

Abstract
Cornelia de Lange Syndrome (CdLS) encompasses a broad spectrum of phenotypes characterized by distinctive craniofacial abnormalities, limb malformations, growth retardation, and intellectual disability. CdLS spectrum disorders are referred to as cohesinopathies, with ∼70% of patients having a mutation in a gene encoding a core cohesin protein (SMC1A, SMC3, or RAD21) or a cohesin regulatory protein (NIPBL or HDAC8). Notably, the regulatory function of HDAC8 in cohesin biology has only recently been discovered. This Zn2+‐dependent hydrolase catalyzes the deacetylation of SMC3, a necessary step for cohesin recycling during the cell cycle. To date, 23 different missense mutants in the gene encoding HDAC8 have been identified in children with developmental features that overlap those of CdLS. Enzymological, biophysical, and structural studies of CdLS HDAC8 protein mutants have yielded critical insight on compromised catalysis in vitro. Most CdLS HDAC8 mutations trigger structural changes that directly or indirectly impact substrate binding and catalysis. Additionally, several mutations significantly compromise protein thermostability. Intriguingly, catalytic activity in many HDAC8 mutants can be partially or fully restored by an N‐acylthiourea activator, suggesting a plausible strategy for the chemical rescue of compromised HDAC8 catalysis in vivo.
Keywords: birth defect, human genetics, protein crystallography, zinc enzyme, lysine deacetylase
Abbreviations
- CdLS
Cornelia de Lange Syndrome
- HDAC8
human histone deacetylase 8
- MD
molecular dynamics
- TM251
N‐(phenylcarbamothioyl)benzamide.
Introduction
Cornelia de Lange Syndrome (CdLS) is a clinically variable disorder of multiple congenital anomalies consisting of distinctive facial abnormalities, limb defects, growth retardation, and intellectual disability, often accompanied by cardiovascular and central nervous system complications.1, 2, 3, 4 The overall prevalence of CdLS is estimated to be 1.6‐2.2/100,000 births, but this may be an underestimate since mild cases are difficult to recognize.5 Facial features characteristic of CdLS are especially prominent in children, and computerized facial recognition approaches have been developed to aid diagnosis.6, 7
CdLS spectrum disorders have also been referred to as cohesinopathies, since most cases result from genetic defects in cohesin structural or regulatory proteins. Cohesin is a highly‐regulated, multiprotein assembly that ensures cohesion of sister chromatids during the cell cycle, and consists of four core protein components: SMC1A and SMC3 each consist of a long antiparallel coiled‐coil with a hinge domain at one end and an ATPase domain at the other end (SMC1A and SMC3 associate through their hinge domains); RAD21, which binds to the ATPase domains of SMC1A and SMC3; and STAG proteins that associate with RAD21 (Fig. 1).8, 9 The quaternary structure of cohesin is analogous to a hinged bracelet, in which SMC1A and SMC3 correspond to the two halves of the bracelet and RAD21 corresponds to the clasp that locks the bracelet closed. In view of this ring‐like quaternary structure, one accepted model is that cohesin encircles and topologically entraps sister chromatids.9, 10, 11, 12 Human developmental disorders that include and overlap with CdLS have been found to be caused by mutations in genes encoding cohesin structural proteins SMC1A, SMC3, and RAD21, as well as the regulatory proteins NIPBL and the zinc‐dependent deacetylase HDAC8.4
Figure 1.
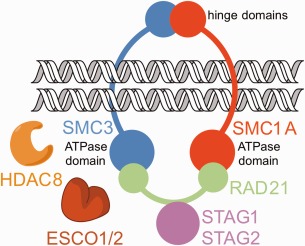
Cohesin. The cohesin complex consists of four main protein components that encircle sister chromatids during cell division: Structural Maintenance of Chromosomes proteins SMC1A and SMC3, Radiation‐Sensitive Mutant 21 protein RAD21, and Stromal Antigen protein(s) STAG1 or STAG2. ESCO1 and ESCO2 catalyze the acetylation of tandem lysine residues K105 and K106 in SMC3, and HDAC8 catalyzes the deacetylation of these lysine residues to ensure efficient cohesin recycling.
Although NIPBL and HDAC8 are not core components of the cohesin complex, these proteins play critical roles in regulating cohesin function. NIPBL is responsible for loading chromatin into the ring‐like cohesin complex during G1 phase.9, 10 Subsequently, cohesion of sister chromatids is established during S phase upon acetylation of SMC3 at tandem lysine residues K105 and K106, catalyzed by the N‐acetyltransferases ESCO1 and ESCO2.13, 14, 15, 16 Recent evidence suggests that acetylation blocks activity at one of the ATPase sites in cohesin that otherwise would facilitate ring opening,17, 18 and the accessory protein Pds5 prevents SMC3 deacetylation.19 It is believed that one role of SMC3 acetylation is to prevent RAD21 dissociation, hence locking sister chromatids within the cohesin ring.20 Dissociation of the cohesin complex is initiated during prophase and completed during anaphase, mediated in part by the cysteine protease separase which cleaves RAD21.9, 11, 21 Cohesin ring opening is a key step in the separation of sister chromatids in cell division.
HDAC8 plays a key role in cohesin function by catalyzing the deacetylation of K105 and K106 of SMC3. In addition to playing roles in regulating dissociation of the cohesin complex, this deacetylation is essential for cohesin recycling for the next cell cycle.22 The specific reaction catalyzed is the hydrolysis of the side chain of trans‐N6‐acetyl‐L‐lysine to yield products L‐lysine and acetate. In recent years, a number of children with clinical features resembling those of CdLS have been found to have loss‐of‐function missense or nonsense mutations in the gene encoding HDAC8 (the gene is designated “HDAC8”).22, 23, 24, 25 To date, 23 different missense mutations in HDAC8 have been reported that yield mutant enzymes exhibiting partial or complete loss of catalytic activity (Fig. 2).22, 23, 24, 25 Ten of these mutant enzymes have been characterized at the molecular level in terms of their three‐dimensional structure, thermostability, and catalytic activity.26, 27 These studies have provided a critical first view of HDAC8 dysfunction in human disease.
Figure 2.
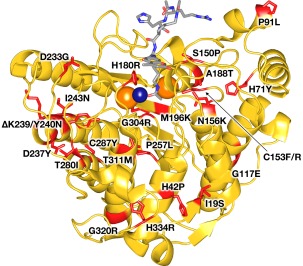
CdLS HDAC8 mutations. A total of 23 different missense mutants have been identified to date in children diagnosed with Cornelia de Lange Syndrome spectrum disorders. Mutations (red) are mapped onto the crystal structure of the Y306F HDAC8‐substrate complex (PDB 2V5W). The active site Zn2+ ion is a dark blue sphere, and 2 K+ ions that regulate HDAC8 activity are orange spheres. The bound substrate is a stick‐figure color‐coded as follows: C = gray, N = blue, O = red.
Here, we review the structure and catalytic mechanism of HDAC8, followed by a discussion of representative mutants to illustrate the range of functional consequences resulting from various mutations. We conclude with discussion of a small molecule activator capable of rescuing catalytic activity in most CdLS HDAC8 mutants studied to date.
Structure and Catalytic Mechanism of HDAC8
The X‐ray crystal structure of HDAC8 was first reported independently by Somoza et al.28 and Vannini and colleagues29 in 2004, revealing an 8‐stranded α/β fold as illustrated in Figure 2. Surprisingly, this fold had first been observed in the crystal structure of the binuclear manganese metalloenzyme arginase, reported 8 years prior by Kanyo and colleagues.30, 31, 32 Arginase catalyzes the hydrolysis of the guanidinium side chain of L‐arginine to form products L‐ornithine and urea. Although the amino acid sequences of arginases and HDACs have diverged significantly (human arginase I and HDAC8 share approximately 12% sequence identity), the conservation of a common fold as well as a metal binding site (the Zn2+ site of HDAC8 corresponds to the site of arginase) suggests divergence from a common metalloenzyme ancestor.32, 33, 34 This structural relationship was anticipated based on the structure determination of a bacterial histone deacetylase‐like protein in 1999 by Finnin et al., which similarly exhibited the arginase fold despite insignificant amino acid sequence identity.35
A summary of the catalytic mechanism of HDAC8 is found in Figure 3. In the structure of native unliganded HDAC8, the Zn2+ coordination polyhedron adopts 5‐coordinate square pyramidal geometry and includes two solvent molecules,36, 37 one of which must be displaced by the side chain carbonyl of acetyllysine (Fig. 4). In comparison, the crystal structure of the unliganded form of the related isozyme HDAC6 (catalytic domain 2) reveals only a single solvent molecule coordinated to Zn2+ in an incomplete square pyramidal coordination polyhedron, with a vacant coordination site ready to accommodate the substrate carbonyl.38 These observations suggest that the Zn2+‐bound solvent molecule closest to Y306 in HDAC8 is coordinated more weakly so as to enable its displacement by the substrate carbonyl.
Figure 3.
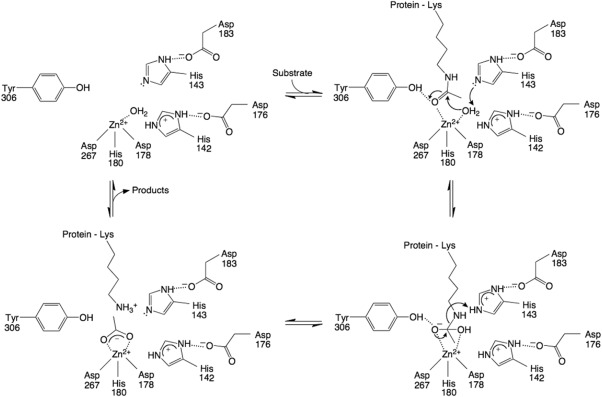
Proposed mechanism of HDAC8. The Zn2+ ion and Y306 orient and activate the substrate carbonyl for nucleophilic attack by a Zn2+‐bound water molecule, assisted by general base H143. The tetrahedral intermediate and its flanking transition states are stabilized by Zn2+ and hydrogen bond interactions with H142, H143, and Y306; H142 remains protonated throughout catalysis and serves as an electrostatic catalyst. H143 subsequently serves as a general acid catalyst to protonate the leaving amino group to enable the collapse of the tetrahedral intermediate. Reprinted with permission from ref. 40. Copyright 2016, American Chemical Society.
Figure 4.
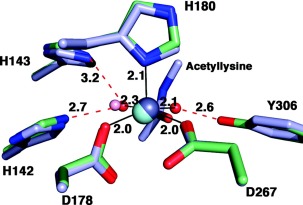
Zn2+ coordination polyhedron in HDAC8. The active site Zn2+ ion of HDAC8 is coordinated with square pyramidal geometry: D178, D267, and two solvent molecules occupy equatorial positions, and H180 occupies the apical position, as observed in the unliganded active site of D101L HDAC8 (PDB 3EW8). Atoms are color coded as follows: C= light green, N = blue, O = red, Zn2+ = gray sphere, water molecules = red spheres. Metal coordination and hydrogen bond interactions are shown as solid black and dotted red lines, respectively. Coordinates of the Y306F HDAC8‐substrate complex are superimposed (PDB 2V5W), with atoms color coded similarly except that C = light blue, Zn2+ = cyan sphere, and water = pink sphere. Note that the substrate carbonyl displaces one of the Zn2+‐bound solvent molecules and accepts a hydrogen bond from Y306. Reprinted with permission from ref 36. Copyright 2008, American Chemical Society.
Upon substrate binding, the scissile carbonyl coordinates to the Zn2+ ion and accepts a hydrogen bond from Y306, as revealed in crystal structures of H143A HDAC8 and Y306F HDAC8 complexed with an intact peptide substrate (the H143A and Y306F mutations sufficiently inactivate the enzyme to enable crystallization with an unreacted substrate).36, 39 These interactions orient and polarize the carbonyl for nucleophilic attack by a Zn2+‐bound water molecule, assisted by general base H143.40 This is the rate‐determining step of catalysis based on the rate acceleration observed for a substrate containing an activated trifluoroacetyllysine substrate.41 When Zn2+ is substituted with Co2+ or Fe2+, the KM values vary significantly, indicating a significant substrate‐metal coordination interaction in the precatalytic enzyme‐substrate complex.42
By analogy to intermolecular interactions observed in transition state analogue complexes with the related isozyme HDAC6,38 the tetrahedral intermediate and its flanking transition states in the HDAC8 mechanism are stabilized by Zn2+ coordination and hydrogen bond interactions with Y306, H142, and H143. Furthermore, H142 plays an important role as an electrostatic catalyst and must remain protonated for maximal catalytic activity.40, 43 H142 donates a hydrogen bond to D176, which in turn coordinates to a monovalent cation; K+ binds with higher affinity than Na+ and is likely to be the predominant metal ion bound in vivo. Monovalent cation coordination by D176 inhibits catalysis by depressing the pKa of H142, which results in the neutral imidazole form of the histidine side chain (when in the neutral state, H142 is unable to provide electrostatic stabilization of the transition state).40 A second monovalent cation site is located more than 20 Å away from the Zn2+ and activates catalysis, presumably by stabilizing the active enzyme conformation.43
The crystal structures of H143A HDAC8 and Y306F HDAC8 complexed with intact peptide substrates36, 39 show that H143 is closer to the amino leaving group that must be protonated to facilitate the collapse of the tetrahedral intermediate. Therefore, H143 serves as a single general base‐general acid in the HDAC8 mechanism.40 By analogy with the crystal structure of the HDAC6‐acetate complex,38 the acetate coproduct of HDAC8 is initially coordinated to Zn2+ with bidentate coordination geometry, making hydrogen bonds with Y306, H142, and H143. Acetate may subsequently exit the active site through an internal channel,44 so acetate release does not necessarily require release of the deacetylated protein substrate.
The chemical requirements for the acceleration of amide hydrolysis are similar regardless of whether the scissile amide linkage is in the protein main chain or a side chain, so it is perhaps not surprising that the catalytic mechanism of HDAC8 illustrated in Figure 3 is reminiscent of mechanisms first proposed for the Zn2+‐dependent peptidases thermolysin and carboxypeptidase A.45, 46 Specifically, a Zn2+‐bound solvent molecule serves as a catalytic nucleophile, and a glutamate (E241 in thermolysin and E270 in carboxypeptidase A) serves as a single general base‐general acid. However, while the carbonyl of the scissile peptide linkage is proposed to coordinate to Zn2+ in the precatalytic enzyme‐substrate complex with thermolysin,45 a non‐zinc binding site is proposed for the precatalytic enzyme‐substrate complex with carboxypeptidase A; metal coordination is believed to occur as the transition state for nucleophilic attack is approached.46 This is consistent with the relative invariance of KM when the active site Zn2+ ion is substituted by other transition metal ions.47 Possibly, the degree of C = O—Zn2+ coordination in the precatalytic enzyme‐substrate complex varies depending on the specific enzyme‐substrate pair. Regardless, strong inner‐sphere Zn2+ coordination is a hallmark of transition state stabilization by a zinc hydrolase.
Clinical Features Resulting from Mutations in HDAC8
In several international efforts over the past few years to understand the effects of HDAC8 mutations, approximately 60 individuals have been reported, comprising approximately 4% of patients having a CdLS‐like clinical presentation.22, 23, 24, 25 Although the facial features of these patients certainly overlap with those of patients with typical CdLS caused by NIPBL mutations, there are some notable and qualitatively different features in individuals with HDAC8 mutations. The most distinct differences likely derive from abnormal skull formation, also observed in the Hdac8 mouse knockout, which demonstrates extreme growth failure and markedly delayed closure of skull sutures.48 Many individuals with HDAC8 mutations exhibit delayed closure of the anterior fontanelle to result in hypertelorism, telecanthus and/or forehead nevus flammeus. Furthermore, several additional facial features, including hooding of eyelids and dental anomalies, are distinctive in the HDAC8 patient cohort. These findings, in combination with a more well‐formed philtrum and upper lip, suggest that the face may be useful in defining a clinically recognizable subgroup of individuals. In addition to the features above, there are also quantitative differences between individuals with HDAC8 mutations and individuals with other causes of a CdLS‐like phenotype. Specifically, subjects with HDAC8 mutations have fewer structural anomalies and less growth retardation. These individuals also display a happy, cheerful demeanor.23 In addition to a CdLS‐like phenotype, the identification of HDAC8 mutations by genomic testing in a number of individuals without a prior diagnosis of CdLS reinforces that this phenotype differs from typical CdLS and may ultimately have wider ranging features than those described for patients reported to date.
Can Clinical Severity Be Correlated with Functional Effects of HDAC8 Mutations?
Of the nearly 60 patients reported to date, nearly half have HDAC8 mutations that are genetic loss of function (i.e. chromosomal microdeletions, nonsense, splice site or frameshift mutations). In contrast, 32 individuals with 23 unique missense mutations have been reported. Enzymatic assays of all missense mutations demonstrate reduced or absent activity, providing important insight regarding the biological function of HDAC8 catalysis and human developmental disorders.22, 23, 24, 25, 26, 27
However, understanding genotype‐phenotype correlations for HDAC8 mutations presents several challenges. Specifically, these include the relatively small numbers of patients as well as the X‐linked nature of the HDAC8 gene. Located on the long arm of the X chromosome at Xq13.1, HDAC8 is among the majority of genes on the X‐chromosome subject to inactivation in females. This results in only one allele of HDAC8, either mutant or wild‐type, being expressed in most cells of the body. Furthermore, expression of a specific allele in a specific cell type is largely random, leading to a nonuniform effect of a mutation in females that is largely ameliorated by the expression of the normal allele.
Random X‐inactivation accordingly leads to limitations in the correlation of clinical severity and molecular activity in females, who represent approximately two‐thirds of reported patients. However, some correlations can be observed. In the 43 females reported to date, just over half (22) have missense mutations and the remainder are presumed to have complete loss‐of‐function mutations (microdeletion, nonsense and frameshift mutations). This is in contrast to males, where the moderating effect of X‐inactivation is not a factor: in the 12 male probands reported to date, 10 have missense mutations, one has a clear loss of function microdeletion, and one has a functionally uncharacterized +5 splice site mutation that is presumed to be a weak loss of function mutation. Since loss of function mutations are more prevalent in females, in whom the mutant allele is only partially expressed, this suggests that the severe clinical phenotype of patients is due to the net enzymatic activity of HDAC8 in a given tissue. Since intellectual disability is a large component of this diagnosis, we suspect that females with unfavorable lyonization in the brain would display a more affected phenotype. However, additional investigation is required to test this model.
While the direct correlation of clinical severity with catalytic activity or thermostability of CdLS HDAC8 misssense mutants is challenging, a weak correlation is observed for males: few individuals have HDAC8 mutants exhibiting less than 25% enzymatic activity, presumably because HDAC8 dysfunction increases fetal lethality in males; males with milder phenotypes typically have HDAC8 mutants exhibiting more than 75% enzymatic activity.22, 23, 26, 27
Impact of HDAC8 Mutations on Structure and Catalysis
To date, the structures of 8 HDAC8 missense mutants have been studied using X‐ray crystallography (P91L, G117E, C153F, A188T, D233G, I243N, T311M, and H334R), and two additional mutants (H180R and G304R) have been studied using molecular modeling and molecular dynamics (MD) simulations.26, 27 In general, the impact on catalytic function is most severe for amino acid substitutions that are closest to the active site. Here, we review the structural and functional consequences of six selected mutants, ranging from the active site mutation H180R to the peripheral loop mutation H334R, to illustrate the structural basis of HDAC8 dysfunction in CdLS spectrum disorders.
H180R HDAC8
The imidazole group of H180 coordinates to the active site Zn2+ ion; the H180R substitution is the only CdLS missense mutation characterized to date that involves a known catalytic residue.22, 23, 24, 25 Substitution of the larger, positively charged side chain of arginine for that of histidine at this position destabilizes Zn2+ binding through the deletion of a metal ligand and results in the complete loss of catalytic activity. Notably, this amino acid substitution also causes a substantial decrease in thermostability (ΔT m = −5.8˚C).27
Since H180R HDAC8 did not yield suitable crystals for structure determination, molecular modeling and MD simulations were used to acquire functional insight on the H180R substitution. MD simulations revealed that the R180 side chain not only destabilizes the Zn2+ binding site, but it also occupies the acetyllysine binding groove regardless of whether Zn2+ is coordinated by the remaining protein ligands D178 and D267 [Fig. 5(A)].27 Specifically, the guanidinium group of R180 resides at the approximate location of the substrate acetyllysine Cα atom, sterically blocking substrate and inhibitor binding. This is consistent with the observation that incubation with the inhibitor M344 does not enhance the thermostability of H180R HDAC8 as it does for wild‐type HDAC8.27
Figure 5.
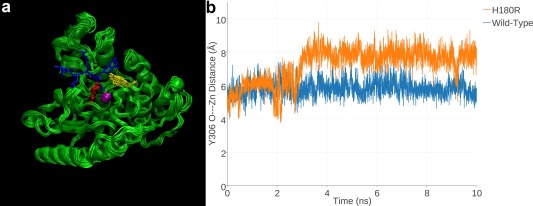
H180R HDAC8. (a) MD simulation of H180R HDAC8 (superimposed 1‐ns snapshots from a 10‐ns simulation). For reference, a tetrapeptide substrate (blue) as bound to H143A HDAC8 (PDB 3EWF) is superimposed to indicate the location of the active site. Zn2+ is a magenta sphere, R180 is red, and Y306 is yellow. The R180 side chain partially overlaps with the scissile acetyllysine side chain of the substrate, indicating that the mutant side chain sterically blocks substrate binding. (b) In a 10‐ns MD simulation of H180R HDAC8, Y306 fluctuates away from the “in” conformation required for catalysis; in a 10‐ns simulation of wild‐type HDAC8, Y306 remains in its catalytically competent conformation. Reprinted with permission from ref. 27. Copyright 2015, American Chemical Society.
The steric bulk of R180 also pushes the side chain of Y306 ∼2 Å away from the catalytically required “in” conformation [Fig. 5(B)]. Thus, even if a substrate were capable of binding to H180R HDAC8, the steric bulk of R180 would prevent Y306 from activating the scissile carbonyl of the substrate acetyllysine residue for nucleophilic attack by Zn2+‐bound water.
G304R HDAC8
G304 is located ∼4 Å away from the active site Zn2+ ion and resides in the glycine‐rich loop G302GGGY that connects β‐strand 8 to helix 1. Notably, this loop contains the catalytically obligatory residue Y306, so it is interesting to note that G302‐G303 are partially conserved and G304‐G305 are strictly conserved among HDACs and HDAC‐related enzymes. Possibly, these glycine residues confer some degree of flexibility so as to facilitate the transition of Y306 between “in” and “out” conformations in an induced‐fit substrate binding mechanism, as sometimes observed in the crystal structures of the related deacetylase acetylpolyamine amidohydrolase.49, 50 G304R HDAC8 is completely inactive and exhibits a substantial decrease in thermostability (ΔT m = −6.8˚C).27
Since G304R HDAC8 did not yield suitable crystals for structure determination, molecular modeling and MD simulations were employed to gain functional insight on the G304R substitution. MD simulations of G304R HDAC8 showed that the R304 side chain protrudes into the substrate binding site [Fig. 6(A)].27 This mutation, too, triggers an approximately 2 Å shift of Y306 away from its position in wild‐type HDAC8 [Fig. 6(B)]. Therefore, even if R304 were to move away to permit substrate binding, this movement would also cause Y306 to shift further away from its catalytically required “in” conformation. As a result, the G304R substitution appears to disable HDAC8 by both the steric blockage of substrate binding and the disruption of the catalytically required Y306 conformation.
Figure 6.
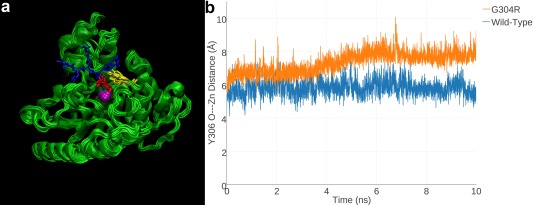
G304R HDAC8. (a) MD simulation of G304R HDAC8 (superimposed 1‐ns snapshots from a 10‐ns simulation). For reference, a tetrapeptide substrate (blue) as bound to H143A HDAC8 (PDB 3EWF) is superimposed to indicate the location of the active site. Zn2+ is a magenta sphere, R180 is red, and Y306 is yellow. The R304 side chain partially overlaps with the scissile acetyllysine side chain of the substrate, indicating that the mutant side chain sterically blocks substrate binding. (b) In a 10‐ns MD simulation of G304R HDAC8, Y306 fluctuates away from the “in” conformation required for catalysis; in a 10‐ns simulation of wild‐type HDAC8, Y306 remains in its catalytically competent conformation. Reprinted with permission from ref. 27. Copyright 2015, American Chemical Society.
C153F HDAC8 and C153R HDAC8
C153 is located ∼8 Å from the active site Zn2+ ion, and C153F HDAC8 exhibits 2% residual activity with only a modest decrease in thermostability (ΔT m = −1.9˚C).26 Being larger than the thiol side chain of C153, the phenyl side chain of F153 must adopt an alternative conformation to fit in its protein environment, as revealed in the 2.24 Å resolution crystal structure of C153F HDAC8 [Fig. 7(A)].26 Moreover, the bulky F153 side chain sterically locks the indole side chain of W141 in the “in” conformation, oriented toward the active site. In wild‐type HDAC8, W141 is in equilibrium between “in” and “out” conformations; only when this side chain is in the “out” conformation is the proposed internal acetate release channel44 open to facilitate product release [Fig. 7(B), 7(C)]. The permanent blockage of the product release channel by W141 presumably accounts for the significant loss of catalytic activity in C153F HDAC8.
Figure 7.
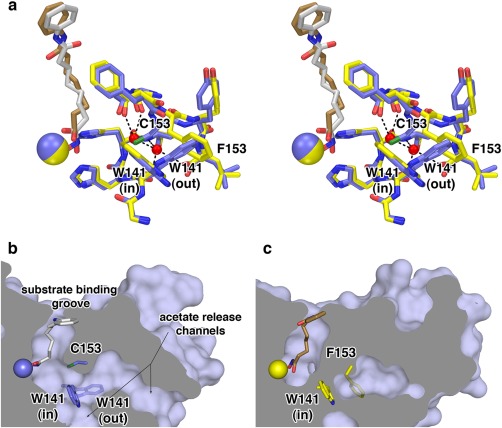
C153F HDAC8. (a) Superimposition of the HDAC8 C153F‐SAHA complex (C = yellow, N = blue, O = red, S = green, Zn2+ = yellow sphere, SAHA = tan) and the wild‐type HDAC8‐SAHA complex (PDB 1T69) (C = blue, Zn2+ = blue sphere, SAHA = gray). Water molecules (red spheres) occupy the space previously occupied by the C153 side chain. The F153 side chain sterically locks W141 in the “in” conformation. (b) The solvent‐accessible surface of wild‐type HDAC8 shows that the W141 side chain, which is in equilibrium between the “in” and “out” conformations, serves as a gate for acetate release channels. When W141 is in the “out” conformation, the channels are open. (c) Corresponding view of C153F HDAC8 shows that when W141 is locked in the “in” conformation by the F153 side chain, the acetate release channels are completely blocked. Reprinted with permission from ref. 26. Copyright 2014, American Chemical Society.
Recently, another HDAC8 missense mutation has been reported at this position, C153R,25 but the mutant enzyme has not yet been characterized. Based on the observations with C153F HDAC8, however, the bulky R153 side chain is similarly expected to lock W141 in the “in” conformation, resulting in blockage of the product release channel and loss of catalytic activity. To date, C153 is the only residue in HDAC8 to be characterized with multiple CdLS mutations. The cysteine at this position is strictly conserved across all metal‐dependent deacetylases that adopt the arginase‐deacetylase fold.
I243N HDAC8
I243 is located on helix F, ∼18 Å away from the active site Zn2+ ion. The side chain of I243 is buried in a hydrophobic region between helix F, helix H3, and β‐strand 6. Intriguingly, I243N HDAC8 exhibits 41% residual activity compared to wild‐type HDAC8, indicating only a relatively modest loss of catalytic function.26 However, this amino acid substitution causes the largest reduction in thermostability observed to date for a CdLS HDAC8 mutant (ΔT m = −9.5˚C). The loss of stability is undoubtedly due to the replacement of a nonpolar side chain with a polar side chain in the hydrophobic interior of the protein, as revealed in the 2.37 Å resolution crystal structure of this mutant [Fig. 8(A)]. Neighboring residues move to accommodate the mutation, resulting in a slight shift of helix H3. The 2.05 Å resolution crystal structure of I243N/Y306F HDAC8 complexed with an intact tetrapeptide substrate reveals less pronounced structural changes in helix H3, and an essentially identical enzyme‐substrate binding mode to that observed for substrate binding to Y306F HDAC8 [Fig. 8(B)].
Figure 8.
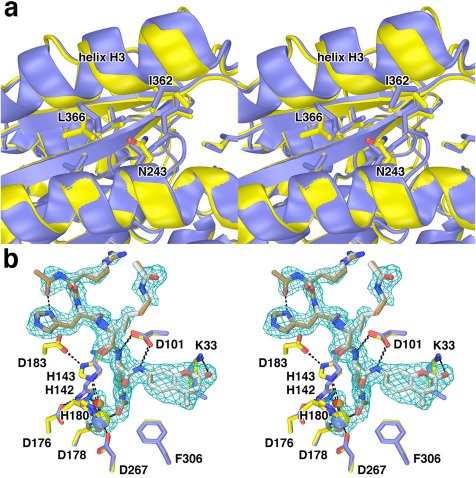
I243N HDAC8. (a) Comparison of the I243N HDAC8‐SAHA complex (yellow, C = yellow, N = blue, O = red) with the wild‐type HDAC8‐SAHA complex (blue; PDB 1T69). Selected residues are indicated. Helix H3 shifts by 0.3–1.6 Å as a result of the mutation. (b) Comparison of the I243N/Y306F HDAC8‐substrate complex (C = yellow (protein) or tan (substrate), N = blue, O = red, Zn2+ = yellow sphere) and the Y306F HDAC8‐substrate complex (C = blue (protein) or gray (substrate), N = blue, O = red, Zn2+ = blue sphere) (PDB 2V5W). Water molecules are indicated as red or orange spheres, respectively. Metal coordination and hydrogen bond interactions are shown as solid black and dashed black lines, respectively. The simulated annealing omit map is contoured at 3.0σ and shows a nearly fully ordered tetrapeptide substrate bound in the active site of I243N/Y306F HDAC8. Reprinted with permission from ref. 26. Copyright 2014, American Chemical Society.
Intriguingly, I243N is located in an apparent “hotspot” of 5 mutations that cluster in the vicinity of helices F and G (Fig. 2). Other mutations found in this region of tertiary structure include D237Y, ΔK239/Y240N, T280I, and C287Y.23, 44 Further studies of these mutants may yield new clues regarding the influence of this region on HDAC8 function. It is interesting to note that the study of disease‐linked mutants sometimes provides novel structure‐function insight for residues or regions of the HDAC8 structure that might not otherwise be targeted for rational mutagenesis studies, such as this mutational hotspot.
H334R HDAC8
Of all HDAC8 mutants studied, H334 is most distant from the active site, ∼25 Å away from the catalytic Zn2+ ion. H334R HDAC8 retains nearly full activity (91%) but exhibits significantly decreased thermostability (ΔT m = −7.0˚C) compared to the wild‐type enzyme.26 The 1.98 Å resolution crystal structure of H334R HDAC8 is generally similar to that of wild‐type HDAC8, and the substrate binding conformation is essentially identical in H334R/Y306F HDAC8 and Y306F HDAC8 [Fig. 9(A)]. Even so, interesting local structural changes are observed in the vicinity of the mutation [Fig. 9(B)]. The bulkier R344 side chain causes the polypeptide backbone to flip so that the R344 guanidinium group is oriented toward solvent. Consequently, D333 flips away from its solvent‐oriented position to form hydrogen bonds with S43, the backbone NH groups of E335 and F336, and an internal water molecule. Since the catalytic activity of H334R HDAC8 is roughly comparable to that of the wild‐type enzyme, the compromised biological function of H334R HDAC8 in CdLS spectrum disorders may largely result from its diminished thermostability.
Figure 9.
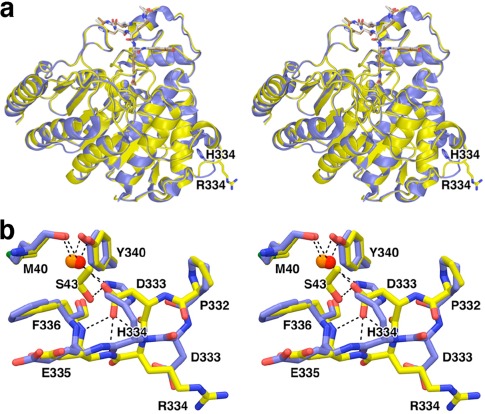
H334R HDAC8. (a) Superposition of the H334R/Y306F HDAC8‐substrate complex (yellow) and the Y306F HDAC8‐substrate complex (blue; PDB 2V5W). (b) Close‐up view of local structural changes resulting from the H334R substitution; hydrogen bond interactions are shown as dashed black lines. Atomic color codes are as follows: C = yellow, N = blue, O = red, S = green, water molecule = red sphere. Reprinted with permission from ref. 26. Copyright 2014, American Chemical Society.
Rescue of HDAC8 Catalysis in CdLS Spectrum Mutants
It has been reported that N‐acylthiourea derivatives serve as selective activators of low‐activity HDAC8,51, 52 and it appears that activators such as N‐(phenylcarbamothioyl)benzamide (designated “TM251”) are best suited for restoring catalytic activity to compromised HDAC8 mutants such as those identified in CdLS.26, 27 The catalytic activity of all but two of the ten CdLS HDAC8 mutants studied to date can be partially or fully activated by TM251 in dose‐dependent fashion, and the catalytic activity of five CdLS HDAC8 mutants can be restored to wild‐type levels (Fig. 10). Only H180R HDAC8 and G304R HDAC8 do not respond to TM251, presumably due to the steric blockage of the substrate binding site by the bulky arginine side chain introduced by each mutation. Additionally responsible for the obliteration of catalysis in H180R HDAC8 is the loss of the catalytic Zn2+ ion due to the loss of metal ligand H180, as well as fluctuations of Y306 away from its catalytically required conformation.27
Figure 10.
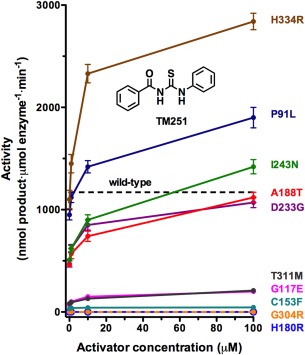
Rescue of catalysis in CdLS HDAC8 mutants by TM251. The activity level for wild‐type HDAC8 in the absence of activator is indicated by a dashed line. The catalytic activities of several mutants can be restored to wild‐type levels in dose‐dependent fashion by TM251. All but the catalytically dead mutants H180R and G304R exhibit at least some tendency for activation. Reprinted with permission from ref. 27. Copyright 2015, American Chemical Society.
In the absence of the crystal structure of a complex between HDAC8 and an N‐acylthiourea activator, the precise mode of action on catalytic activity remains undetermined. However, Srivastava and colleagues53 use enzymological measurements and molecular modeling approaches to suggest that TM251 binds near the active site of HDAC8, potentially stabilizing active site loops that are important for the binding of substrates and inhibitors. Significant conformational differences in loops flanking the HDAC8 active site, particularly the L1 and L2 loops, have been observed in several crystal structures of HDAC8‐inhibitor complexes,28, 29, 36, 54, 55 and such flexibility presumably accommodates the binding of diverse substrates in the HDAC8 active site.
In HDAC8 mutants, it is possible that the flexibility of active site loops is compromised through compensatory structural changes that propagate through the protein scaffolding. Even so, the binding of small molecule activators such as TM251 may help to stabilize these active site loops in a catalytically competent conformation. Hence, unless a CdLS spectrum mutation completely eradicates chemical functionality required for catalysis (e.g., a Zn2+ binding residue such as H180), or introduces a steric blockage to the active site so as to prevent substrate binding, catalysis may be partially or fully rescued by small molecule activators. Accordingly, compounds such as TM251 may ultimately be useful for the clinical management of CdLS spectrum patients diagnosed with HDAC8 mutations. Future studies in our laboratories will continue to probe the molecular basis of HDAC8 dysfunction with the ultimate goal of exploring possible molecular approaches for the treatment of Cornelia de Lange Syndrome spectrum disorders.
Contributor Information
Matthew A. Deardorff, deardorff@email.chop.edu
David W. Christianson, Email: chris@sas.upenn.edu
References
- 1. Liu J, Krantz ID (2009) Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet 76:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dorsett D, Krantz ID (2009) On the molecular etiology of Cornelia de Lange syndrome. Ann NY Acad Sci 1151:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J, Baynam G (2010) Cornelia de Lange syndrome. Adv Exp Med Biol 685:111–123. [PubMed] [Google Scholar]
- 4. Boyle MI, Jespersgaard C, Brøndum‐Nielsen K, Bisgaard A‐M, Tümer Z (2015) Cornelia de Lange syndrome. Clin Genet 88:1–12. [DOI] [PubMed] [Google Scholar]
- 5. Barisic I, Tokic V, Loane M, Bianchi F, Calzolari E, Garne E, Wellesley D, Dolk H, EUROCAT Working Group (2008) Descriptive epidemiology of Cornelia de Lange syndrome in Europe. Am J Med Genet a 146A:51–59. [DOI] [PubMed] [Google Scholar]
- 6. Rohatgi S, Clark D, Kline AD, Jackson LG, Pie J, Siu V, Ramos FJ, Krantz ID, Deardorff MA (2010) Facial diagnosis of mild and variant CdLS: insights from a dysmorphologist survey. Am J Med Genet A 152:1641–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Chan CY, Kayserili H, Avci S, Hennekam RCM, Lampe AK, Redeker E, Homfray T, Ross A, Smeland MF, Mansour S, Parker MJ, Cook JA, Splitt M, Fisher RB, Fryer A, Magee AC, Wilkie A, Barnicoat A, Brady AF, Cooper NS, Mercer C, Deshpande C, Bennett CP, Pilz DT, Ruddy D, Cilliers D, Johnson DS, Josifova D, Rosser E, Thompson EM, Wakeling E, Kinning E, Stewart F, Flinter F, Girisha KM, Cox H, Firth HV, Kingston H, Wee JS, Hurst JA, Clayton‐Smith J, Tolmie J, Vogt J, Tatton‐Brown K, Chandler K, Prescott K, Wilson L, Behnam M, McEntagart M, Davidson R, Lynch S‐A, Sisodiya S, Mehta SG, McKee SA, Mohammed S, Holden S, Park S‐M, Holder SE, Harrison V, McConnell V, Lam WK, Green AJ, Donnai D, Bitner‐Glindzicz M, Donnelly DE, Nellåker C, Taylor MS, FitzPatrick DR (2014) Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS‐like phenotypes with observed and predicted levels of mosaicism. J Med Genet 51:659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Losada A (2008) The regulation of sister chromatid cohesion. Biochim Biophys Acta 1786:41–48. [DOI] [PubMed] [Google Scholar]
- 9. Nasmyth K, Haering CH (2009) Cohesin: its roles and mechanisms. Annu Rev Genet 43:525–558. [DOI] [PubMed] [Google Scholar]
- 10. Gruber S, Haering CH, Nasmyth K (2003) Chromosomal cohesin forms a ring. Cell 112:765–777. [DOI] [PubMed] [Google Scholar]
- 11. Remeseiro S, Losada A (2013) Cohesin, a chromatin engagement ring. Curr Opin Cell Biol 25:63–71. [DOI] [PubMed] [Google Scholar]
- 12. Gligoris TG, Scheinost JC, Bürmann F, Petela N, Chan KL, Beckouët F, Gruber S, Nasmyth K, Löwe J (2014) Closing the cohesin ring: structure and function of its Smc3‐kleisin interface. Science 346:963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, Zhang P, Kim ST, Pan X, Qin J (2008) Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell 31:143–151. [DOI] [PubMed] [Google Scholar]
- 14. Ben‐Shahar TR, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F (2008) Eco1‐dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321:563–566. [DOI] [PubMed] [Google Scholar]
- 15. Unal E, Heidinger‐Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE (2008) A molecular determinant for the establishment of sister chromatid cohesion. Science 321:566–569. [DOI] [PubMed] [Google Scholar]
- 16. Beckouët F, Hu B, Roig MB, Sutani T, Komata M, Uluocak P, Katis VL, Shirahige K, Nasmyth K (2010) An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell 39:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elbatsh AM, Haarhuis JH, Petela N, Chapard C, Fish A, Celie PH, Stadnik M, Ristic D, Wyman C, Medema RH, Nasmyth K, Rowland BD (2016) Cohesin releases DNA through asymmetric ATPase‐driven ring opening. Mol Cell 61:575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beckouët F, Srinivasan M, Roig MB, Chan KL, Scheinost JC, Batty P, Hu B, Petela N, Gligoris T, Smith AC, Strmecki L, Rowland BD, Nasmyth K (2016) Releasing activity disengages cohesin's Smc3/Scc1 interface in a process blocked by acetylation. Mol Cell 61:563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan K‐L, Gligoris T, Upcher W, Kato Y, Shirahige K, Nasmyth K, Beckouët F (2013) Pds5 promotes and protects cohesin acetylation. Proc Natl Acad Sci USA 110:13020–13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan K‐L, Roig MB, Hu B, Beckouët F, Metson J, Nasmyth K (2012) Cohesin's DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 150:961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uhlmann F, Lottspeich F, Nasmyth K (1999) Sister‐chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 400:37–42. [DOI] [PubMed] [Google Scholar]
- 22. Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi‐Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen‐Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaiser FJ, Ansari M, Braunholz D, Gil‐Rodríguez MC, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillén‐Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I, Care4Rare Canada Consortium, Mohammed S , Moran E, Mortier GR, Moser J‐AS, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Pérez‐Aytés A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC, University of Washington Center for Mendelian Genomics , Hakonarson H, Quintero‐Rivera F, Wierzba J, Musio A, Gillessen‐Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pié J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA (2014) Loss of function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange Syndrome‐like features, ocular hypertelorism, large fontanelle and X‐linked inheritance. Hum Mol Genet 23:2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng L, Zhou D, Zhang Z, Liu Y, Yang Y (2014) Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J Hum Genet 59:536–539. [DOI] [PubMed] [Google Scholar]
- 25. Parenti I, Gervasini C, Pozojevic J, Wendt KS, Watrin E, Azzollini J, Braunholz D, Buiting K, Cereda A, Engels H, Garavelli L, Glazar R, Graffmann B, Larizza L, Lüdecke HJ, Mariani M, Masciadri M, Pié J, Ramos FJ, Russo S, Selicorni A, Stefanova M, Strom TM, Werner R, Wierzba J, Zampino G, Gillessen‐Kaesbach G, Wieczorek D, Kaiser FJ (2016) Expanding the clinical spectrum of the 'HDAC8‐phenotype'—implications for molecular diagnostics, counseling and risk prediction. Clin Genet 89:564–573. [DOI] [PubMed] [Google Scholar]
- 26. Decroos C, Bowman CM, Moser J‐AS, Christianson KE, Deardorff MA, Christianson DW (2014) Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem Biol 9:2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Decroos C, Christianson NH, Gullett LE, Bowman CM, Christianson KE, Deardorff MA, Christianson DW (2015) Biochemical and structural characterization of HDAC8 mutants associated with Cornelia de Lange Syndrome spectrum disorders. Biochemistry 54:6501–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12:1325–1334. [DOI] [PubMed] [Google Scholar]
- 29. Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, D, Marco S (2004) Crystal structure of a eukaryotic zinc‐dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA 101:15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanyo ZF, Scolnick LR, Ash DE, Christianson DW (1996) Structure of a unique binuclear manganese cluster in arginase. Nature 383:554–557. [DOI] [PubMed] [Google Scholar]
- 31. Ash DE, Cox JD, Christianson DW, Arginase: a binuclear manganese metalloenzyme In: Sigel A, Sigel H, Eds. (1999) Manganese and its role in biological processes, Vol. 37 of Metal Ions in Biological Systems, New York: M. Dekker, pp 407–428. [PubMed] [Google Scholar]
- 32. Christianson DW (2005) Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res 38:191–201. [DOI] [PubMed] [Google Scholar]
- 33. Dowling DP, Di Costanzo L, Gennadios HA, Christianson DW (2008) Evolution of the arginase fold and functional diversity. Cell Mol Life Sci 65:2039–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lombardi PM, Cole KA, Dowling DP, Christianson DW (2011) Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol 21:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finnin MS, Donigan JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Nature 401:188–193. [DOI] [PubMed] [Google Scholar]
- 36. Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW (2008) Structural studies of human histone deacetylase 8 and its site‐specific variants complexed with substrate and inhibitors. Biochemistry 47:13554–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dowling DP, Gattis SG, Fierke CA, Christianson DW (2010) Structures of metal‐substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 49:5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hai Y, Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol 12:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8‐substrate complex. EMBO Rep 8:879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gantt SML, Decroos C, Lee MS, Gullett LE, Bowman CM, Christianson DW, Fierke CA (2016) General base‐general acid catalysis in human histone deacetylase 8. Biochemistry 55:820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riester D, Wegener D, Hildmann C, Schwienhorst A (2004) Members of the histone deacetylase superfamily differ in substrate specificity towards small synthetic substrates. Biochem Biophys Res Commun 324:1116–1123. [DOI] [PubMed] [Google Scholar]
- 42. Gantt SL, Gattis SG, Fierke CA (2006) Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45:6170–6178. [DOI] [PubMed] [Google Scholar]
- 43. Gantt SL, Joseph CG, Fierke CA (2010) Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem 285:6036–6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haider S, Joseph CG, Neidle S, Fierke CA, Fuchter MJ (2011) On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg Med Chem Lett 21:2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Matthews BW (1988) Structural basis of the action of thermolysin and related zinc peptidases. Acc Chem Res 21:333–340. [Google Scholar]
- 46. Christianson DW, Lipscomb WN (1989) Carboxypeptidase A. Acc Chem Res 22:62–69. [Google Scholar]
- 47. Auld DS, Holmquist B (1974) Carboxypeptidase A. Differences in the mechanisms of ester and peptide hydrolysis. Biochemistry 13:4355–4361. [DOI] [PubMed] [Google Scholar]
- 48. Haberland M, Mokalled MH, Montgomery RL, Olson EN (2009) Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev 23:1625–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lombardi PM, Agnell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW (2011) Structure of a prokaryotic polyamine deacetylase reveals evolutionary functional relationships with eukaryotic histone deacetylases. Biochemistry 50:1808–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Decroos C, Christianson DW (2015) Design, synthesis and evaluation of polyamine deacetylase inhibitors, and high‐resolution crystal structures of their complexes with acetylpolymine amidohydrolase. Biochemistry 54:4692–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh RK, Mandal T, Balsubramanian N, Viaene T, Leedahl T, Sule N, Cook G, Srivastava DK (2011) Histone deacetylase activators: N‐acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase‐8 on a fluorescent substrate. Bioorg Med Chem Lett 21:5920–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toro TB, Pingali S, Nguyen TP, Garrett DS, Dodson KA, Nichols KA, Haynes RA, Payton‐Stewart F, Watt TJ (2016) KDAC8 with high basal velocity is not activated by N‐acetylthioureas. PLoS ONE 11:e0146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singh RK, Cho K, Padi SKR, Yu J, Haldar M, Mandal T, Yan C, Cook G, Guo B, Mallik S, Srivastava DK (2015) Mechanism of N‐acylthiourea‐mediated activation of human histone deacetylase 8 (HDAC8) at molecular and cellular levels. J Biol Chem 290:6607–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW (2011) Structural basis of the antiproliferative activity of largazole, a despsipeptide inhibitor of the histone deacetylases. J Am Chem Soc 133:12474–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Decroos C, Clausen DJ, Haines BE, Wiest O, Williams RM, Christianson DW (2015) Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry 54:2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]