Pommer A, Kleanthous C, Bouchcinsky CA, Hemmings AM (2002) Structural aspects of the inhibition of DNase and rRNase colicins by their immunity proteins. Biochimie 84:439–446. [DOI] [PubMed] [Google Scholar]
- 30. Papadakos G, Wojdyla JA, Kleanthous C (2012) Nuclease colicins and their immunity proteins. Q Rev Biophys 45:57–103. [DOI] [PubMed] [Google Scholar]
- 31. Meenan NA, Sharma A, Fleishman SJ, Macdonald CJ, Morel B, Boetzel R, Moore GR, Baker D, Kleanthous C (2010) The structural and energetic basis for high selectivity in a high‐affinity protein‐protein interaction. Proc Natl Acad Sci USA 107:10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Czene A, Tóth E, Németh E, Otten H, Poulsen JCN, Christensen HEM, Rulíšek L, Nagata K, Larsen S, Gyurcsik B (2014) A new insight into the zinc‐dependent DNA‐cleavage by the colicin E7 nuclease: a crystallographic and computational study. Metallomics 6:2090–2099. [DOI] [PubMed] [Google Scholar]
- 33. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 34. Micsonai A, Wien F, Kernya L, Lee YH, Goto Y, Réfrégiers M, Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci USA 112:E3095–E3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89:392–400. [DOI] [PubMed] [Google Scholar]
- 36. Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1:2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260. [DOI] [PubMed] [Google Scholar]
- 38. Böhm G, Muhr R, Jaenicke R (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng 5:191–195. [DOI] [PubMed] [Google Scholar]
- 39. Norden B, Kubista M, Kurucsev T (1992) Linear dichroism spectroscopy of nucleic acids. Q Rev Biophys 25:51–170. [DOI] [PubMed] [Google Scholar]
- 40. Sakai‐Kato K, Umezawa Y, Mikoshiba K, Aruga J, Utsunomiya‐Tate N (2009) Stability of folding structure of Zic zinc finger proteins. Biochem Biophys Res Commun 384:362–365. [DOI] [PubMed] [Google Scholar]
- 41. Negi S, Imanishi M, Sasaki M, Tatsutani K, Futaki S, Sugiura Y (2011) An arginine residue instead of a conserved leucine residue in the recognition helix of the finger 3 of Zif268 stabilizes the domain structure and mediates DNA binding. Biochemistry 50:6266–6272. [DOI] [PubMed] [Google Scholar]
- 42. Rich AM, Bombarda E, Schenk AD, Lee PE, Cox EH, Spuches AM, Hudson LD, Kieffer B, Wilcox DE (2012) Thermodynamics of Zn2+ binding to Cys2His2 and Cys2HisCys zinc fingers and a Cys4 transcription factor site. J Am Chem Soc 134:10405–10418. [DOI] [PubMed] [Google Scholar]
- 43. Worth CL, Preissner R, Blundell TL (2011) SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res 39:W215–W222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pires DEV, Ascher DB, Blundell TL (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42:W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gyurcsik B, Czene A (2011) Towards artificial metallonucleases for gene therapy: recent advances and new perspectives. Future Med Chem 3:1935–1966. [DOI] [PubMed] [Google Scholar]
- 46. Németh E, Schilli GK, Nagy G, Hasenhindl C, Gyurcsik B, Oostenbrink C (2014) Design of a colicin E7 based chimeric zinc‐finger nuclease. J Comput Aided Mol Des 28:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maffei M, Arbesú M, Le Roux A‐L, Amata I, Roche S, Pons M (2015) The SH3 domain acts as a scaffold for the N‐terminal intrinsically disordered regions of c‐Src. Structure 23:893–902. [DOI] [PubMed] [Google Scholar]
- 48. Wang X, Mahoney BJ, Zhang M, Zintsmaster JS, Peng JW (2015) Negative regulation of peptidyl‐prolyl isomerase activity by interdomain contact in human Pin1. Structure 23:2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Forman‐Kay JD, Mittag T (2013) From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure 21:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM (2014) Classification of intrinsically disordered regions and proteins. Chem Rev 114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ochi T, Wu Q, Chirgadze DY, Grossmann JG, Bolanos‐Garcia VM, Blundell TL (2012) Structural insights into the role of domain flexibility in human DNA ligase IV. Structure 20:1212–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gao M, Zhou H, Skolnick J (2015) Insights into disease‐associated mutations in the human proteome through protein structural analysis. Structure 23:1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Joerger AC, Bauer MR, Wilcken R, Baud MGJ, Harbrecht H, Exner TE, Boeckler FM, Spencer J, Fersht AR (2015) Exploiting transient protein states for the design of small‐molecule stabilizers of mutant p53. Structure 23:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nadvi NA, Michie KA, Kwan AH, Guss JM, Trewhella J (2016) Clinically linked mutations in the central domains of cardiac myosin‐binding protein C with distinct phenotypes show differential structural effects. Structure 24:105–115. [DOI] [PubMed] [Google Scholar]
- 55. Chiang CH, Grauffel C, Wu LS, Kuo PH, Doudeva LG, Lim C, Shen CK, Yuan HS (2016) Structural analysis of disease‐related TDP‐43 D169G mutation: linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci Rep 6:21581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bukowska MA, Grütter MG (2013) New concepts and aids to facilitate crystallization. Curr Opin Struct Biol 23:409–416. [DOI] [PubMed] [Google Scholar]
- 57. Kumar A, Balbach J (2015) Real‐time protein NMR spectroscopy and investigation of assisted protein folding. Biochim Biophys Acta 1850:1965–1972. [DOI] [PubMed] [Google Scholar]
- 58. Park SJ, Kostic M, Dyson HJ (2011) Dynamic interaction of Hsp90 with its client protein p53. J Mol Biol 411:158–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carr S, Walker D, James R, Kleanthous C, Hemmings AM (2000) Inhibition of a ribosome‐inactivating ribonuclease: the crystal structure of the cytotoxic domain of colicin E3 in complex with its immunity protein. Structure 8:949–960. [DOI] [PubMed] [Google Scholar]
- 60. Soelaiman S, Jakes K, Wu N, Li C, Shoham M (2001) Crystal structure of colicin E3: implications for cell entry and ribosome inactivation. Mol Cell 8:1053–1062. [DOI] [PubMed] [Google Scholar]
- 61. Ng CL, Lang K, Meenan NAG, Sharma A, Kelley AC, Kleanthous C, Ramakrishnan V (2010) Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nat Struct Mol Biol 17:1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Buchner J, Kessler H (2014) Protein folding by interaction. Structure 22:936–937. [DOI] [PubMed] [Google Scholar]
- 63. Flanagan JM, Kataoka M, Shortle D, Engelman DM (1992) Truncated staphylococcal nuclease is compact but disordered. Proc Natl Acad Sci USA 89:748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bejar CM, Ballicora MA, Iglesias AA, Preiss J (2006) ADPglucose pyrophosphorylase's N‐terminus: structural role in allosteric regulation. Biochem Biophys Res Commun 343:216–221. [DOI] [PubMed] [Google Scholar]
- 65. Tompa P, Csermely P (2004) The role of structural disorder in the function of RNA and protein chaperones. Faseb J 18:1169–1175. [DOI] [PubMed] [Google Scholar]
- 66. MacRaild CA, Richards JS, Anders RF, Norton RS (2016) Antibody recognition of disordered antigens. Structure 24:148–157. [DOI] [PubMed] [Google Scholar]
- 67. Huxford T, Ghosh G (2009) A structural guide to proteins of the NF‐kappaB signaling module. Cold Spring Harb Perspect Biol 1:a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR (2008) Structure of tumor suppressor p53 and its intrinsically disordered N‐terminal transactivation domain. Proc Natl Acad Sci USA 105:5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ithuralde RE, Turjanski AG (2016) Phosphorylation regulates the bound structure of an intrinsically disordered protein: the p53‐TAZ2 case. PLoS One 11:e0144284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ganguly D, Chen J (2015) Modulation of the disordered conformational ensembles of the p53 transactivation domain by cancer‐associated mutations. PLoS Comput Biol 11:e1004247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walker PA, Leong LE‐C, Ng PW, Tan SH, Waller S, Murphy D, Porter AG (1994) Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Nat Biotechnol 12:601–605. [DOI] [PubMed] [Google Scholar]
- 72. Sambrook J, Russel DW (2006) Condensed protocols from molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 73. Miles AJ, Hoffmann SV, Tao Y, Janes RW, Wallace BA (2007) Synchrotron radiation circular dichroism (SRCD) spectroscopy: new beamlines and new applications in biology. J. Spectroscopy 21:245–255. [Google Scholar]
- 74. Miles AJ, Janes RW, Brown A, Clarke DT, Sutherland JC, Tao Y, Wallace BA, Hoffmann SV (2008) Light flux density threshold at which protein denaturation is induced by synchrotron radiation circular dichroism beamlines. J Synchrotron Radiat 15:420–422. [DOI] [PubMed] [Google Scholar]
- 75. Marrington R, Dafforn T, Halsall D, Rodger A (2004) Micro‐volume Couette flow sample orientation for absorbance and fluorescence linear dichroism. Biophys J 87:2002–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]